Introduction
Fetal growth restriction (FGR), the second primary
cause of perinatal mortality, is a clinical entity that affects
5–10% of gestations (1). FGR has
multiple heterogeneous causes, including maternal, fetal and
placental factors (2). Effective
treatments for FGR have not been proposed, apart from the
interruption of pregnancy (3).
Consequently, early diagnosis and prevention is of importance for
patients with FGR, which may permit the etiological identification
and adequate monitoring of fetal vitality, minimizing the risks
associated with prematurity and intrauterine hypoxia (1,4).
Therefore, the identification of biological markers for the early
diagnosis and detection of FGR is required, in order to elucidate
the molecular mechanism underlying FGR.
At present, numerous diseases have been attributed
to the differential expression of genes compared with normal
controls [differentially expressed genes (DEGs)] (5). However, genes frequently do not
function individually; rather, they interact with other genes. A
network-based approach is able to extract informative and notable
genes via biological molecular networks, including the
co-expression network (CEN), rather than focusing on individual
genes (6,7). Providing that gene connections are
based on guilt-determination, predictions of their gene functions
may be conducted utilizing guilt-by-association (GBA) method
(8). The GBA is a basic element
for predicting gene function, and typically uses the interactions
between any two genes for the purpose of investigation the role of
novel genes in gene function categories.
Therefore, the present study took Gene Ontology (GO)
annotations and gene expression data as study objectives, and
integrated the network approach with the GBA method, termed the
network-based gene function inference method, for the purpose of
predicting the optimal gene functions for FGR. These gene functions
may be potential biomarkers for the early detection and targeted
treatment of FGR.
Materials and methods
Network-based gene function inference
method
The network-based gene function inference method was
comprised of four steps: i) Identifying DEGs between patients with
FGR and normal controls using Limma based on gene expression data;
ii) constructing the CEN dependent on DEGs using the Spearman
correlation coefficient (SCC); iii) collecting GO data for FGR on
the basis of a known confirmed database and DEGs; and iv)
predicting gene functions using the GBA algorithm, for which the
area under the receiver operating characteristic curve (AUC) was
calculated for each GO term. An AUC of 0.5 represents
classification at chance levels, while an AUC of 1.0 represents a
perfect classification. In the gene function prediction literature,
AUC >0.7 is considered to be optimal (9). Therefore, GO terms with AUC >0.7
were defined as optimal gene functions for patients with FGR in the
present study.
Identifying DEGs
Gene expression data [GSE24129 (2)] for human FGR were downloaded from the
Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo) using the accession number.
GSE24129 was deposited on an Affymetrix Human Gene 1.0 ST Array
(Affymetrix; Thermo Fisher Scientific, Inc., Waltham, MA, USA), and
comprised normotensive pregnancies with or without FGR. The 8
examples with FGR were attributed to the case group, whereas 8
cases without FGR were denoted as normal controls. In order to
control the quality of GSE24129, standard pretreatments were
performed, which included background correction, normalization,
probe matching and summarization (10–12).
Following conversion of pretreated data at the probe level into
gene symbols and removal of duplications, 14,398 genes were
obtained for FGR in total.
Subsequently, DEGs between the FGR samples and
normal controls were identified using the Limma package (13). The lmFit function implemented in
Limma was utilized to perform empirical Bayes statistics and false
discovery rate calibration of the P-values on the data (14,15).
Only genes which met the thresholds of P<0.05 and
|log2Fold Change| >2 were defined as DEGs across FGR
patients and normal controls.
Constructing CEN
Cytoscape is an open source software project for
integrating biomolecular interactions with high-throughput
expression data and other molecular states into a unified
conceptual network (16).
Therefore, the DEGs were inputted into the Cytoscape software to
visualize the CEN. In order to further evaluate the cooperated
strength for each interaction in the CEN, the SCC method was
utilized (17). SCC is a measure
of the correlation between two variables, giving a value between −1
and +1 inclusive. If the SCC analysis returned a positive value,
this indicated a positive linear correlation between two genes;
otherwise, a negative correlation was indicated. For an interaction
between gene i and j, the absolute SCC value was denoted as its
weight value. The SCC was computed as follows:
SCC=1n-1∑k=1n(g(i,k)-ǵ(i)σ(i))·(g(j,k)-ǵ(j)σ(j))
Where n was the number of samples in the gene
expression data; g(i, k) or g(j, k) was
the expression level of gene i or j in the sample
k under a specific condition; and g(i) or g(j)
represented the mean expression level of gene i or
j.
Recruiting GO annotation data
In the present study, the GO annotations were
recruited from the GO Consortium (geneontology.org) (18). There were 19,003 terms and 18,402
genes in total for human beings. Notably, only one category
(biological process) of GO was selected to be the study objective.
In subsequent steps, the GO structure was diffused, and filtered
for GO terms on size ranging from 20 to 1,000 genes after excluding
those inferred from electronic annotation, a range that generally
gives stable performance (8,9). In
addition, to make ensure that the GO terms correlated closely to
FGR, if a GO term had a number of DEGs <20, it was removed.
Therefore, only GO terms including ≥20 DEGs were reserved. A total
of 109 GO terms involved in 115 DEGs remained to be used in the
following analyses.
Predicting gene function
As mentioned above, the GBA method was employed to
predict the important gene function in the progression of FGR.
Taking the GO functional annotations, a multi-functionality score
(MFS) was assigned to each gene i in the CEN (8):
MFS(i)=∑χ∨i∈GOχ1Num¿χ*Numoutχ
Where Numinx was the number of
genes within GO group x, whose weighting had the effect of
giving contribution to a GO group; and Numoutx
was the number of genes outside the GO group x in the CEN,
whose weighting provided a corresponding weight to genes outside
the GO group. Therefore, as the only gene outside a large GO group,
the score of the only gene within a GO group was added to the score
of another gene. Notably, weighting referred to the impact of
measuring connectivity in a group through the number of
contributions of the gene to that GO group. A 3-fold
cross-validation was applied to determine an MFS ranked list score
for genes as to how well they fitted with the known gene set, and
computed the AUC values for assessing the classification
performances between FGR samples and normal controls. To the best
of our knowledge, AUC has been introduced as a better measure for
evaluating the predictive ability of machine learning in support
vector machine (SVM) models compared with assessing the clinical
classification performance (19).
Consequently, the AUC values for GO terms were obtained, and terms
with AUC >0.7 were identified to be optimal gene functions.
Results
DEGs and GO terms
In the present study, a total of 14,398 genes were
obtained from the gene expression data following standard
preprocessing. Based on these genes, 115 DEGs between FGR patients
and normal controls were identified using the Limma package under
the thresholds of P<0.05 and |log2FoldChange| >2.
As presented in Table I, all DEGs
were ranked in ascending order of their P-values and the regulation
directions were labeled; 58 were upregulated and 57 were
downregulated. The most significant 5 DEGs were transmembrane
protein 136 (P=4.09×10−5; downregulated), acid
phosphatase, prostate (P=5.42×10−5; downregulated),
protein tyrosine phosphatase, non-receptor type 3
(P=6.05×10−5; downregulated), thrombospondin 1
(P=7.68×10−5; upregulated) and potassium two pore domain
channel subfamily K member 17 (P=1.32×10−4;
downregulated).
 | Table I.Differentially-expressed genes for
fetal growth restriction. |
Table I.
Differentially-expressed genes for
fetal growth restriction.
| Gene | Direction |
|---|
| TMEM136 | Down |
| ACPP | Down |
| PTPN3 | Down |
| THBS1 | Up |
| KCNK17 | Down |
| TCN2 | Up |
| EDN1 | Up |
| NNAT | Up |
| ZNF429 | Down |
| TMEM168 | Down |
| SLA | Up |
| F5 | Down |
| TNNT3 | Up |
| P3H2 | Down |
|
CATSPERB | Down |
| BTNL9 | Up |
|
NAALADL2 | Down |
| GPER1 | Up |
| RPS6KA6 | Down |
| APLN | Down |
| PGAP1 | Down |
| CTGF | Up |
| DHCR24 | Down |
| C1GALT1 | Down |
| SOD1 | Down |
| FBN2 | Down |
|
HIST1H1T | Down |
| ADGRA3 | Down |
| SLC41A2 | Down |
| LOX | Up |
| CCDC125 | Down |
| FAM234B | Down |
| SLC20A1 | Down |
| ACSL1 | Up |
| PLAC1 | Down |
| CYR61 | Up |
| GSTA3 | Down |
| LGALS9B | Up |
| GABRA4 | Down |
| DFNA5 | Down |
| QPCT | Up |
| DDX60L | Down |
| MSL3P1 | Down |
| ABCG2 | Down |
| ADGRL3 | Down |
| ALDH7A1 | Down |
| AGL | Down |
| CD68 | Up |
| TFDP2 | Down |
| LEP | Up |
| VWF | Up |
| ERV3-1 | Down |
| CTSV | Down |
| C1QA | Up |
| BHLHE40 | Up |
| ZC2HC1A | Down |
| FAM26D | Down |
| SH3TC2 | Down |
| TIMP1 | Up |
| SLC38A9 | Down |
| LRP2 | Down |
| DSC3 | Down |
| TGFBI | Up |
| LGR5 | Down |
| GALNT11 | Down |
| SEL1L3 | Down |
| OR4F16 | Down |
| OR4F21 | Down |
| LAPTM5 | Up |
| MET | Down |
| DUSP1 | Up |
| NPR3 | Up |
| PLA2G2A | Down |
| CHI3L1 | Up |
| CRH | Up |
| ERAP2 | Down |
| C1QB | Up |
| EXTL2 | Down |
| PSMB9 | Up |
| CXCL9 | Up |
| CLDN1 | Up |
| IFI44L | Up |
| LGALS13 | Down |
|
HLA-DQA1 | Up |
| CXCL10 | Up |
| TAP1 | Up |
| BCL6 | Up |
| GBP5 | Up |
| FPR3 | Up |
|
HLA-DQB1 | Up |
| WNT2 | Down |
| HTRA1 | Up |
|
KRTAP26-1 | Up |
| FSTL3 | Up |
| SLAMF7 | Up |
|
HLA-DQA2 | Up |
| HTRA4 | Up |
| CGB2 | Up |
| SLC27A2 | Down |
| CCL8 | Up |
|
HLA-DPB1 | Up |
| ANKRD22 | Up |
| CGB3 | Up |
| CP | Up |
| CGB1 | Up |
| CGB5 | Up |
| HLA-DMA | Up |
| CGB7 | Up |
| ALAS2 | Up |
| AOC1 | Down |
| FCGR3A | Up |
| HLA-DRA | Up |
| LPL | Up |
| USP9Y | Down |
| LYZ | Up |
In addition, 19,003 GO terms and 18,402 genes
associated with the biological process category of GO were
collected from the GO Consortium. By removing terms with gene sizes
not in the range 20–1,000 and intersected DEGs <20, 109 GO terms
including 115 DEGs were reserved. In order to illustrate the
details of the GO annotations more clearly, a DEG enriched in one
term was assigned a value of 1; otherwise, the value for the DEG in
the GO term=0. The results are presented in Fig. 1, in which the yellow squares refer
to 0 and red squares refer to 1.
CEN
For the purpose of further investigating the
biological activities of DEGs, a CEN with 115 nodes and 6,555
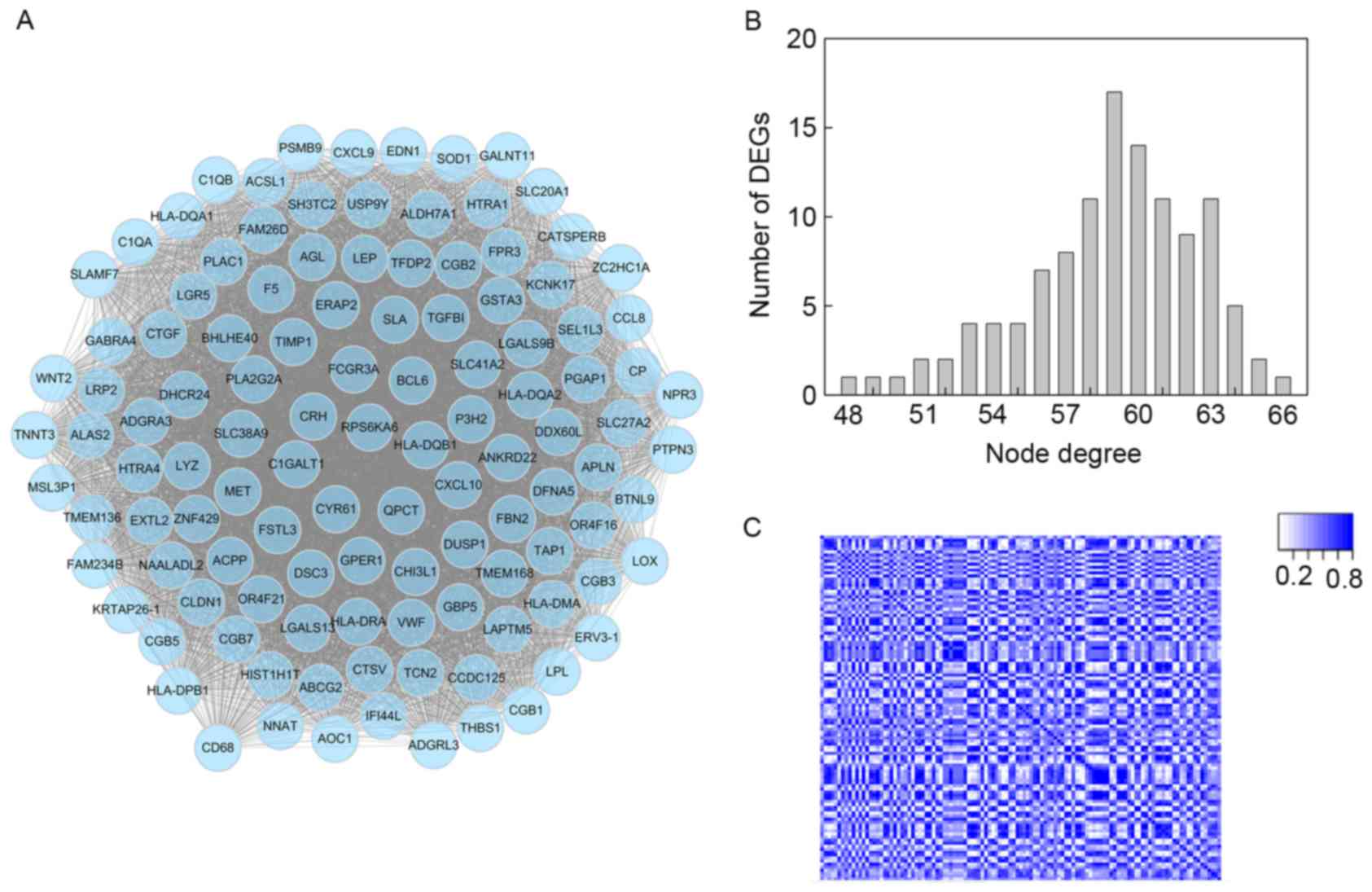
interactions for FGR was visualized using Cytoscape (Fig. 2A), which indicated that all DEGs
were mapped to the CEN. In particular, the topological degree for
each node was calculated by the sum of the nodes to which it was
connected directly, and the degree distribution is presented in
Fig. 2B. It was observed that the
degrees for a large number of DEGs (~55%) ranged between 56 and 60,
and the trend was approximately normally-distributed. Specifically,
ankyrin repeat domain 22 possessed the highest degree of 65. Apart
from the number of connections for each node, the interaction
strength is a parameter that has been used to evaluate interactions
in the CEN. Consequently, a weight was attributed to each edge
using SCC analysis (data not shown). The heatmap for weights in the
CEN is presented in Fig. 2C. In
the figure, squares represent edges in the CEN. Darker squares
indicate larger weight values. Notably, a clear linear correlation
was revealed among interactions, suggesting that the CEN exhibited
good network scale properties.
Optimal gene functions
Prediction of gene function was performed using the
GBA method, based on the integration between GO terms and the CEN.
For each gene in a GO term, the MFS was computed. A high MFS
indicated the possibility of a more optimal gene function.
Therefore, all genes were ranked in descending order of the MFS and
3-fold cross-validation was performed to calculate the AUC for the
GO terms, with the aim of classifying patients with FGR and normal
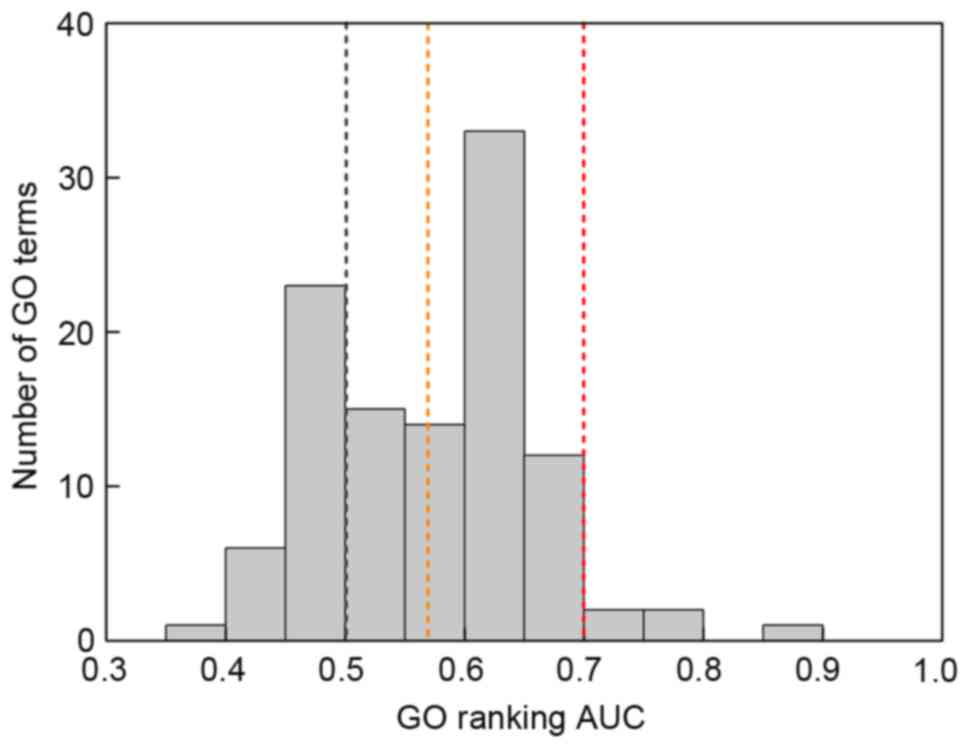
controls. The AUC distribution among GO terms is illustrated in
Fig. 3. The AUC for the majority
of GO terms fell within the range 0.4–0.7, particularly 0.6–0.65.
When AUC was used as a predictor of GO category membership, 78 GO
terms of AUC >0.5 were obtained. It was noted that this single
ranking of genes gave a mean AUC of 0.57 across all GO terms
tested. In addition, 5 of the 78 GO terms had an AUC >0.7 and
were denoted as optimal gene functions (Table II): Defense response (GO:0006952;
AUC=0.861), immune system process (GO:0002376; AUC=0.789), response
to stress (GO:0006950; AUC=0.759), cellular response to chemical
stimulus (GO:0070887; AUC=0.724) and positive regulation of
biological process (GO:0048518; AUC=0.720).
| Table II.GO terms with AUC >0.5. |
Table II.
GO terms with AUC >0.5.
| Ranking | ID | GO term | AUC |
|---|
| 1 | GO:0006952 | Defense
response | 0.861 |
| 2 | GO:0002376 | Immune system
process | 0.789 |
| 3 | GO:0006950 | Response to
stress | 0.759 |
| 4 | GO:0070887 | Cellular response
to chemical stimulus | 0.724 |
| 5 | GO:0048518 | Positive regulation
of biological process | 0.720 |
| 6 | GO:0044459 | Plasma membrane
part | 0.688 |
| 7 | GO:0005615 | Extracellular
space | 0.687 |
| 8 | GO:0010033 | Response to organic
substance | 0.685 |
| 9 | GO:0048522 | Positive regulation
of cellular process | 0.681 |
| 10 | GO:0048583 | Regulation of
response to stimulus | 0.681 |
| 11 | GO:0050896 | Response to
stimulus | 0.679 |
| 12 | GO:0048584 | Positive regulation
of response to stimulus | 0.678 |
| 13 | GO:0044237 | Cellular metabolic
process | 0.676 |
| 14 | GO:0065009 | Regulation of
molecular function | 0.675 |
| 15 | GO:0007166 | Cell surface
receptor linked signal transduction | 0.671 |
| 16 | GO:0044249 | Cellular
biosynthetic process | 0.666 |
| 17 | GO:0042221 | Response to
chemical | 0.652 |
| 18 | GO:0050794 | Regulation of
cellular process | 0.647 |
| 19 | GO:0005488 | Binding | 0.646 |
| 20 | GO:0032991 | Macromolecular
complex | 0.644 |
| 21 | GO:0043169 | Cation binding | 0.643 |
| 22 | GO:0048523 | Negative regulation
of cellular process | 0.640 |
| 23 | GO:1901576 | Organic substance
biosynthetic process | 0.638 |
| 24 | GO:0080090 | Regulation of
primary metabolic process | 0.633 |
| 25 | GO:0044421 | Extracellular
region part | 0.633 |
| 26 | GO:0031988 | Membrane-bounded
vesicle | 0.633 |
| 27 | GO:0031224 | Intrinsic component
of membrane | 0.632 |
| 28 | GO:0065007 | Biological
regulation | 0.632 |
| 29 | GO:0044700 | Single organism
signaling | 0.630 |
| 30 | GO:0065010 | Extracellular
membrane-bounded organelle | 0.630 |
| 31 | GO:0048519 | Negative regulation
of biological process | 0.627 |
| 32 | GO:0051716 | Cellular response
to stimulus | 0.627 |
| 33 | GO:0005886 | Plasma
membrane | 0.626 |
| 34 | GO:0005576 | Extracellular
region | 0.625 |
| 35 | GO:0003824 | Catalytic
activity | 0.623 |
| 36 | GO:0048869 | Cellular
developmental process | 0.623 |
| 37 | GO:0005783 | Endoplasmic
reticulum | 0.622 |
| 38 | GO:0031982 | Vesicle | 0.622 |
| 39 | GO:0044260 | Cellular
macromolecule metabolic process | 0.621 |
| 40 | GO:0007154 | Cell
communication | 0.619 |
| 41 | GO:1903561 | Extracellular
vesicle | 0.614 |
| 42 | GO:0023051 | Regulation of
signaling | 0.610 |
| 43 | GO:0031323 | Regulation of
cellular metabolic process | 0.608 |
| 44 | GO:0060255 | Regulation of
macromolecule metabolic process | 0.607 |
| 45 | GO:0023052 | Signaling | 0.606 |
| 46 | GO:0009058 | Biosynthetic
process | 0.605 |
| 47 | GO:0043234 | Protein
complex | 0.603 |
| 48 | GO:0044425 | Membrane part | 0.603 |
| 49 | GO:0071840 | Cellular component
organization or biogenesis | 0.601 |
| 50 | GO:0043230 | Extracellular
organelle | 0.600 |
| 51 | GO:0043226 | Organelle | 0.599 |
| 52 | GO:0051171 | Regulation of
nitrogen compound metabolic process | 0.592 |
| 53 | GO:0043227 | Membrane-bound
organelle | 0.592 |
| 54 | GO:0005515 | Protein
binding | 0.590 |
| 55 | GO:0008152 | Metabolic
process | 0.589 |
| 56 | GO:0044765 | Single-organism
transport | 0.576 |
| 57 | GO:0043167 | Ion binding | 0.573 |
| 58 | GO:0065008 | Regulation of
biological quality | 0.573 |
| 59 | GO:0043229 | Intracellular
organelle | 0.567 |
| 60 | GO:0016021 | Integral component
of membrane | 0.558 |
| 61 | GO:0006810 | Transport | 0.558 |
| 62 | GO:0051179 | Localization | 0.555 |
| 63 | GO:0050789 | Regulation of
biological process | 0.550 |
| 64 | GO:0009966 | Regulation of
signal transduction | 0.548 |
| 65 | GO:0008150 | Biological
process | 0.546 |
| 66 | GO:0005623 | Cell | 0.544 |
| 67 | GO:0071944 | Cell periphery | 0.536 |
| 68 | GO:0019222 | Regulation of
metabolic process | 0.535 |
| 69 | GO:0043170 | Macromolecule
metabolic process | 0.534 |
| 70 | GO:0051234 | Establishment of
localization | 0.532 |
| 71 | GO:0007165 | Signal
transduction | 0.527 |
| 72 | GO:1901360 | Organic cyclic
compound metabolic process | 0.514 |
| 73 | GO:0031090 | Organelle
membrane | 0.512 |
| 74 | GO:0016043 | Cellular component
organization | 0.511 |
| 75 | GO:0044710 | Single-organism
metabolic process | 0.511 |
| 76 | GO:0019538 | Protein metabolic
process | 0.510 |
| 77 | GO:0034641 | Cellular nitrogen
compound metabolic process | 0.509 |
| 78 | GO:0010646 | Regulation of cell
communication | 0.507 |
Discussion
Co-expression analysis dependent on networks has
been used widely due to its good statistical confidence for
individual connections, overlap with protein interactions, and
mathematical convenience (20). In
addition, the criterion in a CEN is generally divided into two
types: Hard thresholding, which produces less robust results
(21) and soft thresholding.
Specifically, soft thresholding works well in network analysis
(22) by combining greater
sparsity with similarity to the original correlation matrix
(23), for example in a weighted
CEN. Pearson's correlation coefficient (PCC) is the most widely
used measure for co-expression analysis. SCC is a nonparametric
(distribution-free) rank statistical measure of a monotone
association that is used when the distribution of data makes PCC
undesirable or misleading (24).
Therefore, in the present study, SCC was implemented to weight the
CEN which was constructed dependent on DEGs for FGR, and its weight
distribution suggested that the CEN had good scale network
properties. There were 115 nodes and 6,555 interactions in the CEN,
which was prepared for subsequent analysis.
Previously, various methods have been produced to
expand the scale of the GBA to indirect connections, including
weighting indirect connections by local topology, network
propagation and topological overlap (23,25,26).
The majority of these methods refer to improvement over GBA between
direct connections, although they tend to perform comparably and
only slightly better than direct GBA (27). The present study integrated the GBA
method with CEN-associated analysis to further explore direct and
indirect optimal gene functions for FGR, based on GO annotations
and gene expression data. A network based-GBA or extended-GBA
approach may facilitate the exhaustive examination of issues (due
to being less subject to fine-tuning) compared with simple GBA. In
the present study, an MFS was assigned to each gene enriched in the
GO term. Ranking genes by AUC based on MFS was demonstrated to be a
means of obtaining good performance from a gene function prediction
algorithm, which validated the feasibility and confidence of the
network-based GBA method. The results of the present study
demonstrated that 78 GO terms had a good classification performance
with an AUC >0.5; 5 of the GO terms had an AUC >0.7 and were
defined as optimal gene functions, which included defense response,
immune system process, response to stress, cellular response to
chemical stimulus and positive regulation of biological
process.
Specifically, defense response refers to reactions
triggered in response to the presence of a foreign body or the
occurrence of an injury, and results in restriction of damage to
the organism attacked or prevention/recovery from the infection
caused by the attack (28).
Therefore, it is reasonable to infer that alterations of defense
response caused by certain unexplained and unknown reasons in
pregnancy may lead to the occurrence of FGR. In addition, immune
system process includes any process involved in the development or
functioning of the immune system, and is an organismal system which
produces calibrated responses to potential internal or invasive
threats (29). The immune system
is a host defense system comprising a number of biological
structures and processes within an organism that protect against
disease, and disorders of the immune system may lead to autoimmune
diseases, inflammatory diseases and cancer (30). It had been demonstrated that
cytokines drive the innate immune response, and they are logical
candidates for the disruption of fetal brain development (31). Therefore, immune system process was
observed to be correlated to the progression of FGR. Regarding
cellular response to chemical stimulus, this gene function
comprises any process that results in a change in state or activity
of a cell (e.g. movement, secretion, enzyme production or gene
expression) as a result of a chemical stimulus (32). Therefore, pregnant women are
recommended to be alert to the possibility of chemical stimuli
within their food and water intake.
In conclusion, the present study identified 5
optimal gene functions in the process of FGR. The present findings
may provide insights into the pathological mechanism underlying
FGR, and provide potential biomarkers for the early detection and
targeted treatment of this disease. However, the potential
interactions between the 5 GO terms remain to be elucidated in
future studies.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KY made substantial contributions to the design of
the present study and drafted the paper. JD conducted literature
searching for the paper. LL and MP conducted data analysis and
manuscript revised the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nardozza LM, Araujo Júnior E, Barbosa MM,
Caetano AC, Lee DJ and Moron AF: Fetal growth restriction: Current
knowledge to the general Obs/Gyn. Arch Gynecol Obstet. 286:1–13.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nishizawa H, Ota S, Suzuki M, Kato T,
Sekiya T, Kurahashi H and Udagawa Y: Comparative gene expression
profiling of placentas from patients with severe pre-eclampsia and
unexplained fetal growth restriction. Reprod Biol Endocrinol.
9:1072011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Veiby G, Daltveit AK, Engelsen BA and
Gilhus NE: Fetal growth restriction and birth defects with newer
and older antiepileptic drugs during pregnancy. J Neurol.
261:579–588. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Figueras F and Gratacós E: Update on the
diagnosis and classification of fetal growth restriction and
proposal of a stage-based management protocol. Fetal Diagn Ther.
36:86–98. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao J, Yang TH, Huang Y and Holme P:
Ranking candidate disease genes from gene expression and protein
interaction: A Katz-centrality based approach. PLoS One.
6:e243062011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu ZP, Wang Y, Zhang XS and Chen L:
Network-based analysis of complex diseases. IET Syst Biol. 6:22–23.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen L, Wang RS and Zhang XS:
Reconstruction of Gene Regulatory NetworksBiomolecular Networks.
John Wiley & Sons, Inc; pp. 47–87. 2009, View Article : Google Scholar
|
|
8
|
Gillis J and Pavlidis P: The impact of
multifunctional genes on ‘guilt by association’ analysis. PLoS One.
6:e172582011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gillis J and Pavlidis P: The role of
indirect connections in gene networks in predicting function.
Bioinformatics. 27:1860–1866. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Irizarry RA, Bolstad BM, Collin F, Cope
LM, Hobbs B and Speed TP: Summaries of Affymetrix GeneChip probe
level data. Nucleic Acids Res. 31:e152003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bolstad BM, Irizarry RA, Astrand M and
Speed TP: A comparison of normalization methods for high density
oligonucleotide array data based on variance and bias.
Bioinformatics. 19:185–193. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bolstad B: affy: Built-in processing
methods. Journal. 2013.
|
|
13
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: Limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Datta S, Satten GA, Benos DJ, Xia J,
Heslin MJ and Datta S: An empirical bayes adjustment to increase
the sensitivity of detecting differentially expressed genes in
microarray experiments. Bioinformatics. 20:235–242. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Reiner A, Yekutieli D and Benjamini Y:
Identifying differentially expressed genes using false discovery
rate controlling procedures. Bioinformatics. 19:368–375. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Szmidt E and Kacprzyk J: The Spearman rank
correlation coefficient between intuitionistic fuzzy sets. Journal.
276–280. 2010.
|
|
18
|
Gene Ontology Consortium: Gene ontology
consortium: Going forward. Nucleic Acids Res. 43:(Database Issue).
D1049–D1056. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang J and Ling CX: Using AUC and
accuracy in evaluating learning algorithms. IEEE Transact Know Data
Eng. 17:299–310. 2005. View Article : Google Scholar
|
|
20
|
Ma S, Shah S, Bohnert HJ, Snyder M and
Dinesh-kumar SP: Incorporating motif analysis into gene
co-expression networks reveals novel modular expression pattern and
new signaling pathways. PLoS Genet. 9:e10038402013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang B and Horvath S: A general framework
for weighted gene co-expression network analysis. Stat Appl Genet
Mol Biol. 4:Article17. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Horvath S and Dong J: Geometric
interpretation of gene coexpression network analysis. PLoS Comput
Biol. 4:e10001172008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yip AM and Horvath S: Gene network
interconnectedness and the generalized topological overlap measure.
BMC Bioinformatics. 8:222007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hauke J and Kossowski T: Comparison of
values of pearson's and spearman's correlation coefficients on the
same sets of data. Quaestiones Geographicae. 30:87–93. 2011.
View Article : Google Scholar
|
|
25
|
Chua HN, Sung WK and Wong L: Exploiting
indirect neighbours and topological weight to predict protein
function from protein-protein interactions. Bioinformatics.
22:1623–1630. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Weston AD and Hood L: Systems biology,
proteomics, and the future of health care: Toward predictive,
preventative, and personalized medicine. J Proteome Res. 3:179–196.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Peña-Castillo L, Tasan M, Myers CL, Lee H,
Joshi T, Zhang C, Guan Y, Leone M, Pagnani A, Kim WK, et al: A
critical assessment of Mus musculus gene function prediction using
integrated genomic evidence. Genome Biol. 9 Suppl 1:S22008.
View Article : Google Scholar
|
|
28
|
Grice EA, Snitkin ES, Yockey LJ and
Bermudez DM: NISC Comparative Sequencing Program, Liechty KW and
Segre JA: Longitudinal shift in diabetic wound microbiota
correlates with prolonged skin defense response. Proc Natl Acad Sci
USA. 107:14799–14804. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Osborn O and Olefsky JM: The cellular and
signaling networks linking the immune system and metabolism in
disease. Nat Med. 18:363–374. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Finn OJ: Immuno-oncology: Understanding
the function and dysfunction of the immune system in cancer. Ann
Oncol. 23 Suppl 8:viii6–viii9. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Smith SE, Li J, Garbett K, Mirnics K and
Patterson PH: Maternal immune activation alters fetal brain
development through interleukin-6. J Neurosci. 27:10695–10702.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fenzl SC, Hirsch T and Wolfbeis OS:
Photonic crystals for chemical sensing and biosensing. Angew Chem
Int Ed Engl. 53:3318–3335. 2014. View Article : Google Scholar : PubMed/NCBI
|