Introduction
During the progression of atherosclerosis, oxidized
low-density lipoprotein (oxLDL) initiates endothelial dysfunction
and increases the expression of vascular cell adhesion molecule-1
(VCAM-1), P-selectin, and chemokine monocyte chemoattractant
protein-1 (MCP-1) that recruits monocytes, T-lymphocytes and
platelets. Monocytes are stimulated by monocyte-colony stimulating
factor (M-CSF), and pro-inflammatory chemokines and cytokines,
which differentiate them into macrophages. Macrophages then engulf
oxLDL by receptor-mediated phagocytosis and transform themselves
into lipid-laden foam cells. Accumulated foam cells and immune
cells in the vessel wall develop an identifiable pathological
condition called, the ‘fatty streak’. This first symptom represents
asymptomatic and non-stenotic plaque, and is the early sign for the
development of atherosclerosis. Subsequently, the smooth muscle
cells (SMCs) triggered by cytokines, chemokines, and growth
factors, gain the ability to proliferate and migrate to develop a
fibrous cap covering the atherosclerotic core. In this advanced
pathological stage of atherosclerosis, matrix degradation,
cytotoxic T-cells and cholesterol crystals accumulate to form a
lipid-rich necrotic core. In the very late stage of
atherosclerosis, apoptosis in the SMCs of the fibrous cap occurs
which causes plaque instability and thrombosis. As a result, distal
embolism occurs which leads to blockage in the cerebral arteries.
Thus, atherosclerosis in coronary arteries and in carotid arteries
results in coronary heart disease and ischemia in the brain which
in severe conditions may cause significant morbidity and mortality
(1). During the progression of
atherosclerosis from ‘fatty streak’ to atheroma development,
changes in the gene expression in different cell types have been
reported. Below, we describe the de novo epigenetic changes,
specifically histone modifications which were reported in different
cell types that are involved in the development of
atherosclerosis.
Histone methylation
DNA normally remains coiled to the histones proteins
(H1, H2A, H2B, H3 and H4) in eukaryotes systems. Tight control over
gene expression occurs accurately and effectively via chromatin
compaction or signaling, or by activating other signaling events
such as phosphorylation, acetylation, ubiquitination and
methylation on histones. These modifications on the histone
proteins are collectively referred to as histone post-translational
modifications (PTMs). Since these histone modifications have been
identified to either induce or inhibit transcription, their role in
regulating gene expression, which is specific to each type of
modification on different histones, has been well studied (2,3).
Histone proteins are predominantly methylated on
arginine and lysine residues. Multiple modifications can occur on
these histone proteins such as mono-methylation (me1),
di-methylation (me2) or tri-methylation (me3). The Su(var)3-9,
Enhancer-of-zeste and Trithorax domain containing proteins
(SET-domain-containing proteins), Disruptor of telomeric silencing
1-like (DOT1-like proteins), and Calmodulin-lysine
N-methyltransferase are three families of proteins which
transfer methyl groups from S-adenosyl methionine to
histones. On the contrary, the two family of proteins which
function as demethylases have also been identified which include
amine oxidases and jumonji C (JmjC)-domain-containing proteins, and
iron-dependent dioxygenases (4).
Histone methylation as a prognostic
marker in atherosclerosis
Methylation on H3K4 has been correlated with
stage-specific progression of atherosclerosis mediated by GCN5L and
MYST1, MLL2/4 proteins (3). In
ApoE-/-mice, high levels of homocysteine (Hcy) induced EZH2
expression was detected which led to H3 at lysine 27 (H3K27)
tri-methylation (5). The polycomb
complex protein, BMI-1, is one of the proteins in PRC1-like
complex, which is required to inhibit the expression of target
genes through histone modification such as mono-ubiquitination on
H2AK119. As part of PRC1 complex, BMI-1 promotes E3
ubiquitin-protein ligase activity (6). Global increase in trimethylation of
H3K27 was observed in atherosclerotic plaques at late stage in the
pathology. However, decreased trimethylation of H3K27 was not
associated with either BMI-1, histone methyltransferase EZH2, or
histone demethylase JMJD3, which targets H3K27 (7). Increased expression of H2AK119Ub and
H2BK120Ub has been reported in diabetic cardiomyopathy, and
treatment with Esculetin was identified to mitigate the
renin-angiotensin system, oxidative stress (Keap1) and cell
proliferation (Ki67) via H2A/H2B ubiquitination, which eventually
attenuated metabolic alterations, hypertension, cardiomyocytes
hypertrophy, and fibrosis in the hearts of type 2 diabetic rats
(8). The S-Adenosyl homocysteine
(SAH) level in the plasma holds a strong correlation with the size
of atherosclerotic lesion and in the pathogenicity of
hyperhomocysteinemia during atherosclerosis. The increased levels
of SAH in plasma was reported to promote expression of
glucose-regulated protein-78 and CEBP-homologous protein, which are
indicators of endoplasmic reticulum stress resulting from the
repression of trimethylation on histone H3 at lysine 9 (H3K9)
(9). The histone-lysine
N-methyltransferase (MLL5) activity, which serves an important role
in cell cycle regulation, and SNP variations at this locus, have
been attributed with coronary artery disease. The dominant genotype
of rs12671368 and the recessive genotype of rs2192932 were found to
provide protective effects in a study that was carried out on
Chinese Han population (10).
Monocytes and macrophages
One of the primary functions of G9a is to
mono-methylate and di-methylate H3K9 by forming G9a-GLP (G9a-like
protein) complex, which plays a critical role in the regulation of
gene expression in monocytes (11). In hyperhomocysteinemic ApoE-/-mice
that were treated with methionine, decreased expression of G9a as
well as H3K9 di-methylation was reported to promote apoptosis in
the macrophages affecting the foam cell formation and also plaque
stability (12). Inhibiting
trimethylation of H3K4me3 and H3K27me3 was found tightly associated
with increased expression of specific marker genes such as KLF4,
IRF8, HOXA and FOXO which stimulate monocyte-into-phagocyte (MP)
differentiation (13). In
monocytes, reduced methylation on H3K9 and H3K27 was observed in
inflammatory cells (14).
Monocytes typically gets reprogramed into pro-inflammatory
phenotype by oxLDL, and this was found to increase during aerobic
glycolysis. Analysis of the monocytes from patients with
asymptomatic and symptomatic carotid plaques revealed that
monocytes from patients with symptomatic carotid plaque expressed
more pro-inflammatory cytokines after stimulating with LPS. Lower
levels of H3K4me3 and H3K27me3 were found on the promotor region of
TNF-α. At the same time, reduced glycolytic rate limiting enzymes
hexokinase 2 and
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 expressions
were detected in the carotid plaques of symptomatic compared to
asymptomatic patients with carotid stenosis (15). The suppressor of variegation 3–9
homologue1 (SUV39H1) is a histone methyltransferase which mediates
trimethylation at H3K9, and acetylation at lysine residue 266 was
found to decrease its activity. SIRT1 was reported to recruit and
deacetylate SUV39H1, and this deacetylation of SUV39H1 contributes
to the increased functional activity of SUV39H1 thereby, the levels
of H3K9me3 will get elevated (16). In macrophages, high-glucose
stimulation reduced trimethylation on H3K9 which was mediated
through SUV39H1. This further led to increased expression of the
inflammatory cytokines such as IL-6, IL-12, p40, MIP-1α, and MIP-1β
(17). In peripheral blood
monocytes (PBM) of patients with type 2 diabetes mellitus (T2DM),
Set7 induced mono-methylation at H3K4 was found increased at the
promotor of NF-κB p65, leading to the activation of NF-κB-dependent
oxidative and inflammatory signaling. This in turn elevated the
expression levels of the downstream pro-inflammatory genes ICAM-1
and MCP-1. Set7 was also found associated with the oxidative marker
8-isoPGF2α and brachial artery flow-mediated dilation (FMD)
(18). The oxLDL, TLR-2 and TLR-4
agonists can induce human monocyte differentiation into foam cells
by stimulating trimethylation of H3K4 at the promotor regions of
different genes such as IL-6, IL-18, TNF-α, MCP-1, MMP2, MMP9,
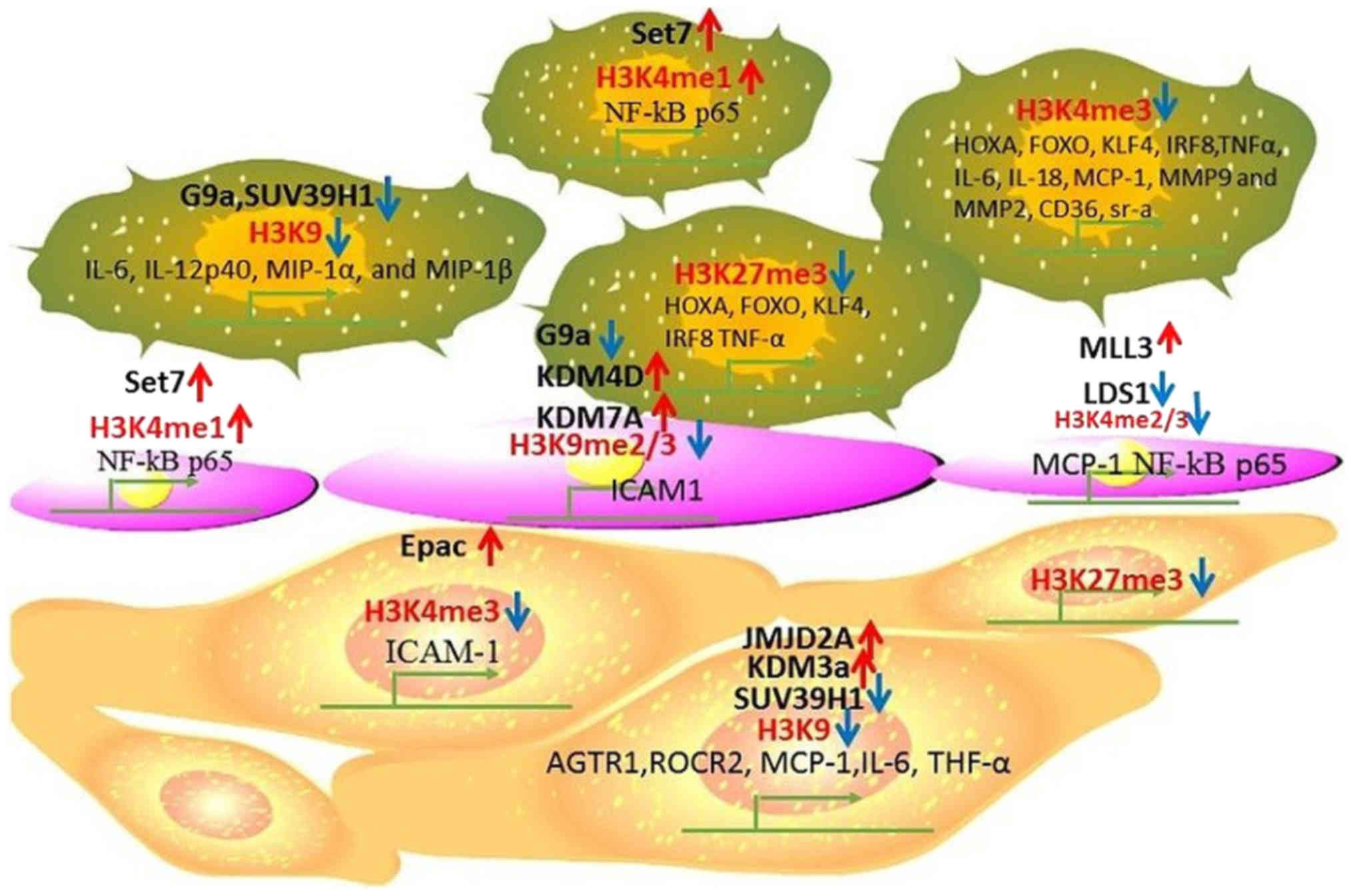
CD36, and SR-A (19). In Table I, we summarize histone
modifications occurring on different genes in monocytes and
macrophages that are regulated during atherosclerosis.
 | Table I.Histone methylation markers in
monocytes and macrophages in atherosclerosis. |
Table I.
Histone methylation markers in
monocytes and macrophages in atherosclerosis.
| Epigenetic
mediator | Histone
modification | Expression in
atherosclerosis | Target genes |
|---|
| G9a, SUV39H1 | H3K9 | Decreased | IL-6, IL-12p40,
MIP-1α, and MIP-1β |
| Set7 | H3K4me1 | Increased | NF-κB p65 |
|
| H3K4me3 | Decreased | HOXA, FOXO, KLF4,
IRF8, TNFα, IL-6, IL-18, MCP-1, MMP9, MMP2, CD36 and SR-A |
|
| H3K27me3 | Decreased | HOXA, FOXO, KLF4,
IRF8 and TNF-α |
Endothelial cell
TNF-α stimulates the expression of ICAM1 which
causes leukocyte adhesion to endothelial cells via reducing
methylation of H3K9 and H3K27 residues. Overexpression of G9a or
inhibition of KDM4D can increase the expression of H3K9me2 and
reduce adhesion of leukocytes to endothelial cells (20). In endothelial cells, SAH which is a
potent inhibitor of S-adenosylmethionine-dependent
methyltransferases, was found to induce endothelial cell activation
by increasing the expression of adhesion molecules and cytokines,
by stimulating NF-κB pathways. SAH reduced the expression of EZH2
and tri-methylation of H3K27 (21). In endothelial cells, TNF-α
increased the expression of histone demethylases KDM1B and KDM7A
which further induced H3K9me2 di-methylation. KDM7A mediated
upregulation of ICAM1 was found associated with decreased
expression of the transcription factor EB which positively
regulates the activity of lysosome formation (22). Arginine methyltransferase
inhibitors, AMI-5 and AMI-1, inhibit migration of endothelial cells
to prevent pathological angiogenesis by stimulating the formation
of heterochromatin. This was reported to be mediated through
downregulation of arginine and lysine histone methyltransferases
(HMTs) (23). PHD finger protein 8
(PHF8) was shown to induce endothelial cell migration and formation
of capillary-like structures by decreasing di-methylation of E2F4,
suggesting its functions as a repressor (24). In human umbilical vein cell line
EA.hy926, elevated di- and tri-methylation (H3K4me2/3) on MCP-1
promotor was found regulated by increased H3K4 histone
methyltransferase MLL3, menin and SET7 along with a concomitant
decrease in the demethylase LSD1 expression in high glucose
conditions. Puerarin was also reported to decrease the expression
of MCP-1 by reversing the expression of methyltransferases and
demethylases (25). During the
progression of atherosclerosis, a subset of dendritic cells also
takes up the lipid molecules and this leads to the formation of
foam cells. Elevated cholesterol levels in plasma indicates the
onset of atherosclerotic phenotype in T-cells compared to
anti-atherosclerotic phenotype in normal group (26). In human aortic endothelial cells
(HAECs), Set7 was found to stimulate NF-κB-dependent oxidative and
inflammatory signaling by mono-methylation of H3K4 at its promotor
(18). In Endothelial cells, EZH2
was reported to be induced by LDL. This was observed to be mediated
through the transcription activator, myocyte enhancing factor-2,
which decreased the expression of KLF2. The decreased expression of
KLF2 was found to affect the expression of different cellular
regulators of atherosclerosis such as thrombomodulin, endothelial
NO synthase, and plasminogen activator inhibitor-1, which promote
accumulation of platelets on endothelial cells (27). ‘Hyperglycemic memory’ is a
condition which induces deleterious effects in patients even after
more than five years of the onset of glycemic control. In bovine
aortic endothelial cells, hyperglycemia induced expression of
NF-κB-p65 was found to upregulate the expression of H3K4m1, and
this was associated with the reduced expression of di- and
tri-methylation of histone H3 (H3K9m2 and H3K9m3). This reduction
in histone methylation was identified as mediated through histone
demethylase LSD1 (28,29). Hyperglycemia also stimulated the
expression of p65 in aortic endothelial cells, which induced the
expression of MCP-1 and VCAM-1. This suggests that by decreasing
the levels of mitochondrial superoxide production or
superoxide-induced α-oxoaldehydes, inflammation under hyperglycemic
condition can be prevented (29).
In Table II, we summarized the
histone modifications occurring exclusively in the endothelial
cells during atherosclerosis, affecting different target gene
loci.
| Table II.Histone methylation markers in
endothelial cells in atherosclerosis. |
Table II.
Histone methylation markers in
endothelial cells in atherosclerosis.
| Epigenetic
mediator | Histone
modification | Expression in
atherosclerosis | Target genes |
|---|
| G9a | H3K9m2 | Decreased | ICAM1 |
| KDM4D | H3K9m2 | Increased | ICAM1 |
| KDM7A | H3K9m2 | Increased | ICAM1 |
| PHF8 | Demethylation | Increased | E2F4 |
| MLL3 | H3K4m2, H3K4m3 | Increased | MCP-1, p65 |
| LDS1 | H3K4m2, H3K4m3
H3K9m2, H3K9m3 | Decreased | MCP-1, p65 |
| SET7 | H3K4m1 | Increased | NF-κB |
| EZH2 |
| Increased | KLF2 |
Vascular SMCs
Under in vitro conditions, high glucose
increases the expression of KDM3a, causing reduction in H3K9
di-methylation at AGTR1 and ROCK2 loci. This indicates that the
proliferation and migration of vascular SMCs (VSMCs) is enhanced
through activation of Rho/ROCK and AngII/AGTR1 pathways (30). Increased histone acetylation at
H3K9 and H3K27 led to a parallel decrease in the methylation at
H3K9 and H3K27 which was found in the advanced atherosclerotic
lesions (14). VSMCs upon
treatment with Acrolein, which is an air pollutant and consists of
cigarette smoke, was identified to induce histone acetylation at
H3K9 and H3K56. Simultaneous decrease in the histone
tri-methylation at H3K9 was also observed. Co-culturing with
N-acetyl cysteine has been shown to inhibit the toxicity of
acrolein. In VSMCs this was correlated with significant decrease in
histone acetylation at H3K9. At the same time, other histone
modifications were also observed such as di-methylation at H3K4,
and phosphorylation and di-methylation at H3S10 (31). In VSMCs, treatment with
Roflumilast, which is a PDE4 inhibitor, was reported to affect
TNF-α functions through VCAM-1 by promoting the expression of the
cyclic AMP effector, Epac. Upregulation of Epac subsequently
reduced histone di-methylation H3K4me2 at VCAM-1 and promoted its
expression. This epigenetic action of Roflumilast on VCAM-1 can be
reverted by treatment with an HDAC inhibitor (32). High-glucose conditions were also
reported to increase JMJD2A expression in the VSMCs of the rat
carotid arteries which enhanced cell proliferation. Tri-methylation
of H3K9 was inversely correlated with JMJD2A expression in VSMCs of
the balloon-injured arteries of diabetic rats where histone
methylation suppressed the expression of MCP-1 and IL-6. Thus,
inhibition of JMJD2A appears to be a potential treatment option to
prevent inflammation in VSMCs (33). Moreover, global reduction in the
levels of H3K27 in advanced atherosclerotic plaques did not hold
any correlation with the expression of histone methyltransferase,
EZH2, which is the catalytic component of the polycomb repressive
complex 2 (PRC2). Further, BMI1 which consists of PRC1 complex,
promoted tri-methylation of H3K27 and JMJD3 which is a histone
demethylase that targets H3K27. The reduction in H3K27Me3
tri-methylation in the tunica media also plays an important role in
the differentiation and proliferation of VSMCs in atherosclerosis
(7). Decreased expression of
Suv39h1 would lead to reduced levels of H3K9me3 in VSMCs in type 2
diabetic db/db mice when compared with control b/+ cells,
suggesting that VSMCs are hypersensitive to TNF-α stimulation.
Moreover, the corepressor HP1α was also reported to be decreased in
db/db cells (34).
Upregulation of major histocompatibility complex II
(MHC II) molecules in VSMCs, which recruit T lymphocyte and
stimulate the expression of inflammatory cytokines, has been well
characterized in the pathogenesis of atherosclerosis. By
stimulating A2b adenosine signaling pathway, PCAF/GCN5 which are
histone H3K9 acetyltransferases, and WDR5 which consists of H3K4
methyltransferase complex, prevents their recruitment at the
promotor region of CIITA transcription, thereby inhibiting the
expression of CIITA in a STAT1-dependent manner (35). Specific methylation patterns in
histone proteins that have been reported in the VSMCs during
atherosclerosis was summarized in Table III.
| Table III.Histone methylation markers in
vascular smooth muscle cells in atherosclerosis. |
Table III.
Histone methylation markers in
vascular smooth muscle cells in atherosclerosis.
| Epigenetic
mediator | Histone
modification | Expression in
atherosclerosis | Target genes |
|---|
| KDM3a | H3K9 | Increased | AGTR1, ROCR2 |
| Epac | H3K4m3 | Increased | ICAM-1 |
| JMJD2A | H3K9 | Increased | MCP-1, IL-6 |
| EZH2 | H3K27 | Increased | – |
| SUV39H1 | H3K9m3 | Decreased | TNF-α |
In utero programming and postnatal
histone methylation in atherosclerosis
Maternal hypercholesterolemia could have direct
impact in inducing atherosclerosis in the offspring as reported in
ApoE-/-Leiden mice (36). Compared
with the offspring from wild-type mothers, the offspring from
ApoE-/-mothers showed differences in histone tri-methylation in
endothelial cells and VSMCs, which caused different responses to
high cholesterol diet. This study highlights the importance of
in-utero programming and postnatal histone modification in
atherosclerosis which if addressed in early stages may help in
treating the disease (37). In
Fig. 1, we summarize the histone
methylation in different cell types and their effects on regulating
gene expression during atherosclerosis.
Histone acetylation
Histone acetylation is regulated by two key enzymes,
histone acetyltransferases (HATs) and histone deacetylases (HDAC).
By transferring an acetyl group to lysine side chains of histone
proteins, HATs weaken the binding between histone and DNA. On the
contrary, HDACs remove the acetyl group on the histones to increase
the interaction between histone and DNA. HATs are further
categorized into two groups, type-A and type-B (38). HDACs regulate transcription and
cell cycle progression by deacetylating lysine residues located on
the N-terminal of the core histone proteins (H2A, H2B, H3 and H4).
HDACs are further classified into four Classes I, II, III and IV.
Classes I, II and IV represent ‘classical’ HDACs which include 11
genes, and in Class III, 7 genes are included which are named as
sirtuins (39). Based on the type
of modification occurring on the histones, gene expression is
either favored or inhibited. This characteristic feature of the
PTMs on histones and their role in transcriptional regulation is
developmental and tissue specific. However, such histone
modifications, to regulate gene expression, are frequently observed
in many pathological conditions. Such modifications occurring in
atherosclerosis are discussed below.
Histone acetylation as an indicative
marker in atherosclerosis
In the heart tissues of New Zealand white rabbits
that were treated with high cholesterol diet and atorvastatin, the
mRNA levels of ACE2 was reported to be elevated when compared with
control high cholesterol diet. Notably, atorvastatin was identified
to promote histone H3 acetylation (H3-Ac) at the promoter region of
ACE2 (40). High plasma levels of
Hcy was identified not only to increase abnormal DNA methylation in
the cells but also elevates the levels of N-methyl-d-aspartate
receptor-1 (NMDAR1), DNA (cytosine-5)-methyltransferase-1 (DNMT1)
and MMP-9, via acetylation on H3K9 and by inhibiting the expression
of HDAC1. These regulatory proteins were identified as possible
targets that have high potential to cause heart failure as reported
in cardiomyocytes (41).
Monocytes and macrophages
The ‘Silent mating type information regulator two
homologue one’ (SIRT1), which belongs to class III HDAC, requires
Nicotinamide adenine dinucleotide (NAD+) as a cofactor for its
functional activity. The role of SIRT1 has been implicated in many
cellular processes such as in cell cycle progression, inflammation,
DNA damage, apoptosis, autophagy, aging as well as in metabolic
disease (42). SIRT1 mediates
transfer of the acetyl group from proteins to the targeting
co-substrate by removing the nicotinamide ribosyl bond of NAD+. In
the peripheral blood mononuclear cells (PBMCs) of patients with
T2DM, increased transcription of HDAC3 was reported. SIRT1, which
was identified as a protective marker of cardiovascular diseases,
was found reduced in PBMCS of these T2DM patients. In addition, the
pro-inflammatory markers TNF-α, IL-6, MCP-1, IL1-β, NFκB, TLR2, and
TLR4 were also reported to be upregulated in the plasma of the T2DM
patients. Further, DBC1 (deleted in breast cancer 1), which
represses HDAC3, was also reported to be decreased in the PBMCs of
patients with T2DM (43). Myeloid
HDAC3 deficiency was reported to have plaque stabilization effect
by increasing the deposition of collagen, HDAC3 expression can
potentially induce macrophage polarization, favoring the formation
M2 phenotypes. These M2 macrophages by secreting TGFβ1 were shown
to initiate VSMCs to secrete excess collagen (44,45)
Under hypercholesterolemic conditions, the orphan nuclear receptor
NR4A1, which can effectively reduce IL-6 and MCP-1 expression, was
found increased due to the recruitment of p300 acetyltransferase.
As a result, HDAC7 (histone deacetylase 7) was found repressed
which contributes to the expression of NR4A1 in human monocytes,
THP-1 and U937 (46). The histone
deacetylase 9, HDAC9, was reported to be associated with MMP12
expression in the pro-inflammatory macrophages M2 and M4, in the
advanced human plaques. However, the presence of both these types
of macrophages could not be correlated with genes regulating plaque
stabilization or thrombosis (47).
HDAC9 deficient cells were identified to express increased levels
of ABCA1, ABCG1, and PPAR-γ. This was identified to be mediated
through upregulation of H3 and H4 acetylation, preventing the
cholesterol efflux. Moreover, HDAC9 knockout macrophages were
identified to be secreting lower levels of pro-inflammatory
cytokines and showed prominent expression of M2 phenotype markers,
and at the same time, it simultaneously decreased the expression of
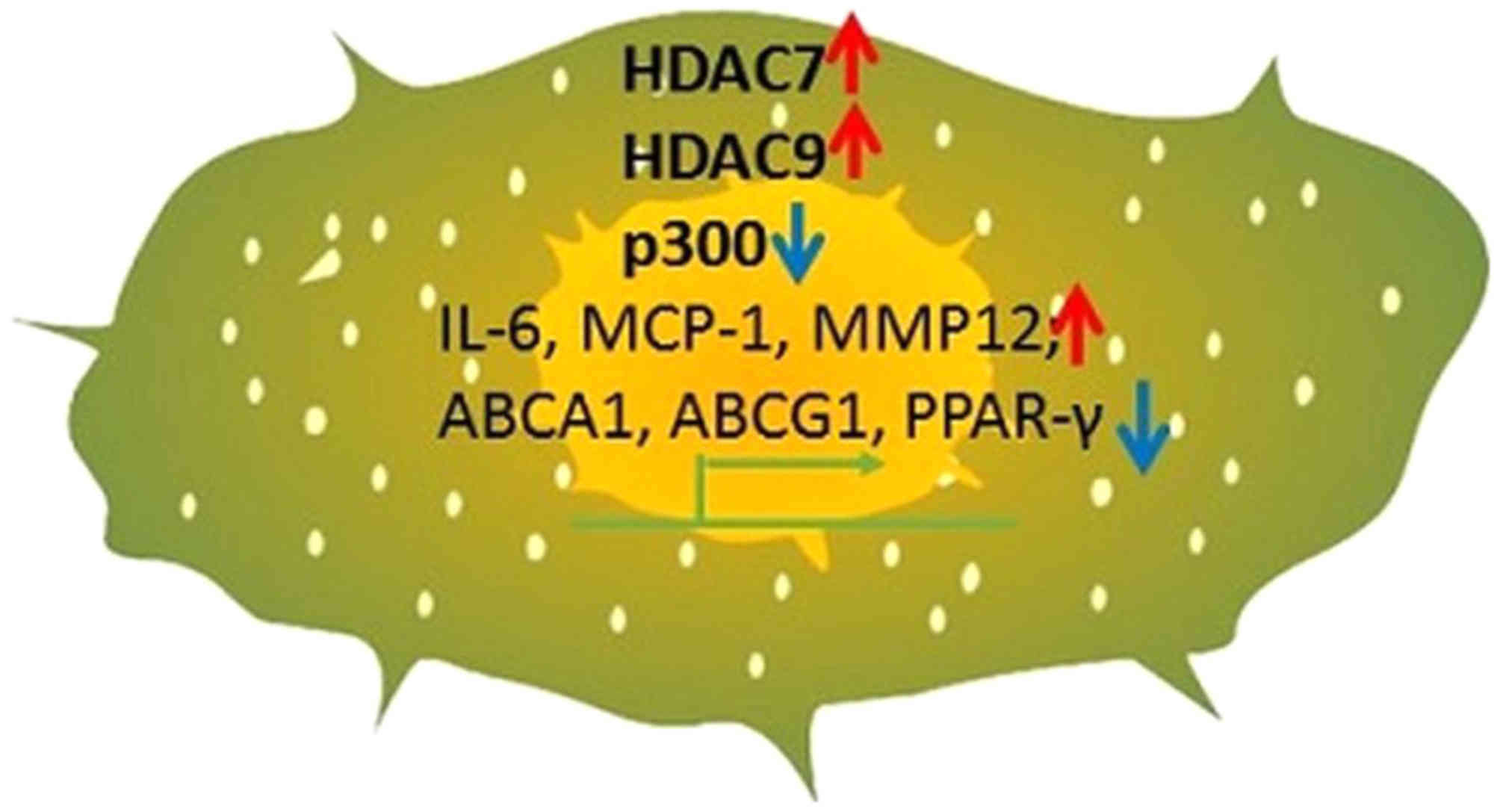
M1 markers (48,49). In Table IV, we summarized acetylation
events on different residues of histones in the above monocytes and
macrophages occurring during atherosclerosis. The same was
represented in Fig. 2.
| Table IV.Histone acetylation markers in
monocytes and macrophages in atherosclerosis. |
Table IV.
Histone acetylation markers in
monocytes and macrophages in atherosclerosis.
| Enzyme | Expression in
atherosclerosis | Target genes |
|---|
| HDAC7 | Increased | IL-6, MCP-1 |
| P300 | Decreased | Transcription
regulation |
| HDAC9 | Increased | MMP12; ABCA1,
ABCG1, PPAR-γ |
Other cell types
The protein ‘General Control Non-repressible 5’
(GCN5)-related N-acetyltransferase (GNAT), mediates transfer of the
acyl group from acyl coenzyme A (acyl-CoA) to different proteins
substrates which are involved in transcription, cell proliferation,
antioxidant functions, antibiotic resistance and detoxification
(50). The p300/CREB-binding
protein (CBP) which is a HAT, acetylates non-histone proteins and
controls transcription and DNA repair mechanisms in the cells
(51). When compared between the
cardiac mesenchymal stem cells from normoglycemic subjects, cardiac
mesenchymal stem cells from type 2 diabetic patients had decreased
levels of H3K9Ac and H3K14Ac acetylations, which are caused
primarily due to the reduced activity of GCN5-related
N-acetyltransferases (GNAT) p300/CBP-associated factor. Also its
isoform, GCN5a, decreased proliferation and differentiation of
MSCs. Moreover treatment with the HAT activator,
pentadecylidenemalonate 1b (SPV106) not only recovered the levels
of H3K9Ac and H3K14Ac acetylation, but also decreased CpG
hypermethylation in the genomic DNA, suggesting the involvement of
two different epigenetic mechanisms regulating at the same time in
these cells (52).
Targeting histone methylation and histone
acetylation in the treatment of atherosclerosis
As cell specific histone modifications have been
identified during atherosclerosis, targeting the enzymes which
regulate these de novo changes can be a promising strategy
for the treatment of atherosclerosis. This strategy is favored by
two important features. Frist, like all epigenetic therapies,
targeting histone modification alone do not affect the genetic
component in the cells. Second, advances in the structural biology
greatly assists the researchers to develop unique compounds that
can specifically target the desired histone modification, such as
the DNMT inhibitor (2′-deoxy-5-azacytidine, DAC) which targets H3K9
dimethylation, and the HDAC inhibitor TSA, which targets H3K27
trimethylation (53). Since any
given histone modification is not unique for a specific disease and
as they can be observed in different pathologies, drugs targeting a
specific histone modification can be repurposed for other disease
conditions. In addition, use of these specific histone targeting
drugs as combination therapy with other therapeutic drugs appears
to be more promising.
Treatment strategies targeting histone
methylation
The selective inhibitor, 3-deazaneplanocin A (DZNep)
prevents methylation at lysine 27 on histone H3 (H3K27me3) and
lysine 20 on histone H4 (H4K20me3) is currently used in the
treatment of cancer (54).
Upregulation of Lysine methyltransferases (KMTs) plays an important
role in the differentiation of monocytes into immature dendritic
cells (iDCs). Two of the know drugs that inhibit histone
methylation, BIX-01294 which is KMT1c inhibitor, and DZNep,
prevents global KMT activity were found to inhibit monocytes
differentiating into iDCs (55).
Treatment strategies targeting histone
acetylation
HDAC1 expression was identified to decrease
acetylation of H3K9ac which induces accumulation of total
cholesterol, free cholesterol, and triglycerides in foam cells, as
seen in the aorta of ApoE-/-mice, which was abrogated by
upregulation of the microRNA miR-34a (56). The compound
4-hydroperoxy-2-decenoic acid ethyl ester (4-HPO-DAEE) isolated
from the royal jelly was found to acetylate histone H3 and H4 at
the proximal promoter region of EC-SOD (extracellular superoxide
dismutase) in THP-1 cells, which is considered as a potential
target for the treatment of atherosclerosis (57). Treatment with histone deacetylase
inhibitor, valproic acid, was shown to promote the release of
endogenous t-PA in males with vascular disease (58). Several HDAC inhibitors are used in
the current standard of care. The known synthetic HDAC inhibitors
which inhibit Class III HDACs include; dihydrocoumarin,
naphthopyranone, 2-hydroxynaphaldehydes and other
sirtuin-inhibiting agents. Class I, II, and IV HDAC inhibitors were
grouped as hydroxamates such as trichostatin A (TSA), vorinostat
(suberoylanilide hydroxamic acid; SAHA), belinostat (PXD101),
panobinostat (LBH589), LAQ824, peptide inhibitors such as cyclic
tetrapeptides (trapoxin B) and depsipeptides, benzamides
(entinostat (MS-275), mocetinostat (MGCD0103), CI994, aliphatic
acid components (phenylbutyrate, valproic acid), and electrophilic
ketones (51). Some of the plant
polyphenols such as curcumin (diferuloylmethane) and resveratrol
were identified as naturally available HDAC inhibitors.
Future directions in targeting histone
methylation and histone acetylation in the treatment of
atherosclerosis
Several histone modifications regulating the
off-target drug effects have been reported which indicates their
additional benefits. Both Hydralazine and Osalazine which are used
to treat hypertension can be repurposed for treating neoplasia as
their treatment can restore the functions of tumor suppressor
genes. As a histone-deacetylase inhibitor, Romidepsin when used in
endometriosis, efficiently induced apoptosis in the epithelial
cells of endometrium. However, off-targets effects on histone
modification can potentially exert negative effects in the cells.
Similarly, the histone deacetylase inhibitor, valproic acid which
is prescribed for epilepsy treatment, was identified as a causative
factor for spina bifida in the growing embryos (59). Thus, for future considerations,
specific mechanisms of histone modifications should be determined
in patients for prognosis to avoid adverse drug effects.
DNA methylation intercepts histone
modifications
During the early developmental stages of the embryo,
unmethylated CpG islands were found to induce the expression of
DNMT3A and DNMT3B. At the same time, DNMT3L interacts with the
chromatin and targets H3K4. G9a was found to methylate H3K9 and
recruit heterochromatin protein 1, DNMT3A and DNMT3B during stem
cell production (60). Existence
of such epigenetic cross connections may also lead to de novo gene
regulations which can be critically involved in the disease
progression.
Non-coding RNA and histone
modification
Another key epigenetic mechanism that was found to
intercept histone modifications is the expression of long
non-coding RNAs. Under hypoxic conditions, upregulated expression
of HDAC3 was reported to inhibit the expression of long non-coding
RNA ‘lncRNA-LET’ (61). During the
phenotype switch in VSMCs, the long non-coding RNA ‘taurine
up-regulated gene-1’ (TUG1), was found to form a hetero-complex
with EZH2 to methylate α-actin. As a result, the levels of α-actin
were found decreased, and simultaneously F-actin was elevated. This
imbalance was presumed to be a causative factor for the phenotype
switch in the VSMCs which change from contractile to synthetic
phenotype (62). In lung cancer,
the long non-coding RNA, MALAT1, was reported to upregulate the
expression of histone demethylase JMJD1A, which was known to
promote cell migration and invasion (63).
Hyperlipidemia and hypertension
Several factors such as hyperlipidemia,
hypertension, diabetes and also diet play an important role in the
development and progression of atherosclerosis. The onset of de
novo molecular mechanisms which are discussed above indicates
epigenetic mechanisms to be critically involved in the
pathogenesis. It was reported that high-fat diet decreases ATP
citrate-lyase levels as well as acetyl-CoA and/or the ratio of
acetyl-CoA:CoA in white adipose tissue and pancreas, leading to
inhibition of H3K23 acetylation in mice fed with high-fat diet
(64). Dyslipidemia can cause
monocyte priming such as increase in the adhesion of monocytes.
Chemotaxis in the monocytes was promoted due to reduction in the
H3K27 acetylation in non-human primates (65). H3K4 methylation of lysine-specific
demethylase-1 (LSD1), H3K6 methylation of dehydrogenase 2
(HSD11B2), H3K79 methylation on epithelial sodium channel subunit α
(SCNN1A) were also shown to be associated with arterial
hypertension (66).
Clinical significance of HDAC
inhibitors for the treatment of atherosclerosis
Panobinostat is a non-specific HDAC inhibitor that
has been shown to have nanomolar potency, and can effectively
inhibit Class I, II and IV HDACs. It induces apoptosis primarily by
activating caspases and inducing PARP cleavage. In a phaseI/II
clinical trial on 15 HIV positive patients, treatment with an HDAC
inhibitor, panobinostat, was found to lower cardiovascular
biomarkers such as CRP, LDL receptor, MCP1, E-Selectin, and HMGB1
in addition to lowering several inflammatory markers (67).
Trichostatin A, is a nonspecific inhibitor of Class
I and Class II HDACs. Although it has been identified to prevent
inflammatory markers such as IL-1b and IL-6, and have significant
anti-tumor properties, it appears to have a more deleterious role
in atherosclerosis. Treatment with Trichostatin A was found to
promote proatherogenic markers such as TNF-α, SRA, CD36, eNOS and
VCAM-1 (68). However,
Trichostatin A was found to inhibit p21 mediated proliferation of
VSMCs (69). These reports suggest
that Trichostatin A may have a role in preventing neointimal
hyperplasia or maintaining plaque stability. Trichostatin A
treatment upregulates the expression of Arginase 2 enzyme which
promotes eNOS expression, inflammation and cell proliferation
during progression of atherosclerosis. Earlier, it was shown that
HDAC2 directly binds to the Arginase 2 promoter and inhibits its
expression. In the same study, treatment with the HDAC2 specific
inhibitor ‘mocetinostat’, was shown to enhance oxLDL induced
endothelial dysfunction (70).
Though certain HDAC inhibitors have shown promising
results in treating cancer and other diseases, the same had a
different role in atherosclerosis as identified above. However,
there is no detailed information available yet on the clinical
trials using the HDAC inhibitors for the treatment of
atherosclerosis in humans. Additional studies emphasizing the
specific role of each HDAC and their effects on different cell
types during the progression of atherosclerosis are needed. The
ultimate outcome of such studies should substantiate the need for
extensive evaluation of HDAC inhibitors through clinical trials
which are currently lacking.
Technological advancements and
possible applications
Improvements in sequencing strategies, mass
spectrometry, Chromatin immunoprecipitation, microarray and other
novel techniques have contributed to substantial progress in health
science research. Increasing applications of these technological
advancements would have positive effects in the development of
treatment strategies that target de novo epigenetic changes in
different disease conditions including atherosclerosis.
Time-resolved NMR spectroscopy was innovatively developed to
characterize asymmetric histone PTMs (71). Chromatin immunoprecipitation in
conjunction with sequencing methodologies are typically used for
mapping histone modifications. Through inclusion of micrococcal
nuclease (MNase) digestion and barcoding, disease specific
signatures can be identified at the single-cell level. Moreover,
single-cell DamID can also be performed for genome-wide analysis of
histone modifications using a cell line expressing Escherichia coli
deoxyadenosine methylase (Dam) and combining it with specific
histone readers or modifiers (72).
Acknowledgements
Not applicable.
Funding
The present review was developed with the funding
support from ‘LB692-Nebraska Tobacco Settlement Biomedical Research
Development New Initiative Grants’.
Availability of data and materials
Not applicable.
Authors' contributions
WJ wrote the manuscript and performed the literature
review. CSB developed the format of the review and corrected the
manuscript. DKA assisted with the scientific content of the review
and edited the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Seneviratne A, Hulsmans M, Holvoet P and
Monaco C: Biomechanical factors and macrophages in plaque
stability. Cardiovasc Res. 99:284–293. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Karlić R, Chung HR, Lasserre J, Vlahovicek
K and Vingron M: Histone modification levels are predictive for
gene expression. Proc Natl Acad Sci USA. 107:2926–2931. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dong X and Weng Z: The correlation between
histone modifications and gene expression. Epigenomics. 5:113–116.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Greer EL and Shi Y: Histone methylation: A
dynamic mark in health, disease and inheritance. Nat Rev Genet.
13:343–357. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xiaoling Y, Li Z, ShuQiang L, Shengchao M,
Anning Y, Ning D, Nan L, Yuexia J, Xiaoming Y, Guizhong L and
Yideng J: Hyperhomocysteinemia in ApoE-/-Mice leads to
overexpression of enhancer of zeste homolog 2 via miR-92a
regulation. PLoS One. 11:e01677442016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li Z, Cao R, Wang M, Myers MP, Zhang Y and
Xu R: Structure of a Bmi-1-ring1B polycomb group ubiquitin ligase
complex. J Biol Chem. 281:20643–20649. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wierda RJ, Rietveld IM, van Eggermond MC,
Belien JA, van Zwet EW, Lindeman JH and van den Elsen PJ: Global
histone H3 lysine 27 triple methylation levels are reduced in
vessels with advanced atherosclerotic plaques. Life Sci. 129:3–9.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kadakol A, Malek V, Goru SK, Pandey A and
Gaikwad AB: Esculetin reverses histone H2A/H2B ubiquitination, H3
dimethylation, acetylation and phosphorylation in preventing type 2
diabetic cardiomyopathy. J Funct Foods. 17:127–136. 2015.
View Article : Google Scholar
|
|
9
|
Xiao Y, Huang W, Zhang J, Peng C, Xia M
and Ling W: Increased plasma S-adenosylhomocysteine-accelerated
atherosclerosis is associated with epigenetic regulation of
endoplasmic reticulum stress in apoE-/-mice. Arterioscler Thromb
Vasc Biol. 35:60–70. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yuan Q, Xie X, Fu Z, Ma X, Yang Y, Huang
D, Liu F, Dai C and Ma Y: Association of the histone-lysine
N-methyltransferase MLL5 gene with coronary artery disease in
Chinese Han people. Meta Gene. 2:514–524. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen WL, Sun HP, Li DD, Wang ZH, You QD
and Guo XK: G9a-an appealing antineoplastic target. Curr Cancer
Drug Targets. 17:555–568. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cong G, Yan R, Huang H, Wang K, Yan N, Jin
P, Zhang N, Hou J, Chen D and Jia S: Involvement of histone
methylation in macrophage apoptosis and unstable plaque formation
in methionine-induced hyperhomocysteinemic ApoE-/-mice. Life Sci.
173:135–144. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zheng QF, Wang HM, Wang ZF, Liu JY, Zhang
Q, Zhang L, Lu YH, You H and Jin GH: Reprogramming of histone
methylation controls the differentiation of monocytes into
macrophages. FEBS J. 284:1309–1323. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Greißel A, Culmes M, Burgkart R,
Zimmermann A, Eckstein HH, Zernecke A and Pelisek J: Histone
acetylation and methylation significantly change with severity of
atherosclerosis in human carotid plaques. Cardiovasc Pathol.
25:79–86. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bekkering S, van den Munckhof I, Nielen T,
Lamfers E, Dinarello C, Rutten J, de Graaf J, Joosten LA, Netea MG,
Gomes ME and Riksen NP: Innate immune cell activation and
epigenetic remodeling in symptomatic and asymptomatic
atherosclerosis in humans in vivo. Atherosclerosis. 254:228–236.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vaquero A, Scher M, Erdjument-Bromage H,
Tempst P, Serrano L and Reinberg D: SIRT1 regulates the histone
methyl-transferase SUV39H1 during heterochromatin formation.
Nature. 450:4402007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li MF, Zhang R, Li TT, Chen MY, Li LX, Lu
JX and Jia WP: High glucose increases the expression of
inflammatory cytokine genes in macrophages through H3K9
methyltransferase mechanism. J Interferon Cytokine Res. 36:48–61.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Paneni F, Costantino S, Battista R,
Castello L, Capretti G, Chiandotto S, Scavone G, Villano A, Pitocco
D, Lanza G, et al: Adverse epigenetic signatures by histone
methyltransferase Set7 contribute to vascular dysfunction in
patients with type 2 diabetes. Circ Cardiovasc Genet. 8:150–158.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bekkering S, Quintin J, Joosten LA, van
der Meer JW, Netea MG and Riksen NP: Oxidized low-density
lipoprotein induces long-term proinflammatory cytokine production
and foam cell formation via epigenetic reprogramming of
monocytessignificance. Arterioscler Thromb Vasc Biol. 34:1731–1738.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Choi J, Yoon S, Kim S and Jo Ahn S: KDM4B
histone demethylase and G9a regulate expression of vascular
adhesion proteins in cerebral microvessels. Sci Rep. 7:450052017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Barroso M, Kao D, Blom HJ, De Almeida
Tavares I, Castro R, Loscalzo J and Handy DE:
S-adenosylhomocysteine induces inflammation through NFκB: A
possible role for EZH2 in endothelial cell activation. Biochem
Biophys Acta. 1862:82–92. 2016.PubMed/NCBI
|
|
22
|
Choi J and Jo SA: KDM7A histone
demethylase mediates TNF-α-induced ICAM1 protein upregulation by
modulating lysosomal activity. Biochem Biophys Res Commun.
478:1355–1362. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Balcerczyk A, Rybaczek D, Wojtala M,
Pirola L, Okabe J and El-Osta A: Pharmacological inhibition of
arginine and lysine methyltransferases induces nuclear
abnormalities and suppresses angiogenesis in human endothelial
cells. Biochem Pharmacol. 121:18–32. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gu L, Hitzel J, Moll F, Kruse C, Malik RA,
Preussner J, Looso M, Leisegang MS, Steinhilber D, Brandes RP and
Fork C: The histone demethylase PHF8 is essential for endothelial
cell migration. PLoS One. 11:e1466452016. View Article : Google Scholar
|
|
25
|
Han P, Gao D, Zhang W, Liu S, Yang S and
Li X: Puerarin suppresses high glucose-induced MCP-1 expression via
modulating histone methylation in cultured endothelial cells. Life
Sci. 130:103–107. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zernecke A: Dendritic cells in
atherosclerosis. Arterioscler Thromb Vasc Biol. 35:763–770. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kumar A, Kumar S, Vikram A, Hoffman TA,
Naqvi A, Lewarchik CM, Kim YR and Irani K: Histone and DNA
methylation-mediated epigenetic downregulation of endothelial
kruppel-like factor 2 by low-density lipoprotein cholesterol.
Arterioscler Thromb Vasc Biol. 33:1936–1942. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brasacchio D, Okabe J, Tikellis C,
Balcerczyk A, George P, Baker EK, Calkin AC, Brownlee M, Cooper ME
and El-Osta A: Hyperglycemia induces a dynamic cooperativity of
histone methylase and demethylase enzymes associated with
gene-activating epigenetic marks that coexist on the lysine tail.
Diabetes. 58:1229–1236. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
El-Osta A, Brasacchio D, Yao D, Pocai A,
Jones PL, Roeder RG, Cooper ME and Brownlee M: Transient high
glucose causes persistent epigenetic changes and altered gene
expression during subsequent normoglycemia. J Exp Med.
205:2409–2417. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen J, Zhang J, Yang J, Xu L, Hu Q, Xu C,
Yang S and Jiang H: Histone demethylase KDM3a, a novel regulator of
vascular smooth muscle cells, controls vascular neointimal
hyperplasia in diabetic rats. Atherosclerosis. 257:152–163. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yousefipour Z, Newaz MA, Esmaeilli M and
Ranganna K: [PP.36.07] Modification of histone induced by acrolein
in rat vascular smooth muscle cells. J Hypertens. 34:e3372016.
View Article : Google Scholar
|
|
32
|
Lehrke M, Kahles F, Makowska A, Tilstam
PV, Diebold S, Marx J, Stöhr R, Hess K, Endorf EB, Bruemmer D, et
al: PDE4 inhibition reduces neointima formation and inhibits VCAM-1
expression and histone methylation in an Epac-dependent manner. J
Mol Cell Cardiol. 81:23–33. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qi H, Jing Z, Xiaolin W, Changwu X,
Xiaorong H, Jian Y, Jing C and Hong J: Histone demethylase JMJD2A
inhibition attenuates neointimal hyperplasia in the carotid
arteries of balloon-injured diabetic rats via transcriptional
silencing: Inflammatory gene expression in vascular smooth muscle
cells. Cell Physiol Biochem. 37:719–734. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Villeneuve LM, Reddy MA, Lanting LL, Wang
M, Meng L and Natarajan R: Epigenetic histone H3 lysine 9
methylation in metabolic memory and inflammatory phenotype of
vascular smooth muscle cells in diabetes. Proc Natl Acad Sci USA.
105:9047–9052. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xia J, Fang M, Wu X, Yang Y, Yu L, Xu H,
Kong H, Tan Q, Wang H, Xie W and Xu Y: A2b adenosine signaling
represses CIITA transcription via an epigenetic mechanism in
vascular smooth muscle cells. Biochim Biophys Acta. 1849:665–676.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tarling EJ, Ryan KJ, Austin R, Kugler SJ,
Salter AM and Langley-Evans SC: Maternal high-fat feeding in
pregnancy programs atherosclerotic lesion size in the ApoE*3 Leiden
mouse. J Dev Orig Health Dis. Feb 2–2016.(Epub ahead of print).
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Alkemade FE, van Vliet P, Henneman P, van
Dijk KW, Hierck BP, van Munsteren JC, Scheerman JA, Goeman JJ,
Havekes LM, Gittenberger-de Groot AC, et al: Prenatal exposure to
apoe deficiency and postnatal hypercholesterolemia are associated
with altered cell-specific lysine methyltransferase and histone
methylation patterns in the vasculature. Am J Pathol. 176:542–548.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bannister AJ and Kouzarides T: Regulation
of chromatin by histone modifications. Cell Res. 21:381–395. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Witt O, Deubzer HE, Milde T and Oehme I:
HDAC family: What are the cancer relevant targets? Cancer Lett.
277:8–21. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tikoo K, Patel G, Kumar S, Karpe PA,
Sanghavi M, Malek V and Srinivasan K: Tissue specific up regulation
of ACE2 in rabbit model of atherosclerosis by atorvastatin: Role of
epigenetic histone modifications. Biochem Pharmacol. 93:343–351.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chaturvedi P, Kalani A, Givvimani S, Kamat
PK, Familtseva A and Tyagi SC: Differential regulation of DNA
methylation versus histone acetylation in cardiomyocytes during
HHcy in vitro and in vivo: An epigenetic mechanism. Physiol
Genomics. 46:245–255. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kumar A and Chauhan S: How much successful
are the medicinal chemists in modulation of SIRT1: A critical
review. Eur J Med Chem. 119:45–69. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sathishkumar C, Prabu P, Balakumar M,
Lenin R, Prabhu D, Anjana RM, Mohan V and Balasubramanyam M:
Augmentation of histone deacetylase 3 (HDAC3) epigenetic signature
at the interface of proinflammation and insulin resistance in
patients with type 2 diabetes. Clin Epigenetics. 8:1252016.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hoeksema MA, Gijbels MJ, Van den Bossche
J, van der Velden S, Sijm A, Neele AE, Seijkens T, Stöger JL,
Meiler S, Boshuizen MC, et al: Targeting macrophage Histone
deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med.
6:1124–1132. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Van den Bossche J, Neele AE, Hoeksema MA,
de Heij F, Boshuizen MC, van der Velden S, de Boer VC, Reedquist KA
and de Winther MPJ: Inhibiting epigenetic enzymes to improve
atherogenic macrophage functions. Biochem Biophys Res Commun.
455:396–402. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Xie X, Song X, Yuan S, Cai H, Chen Y,
Chang X, Liang B and Huang D: Histone acetylation regulates orphan
nuclear receptor NR4A1 expression in hypercholesterolaemia. Clin
Sci (Lond). 129:1151–1161. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Oksala NKJ, Seppälä I, Rahikainen R,
Mäkelä KM, Raitoharju E, Illig T, Klopp N, Kholova I, Laaksonen R,
Karhunen PJ, et al: Synergistic expression of histone deacetylase 9
and matrix metalloproteinase 12 in M4 macrophages in advanced
carotid plaques. Eur J Vasc Endovasc. 53:632–640. 2017. View Article : Google Scholar
|
|
48
|
Smith JD: New role for histone deacetylase
9 in atherosclerosis and inflammation. Arterioscler Thromb Vasc
Biol. 34:1798–1799. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cao Q, Rong S, Repa JJ, Clair RS, Parks JS
and Mishra N: Histone deacetylase 9 represses cholesterol efflux
and generation of alternatively activated macrophages in
atherosclerosis development. Arterioscler Thrombosis Vasc Biol.
34:1871–1879. 2014. View Article : Google Scholar
|
|
50
|
Salah Ud-Din IA, Tikhomirova A and
Roujeinikova A: Structure and Functional diversity of GCN5-related
n-acetyltransferases (GNAT). Int J Mol Sci. 17:E10182016.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chistiakov DA, Orekhov AN and Bobryshev
YV: Treatment of cardiovascular pathology with epigenetically
active agents: Focus on natural and synthetic inhibitors of DNA
methylation and histone deacetylation. Int J Cardiol. 227:66–82.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Vecellio M, Spallotta F, Nanni S, Colussi
C, Cencioni C, Derlet A, Bassetti B, Tilenni M, Carena MC, Farsetti
A, et al: The histone acetylase activator pentadecylidenemalonate
1b rescues proliferation and differentiation in the human cardiac
mesenchymal cells of type 2 diabetic patients. Diabetes.
63:2132–2147. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zeidler R, de Freitas Soares BL, Bader A
and Giri S: Molecular epigenetic targets for liver diseases:
Current challenges and future prospects. Drug Discov Today.
22:1620–1636. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Miranda TB, Cortez CC, Yoo CB, Liang G,
Abe M, Kelly TK, Marquez VE and Jones PA: DZNep is a global histone
methylation inhibitor that reactivates developmental genes not
silenced by DNA methylation. Mol Cancer Ther. 8:1579–1588. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wierda RJ, Goedhart M, van Eggermond MC,
Muggen AF, Miggelbrink XM, Geutskens SB, van Zwet E, Haasnoot GW
and van den Elsen PJ: A role for KMT1c in monocyte to dendritic
cell differentiation. Hum Immunol. 76:431–437. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhao Q, Li S, Li N, Yang X, Ma S, Yang A,
Zhang H, Yang S, Mao C, Xu L, et al: miR-34a targets
HDAC1-regulated H3K9 acetylation on lipid accumulation induced by
homocysteine in foam cells. J Cell Biochem. 118:4617–4627. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Makino J, Ogasawara R, Kamiya T, Hara H,
Mitsugi Y, Yamaguchi E, Itoh A and Adachi T: Royal jelly
constituents increase the expression of extracellular superoxide
dismutase through histone acetylation in monocytic thp-1 cells. J
Nat Prod. 79:1137–1143. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Svennerholm K, Haney M, Biber B, Ulfhammer
E, Saluveer O, Larsson P, Omerovic E, Jern S and Bergh N: Histone
deacetylase inhibition enhances tissue plasminogen activator
release capacity in atherosclerotic man. PLoS One. 10:e1211962015.
View Article : Google Scholar
|
|
59
|
Anderson SJ, Feye KM, Schmidt-McCormack
GR, Malovic E, Mlynarczyk GSA, Izbicki P, Arnold LF, Jefferson MA,
de la Rosa BM, Wehrman RF, et al: Off-target drug effects resulting
in altered gene expression events with epigenetic and
Quasi-Epigenetic origins. Pharmacol Res. 107:229–233. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cedar H and Bergman Y: Linking DNA
methylation and histone modification: Patterns and paradigms. Nat
Rev Gene. 10:295–304. 2009. View Article : Google Scholar
|
|
61
|
Yang F, Huo XS, Yuan SX, Zhang L, Zhou WP,
Wang F and Sun SH: Repression of the long noncoding RNA-LET by
histone deacetylase 3 contributes to hypoxia-mediated metastasis.
Mol Cell. 49:1083–1096. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chen R, Kong P, Zhang F, Shu YN, Nie X,
Dong LH, Lin YL, Xie XL, Zhao LL, Zhang XJ and Han M: EZH2-mediated
α-actin methylation needs lncRNA TUG1 and promotes the cortex
cytoskeleton formation in VSMCs. Gene. 616:52–57. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tee AE, Ling D, Nelson C, Atmadibrata B,
Dinger ME, Xu N, Mizukami T, Liu PY, Liu B, Cheung B, et al: The
histone demethylase JMJD1A induces cell migration and invasion by
up-regulating the expression of the long noncoding RNA MALAT1.
Oncotarget. 5:1793–1804. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Carrer A, Parris JLD, Trefely S, Henry RA,
Montgomery DC, Torres A, Viola JM, Kuo Y, Blair IA, Meier JL, et
al: Impact of a high-fat diet on tissue Acyl-CoA and histone
acetylation levels. J Biol Chem. 292:3312–3322. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Short JD, Tavakoli S, Nguyen HN, Carrera
A, Farnen C, Cox LA and Asmis R: Dyslipidemic diet-induced monocyte
‘priming’ and dysfunction in non-human primates is triggered by
elevated plasma cholesterol and accompanied by altered histone
acetylation. Front Immunol. 8:9582017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Friso S, Carvajal CA, Fardella CE and
Olivieri O: Epigenetics and arterial hypertension: The challenge of
emerging evidence. Transl Res. 165:154–165. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Høgh Kølbæk Kjær AS, Brinkmann CR,
Dinarello CA, Olesen R, Østergaard L, Søgaard OS, Tolstrup M and
Rasmussen TA: The histone deacetylase inhibitor panobinostat lowers
biomarkers of cardiovascular risk and inflammation in HIV patients.
AIDS. 29:1195–1200. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Choi JH, Nam KH, Kim J, Baek MW, Park JE,
Park HY, Kwon HJ, Kwon OS, Kim DY and Oh GT: Trichostatin A
exacerbates atherosclerosis in low density lipoprotein
receptor-deficient mice. Arterioscler Thromb Vasc Biol.
25:2404–2409. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Okamoto H, Fujioka Y, Takahashi A,
Takahashi T, Taniguchi T, Ishikawa Y and Yokoyama M: Trichostatin
A, an inhibitor of histone deacetylase, inhibits smooth muscle cell
proliferation via induction of p21 (WAF1). J Atheroscler Thromb.
13:183–191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Pandey D, Sikka G, Bergman Y, Kim JH, Ryoo
S, Romer L and Berkowitz D: Transcriptional regulation of
endothelial arginase 2 by histone deacetylase 2. Arterioscler
Thromb Vasc Biol. 34:1556–1566. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Liokatis S, Klingberg R, Tan S and
Schwarzer D: Differentially isotope-labeled nucleosomes to study
asymmetric histone modification crosstalk by time-resolved NMR
spectroscopy. Angew Chem Int Ed Engl. 55:8262–8265. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Clark SJ, Lee HJ, Smallwood SA, Kelsey G
and Reik W: Single-cell epigenomics: Powerful new methods for
understanding gene regulation and cell identity. Genome Biol.
17:722016. View Article : Google Scholar : PubMed/NCBI
|