Introduction
Gardner's syndrome (GS) is considered to be a
variant form of familial adenomatous polyposis. In the early 1950s,
GS was described by Gardner as a rare genetic disorder with
autosomal dominant inheritance (1). GS affects approximately 10% of
individuals with familial adenomatous polyposis (2). The primary clinical features of the
syndrome are diffuse adenomatous polyposis, multiple osteomas,
dental abnormalities and soft-tissue tumors (3). The intestinal polyps typically
develop at ~20 years of age and have up to a 100% potential for
malignant transformation following 10–20 years, whereas dental
anomalies precede the intestinal polyps and are present in 30–75%
of patients with GS (1,4). The dental anomalies may include
impacted or un-erupted teeth, congenitally missing teeth,
hypercementosis, supernumerary teeth, dentigerous cysts, long and
tapered molar roots, fused molar roots, hypodontia or compound
odontomas (3,5). Osteomas are present in 68–82% of GS
patients and are generally located in the mandible and paranasal
sinuses (6).
A series of mutations in certain genes, including
adenomatous polyposis coli (APC), mutY DNA glycosylase, mutL
homolog (MLH)1, mutS homolog (MSH)2, MSH6, PMS1 homolog (PMS)2,
birt-hogg-dube homolog, serine/threonine kinase 11, bone
morphogenetic protein receptor type 1A, mothers against
decapentaplegic homolog 4 and phosphatase and tensin homolog, have
been reported to be associated with hereditary gastrointestinal
polyposis syndromes (7). At
present, only a few gene mutations associated with GS have been
reported, probably due to fewer patients suffering from this
disease. Namely, mutations in MYH and APC, including various
different mutations in APC, have been associated with GS (8–10).
With the development of next-generation sequencing
technology, whole-exome sequencing is now used to detect exome
variant profiles, and is considered a powerful and cost-effective
tool due to its identification of extensive disease-associated
variations in novel genes (11,12).
Therefore, the present study assessed two members of a Chinese
family with GS using exome sequencing to screen for the
disease-causing gene mutation. From analysis of the exome
sequencing data, the present study first identified an MLH1
missense mutation (NM_000249.3:p.Tyr379Ser/c.1136A>C) in the
family members with GS, which was subsequently confirmed by Sanger
sequencing. The reported mutation may be valuable for the prenatal
and genetic diagnosis of GS.
Materials and methods
Subjects
The present study recruited a Chinese Han family in
which two family members (the father and the son) presented with GS
in August, 2016. The father was 52-years-old, and the son were
26-years-old. The diagnosis of GS was based on clinical features,
family history, cone-beam computed tomography (CBCT), colonoscopy
and pathological examinations. Genomic DNA was extracted from
peripheral venous blood from the two affected individuals using a
QIAamp DNA Blood Mini kit (Qiagen GmbH, Hilden, Germany) following
the manufacturer's protocol. Informed consent was obtained from
each patient involved in the study, and the study protocol was
approved by the Ethics Committee of Guangxi Key Laboratory of
Metabolic Diseases Research (Guilin, China).
Whole-exome sequencing
Genomic DNA was isolated from the peripheral venous
blood of the patients using the QIAamp DNA Blood Mini kit (Qiagen
GmbH) and then used for exome capture using a NimbleGen SeqCap EZ
Human Exome Library v2.0 kit (Roche NimbleGen Inc., Wisconsin, USA)
following the manufacturer's protocol. Random DNA fragmentation was
performed with a Covaris Ultrasonicator system (Covaris Inc.,
Woburn, MA, USA), after which the sizes of the library fragments
were mainly distributed between 150 and 250 bp. An ‘A’ base was
added at the 3′-end of each strand, then ligated to sequencing
adapters, which was followed by ligation-mediated polymerase chain
reaction (PCR) with probe hybridization, amplification and
purification to enrich for targets to sequence. Primers used were
included in the NimbleGen SeqCap EZ Human Exome Library v2.0 kit.
Each resulting qualified captured library then was sequenced on a
BGISEQ-500 sequencing platform (Beijing Genomics Institute,
Guangdong, China) and the desired average sequencing coverage for
each sample was obtained. Raw image files were processed to produce
pair-end reads for each individual using default parameters of base
calling software developed for BGISEQ-500.
Read mapping and variant analysis
Clean data was obtained by raw data filtering, and
the clean data of each sample was mapped to the human reference
genome (GRCh37/HG19) using a Burrows-Wheeler Aligner (BWA V0.7.15)
(13,14). All genomic variations, including
single-nucleotide polymorphisms (SNPs) and insertions/deletions
(InDels) were identified using HaplotypeCaller of GATK (v3.3.0)
(Broad Institute, Cambridge, MA, USA) with the proper filtering
parameters (15,16). Subsequently, the SnpEff tool
(http://snpeff.sourceforge.net/SnpEff\\_manual.html)
was used to perform a series of annotations for variants. Potential
disease-causing mutations were predicted using the sorting
intolerant from tolerant (SIFT) algorithm (17). If the SIFT score was ≤0.05, the
present study predicted this variant to be a deleterious variant.
Data were filtered with several variant databases, including dbSNP
(https://www.ncbi.nlm.nih.gov/projects/SNP/), the 1000
Genomes Project (ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/release),
and the NHLBI-ESP6500 database (http://evs.gs.washington.edu/EVS/). Candidate
mutations were expected to be absent from these databases. The
conservation analysis of amino acid sequences were aligned using
ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/).
Sanger sequencing
Sanger sequencing was used to confirm candidate
mutations in the MLH1 gene identified by exome sequencing. The PCR
primers used were as follows: Forward,
5′-CTTAGTACTGCTCCATTTGGGGA-3′ and reverse,
5′-TTGTTGTATCCCCCTCCAAGC-3′. PCR amplification was performed using
a HEMA 9600 PCR thermo cycler (Technological Innovation Beach, Zhu
Hai, Guangdong, China) with 35 cycles of denaturation at 98°C for
10 sec, annealing at 55°C for 30 sec and extension at 72°C for 30
sec. The PCR reagents, including Taq polymerase, Taq polymerase
buffer, MgCl2 and dNTP mixture were purchased from
Takara Biotechnology, Co., Ltd. (Dalian, China). The PCR products
were sequenced using an ABI Prism 3730 DNA analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). Each
read was compared with the genomic DNA sequence of MLH1, and
nucleotide alterations were numbered according to their position in
MLH1.
Results
Clinical data
Clinical data was collected for the two patients
through patient interviews, medical record extraction, physical
examinations and CBCT imaging.
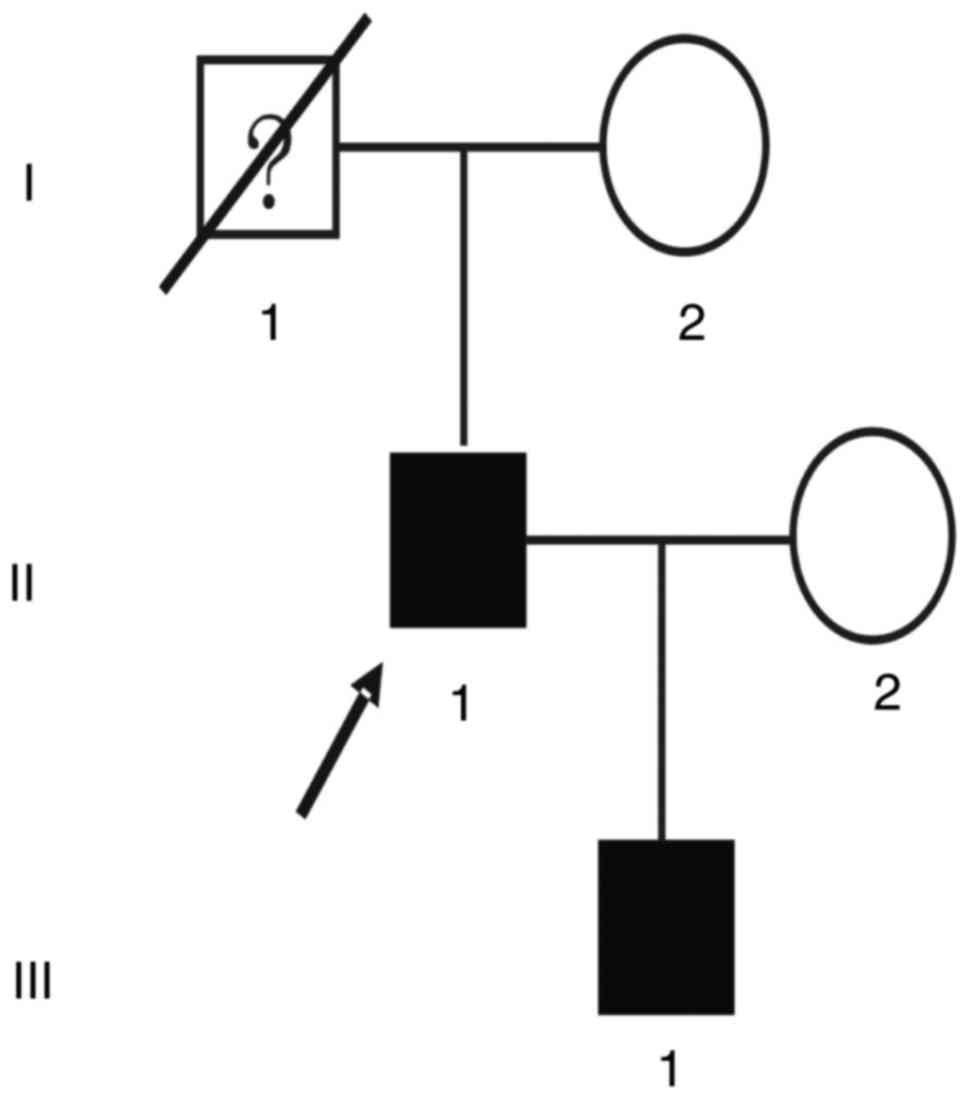
The proband (II:1; Fig.
1) of the GS family, a 52-year-old male patient, presented at
the hospital with swelling and pain in the mandibular region, and
was diagnosed with GS. From his medical history, it was discovered
that the patient had already undergone intestinal polypectomy 3
years ago with recurrence. He had presented with swelling and pain
in the mandibular region 2 years previously, and had undergone
surgical debridement following anti-inflammatory treatment. A
pathological diagnosis of ‘ossified fibromatosis with infection’
was made. The patient continued to present with swelling and
discomfort repeatedly, and underwent curative jaw osteomyelitis
surgery in the other hospital a year prior to the most recent
presentation. Family history revealed that the patient's father had
died from gastrointestinal cancer. At the clinical examination, the
patient demonstrated no abnormal skin pigmentation, skin mass or
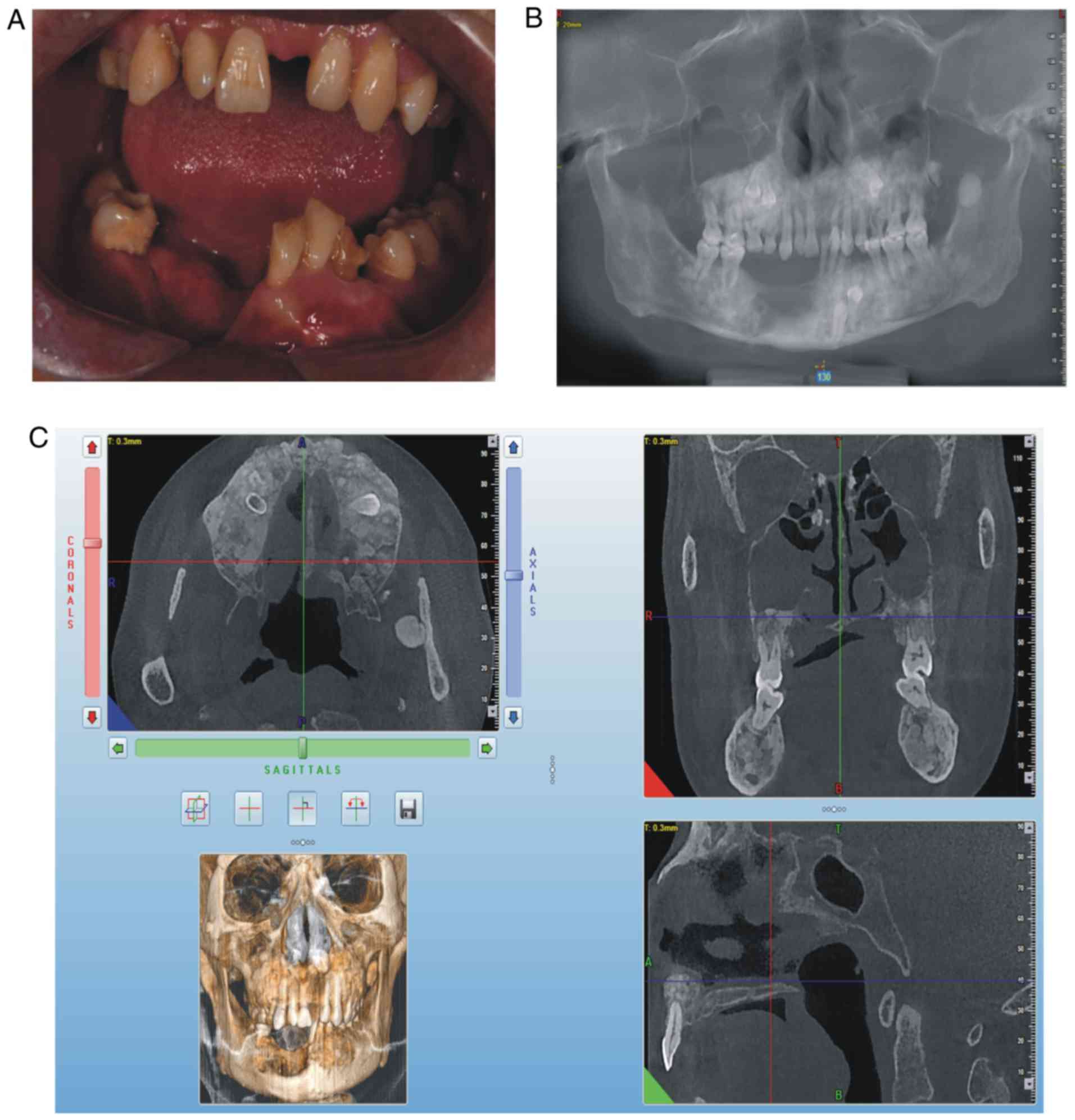
swelling in the liver and spleen. On intraoral examination, it was
observed that the patient's lower teeth on the right side were
missing since surgery, a swelling was present in the right
mandibular buccal side, a swelling with pus was present in the
mucosal layer, the deciduous teeth 54, 55, 65 and 73 were retained
and tooth 34 was unerupted (Fig.
2A). The panoramic radiograph (Fig. 2B) and CBCT revealed that there was
an increase in irregular density in the upper and lower jaw
regions, and the retained deciduous teeth corresponding to
permanent teeth 14, 15, 25, 33 and 34 were affected, and that
multiple convex hyperplastic bone lesions were present in the right
upper and lower jaw and ethmoidal sinus (Fig. 2C).
The son of the proband (III:1; Fig. 1), a 26-year-old male, requested a
clinical assessment due to his father being diagnosed with GS.
Medical history revealed that the son had repeatedly abdominal
fibrous tumors lasting for 10 years and intestinal polyps lasting
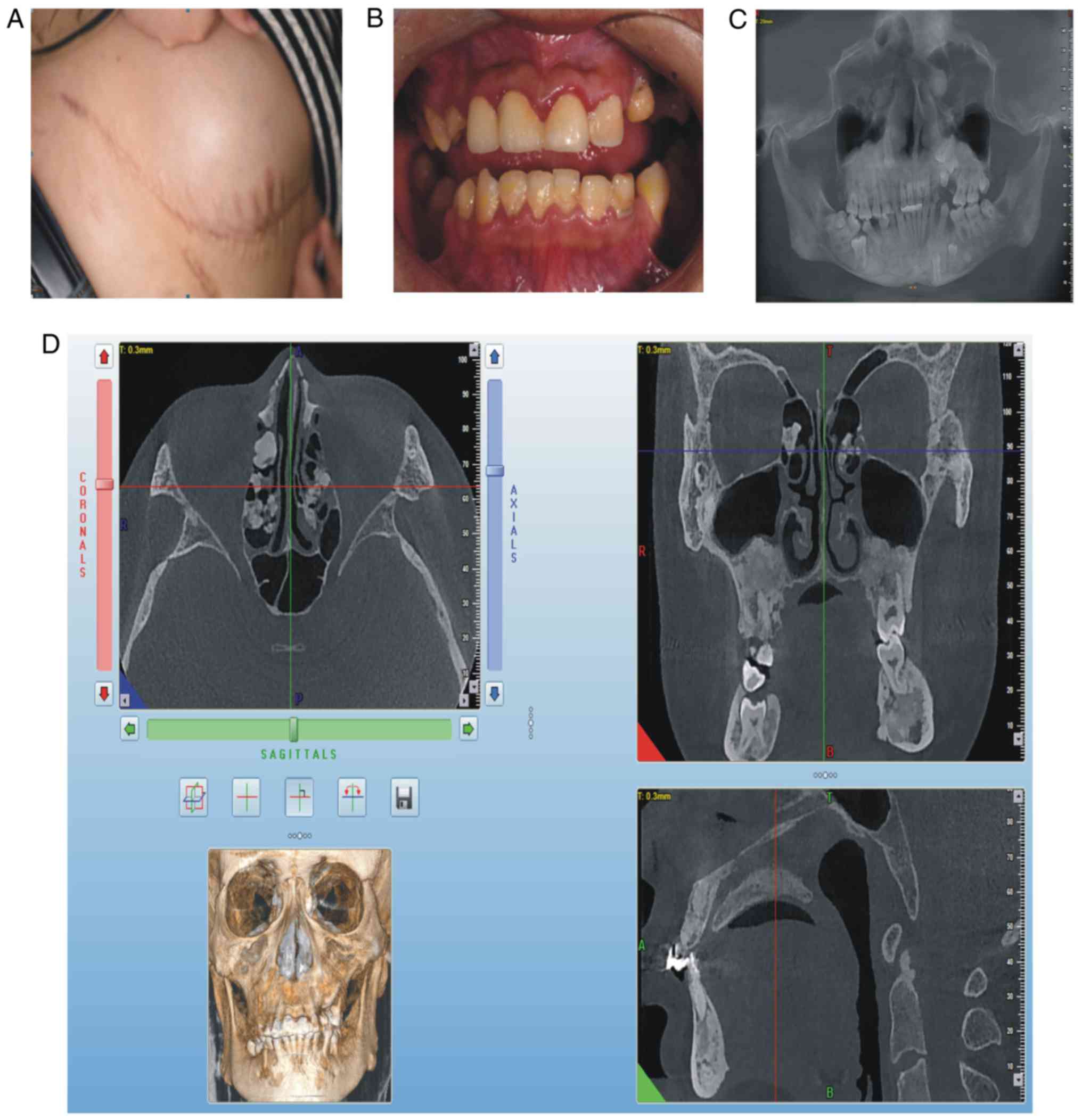
for 3 years. At clinical examination, the patient exhibited a
semicircular elevated region due to a tough, fixed mass with clear
boundaries on the right abdominal skin, ~15 cm in diameter
(Fig. 3A). On intraoral
examination, it was revealed that deciduous teeth 53, 63, 73 and 83
were retained, whereas teeth 25, 35 and 46 were erupted (Fig. 3B). The panoramic radiograph
(Fig. 3C) and CBCT indicated that
permanent teeth 13, 23, 25, 33, 35 and 46 were impacted, that
supernumerary teeth were not present, that there was an increase in
irregular density in the upper and lower jaw regions, and that
multiple convex hyperplastic bone lesions were present in the
ethmoid sinus (Fig. 3D).
Whole-exome sequencing
The present study performed whole-exome sequencing
on both affected individuals (II:1 and III:1) in the GS Chinese
family, for which an average of 31,766.99 Mb raw bases were
screened. Following removal of low-quality reads, an average of
31,753.08 Mb clean reads were obtained. The average GC content was
54.26%. Total clean reads per sample were aligned to the human
reference genome (GRCh37/HG19) using BWA. On average, 99.05% mapped
successfully. The duplicate reads were removed, resulting in an
average of 20,694.12 Mb effective reads. The mean sequencing depth
of target regions was 232.41-fold. On average, per individual
sequenced, 99.86% of targeted bases were covered by at least 1×
coverage while 95.09% of the targeted bases had at least 10×
coverage (Table I). Overall,
112,819 single nucleotide polymorphisms (SNPs) across both
individuals were identified. Of these variants, 96.79% were
represented in dbSNP and 94.31% were annotated in the 1000 Genomes
Project database. The number of novel SNPs was 2,967. The ratio of
transition to transversion was 2.33. Of the total SNPs, 12,736 were
synonymous, 11,716 were missense, 37 were stop-loss, 96 were
stop-gain, 24 were start-loss and 105 were splice site mutations.
In total, 16,829 InDels were detected across all samples. Of these
variants, 76.89% were represented in dbSNP and 57.26% were
annotated in the 1000 Genomes Project database. The number of novel
InDels was 3,422. Of the overall InDels, 344 were frameshift, 4
were stop-loss, 3 were start-loss and 58 were splice site mutations
(Table II).
 | Table I.Summary of whole exome sequencing
data. |
Table I.
Summary of whole exome sequencing
data.
|
| Pedigree ID |
|
|---|
|
|
|
|
|---|
| Parameter | II:1 | III:1 | Average |
|---|
| Raw reads | 613,323,244 | 657,356,178 | 635,339,711 |
| Raw bases (Mb) | 30666.16 | 32867.81 | 31766.99 |
| Clean bases (Mb) | 30652.62 | 32853.54 | 31753.08 |
| GC rate (%) | 53.17 | 55.34 | 54.26 |
| Total effective reads
(Mb) | 19,086.35 | 22,301.89 | 20,694.12 |
| Mapping rate on
genome (%) | 98.97 | 99.13 | 99.05 |
| Average sequencing
depth on target | 199.95 | 264.86 | 232.41 |
| Fraction of target
covered>=1× (%) | 99.98 | 99.73 | 99.86 |
| Fraction of target
covered>=10× (%) | 99.47 | 90.71 | 95.09 |
| Table II.Summary statistics for identified SNPs
and InDels. |
Table II.
Summary statistics for identified SNPs
and InDels.
|
| Pedigree ID |
|
|---|
|
|
|
|
|---|
| Parameter | II:1 | III:1 | Overall |
|---|
| Total number of
SNPs | 99,562 | 91,132 | 112,819 |
| Fraction of SNPs in
dbSNP (%) | 96.18 | 96.40 | 96.79 |
| Fraction of SNPs in
1,000 genomes (%) | 92.45 | 92.56 | 94.31 |
| Novel | 3,258 | 2,764 | 2,967 |
| Ti/Tv | 2.31 | 2.33 | 2.33 |
| Synonymous | 11,149 | 10,999 | 12,736 |
| Missense | 10,424 | 10,179 | 11,716 |
| Splicing | 101 | 90 | 105 |
| Total number of
Indels | 13,968 | 11,050 | 16,829 |
| Frameshift | 274 | 291 | 344 |
| Non-frameshift
Insertion | 138 | 147 | 169 |
| Non-frameshift
Deletion | 149 | 141 | 192 |
| Splicing | 53 | 54 | 58 |
Through read mapping and variant analysis, the
presence of previously known mutations in APC and MYH in the two
patients was ruled out. It was observed that a novel missense
mutation in the MLH1 gene (NM_000249.3:p.Tyr379Ser/c.1136A>C)
was present in both affected individuals, and absent in the dbSNP,
1000 Genomes Project, and NHLBI-ESP6500 databases.
Sanger sequencing
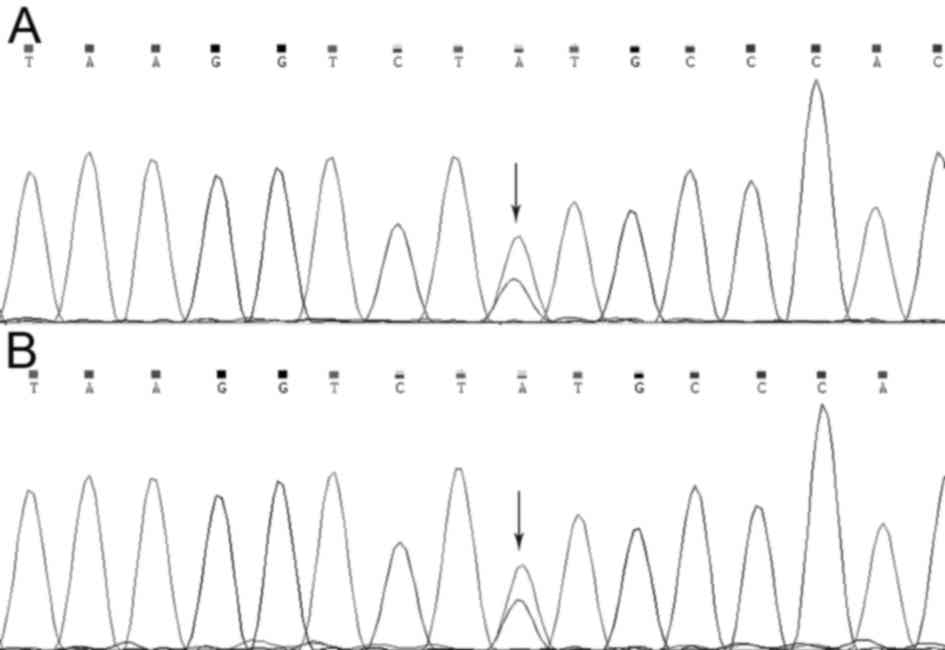
To verify the mutation in the MLH1 gene that was
identified using whole-exome sequencing, Sanger sequencing was used
to examine the MLH1 gene of the two patients (II:1 and III:1).
Using method, it was revealed that a missense mutation in the MLH1
gene (NM_000249.3:p.Tyr379Ser/c.1136A>C) was present in both
affected individuals (Fig. 4).
Therefore, combining the clinical data, whole-exome sequencing
data, and the literature on GS, it was suggested that the MLH1 gene
mutation may be responsible for GS.
Conservation analysis of MLH1
p.Tyr379
MLH1 amino acid sequences from the species Homo
sapiens, Pan troglodytes, Pongo abelii, Mus musculus, Danio rerio,
Solanum lycopersicum and Saccharomyces cerevisiae were
aligned, and found that p.Tyr379 was located within a highly
conserved region of MLH1, and was revealed to be highly conserved
among the different species, which indicated the structural and
functional importance (Fig.
5).
Discussion
GS, an autosomal dominant disease, has high
penetrance and variable expression that may present as intestinal
polyposis or extracolonic manifestations, including osteomas, skin
and soft tissue tumors, dental anomalies and congenital hypertrophy
of the retinal pigment epithelium. The incidence rate of GS ranges
from 1 in 12,000 to 1 in 4,000, depending on the region worldwide
(18).
Intestinal polyps in GS may affect the entire
gastrointestinal tract. They typically start to develop during
puberty and fully emerge between the ages of 20 and 40 years. The
lesions have up to a 100% potential for malignant transformation
generally in the 40–50-year age group (19). Due to the high potential for
transformation to adenocarcinoma, the polyps must be completely
removed in order to effectively prevent colon cancer. Additionally,
desmoid tumors are a common manifestation of GS and are usually
locally aggressive, non-malignant and non-encapsulated (20). These lesions appear in 3.5–5.7% of
GS cases and are three times more common in women (21). Desmoid tumors frequently occur in
the first 3 years following colon surgery in the abdominal wall
and/or intra-abdominal cavity, as observed in the second patient in
the present study, who exhibited desmoid tumors in the
intra-abdominal cavity. GS diagnosis is based on the presence of
osteomas, and three or more lesions are typically present in 26–46%
of patients (22).
In the present study, panoramic radiograph and CBCT
revealed that the two patients had several dental abnormalities
including multiple impacted teeth in the mandible, maxilla and
ethmoidal sinus; however, surgery was performed only in the first
patient (II:1), who had ossifying fibrosis with infection,
resulting in swelling and pain in the mandibular region. The two
patients also had a family history of GS. Therefore, the present
study performed whole-exome sequencing and a large number of
variants in each individual were initially identified. Several
mutations in GS family members were identified by bioinformatic
filtering and segregation analysis of the data. It was concluded
that the same variations in both affected individuals were likely
the cause of GS.
MLH1 is a primary component of DNA mismatch repair
(MMR) protein complexes, and heterodimerizes with PMS2 to form
MutLα. In this way, MMR serves an important role in genome
stability (23,24). MLH1 is located on 3p21.3–23, and is
commonly dysregulated in colon cancer (25). It has been previously reported that
the MLH1 mutation may be responsible for 50% of cases of Lynch
syndrome (also known as hereditary non-polyposis colon cancer,
HNPCC) (26). Acquired defects in
MLH1 have been observed in 13–15% of sporadic colorectal cancers
(CRCs) (27), and the lifetime
risk of contracting CRC is as high as 68% in MLH1 mutation carriers
(28). It has been documented that
there are numerous mutation sites in the MLH1 gene responsible for
HNPCC (28), however whether the
mutation site identified in the present study
(NM_000249.3:p.Tyr379Ser/c.1136A>C) results in HNPCC has not
been reported. Due to this, the present study focused on GS, and
whether the mutation site results in HNPCC will be investigated in
future work. Depending on the feature of the disease, the present
study firstly sequenced MLH1 in peripheral blood from these
patients, and in the future, may sequence MLH1 in tissue samples.
To verify that this MLH1 mutation results in GS syndrome, the
author's will perform further functional analyses in the
future.
In conclusion, the present study screened for
mutations in the MLH1 gene in a Chinese family with GS using
whole-exome sequencing, and the findings were confirmed by Sanger
sequencing. The two affected individuals in the family harbored a
missense mutation in the MLH1 gene
(NM_000249.3:p.Tyr379Ser/c.1136A>C) and shared a number of
symptoms, including osteomas, skin and soft tissue tumors and
dental anomalies. The data also demonstrated that the amino acid
residue of p.Tyr379 was highly conserved among different species.
Therefore, it was predicted that the p.Tyr379 mutation may impact
on the proper function of MLH1 and thus may be associated with the
development of GS in this family. Additionally, the present study
demonstrated that whole-exome sequencing is a time- and
cost-efficient method of screening and identifying gene mutations
in GS. To the best of the author's knowledge, the present results
may be the first to identify the MLH1 missense mutation
(NM_000249.3:p.Tyr379Ser/c.1136A>C) in a Chinese family with GS,
which may aid in determining genetic diagnosis and subsequent
therapeutic regimens for this family.
Acknowledgements
Not applicable.
Funding
The present study was supported by Guangxi Key
Laboratory of Metabolic Diseases Research (grant no. 2016-181h-03),
and by Guangxi Natural Science Foundation (grant no.
2015GXNSFBA139176).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MO made substantial contributions to the design and
data analysis of the present study, ZL participated in experiment
of the present study and data analysis, and drafted the manuscript.
SW made substantial contributions to sample collection and clinical
data analysis. CW participated in DNA sequencing and mutation
analysis, LW, BG, DZ and YZ participated in whole-exome sequencing
data analysis. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Guangxi Key Laboratory of Metabolic Diseases Research.
Written informed consent was obtained from all patients.
Consent for publication
Written informed consent for the publication of any
associated data and accompanying images was obtained from each
participant.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cankaya AB, Erdem MA, Isler SC, Cifter M,
Olgac V, Kasapoglu C and Oral CK: Oral and maxillofacial
considerations in Gardner's Syndrome. Int J Med Sci. 9:137–141.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ramaglia L, Morgese F, Filippella M and
Colao A: Oral and maxillofacial manifestations of Gardner's
syndrome associated with growth hormone deficiency: Case report and
literature review. Oral Surg Oral Med Oral Pathol Oral Radiol
Endod. 103:e30–e34. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ponti G, Tomasi A, Manfredini M and
Pellacani G: Oral mucosal stigmata in hereditary-cancer syndromes:
From germline mutations to distinctive clinical phenotypes and
tailored therapies. Gene. 582:23–32. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Klein OD, Oberoi S, Huysseune A,
Hovorakova M, Peterka M and Peterkova R: Developmental disorders of
the dentition: An update. Am J Med Genet C Semin Med Genet.
163C:318–332. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Singh K, Singh A, Kumar P and Gupta N:
Prosthodontic management of a patient with Gardner's syndrome: A
clinical case report. Dent Res J (Isfahan). 11:276–280.
2014.PubMed/NCBI
|
|
6
|
Madani M and Madani F: Gardner's syndrome
presenting with dental complaints. Arch Iran Med. 10:535–539.
2007.PubMed/NCBI
|
|
7
|
Aretz S: The differential diagnosis and
surveillance of hereditary gastrointestinal polyposis syndromes.
Dtsch Arztebl Int. 107:163–173. 2010.PubMed/NCBI
|
|
8
|
Davies DR, Armstrong JG, Thakker N, Horner
K, Guy SP, Clancy T, Sloan P, Blair V, Dodd C, Warnes TW, et al:
Severe Gardner syndrome in families with mutations restricted to a
specific region of the APC gene. Am J Hum Genet. 57:1151–1158.
1995.PubMed/NCBI
|
|
9
|
Juhn E and Khachemoune A: Gardner
syndrome: Skin manifestations, differential diagnosis and
management. Am J Clin Dermatol. 11:117–122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gu GL, Wang SL, Wei XM and Bai L:
Diagnosis and treatment of Gardner syndrome with gastric polyposis:
A case report and review of the literature. World J Gastroenterol.
14:2121–2123. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang Y, Muzny DM, Reid JG, Bainbridge MN,
Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al:
Clinical whole-exome sequencing for the diagnosis of mendelian
disorders. N Engl J Med. 369:1502–1511. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bamshad MJ, Ng SB, Bigham AW, Tabor HK,
Emond MJ, Nickerson DA and Shendure J: Exome sequencing as a tool
for Mendelian disease gene discovery. Nat Rev Genet. 12:745–755.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinformatics.
26:589–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2010. View Article : Google Scholar
|
|
15
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mckenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The Genome Analysis Toolkit: a MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ng PC and Henikoff S: SIFT: Predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Basaran G and Erkan M: One of the rarest
syndromes in dentistry: Gardner syndrome. Eur J Dent. 2:208–212.
2008.PubMed/NCBI
|
|
19
|
Jaiswal AS, Balusu R and Narayan S:
Involvement of adenomatous polyposis coli in colorectal
tumorigenesis. Front Biosci. 10:1118–1134. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chung J, Namkoong S, Jung KE, Park JW,
Park BC, Cinn YW and Kim MH: A case of gardner's syndrome
associated with desmoid tumor. Ann Dermatol. 22:418–421. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fotiadis C, Tsekouras DK, Antonakis P,
Sfiniadakis J, Genetzakis M and Zografos GC: Gardner's syndrome: A
case report and review of the literature. World J Gastroenterol.
11:5408–5411. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Herford AS, Stoffella E and Tandon R:
Osteomas involving the facial skeleton: A report of 2 cases and
review of the literature. Oral Surg Oral Med Oral Pathol Oral
Radiol. 115:e1–e6. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lu Y, Wajapeyee N, Turker MS and Glazer
PM: Silencing of the DNA mismatch repair gene MLH1 induced by
hypoxic stress in a pathway dependent on the histone demethylase
LSD1. Cell Rep. 8:501–513. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kadyrova LY and Kadyrov FA: Endonuclease
activities of MutLα and its homologs in DNA mismatch repair. DNA
Repair (Amst). 38:42–49. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bronner CE, Baker SM, Morrison PT, Warren
G, Smith LG, Lescoe MK, Kane M, Earabino C, Lipford J and Lindblom
A: Mutation in the DNA mismatch repair gene homologue hMLH1 is
associated with hereditary non-polyposis colon cancer. Nature.
368:258–261. 1994. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ryan E, Sheahan K, Creavin B, Mohan HM and
Winter DC: The current value of determining the mismatch repair
status of colorectal cancer: A rationale for routine testing. Crit
Rev Oncol Hematol. 116:38–57. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hinrichsen I, Ernst BP, Nuber F, Passmann
S, Schäfer D, Steinke V, Friedrichs N, Plotz G, Zeuzem S and
Brieger A: Reduced migration of MLH1 deficient colon cancer cells
depends on SPTAN1. Mol Cancer. 13:112014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cruz-Correa M, Pérez-Mayoral J, Dutil J,
Echenique M, Mosquera R, Rivera-Román K, Umpierre S,
Rodriguez-Quilichini S, Gonzalez-Pons M, Olivera MI, et al:
Hereditary cancer syndromes in Latino populations: Genetic
characterization and surveillance guidelines. Hered Cancer Clin
Pract. 15:32017. View Article : Google Scholar : PubMed/NCBI
|