Introduction
Human adenoviruses (HAdVs), also known as
nonenveloped double-stranded DNA viruses, are members of the
Adenoviridae family with a size ranging from 70 to 100 nm (1). HAdV infections are now recognized as
a significant source of human morbidity and mortality, and affect
patients worldwide and across all age groups. HAdV infections are
readily transmittable and, in some cases, highly contagious.
Currently, more than 84 genotypes, including all previously
characterized serotypes, have been identified and grouped into
seven different species (A-G) (http://hadvwg.gmu.edu/), based on their immunochemical
responses, nucleic acid characteristics, hexon and fiber protein
characteristics, biological properties, and phylogenetic
relationships (2–6). This subdivision also has some
clinical relevance, as distinct adenovirus species show a
preference for specific organs: C, E, and some B species typically
infect the respiratory tract; other B species infect the urinary
tract; species A and F target the gastrointestinal tract; and
species D target the eyes (7,8).
Among the HAdV-associated respiratory diseases, viruses in species
HAdV B (HAdV-3, −7, −11, −14, −16, −21, −34, −35, −50, −55, and
−66), species HAdV C (HAdV-1, −2, −5, and −6), and species HAdV E
(HAdV-4) are recognized as the main pathogens responsible for the
respiratory tract infections (9–12).
Researchers are constantly inventing and optimizing methods to
detect adenovirus so as to avoid the overuse of antibiotics,
improve the level of diagnosis and treatment, and provide a
scientific basis for the prevention and control of HAdV causing
respiratory illness.
Current standard diagnostic approaches rely mainly
on cell culture and immunofluorescence assay (13). Cell culture is considered as gold
standard because of its broad applicability and high specificity.
However, it is time-consuming and usually takes 7–12 days to obtain
a positive culture. Enzyme immunoassays and immunofluorescence give
rapid results, but their relative lack of sensitivity and the
availability of reactive antisera can be limiting factors (14–25).
Advances in conventional reverse transcription-polymerase chain
reaction (PCR) and quantitative PCR (qPCR) assays have greatly
facilitated the etiological study of respiratory infections due to
their higher sensitivity and specificity. These assays can also
reduce labor and cost by detecting more than one pathogen in a
single reaction using multiple probes (26,27).
Single and multiplex conventional and quantitative PCR assays using
species- and type-specific primers/probes have been described for
some HAdVs that cause respiratory tract infection. For example,
Metzgar et al developed a series of single- and multiplex
qPCR assays to detect and discriminate HAdVs implicated in adult
epidemic acute respiratory diseases, including HAdV-B3, E4, B7,
B11, B14, and B21 (28). Lu et
al developed and validated sensitive and type-specific qPCR
assays for detecting and identifying the epidemic-associated
respiratory HAdVs, types B3, E4, B7, B11, B14, B16, and B21, to
facilitate rapid outbreak response (29). Washington et al developed a
multiplexed PCR assay for a similar panel of five HAdVs (HAdV types
B3, E4, B7, B14, and B21) based on Luminex xMAP technology
(30). HAdV types vary in terms of
clinical manifestation and outbreak severity in respiratory
infection. Accurate and rapid identification of HAdV infection in
the respiratory tract not only avoids unnecessary antibiotic
prescription but also prevents or inhibits HAdV-related outbreaks.
Although many multiplex PCR assays have been developed and applied
for detecting HAdV in respiratory tract infections, the assay
covering all the serotypes (C1, C2, B3, E4, C5, C6, B7, B11, B14,
B16, B21, B34, B35, B50, B55, and B66) of HAdV B, C and E has not
been reported.
In this study, a multiplex qPCR assay was developed
to detect HAdV B, C and E by employing four primer pairs and four
virus-specific probes (including internal standard quality
control). The study demonstrated that the aforementioned assay was
a promising method to potentially reduce missed diagnosis or
misdiagnosis in detecting HAdV, which could cause respiratory tract
infections.
Materials and methods
Clinical samples collection
This study was approved by the ethics committee of
Shenzhen Shajing Hospital affiliated to Guangzhou Medical
University (Shenzhen, China) prior to commencement. After informed
written consent from adult patients or legal representatives of
children, throat swab samples, which are determined to be HAdV
negative by D3 Ultra DFA Respiratory Virus Screening & ID kit
(B&C Biological Technology Co., Shanghai, China), were
collected using universal transport media (BD Biosciences, Franklin
Lakes, NJ, USA). From February 2016 to June 2017, 3,160 cases of
adenovirus-negative throat swab samples were collected from 3
districts (Baoan, Futian, Luohu) of Shenzhen.
Nucleic acid extraction
For nucleic acid extraction, all throat swab samples
were centrifuged at 3,000 × g for 5 min at 4°C to collect virus
attached to cells. Nucleic acid was extracted from 200 µl of each
sample using the Nucleic Acid Extraction kit according to the
manufacturer's protocol (Huiyan Biotechnology Company, Shenzhen,
China). Then, 200 µl of the sample was extracted, and nucleic acid
was eluted into a volume of 150 µl. Nucleic acid was then processed
immediately for qPCR.
Primers and TaqMan probe design
Potential primer and probe sequences were selected
after being thoroughly analyzed using the Premier Biosoft software
and the BLAST (National Centre of Biotechnology Information,
National Institutes of Health, Bethesda, MD, USA) program
(http://blast.ncbi.nlm.nih.gov/Blast.cgi). BLAST
analyses were performed to check the specificity of the primers and
the probes against other closely related genome sequences. The
Premier Biosoft software allowed the combination of existing proven
primer and probe sequences with sequence targets to yield
multiplexed assay designs and choose identical cycling parameters.
The primer pairs and probes specific to each virus were grouped in
a multiplex reaction on the basis of the following criteria:
internal primer binding properties for hairpin and primer-dimer
potential, length of the desired amplicon, G-C content, and melting
temperatures (Tm) of the probes and primers. When the parameter
settings were defined, Premier Biosoft software assessed each
multiplex component as it was added to the assay pool, avoiding the
risk of cross-reactivity issues. The primers and TaqMan probes were
purchased from Invitrogen (Thermo Fisher Scientific, Inc.,
Shanghai, China). TaqMan hydrolysis probes were labeled at the
5′-end with the reporter dyes and quenched with Blackhole Quencher
1or 2 (BHQ 1 or 2) at the 3′-end. For more convenience, the method
was somewhat modified and each TaqMan probe was labeled with a
different fluorescent reporter dye (FAM, VIC, ROX, or CY5)
(Table I).
 | Table I.Primers and probes used in this
study. |
Table I.
Primers and probes used in this
study.
| Virus
serotypes | Oligo | Primer/probe
sequence (5′-3′) | 5′ label | 3′ label | Genomic region | Amplicon size
(bp) |
|---|
| HAdV-B | Forward |
CACATGGGAGCCAGGAGT | NO | NO | 1285–1303 | 118 |
|
| Reverse |
RAACATGGCCAGATCGCAC | NO | NO | 1383–1402 |
|
|
| Probe |
TCTGTCCGAGGTCCTGACGGTT | 6-FAM | BHQ1 | 1322–1344 |
|
| HAdV-C | Forward |
YAACCCCTTYTCKGGACCTC | NO | NO | 316–337 | 118 |
|
| Reverse |
CAGTTGCTCTGCCTCTCCA | NO | NO | 375–394 |
|
|
| Probe |
CATTCAGTCGTAGCCGTCCGC | VIC | BHQ1 | 349–370 |
|
| HAdV-E | Forward |
CCTGCATGAAAGTCTTTGTTGTC | NO | NO | 637–660 | 114 |
|
| Reverse |
GTGAAGGTCAGAGACTGGTTG | NO | NO | 730–751 |
|
|
| Probe |
CTGAGATCAGCGACTACTCCGGAC | ROX | BHQ2 | 685–709 |
|
| HAdV-Ref | Forward |
GCTAGTCTCAAGAGTCTGGAAG | NO | NO | NO | 91 |
|
| Reverse |
ACGCCTTCGTCTTGGTGTC | NO | NO | NO |
|
|
| Probe |
CCGTCAGCATCCTCGCATCAAGCA | CY5 | BHQ2 | NO |
|
Quality control/Internal standard
plasmid construction
A 1,000-bp random DNA sequence was generated using
the DNAMAN software (Lynnon Biosoft, Vaudreuil, QC, Canada), and
four bases (A, T, C, and G) accounted for 25%, respectively. The
generated sequence was then synthesized by Takara and introduced
into plasmid pBSK.
Standard curve
Three inactivated HAdV isolates were provided as
gifts by Professor Rong Zhou (State Key Laboratory of Respiratory
Diseases, Guangzhou Medical University, Guangdong, China). These
clinical isolates were sent to Hexin Health Technology Company
(Guangdong, China) and identified using DHI, immunofluorescence,
flow cytometry, and qPCR. The fragments amplified with HAdV primers
were sequenced and compared with HAdV sequences in NCBI (National
Centre of Biotechnology Information, National Institutes of
Health). After comparison, the three clinical isolates were
confirmed as HAdV B3, C2, and E4. DNA of three isolates was
extracted from 200 µl samples as described earlier.
The concentration of three isolates was detected
using the QX100 drop digital PCR system (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). The droplet digital PCR (ddPCR) reaction
mixture consisted of 10 µl of a 2 × ddPCR Master Mix (Bio-Rad
Laboratories, Inc.), 2 µl of HAdV primers/probes, and 5 µl of
sample nucleic acid solution in a final volume of 20 µl. The entire
reaction mixture was loaded into a disposable plastic cartridge
(Bio-Rad Laboratories, Inc.) together with 70 µl of droplet
generation oil (Bio-Rad Laboratories, Inc.) and placed in the
droplet generator (Bio-Rad Laboratories, Inc.). After processing,
the droplets generated from each sample were transferred to a
96-well PCR plate (Eppendorf, Hamburg, Germany). PCR amplification
was carried out on a T100 thermal cycler (Bio-Rad Laboratories,
Inc.) using a thermal profile beginning at 95°C for 2 min, followed
by 40 cycles of 95°C for 10 sec and 60°C for 30 sec, 1 cycle of
95°C for 10 min, and ending at 12°C. After amplification, the plate
was loaded on the droplet reader (Bio-Rad Laboratories, Inc.) and
the droplets from each well of the plate were read automatically at
a rate of 32 wells per hour. ddPCR data were analyzed with Quanta
Soft analysis software (Bio-Rad Laboratories, Inc.), and the
quantification of the target molecule was presented as the number
of copies per ul of PCR mixture.
Then, three HAdV isolates B3, C2, and E4 were
equally mixed, and standard curves for adenovirus were drawn by
performing qPCR on tenfold serial dilution in DNase- and RNase-free
H2O of the corresponding standard virus templates
ranging from 108 to 102 copies (equivalent
genomes)/ml. The standard curves for multiplex qPCR were obtained
by linear regression analysis of the threshold cycle
(CT) value (y axis) vs. the log of the initial copy
number present in each sample dilution (x axis), allowing the
determination of the correlation coefficient (R2). The
amplification efficiencies (E) of reactions were calculated from
the curves using the equation: E=(10(−1/slope)-1) × 100%
(31,32). The resulting curves were used to
determine the number of viral genome copy corresponding to each
virus found in the experimental samples.
Multiplex qPCR
For the multiplex assay, the reaction mixture was
prepared, containing 25 µl of iQ Multiplex Powermix (Bio-Rad
Laboratories, Inc.), 0.5 µl of each primer (final concentrations,
200 nM), and 0.375 µl of each probe (final concentrations, 75 nM).
The final volume of the mixture was adjusted to 30 µl with DNase-
and RNase-free H2O (Nacalai Tesque, Inc., Kyoto, Japan).
Finally, 20 µl of sample DNA was added to the reaction mixture on
ice. qPCR was performed using a Roche Light Cycler 480 (LC480)
(Roche Diagnostics, Basel, Switzerland) with the following
amplification protocol: Pre-denaturation for 2 min at 95°C; 40
cycles of denaturation for 10 sec at 95°C, and annealing and primer
extension for 30 sec at 60°C. The fluorescence emitted by FAM, VIC,
ROX, or CY5 dyes was measured simultaneously and independently at
the end of the annealing step. For a background control, no
template control (NTC, water) was used.
The results were expressed as the quantification
cycle (Cq, cycle at which the fluorescence surpasses the
background level). Lower Cq values corresponded to a
greater amount of initial template and a negative result was
considered to have a Cq value of 40 or more cycles. So
the cutoff point was fixed empirically at a Cq value of
40. The status of the sample (positive or negative) was determined
first using this chosen cutoff to assess the variability of the
observed Cq values. However, the use of such arbitrary
cutoffs was not ideal because they might be either too low
(eliminating valid results) or too high (increasing false-positive
results) (33). Hence, those PCR
products, which were initially identified as adenovirus-positive,
were sent to the Invitrogen Biotechnology Company (Invitrogen,
Shanghai, China) for conventional Sanger sequencing. The nucleotide
sequences of HAdV B, C, and E were screened using the BLAST program
on the NCBI website to determine the sequence origin.
For a good analysis of the presented results, the
performances of the multiplex assays were also assessed using the
confusion matrix that contained information about the two assays.
The following parameters were defined: True positive (TP): The
number of ‘positive’ samples categorized as ‘positive’; false
positive (FP): The number of ‘negative’ samples categorized as
‘positive’; false negative (FN): The number of ‘positive’ samples
categorized as ‘negative’; true negative (TN): The number of
‘negative’ samples categorized as ‘negative’. Based on these four
metrics, the sensitivity [Sn=TP/(TP + FN)] and the specificity
[Sp=TN/(TN + FP)] of the multiplex assay could be evaluated
(33).
Results
Design of primers and probes
In the study of adenovirus infection in the
respiratory tract, most of the literature reported primers and
probes based on the hexon and fiber genes (Table I). HAdV serotypes that could be
detected by designing primers for the hexon gene included HAdV-C1,
C2, B3, E4, C5, C6, B7, B11, B14, B16, B21, B34, and B35. HAdV
serotypes that could be detected by designing primers for the fiber
gene included HAdV-C2, B3, E4, C5, B7, B11, B16, B21, B34, and B35.
As more new adenovirus serotypes (HAdV-B50, B55, and B60) cause
respiratory infections, primers for multiplex qPCR should be able
to detect adenovirus serotypes as comprehensively as possible.
Primers and probes were designed to detect 16 types so as to
achieve the coverage of the main HAdV types that cause the
respiratory tract infections: 3, 7, 11, 14, 16, 21, 34, 35, 50, 55,
and 66 of HAdV species B; 1, 2, 5, and 6 of HAdV species C; 4 of
HAdV species E. HAdVs associated with respiratory tract infections
were subjected to a conservative analysis. Not only hexon and fiber
genes were found to be highly conserved, but E3 genes were also
highly conserved. Unique sets of PCR primers and TaqMan probes were
designed to target highly conserved sequences in each viral hexon,
fiber, and E3 genes. After thorough analysis using the Premier
Biosoft software and the BLAST program, sets of primers and TaqMan
probes designed for the E3 gene were finally selected for
subsequent experiments.
Standard curve and dynamic range of
the multiplex assay
Serial dilutions of mixed adenovirus standards were
tested using multiplex qPCR, and individual standard curves were
constructed from the Cq values (Fig. 1). The multiplex assay could amplify
each plasmid standard between 102 and 108
copies per reaction mixture with a strong linear relationship
between the Cq values and the log10 of the
input number of copies (HAdV B3, r2=0.99957; HAdV
C2, r2=0.99964; HAdV E4,
r2=0.99985). The slopes of the standard curves
were −3.37007 for HAdV B3, −3.37538 for HAdV C2, and −3.43185 for
HAdV E4.
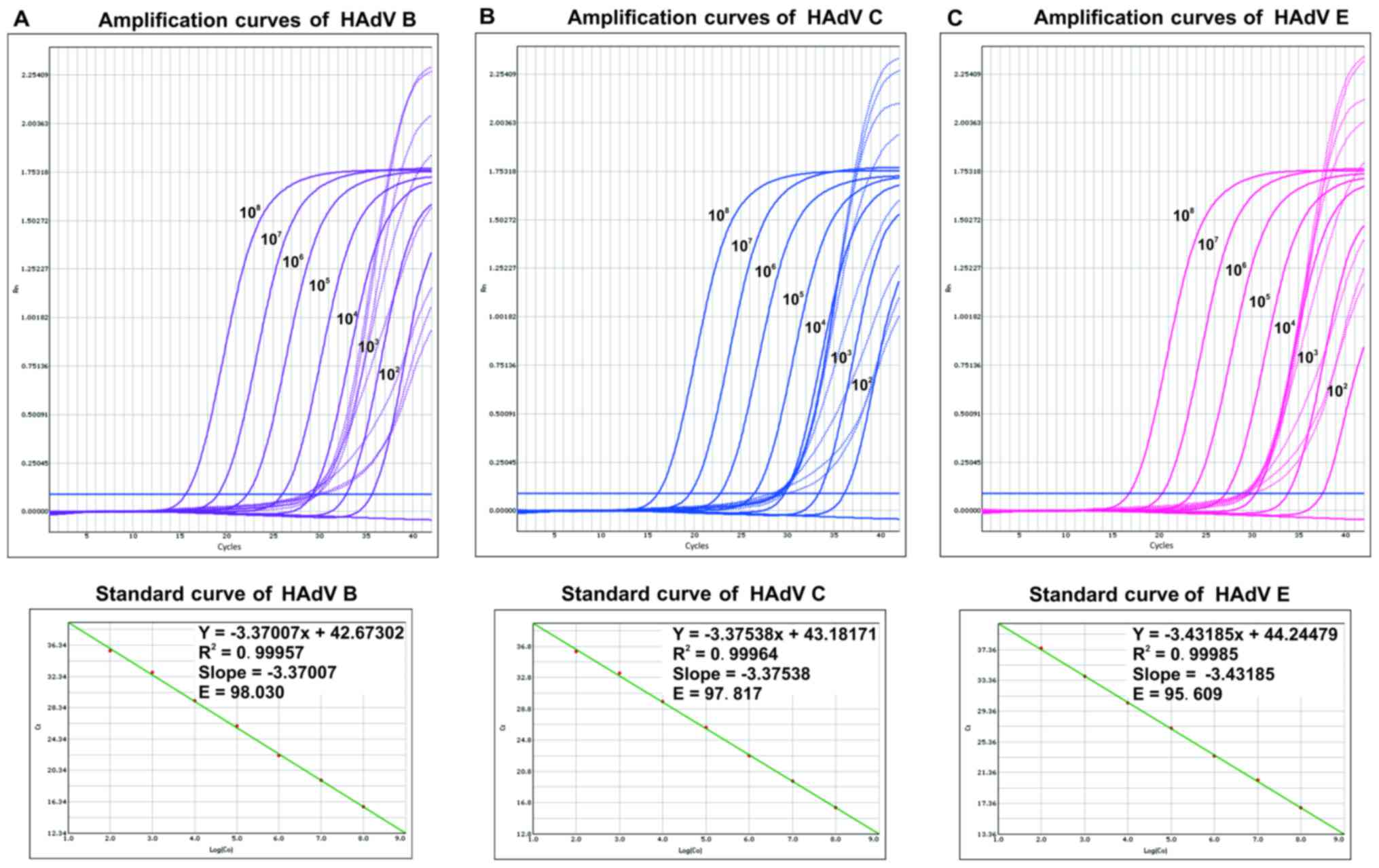 | Figure 1.Amplification plot and standard curve
of adenovirus serotypes (A) B3, (B) C2 and (C) E4 in multiplex qPCR
assay. 10-fold serial dilutions of HAdV serotypes C2, B3, and E4
DNA were used for standard curve construction. DNA concentrations
were (from left to right) 108, 107,
106, 105, 104, 103,
102, and blank. For each dilution, the normalized
fluorescence signal (Rn) was plotted against the PCR cycle number.
A representative amplification plot and a standard curve plot are
shown. qPCR, quantitative polymerase chain reaction; HAdV, human
adenovirus. |
The defined acceptance criteria for accurate and
reproducible quantification were as follows: qPCR
R2 ≥0.98; qPCR E among 90 and 110% (corresponding
to the slope of the regression curve that should be between −3.1
and −3.6). For the multiplex assay, the qPCR E ranged from 80 to
120% (corresponding to the slope of the regression curve that
should be between −3.9 and −2.9) (31). The regression analysis of the
multiplex assay demonstrated that the R2 and the
PCR amplification E were all within the suitable range. The
R2 of the standard curves of each multiplex
reaction were all greater than 0.99 along with the E values ranging
from 95 to 98%, which was considered as acceptable for a multiplex
qPCR assay.
Detection of clinical samples
A total of 3,160 throat swab samples that tested
adenovirus negative by the immunofluorescence assay were retested
using multiplex qPCR assay. The sensitivity and specificity of the
multiplex qPCR assay were compared with those of the
immunofluorescence assay. Further, 2,894 samples were detected as
adenovirus negative, which was consistent with the findings of
immunofluorescence assay. However, 266 samples were detected as
adenovirus positive. The samples were sent for conventional Sanger
sequencing to verify whether 266 adenovirus-positive samples were
TP. Through the BLAST online software sequence alignment, 254
samples were verified as TP and 12 samples were verified as FP
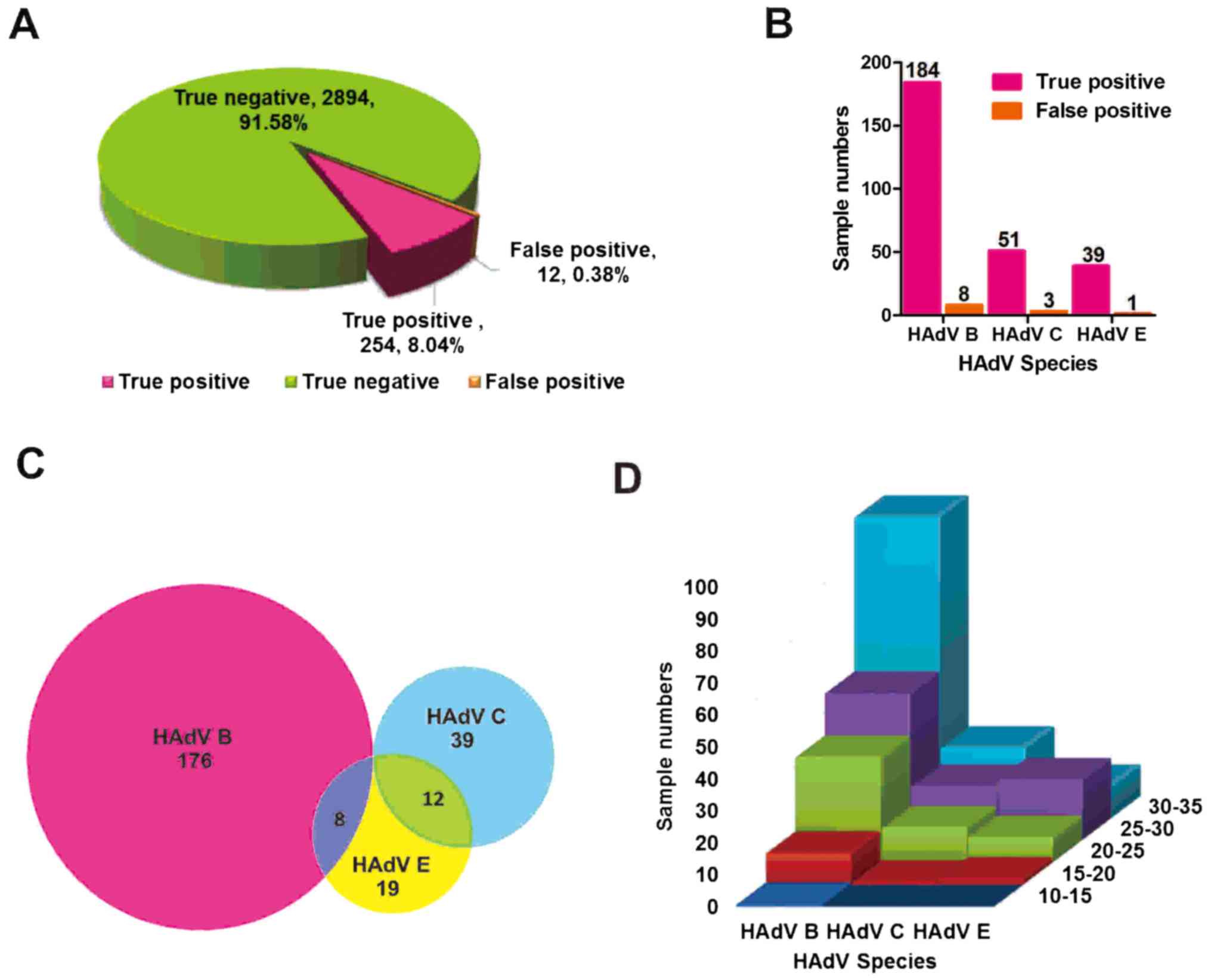
(Fig. 2A). Among the 254 TP
samples, HAdV B was detected in 184 samples, HAdV C was detected in
51 samples, and HAdV E was detected in 39 samples. Among the 12 FP
samples, HAdV B was detected in 8 samples, HAdV C was detected in 3
samples, and HAdV E was detected in 1 sample (Fig. 2B). Co-infections with two HAdVs B
and E were detected in 8 samples and co-infections with HAdVs C and
E in 12 samples. No detection of co-infections of more than two
HAdVs was reported (Fig. 2C).
Briefly, the distribution frequencies of Cq values for a
set of 254 analyzed adenovirus-positive samples is displayed in
Fig. 2D. Further, 66 positive
samples had a Cq value less than 25, 14 a Cq
value less than 20, and one with a Cq value less than 15
in HAdV B species. The aforementioned data indicated that the
sensitivity and the specificity of multiplex qPCR were much better
than those of immunofluorescence assay.
Comparison of the proposed assay with
the reported assays
Multiplex qPCR methods in other studies were used to
detect not only adenovirus but also other respiratory pathogens
(Tables II and III). The multiplex qPCR in this study
was used to detect only HAdV B, C and E. 16 HAdV serotypes
associated with respiratory infections could be detected using
primers and probes in this study. By comparing the number of
detected serotypes, sensitivity and specificity, the more
comprehensive the serotype detection, the higher the sensitivity of
the assay. Therefore, the sensitivity and specificity of this study
were still 100% (254/254) and 99.6% (2894/2906), respectively, in
the large sample size of the respiratory tract.
| Table II.Comparison of the primers of using
multiplex qPCR assay to detect adenovirus serotypes in respiratory
tract infection. |
Table II.
Comparison of the primers of using
multiplex qPCR assay to detect adenovirus serotypes in respiratory
tract infection.
| Adenovirus
species | Serotypes | Target gene | References |
|---|
| B | B1: 3, 7, 16, 21,
50 and 66; B2: 11, 14, 34, 35 and 55 | E3 | This study |
| C | 1, 2, 5 and 6 | E3 |
|
| E | 4 | E3 |
|
| A | 12 | Hexon | Pierce et al
(34), 2012 |
| B | 3, 7, 16, 21 and
34 | Hexon | Popowitch et
al (35), 2013 |
| C | 2, 5 | Hexon | Rand et al
(39), 2011 |
| D | 17, 48 | Hexon | Poritz et al
(40), 2011 |
| F | 40, 41 | Hexon | Heim et al
(46), 2003 |
| E | 4 | Hexon | Loeffelholz et
al (36), 2011 |
| B | 3 | Hexon | Loeffelholz et
al (36), 2011 |
| C | 1,5 | Hexon |
|
| B | 3,7,11, 16, 21, 34
and 35 | Fiber | Xu et al
(37), 2000 |
| C | 2,5 | Fiber |
|
| E | 4 | Fiber |
|
| B | 3,7, 11, 14, 16, 21
and 35 | Hexon E2A
fiber | Metzgar et
al (38), 2005 |
| B | 3, 7 | Hexon | Fujimoto et
al (33), 2004 |
| B | 3,7, 11, 16, 21, 34
and 35 | Hexon | Echavarria et
al (10), 2003 |
| C | 1, 2, 5, 6 | Hexon |
|
| E | 4 | Hexon |
|
| Table III.Different detection methods to detect
the types of pathogens. |
Table III.
Different detection methods to detect
the types of pathogens.
| Assay | The full name | Pathogens
types | References |
|---|
| Multiplex PCR | Multiplex qPCR
assay | HAdV-B3, 7, 11, 14,
16, 21, 34, 35, 50, 55, 66; HAdV-C1, 2, 5, 6; HAdV-E4 | This study |
| IF assay | Immunofluorescence
assay | AdV, influenza
virus A, influenza virus B, PIV1, −2, −3; RSV | This study |
| Film Array RP | The FilmArray
respiratory | AdV; CoV HKU1,
NL63; influenza virus A | Pierce et al
(34), 2012 |
|
| panel multiplexed
nucleic | (H1/2009, H1, H3);
influenza virus B; | Loeffelholz et
al (36), 2011 |
|
| acid amplification
test | MPV; PIV1, −2, −3,
−4; RSV; RhV/EV | Rand et al
(39), 2011 |
| PCR | qPCR | AdV; RSV -A,-B;
influenza virus A | Poritz et al
(40), 2011 |
|
|
| (H1/2009, H1, H3),
influenza virus B; MPV; PIV1, −2, −3; RSV; RhV/EV | Pierce et al
(34), 2012 |
| DFA | Direct fluorescent
antibody | AdV; hMPV; Flu A;
Flu B; PIV1; PIV2; PIV3; RSV | Poritz et al
(40), 2011 |
| xTAG RVP
(xTAG) | The Luminex xTAG
RVP | AdV; influenza
virus A (H1, H3); influenza virus B; MPV; PIV1, −2, −3; RSV (A/B);
RhV/EV | Rand et al
(39), 2011 |
| Prodesse
assays | Prodesse qPCR
assays | Adenovirus; human
metapneumovirus; influenza A virus; influenza B virus;
parainfluenza viruses 1 to 3; respiratory syncytial virus | Loeffelholz et
al (36), 2011 |
Discussion
In this study, a multiplex qPCR assay was developed
that could simultaneously quantify 16 kinds of HAdV serotypes
causing respiratory tract infections. The multiplex qPCR in this
study could not only simultaneously and accurately detect the three
species of HAdV (HAdV B, C and E), but also had a high sensitivity
(100%).
The multiplex qPCR assay to detect throat swab
samples found that 254 patients were adenovirus positive but these
adenovirus-positive patients did not get timely diagnosis and
treatment due to the poor sensitivity of the immunofluorescence
method. Therefore, it is not appropriate to use DFA to detect swab
samples and obtain the epidemiological conclusion of adenovirus
negative. This indicated that the sensitivity of multiplex qPCR was
higher than that of the immunofluorescence assay. Compared with the
traditional cell culture, the whole experiment took only about 1.5
h from the extraction of nucleic acid to qPCR results. This could
not only timely make an accurate diagnosis based on the qPCR result
but also improve work efficiency.
Multiplex PCR is achieved using separate probe and
primer sets for each target sequence, but it requires careful
optimization of assay parameters and primer/probe design. During
qPCR, each amplicon simultaneously amplifies and is independently
detected by different fluorescence spectra. In previous studies,
most of the primers were designed based on the hexon and fiber
genes used to detect adenovirus in the respiratory tract (10,32,34–38).
However, these primers did not fully detect the adenovirus
serotypes that caused respiratory infections. Therefore, in this
study, the highly conserved E3 gene was selected to design primers
and probes. The multiplex qPCR was linear from 102 to
108 copies/ml. The amplification efficiency of HAdV B,
C, and E reached more than 99%. The multiplex qPCR was specific for
three adenovirus species independently, without mutual
cross-reactivity. The results indicated that the E3 gene was
selected to design primers and probes, which not only detected more
adenovirus serotypes but also had good sensitivity and
specificity.
Over the past few years, multiplex qPCR assays have
significantly contributed to the rapid identification of the virus
associated with acute respiratory infections, allowing the quick
adoption of the measures and preventive strategies to avoid the
spread of the disease with high transmissibility (34,35,37,39,40).
The sensitivity of multiplex qPCR is an important consideration in
the development of diagnostic assays; even extremely low adenoviral
levels can cause respiratory infections. Many newly identified
adenovirus serotypes, which are not included in the detection range
of multiplex qPCR, can also cause respiratory infections. Table IV shows that the sensitivity and
specificity of detecting adenovirus in this study were compared
with those in previous multiplex qPCR studies. It was noteworthy
that 100% sensitivity could be achieved for a relatively large
number of samples, compared with 45.8, 54.5, 84.6, and 90% in
previous studies. Multiplex qPCR in this study may be helpful in
reducing missed diagnosis effectively by detecting a wider range of
adenovirus serotypes.
| Table IV.Comparison of the sensitivity and
specificity of using multiplex qPCR assay to detect adenovirus in
respiratory tract infection. |
Table IV.
Comparison of the sensitivity and
specificity of using multiplex qPCR assay to detect adenovirus in
respiratory tract infection.
|
| Sample numbers | Sensitivity | Sensitivity |
|
|---|
|
|
|
|
|
|
|---|
| Sample numbers
detected | (TP)a +/+ | (FP)b +/− | (FN)c −/+ | (TN)d −/− | TP/(TP+FN) (%) | TN/(TN+FP) (%) | References |
|---|
| Multiplex PCR/IF
assay | 254 | 12 | 0 | 2,894 | 254/254 (100) | 2,894/2,906
(99.6) | This study |
| Film Array
RP/PCR | 11 | 0 | 13 |
191 | 11/24
(45.8) | 191/191
(100) | Pierce et al
(34), 2012 |
| Film Array RP/xTAG
RVP | 9 | 0 | 1 |
190 | 9/10
(90) | 190/190
(100) | Rand et al
(39), 2011 |
| Film Array
RP/DFA | 22 | 32 | 4 |
270 | 22/26
(84.6) | 270/302
(89.4) | Poritz et al
(40), 2011 |
| Film Array
RP/Prodesse assays | 6 | 0 | 5 |
181 | 6/11
(54.5) | 181/181
(100) | Loeffelholz et
al (36), 2011 |
In this study, a multiplex qPCR was developed that
could rapidly detect and accurately quantify HAdV B, C, and E in
the respiratory tract with high sensitivity and specificity. All
the serotypes of HAdV B, C, and E in the respiratory tract could be
detected using this assay. The assay simplified the routine service
and decreased the overall costs (41–44).
These benefits had a positive effect on clinical service, allowing
clinicians to tailor patient management or initiate antiviral
therapy more promptly (45).
Acknowledgements
The authors would like to thank Professor Rong Zhou
(State Key Laboratory of Respiratory Diseases, Guangzhou Medical
University) for kindly providing HAdV clinical isolates serotypes
2, 3 and 4. The authors also would like to thank Dr Hongliang Pan
(Shenzhen City Star Huayuan Technology Co., Ltd.) for his help
while collecting throat swab specimens.
Funding
This study was supported by the Shenzhen Science and
Technology Project (grant no. JCYJ20150402095058885), the Dongguan
Social Science and Technology Development Foundation (grant no.
201510515000760), and the Shenzhen Bao'an Science and Technology
Project (grant no. 2015204).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YHD, JKD and HLL conceived and designed the
experiments. YXL, CFM, HJZ, QL, HLL and YT performed the
experiments. YXL, JKD, HLL and RC analyzed the data. YHD and HLL
contributed reagents/materials/analysis tools. YXL and JKD wrote
the paper.
Ethics approval and consent to
participate
The medical ethics committee of the Shenzhen Shajing
Hospital Affiliated to Guangzhou Medical University approved the
present study. All patients provided written consent.
Patient consent for publication
Written informed consent was obtained.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sibanda T and Okoh AI: Assessment of the
incidence of enteric adenovirus species and serotypes in surface
waters in the eastern cape province of South Africa: Tyume River as
a case study. ScientificWorldJournal. 2012:9492162012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jones MS II, Harrach B, Ganac RD, Gozum
MM, Dela Cruz WP, Riedel B, Pan C, Delwart EL and Schnurr DP: New
adenovirus species found in a patient presenting with
gastroenteritis. J Virol. 81:5978–5984. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Walsh MP, Chintakuntlawar A, Robinson CM,
Madisch I, Harrach B, Hudson NR, Schnurr D, Heim A, Chodosh J, Seto
D and Jones MS: Evidence of molecular evolution driven by
recombination events influencing tropism in a novel human
adenovirus that causes epidemic keratoconjunctivitis. PLoS One.
4:e56352009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jeulin H, Salmon A, Bordigoni P and Venard
V: Comparison of in-house real-time quantitative PCR to the
Adenovirus R-Gene kit for determination of adenovirus load in
clinical samples. J Clin Microbiol. 48:3132–3137. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang H, Zheng Y, Deng J, Chen X, Liu P and
Li X: Molecular epidemiology of respiratory adenovirus detection in
hospitalized children in Shenzhen, China. Int J Clin Exp Med.
8:15011–15017. 2015.PubMed/NCBI
|
|
6
|
Zhang Q, Jing S, Cheng Z, Yu Z, Dehghan S,
Shamsaddini A, Yan Y, Li M and Seto D: Comparative genomic analysis
of two emergent human adenovirus type 14 respiratory pathogen
isolates in China reveals similar yet divergent genomes. Emerg
Microbes Infect. 6:e922017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kojaoghlanian T, Flomenberg P and Horwitz
MS: The impact of adenovirus infection on the immunocompromised
host. Rev Med Virol. 13:155–171. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stroparo E, Cruz CR, Mdo Debur C, Vidal
LR, Nogueira MB, Almeida SM, Pereira LA, Rotta I and Raboni SM:
Adenovirus respiratory infection: Significant increase in diagnosis
using PCR comparing with antigen detection and culture methods. Rev
Inst Med Trop Sao Paulo. 52:317–321. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen M, Zhu Z, Huang F, Liu D, Zhang T,
Ying D, Wu J and Xu W: Adenoviruses associated with acute
respiratory diseases reported in Beijing from 2011 to, 2013. PLoS
One. 10:e01213752015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Echavarria M, Sanchez JL, Kolavic-Gray SA,
Polyak CS, Mitchell-Raymundo F, Innis BL, Vaughn D, Reynolds R and
Binn LN: Rapid detection of adenovirus in throat swab specimens by
PCR during respiratory disease outbreaks among military recruits. J
Clin Microbiol. 41:810–812. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu Z, Zhang Y, Xu S, Yu P, Tian X, Wang
L, Liu Z, Tang L, Mao N, Ji Y, et al: Outbreak of acute respiratory
disease in China caused by B2 species of adenovirus type 11. J Clin
Microbiol. 47:697–703. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pellet PE and Roizmann B: Field virology
(5th edition). 2007.
|
|
13
|
Shetty AK, Treynor E, Hill DW, Gutierrez
KM, Warford A and Baron EJ: Comparison of conventional viral
cultures with direct fluorescent antibody stains for diagnosis of
community-acquired respiratory virus infections in hospitalized
children. Pediatr Infect Dis J. 22:789–794. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
De Vos N, Vankeerberghen A, Vaeyens F, Van
Vaerenbergh K, Boel A and De Beenhouwer H: Simultaneous detection
of human bocavirus and adenovirus by multiplex real-time PCR in a
Belgian paediatric population. Eur J Clin Microbiol Infect Dis.
28:1305–1310. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Blaschke AJ, Allison MA, Meyers L,
Rogatcheva M, Heyrend C, Mallin B, Carter M, Lafleur B, Barney T,
Poritz MA, et al: Non-invasive sample collection for respiratory
virus testing by multiplex PCR. J Clin Virol. 52:210–214. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dabisch-Ruthe M, Vollmer T, Adams O,
Knabbe C and Dreier J: Comparison of three multiplex PCR assays for
the detection of respiratory viral infections: Evaluation of xTAG
respiratory virus panel fast assay, RespiFinder 19 assay and
RespiFinder SMART 22 assay. BMC Infect Dis. 12:1632012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Freymuth F, Vabret A, Cuvillon-Nimal D,
Simon S, Dina J, Legrand L, Gouarin S, Petitjean J, Eckart P and
Brouard J: Comparison of multiplex PCR assays and conventional
techniques for the diagnostic of respiratory virus infections in
children admitted to hospital with an acute respiratory illness. J
Med Virol. 78:1498–1504. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu L, Lin XY, Yang ZX, Yao XP, Li GL, Peng
SZ and Wang Y: A multiplex PCR for simultaneous detection of
classical swine fever virus, African swine fever virus, highly
pathogenic porcine reproductive and respiratory syndrome virus,
porcine reproductive and respiratory syndrome virus and
pseudorabies in swines. Pol J Vet Sci. 18:715–723. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang HS, Tsai CL, Chang J, Hsu TC, Lin S
and Lee CC: Multiplex PCR system for the rapid diagnosis of
respiratory virus infection: A systematic review and meta-analysis.
Clin Microbiol Infect pii: S1198-743X. 30649–30653. 2017.
|
|
20
|
Tam Iroh PY, Zhang L and Cohen Z: Impact
of a transition from respiratory virus shell vial to multiplex PCR
on clinical outcomes and cost in hospitalized children. Children
(Basel). 4:pii: E3. 2017.
|
|
21
|
Liu JK, Wei CH, Yang XY, Dai AL and Li XH:
Multiplex PCR for the simultaneous detection of porcine
reproductive and respiratory syndrome virus, classical swine fever
virus, and porcine circovirus in pigs. Mol Cell Probes. 27:149–152.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mahony J, Chong S, Merante F, Yaghoubian
S, Sinha T, Lisle C and Janeczko R: Development of a respiratory
virus panel test for detection of twenty human respiratory viruses
by use of multiplex PCR and a fluid microbead-based assay. J Clin
Microbiol. 45:2965–2970. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mahony JB, Blackhouse G, Babwah J, Smieja
M, Buracond S, Chong S, Ciccotelli W, O'Shea T, Alnakhli D,
Griffiths-Turner M and Goeree R: Cost analysis of multiplex PCR
testing for diagnosing respiratory virus infections. J Clin
Microbiol. 47:2812–2817. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Piewbang C, Rungsipipat A, Poovorawan Y
and Techangamsuwan S: Development and application of multiplex PCR
assays for detection of virus-induced respiratory disease complex
in dogs. J Vet Med Sci. 78:1847–1854. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yue F, Cui S, Zhang C and Yoon KJ: A
multiplex PCR for rapid and simultaneous detection of porcine
circovirus type 2, porcine parvovirus, porcine pseudorabies virus,
and porcine reproductive and respiratory syndrome virus in clinical
specimens. Virus Genes. 38:392–397. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boivin G, Côté S, Déry P, De Serres G and
Bergeron MG: Multiplex real-time PCR assay for detection of
influenza and human respiratory syncytial viruses. J Clin
Microbiol. 42:45–51. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang TG, Li AH, Lyu M, Chen M, Huang F
and Wu J: Detection of respiratory viral and bacterial pathogens
causing pediatric community-acquired pneumonia in Beijing using
real-time PCR. Chronic Dis Transl Med. 1:110–116. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Metzgar D, Gibbins C, Hudson NR and Jones
MS: Evaluation of multiplex type-specific real-time PCR assays
using the LightCycler and joint biological agent identification and
diagnostic system platforms for detection and quantitation of adult
human respiratory adenoviruses. J Clin Microbiol. 48:1397–1403.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu X, Trujillo-Lopez E, Lott L and Erdman
DD: Quantitative real-time PCR assay panel for detection and
type-specific identification of epidemic respiratory human
adenoviruses. J Clin Microbiol. 51:1089–1093. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Washington C, Metzgar D, Hazbón MH, Binn
L, Lyons A, Coward C and Kuschner R: Multiplexed Luminex xMAP assay
for detection and identification of five adenovirus serotypes
associated with epidemics of respiratory disease in adults. J Clin
Microbiol. 48:2217–2222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Broeders S, Huber I, Grohmann L, Berben G,
Taverniers I, Mazzara M, Roosens N and Morisset D: Guidelines for
validation of qualitative real-time PCR methods. Trends Food Sci
Tech. 37:115–126. 2014. View Article : Google Scholar
|
|
32
|
Fujimoto T, Okafuji T, Okafuji T, Ito M,
Nukuzuma S, Chikahira M and Nishio O: Evaluation of a bedside
immunochromatographic test for detection of adenovirus in
respiratory samples, by comparison to virus isolation, PCR, and
real-time PCR. J Clin Microbiol. 42:5489–5492. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Laamiri N, Fällgren P, Zohari S, Ben Ali
J, Ghram A, Leijon M and Hmila I: Accurate detection of avian
respiratory viruses by use of multiplex PCR-Based Luminex
suspension microarray assay. J Clin Microbiol. 54:2716–2725. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pierce VM, Elkan M, Leet M, McGowan KL and
Hodinka RL: Comparison of the Idaho Technology FilmArray system to
real-time PCR for detection of respiratory pathogens in children. J
Clin Microbiol. 50:364–371. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Popowitch EB, O'Neill SS and Miller MB:
Comparison of the Biofire FilmArray RP, Genmark eSensor RVP,
Luminex xTAG RVPv1, and Luminex xTAG RVP fast multiplex assays for
detection of respiratory viruses. J Clin Microbiol. 51:1528–1533.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Loeffelholz MJ, Pong DL, Pyles RB, Xiong
Y, Miller AL, Bufton KK and Chonmaitree T: Comparison of the
FilmArray respiratory panel and prodesse real-time PCR assays for
detection of respiratory pathogens. J Clin Microbiol. 49:4083–4088.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xu W, McDonough MC and Erdman DD:
Species-specific identification of human adenoviruses by a
multiplex PCR assay. J Clin Microbiol. 38:4114–4120.
2000.PubMed/NCBI
|
|
38
|
Metzgar D, Osuna M, Yingst S, Rakha M,
Earhart K, Elyan D, Esmat H, Saad MD, Kajon A, Wu J, et al: PCR
analysis of egyptian respiratory adenovirus isolates, including
identification of species, serotypes, and coinfections. J Clin
Microbiol. 43:5743–5752. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rand KH, Rampersaud H and Houck HJ:
Comparison of two multiplex methods for detection of respiratory
viruses: FilmArray RP and xTAG RVP. J Clin Microbiol. 49:2449–2453.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Poritz MA, Blaschke AJ, Byington CL,
Meyers L, Nilsson K, Jones DE, Thatcher SA, Robbins T, Lingenfelter
B, Amiott E, et al: FilmArray, an automated nested multiplex PCR
system for multi-pathogen detection: Development and application to
respiratory tract infection. PLoS One. 6:e260472011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wada K, Kubota N, Ito Y, Yagasaki H, Kato
K, Yoshikawa T, Ono Y, Ando H, Fujimoto Y, Kiuchi T, et al:
Simultaneous quantification of Epstein-Barr virus, cytomegalovirus,
and human herpesvirus 6 DNA in samples from transplant recipients
by multiplex real-time PCR assay. J Clin Microbiol. 45:1426–1432.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gunson RN, Collins TC and Carman WF:
Practical experience of high throughput real time PCR in the
routine diagnostic virology setting. J Clin Virol. 35:355–367.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ratcliff RM, Chang G, Kok T and Sloots TP:
Molecular diagnosis of medical viruses. Curr Issues Mol Biol.
9:87–102. 2007.PubMed/NCBI
|
|
44
|
Wittwer CT, Herrmann MG, Gundry CN and
Elenitoba-Johnson KS: Real-time multiplex PCR assays. Methods.
25:430–442. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gunson RN, Bennett S, Maclean A and Carman
WF: Using multiplex real time PCR in order to streamline a routine
diagnostic service. J Clin Virol. 43:372–375. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Heim A, Ebnet C, Harste G and
Pring-Åkerblom P: Rapid and quantitative detection of human
adenovirus DNA by real-time PCR. J Med Virol. 70:228–239. 2003.
View Article : Google Scholar : PubMed/NCBI
|