Introduction
Coronary artery disease (CAD) remains one of the
most common causes of morbidity and mortality globally, with an
increased prevalence predicted in the near future (1). The occurrence of CAD events is
primarily due to crosstalk between genetic and environmental
factors (2). A number of risk
factors of CAD have been identified, including smoking, obesity and
family history (3). Available
treatment options primarily include lifestyle alterations, medical
treatment and surgical interventions, which are chosen depending on
comorbidities and the preferences of the individual patient
(4). Prevention and treatment of
this disease remains a daunting task. Therefore, further research
is required to elucidate the underlying biological features for the
development of more adequate therapy for patients.
The rapid prevalence of high-throughput microarray
technologies has facilitated identification of genome variations in
diseases, contributing to a deeper understanding of pathogenesis
and the development of promising biomarkers (5,6).
Such technologies have been applied to investigate the underlying
mechanisms of CAD. Ren et al (7) identified a circulating micro (mi)RNA
signature for CAD using co-expression network analyses on miRNA
array data. In addition, a study of genomic DNA methylation
profiling in patients with CAD was conducted (8). Furthermore, Liu et al
(9) performed a secondary analysis
using a weighted gene co-expression network (WGCNA) on microarray
data of CAD samples (dataset no. GSE23561), which were downloaded
from the Gene Expression Omnibus (GEO) database. It was identified
that the glucose-6-phosphate 1-dehydrogenase, protein S100-A7 and
hypertrophic cardiomyopathy pathways were involved in CAD (9). However, this previous study had the
limitation of sample size (six CAD samples and nine normal
samples).
Therefore, four microarray datasets of CAD from the
GEO database were included in the present study. WGCNA was used to
construct a co-expression network and the highly preserved modules
in the four datasets were determined. The MetaDE method in R
language, which is capable of conducting 12 primary meta-analysis
methods (10), has been utilized
for the detection of differentially expressed genes (DEGs) in a
number of diseases, including gastric cancer (11) and colorectal cancer (12). In the present study, the DEGs with
significant consistency across all four datasets were selected
using the MetaDE method and subsequently compared with the genes in
the highly preserved WGCNA modules. Subsequently, the overlapping
genes were used to construct a protein-protein interaction (PPI)
network, followed by functional analysis of the genes in the
network. The in-depth analysis conducted in the present study may
provide novel insights into the pathogenesis of CAD.
Materials and methods
Microarray data
Microarray data for CAD were searched in NCBI GEO
(http://www.ncbi.nlm.nih.gov/geo/).
Inclusion criteria were: i) The dataset belonged to a gene
expression profile; ii) samples in the dataset were collected from
blood; iii) the samples included patients and healthy controls; iv)
the dataset was based on human gene expression profiles; and v) the
sample number was ≥20. A total of four datasets, GSE12288 (n=222;
CAD, 112; control, 110) (13),
GSE20680 (n=139; CAD, 87; control, 52) (14), GSE20681 (n=198; CAD, 99; control,
99) (15) and GSE42148 (n=24; CAD,
13; control, 11; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE42148)
that met the criteria were included in the present study (Table I).
 | Table I.Information on the four Gene
Expression Omnibus datasets. |
Table I.
Information on the four Gene
Expression Omnibus datasets.
| Accession no. | Platform | Probe no. | Total sample
no. | CAD | Control |
|---|
| GSE12288 | GPL96 | 22283 | 222 | 112 | 110 |
| GSE20680 | GPL4133 | 45220 | 139 | 87 | 52 |
| GSE20681 | GPL4133 | 45220 | 198 | 99 | 99 |
| GSE42148 | GPL13607 | 62976 | 24 | 13 | 11 |
Data preprocessing
Raw data in CEL files from GSE12288 were downloaded
from the Affy platform (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL96)
and subsequently converted into gene symbols under pretreatment.
The median method was used to fill in the missing values, the
MicroArray Suite method to complete background correction, and the
quantiles method to normalize the data, using oligo 1.41.1 in
R3.3.1 language (http://www.bioconductor.org/packages/release/bioc/html/oligo.html)
(16). GSE20680, GSE20681 and
GSE42148 were downloaded from the Agilent platform (Agilent
Technologies, Inc., Santa Clara, CA, USA). Microarray raw data (TXT
files) of the three datasets were log2-transformed using Limma
3.34.0 (https://bioconductor.org/packages/release/bioc/html/limma.html)
to achieve an approximate normal distribution, and were
subsequently standardized using the median normalization method
(17).
WGCNA analysis
WGCNA is a well-established method for constructing
scale-free gene co-expression networks, which is characterized by
the use of soft thresholding (18,19).
GSE12288 was used as the training dataset, while GSE20680, GSE20681
and GSE42148 were the validations sets. Genes with coefficients of
variation <0.1 were discarded. Correlations between the gene
expression in the four datasets were evaluated with the
verboseScatterplot function of the WGCNA package (https://labs.genetics.ucla.edu/horvath/CoexpressionNetwork/Rpackages/WGCNA/).
Weighted gene co-expression networks of the genes in the training
dataset were constructed using the WGCNA method as previously
described (20–22). First, the soft threshold power of β
was set as 18 (scale-free R2=0.9), according to the
scale free topology criterion. The weighted adjacency matrix was
subsequently constructed. Adjacencies and correlations were
transformed into a topological overlap matrix (TOM), followed by
calculating the corresponding dissimilarity (1-TOM). Subsequently,
a hierarchical clustering analysis (23) of genes was performed using 1-TOM as
the distance measure. Modules were detected using dynamic tree cut
algorithm with a minimum module size of 50 and a minimum cut height
of 0.95. Furthermore, module preservation between the training set
and the three validation sets was measured using the module
preservation function of the WGCNA software package. Possible
functions of the preserved module were studied using the user List
Enrichment function of the WGCNA package.
Identification of DEGs with
significant consistency in four datasets
DEGs between CAD samples and healthy controls were
screened in each of the above four datasets using the metaDE method
(10,24) in R language (https://cran.r-project.org/web/packages/MetaDE/).
Heterogeneity was examined across the four datasets to assess the
consistency of gene expression, by calculating tau2 and Qpval
values. A tau2=0 and Qpval >0.05 indicated that the gene was
homogeneous and unbiased. The thresholds for DEG identification
were tau2=0, Qpval >0.05 and false discovery rate <0.05.
Consistency of the DEGs was detected using the MetaDE method, and
the DEGs with significant consistency in the four datasets were
selected for further analysis.
Construction of a PPI network
A PPI network was constructed to evaluate the
interactions between genes in the above network. The common genes
in the preserved modules that were obtained from WGCNA, and the
DEGs with significant consistency, were selected to construct a PPI
network based on three databases: The Biological General Repository
for Interaction Datasets 3.4.153 (http://thebiogrid.org/) (25); the Human Protein Reference Database
Release 9 (http://www.hprd.org/) (26); and STRING 10.5 (https://string-db.org/) (27). The PPIs revealed in at least two of
the three databases were extracted for the PPI network, visualized
using Cytoscape 3.3 software (http://www.cytoscape.org/) (28). In the PPI network, a node
represents a gene; the undirected link between two nodes is an
edge, denoting the interaction between two genes; and the degree of
a node corresponds to the number of interactions of a gene with
other genes in the network.
Gene ontology (GO) functional and
kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment
analyses
In order to elucidate the possible biological roles
of the genes in the PPI network, GO (29) functional and KEGG (30) pathway enrichment analyses were
performed using the Database for Annotation, Visualization and
Integrated Discovery 6.8 software (31). GO terms have three categories,
including biological process (BP), cellular compartment and
molecular function. P<0.05 was considered to indicate a
significant difference for GO terms and KEGG pathways.
Results
Identification of key WGCNA
modules
Following data preprocessing for the four datasets,
the present study attempted to identify CAD-associated
co-expression modules using WGCNA. Using the aforementioned
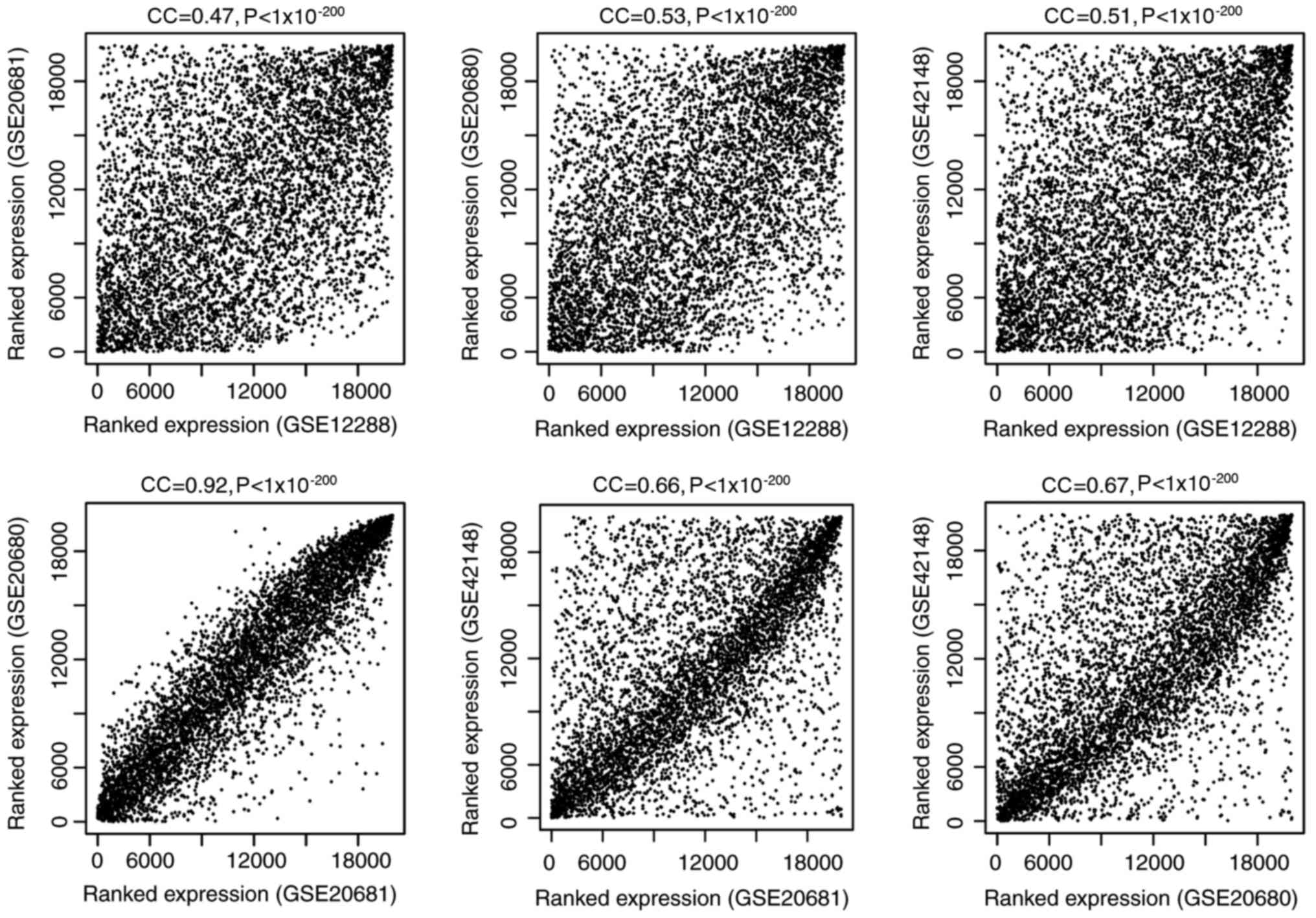
methods, the correlation coefficients (CCs) of genes in the four
datasets were obtained, and the CCs were 0.47–0.92 with P-values
<1×10−200 between any two datasets (Fig. 1), suggesting a good consistency of
the common genes across all datasets.
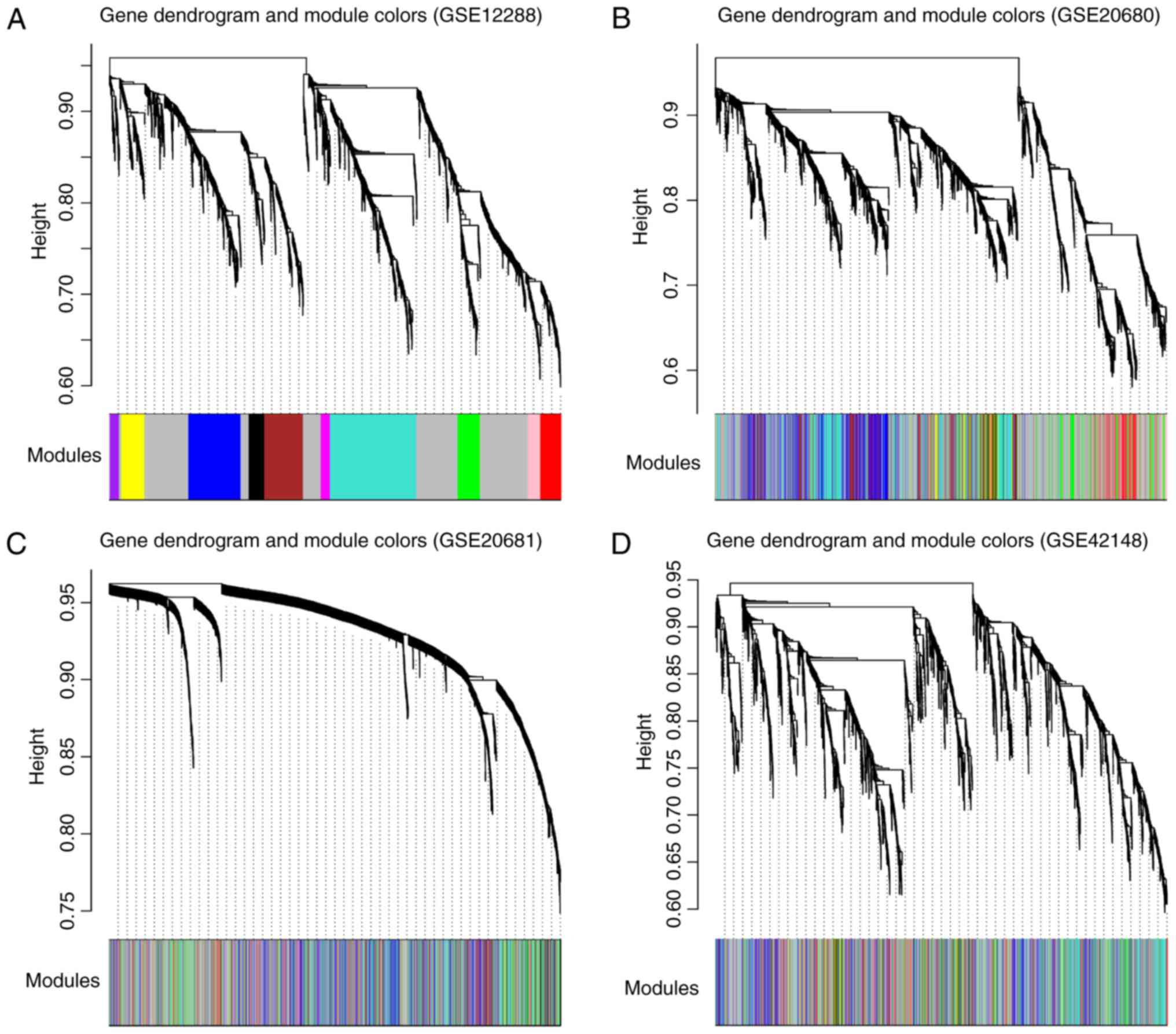
As a result, 11 co-expression modules were
identified for genes in the training set using WGCNA (Fig. 2A). These modules are illustrated in
branches of the dendrogram with different colors. To evaluate the
robustness of these modules in the training set, the modules were
re-constructed in the three validation datasets, GSE20680, GSE20681
and GSE42148, separately (Fig.
2B-D). Genes in the three validation sets were colored,
according to the module color in the training set.
Multi-dimensional scaling of expression data of all genes in these
modules demonstrated that genes in the same module appeared to
cluster together and exhibited a similar expression pattern
(Fig. 3A). Additionally,
hierarchical clustering analysis of these modules in the four
datasets revealed that modules of the same branch tended to have
similar gene expression patterns (Fig.
3B).
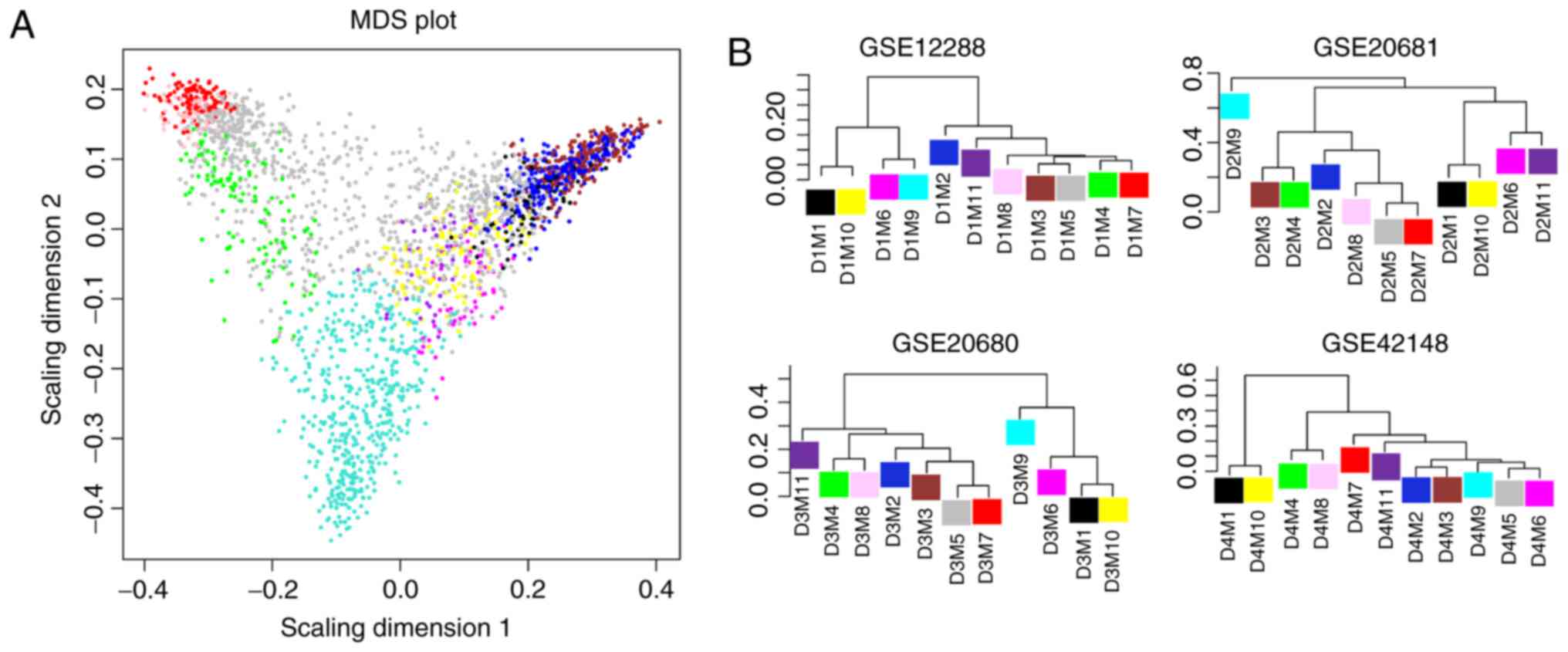 | Figure 3.MDS plot and hierarchical dendrograms
of the four datasets. (A) MDS plot of expression data of genes in
11 modules. (B) Hierarchical clustering tree of 11 modules in the
four datasets. Modules are denoted via a ‘D’ and ‘M’ system: D1,
GSE12288; D2, GSE20681; D3, GSE20680; D4, GSE42148; M1, black; M2,
dark blue; M3, brown; M4, green; M5, grey; M6, magenta; M7, red;
M8, pink; M9, turquoise; M10, yellow; and M11, purple. MDS,
multi-dimensional scaling. |
In total, five of the 11 modules, black, brown,
magenta, turquoise and yellow modules, with Z-scores >5 were
determined to be well preserved across the four datasets, including
873 genes (Table II). This
suggested that the five highly preserved modules may be closely
associated with CAD. Of the five modules, genes in the black module
were significantly linked to ‘response to glucocorticoid stimulus’;
those in the brown module were significantly associated with
‘regulation of transcription’; and those in the magenta module may
be involved in ‘protein localization’ (Table II). Notably, the turquoise and the
yellow module exhibited functions in ‘immune response’ (Table II).
| Table II.Characteristics of weighted gene
co-expression network modules. |
Table II.
Characteristics of weighted gene
co-expression network modules.
| Dataset | Characteristic |
|---|
|
|
|---|
| GSE12288 | GSE20681 | GSE20680 | GSE42148 | Color | Size | Z-score | Module
characterization |
|---|
| D1M1 | D2M1 | D3M1 | D4M1 | Black | 85 | 13.81 | Response to
glucocorticoid stimulus |
| D1M2 | D2M2 | D3M2 | D4M2 | Blue | 285 | 0.58 | Humoral immune
response |
| D1M3 | D2M3 | D3M3 | D4M3 | Brown | 211 | 5.26 | Regulation of
transcription |
| D1M4 | D2M4 | D3M4 | D4M4 | Green | 121 | 0.16 | Regulation of
transcription |
| D1M5 | D2M5 | D3M5 | D4M5 | Grey | 572 | 3.92 | Defense
response |
| D1M6 | D2M6 | D3M6 | D4M6 | Magenta | 51 | 5.66 | Protein
localization |
| D1M7 | D2M7 | D3M7 | D4M7 | Red | 68 | 0.63 | Cell surface
receptor linked signal transduction |
| D1M8 | D2M8 | D3M8 | D4M8 | Pink | 108 | 1.31 |
Second-messenger-mediated signaling |
| D1M9 | D2M9 | D3M9 | D4M9 | Turquoise | 400 | 14.34 | Immune
response |
| D1M10 | D2M10 | D3M10 | D4M10 | Yellow | 126 | 12.54 | Immune
response |
| D1M11 | D2M11 | D3M11 | D4M11 | Purple | 50 | 1.35 | Lymphocyte
activation |
Screening for DEGs with significant
consistency in the four datasets
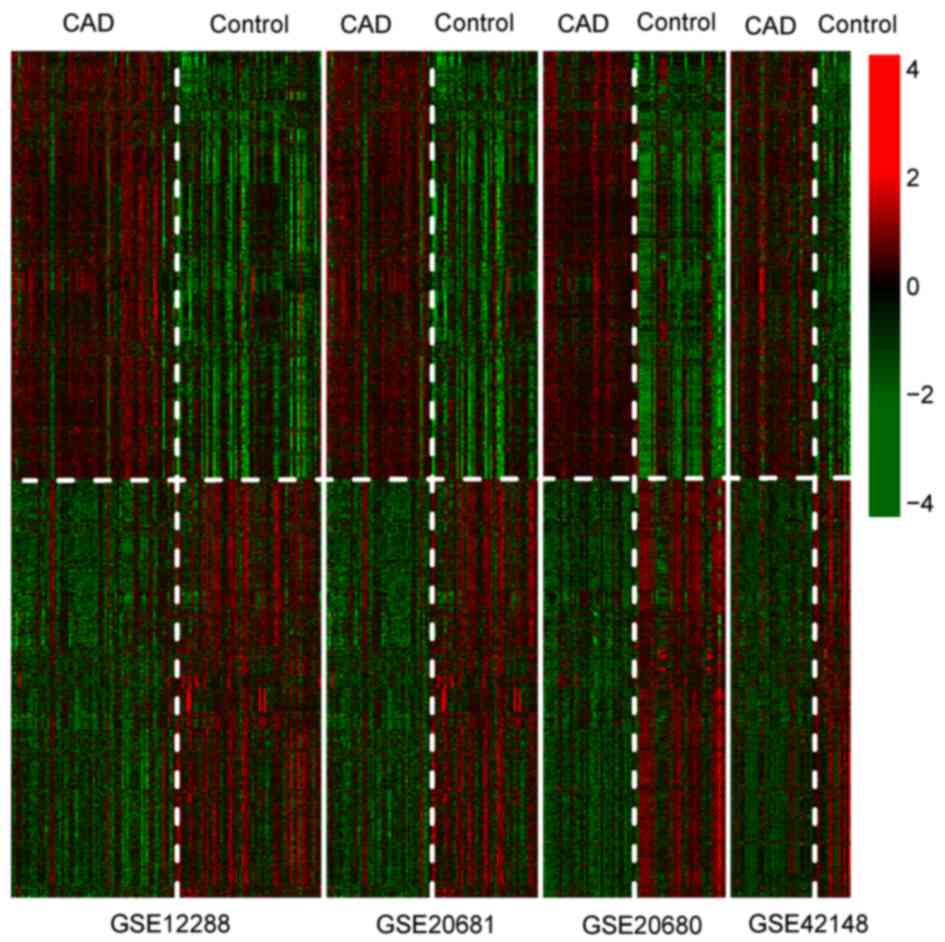
Using the metaDE method, 836 DEGs were identified
with significant consistency across the four datasets. A heatmap
for these DEGs demonstrated that the expression patterns of these
DEGs differed between the CAD and control samples (Fig. 4).
Construction of a PPI network for the
overlapping genes
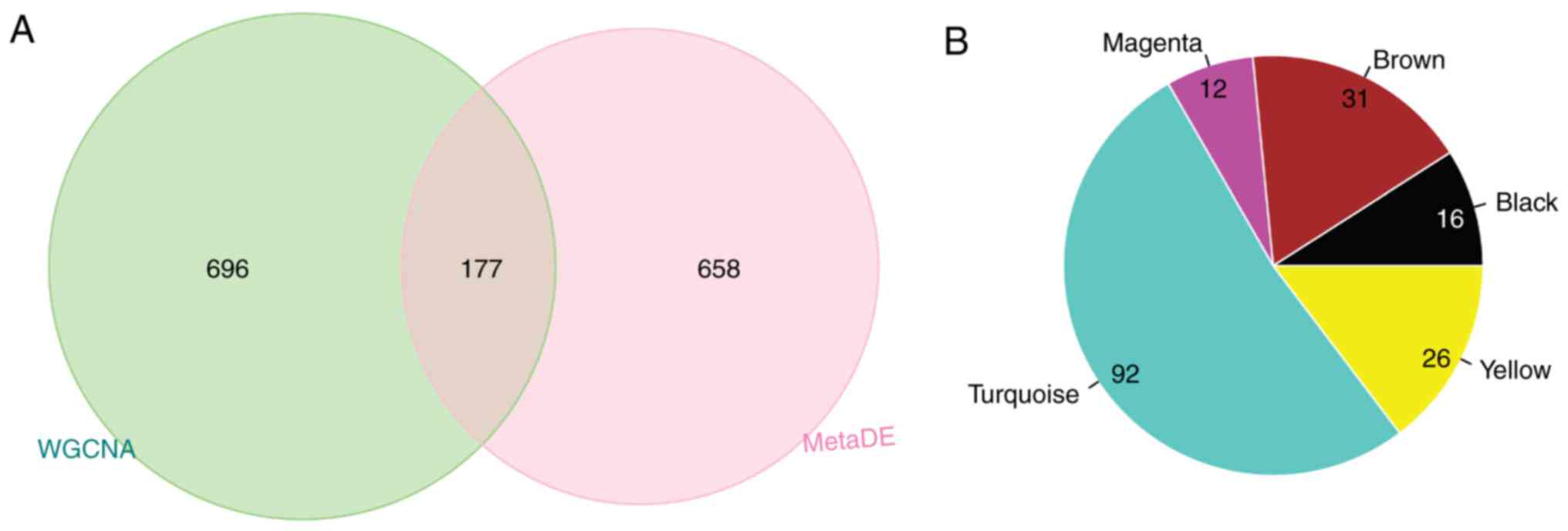
A total of 177 genes were overlapping between WGCNA
modules and the DEGs with significant consistency (Fig. 5A). Of them, 92 genes were included
in the turquoise module, 26 in the yellow module, 16 in the black
module, 31 in the brown module and 12 in the magenta module
(Fig. 5B). Based on the three
databases mentioned above, a PPI network was built with these
overlapping genes. In total, 150 paired PPIs that appeared in at
least two of the three databases were included in the PPI network
(Fig. 6A), which contained 59
downregulated genes and 40 upregulated genes (Fig. 6B). All genes in the network were
ranked in a descending order, according to their degrees. The top
five genes were LCK proto-oncogene, Src family tyrosine kinase
(LCK; degree=15), euchromatic histone lysine
methyltransferase 2 (EHMT2; degree=14), inosine
monophosphate dehydrogenase 2 (IMPDH2; degree=12), protein
phosphatase 4 catalytic subunit (PPP4C; degree=11) and
ζ-chain of T-cell receptor associated protein kinase 70
(ZAP70; degree=11).
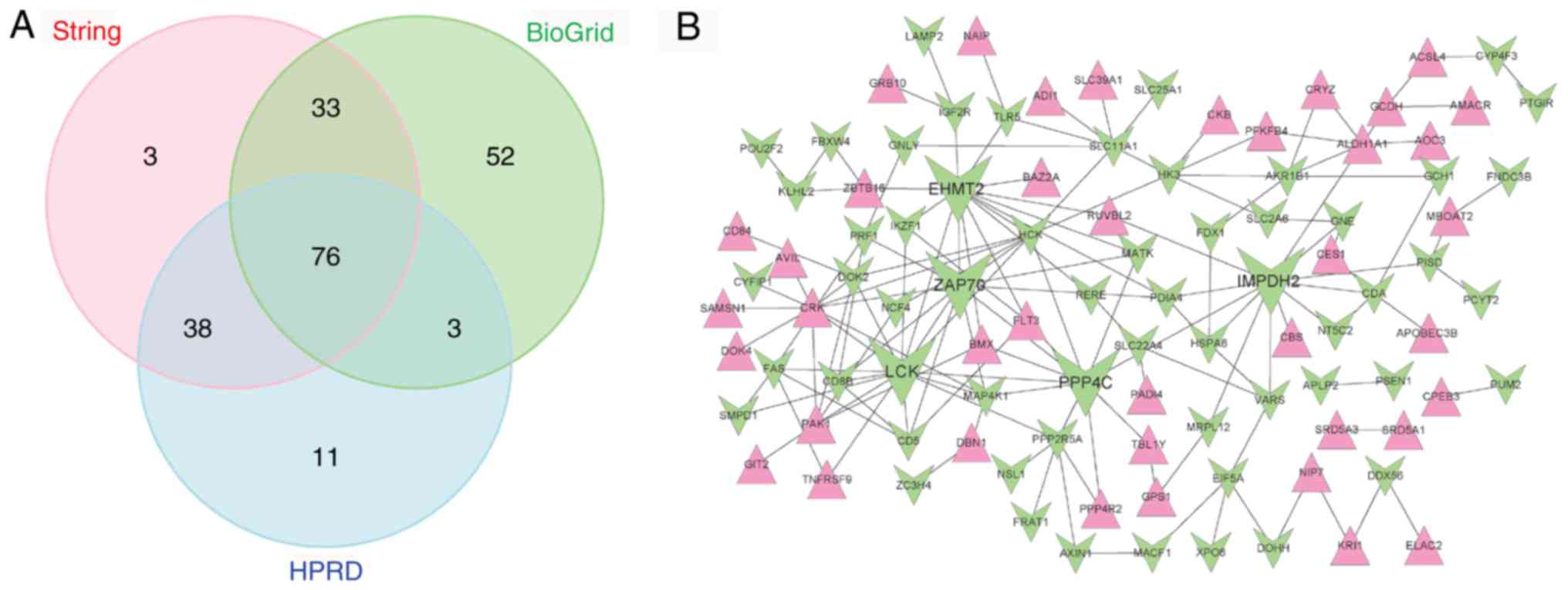 | Figure 6.Construction of the PPI network. (A)
Venn diagram of PPIs obtained from BioGRID, HPRD Release 9 and
STRING. (B) PPI network. A red triangle denotes an upregulated gene
and a green inverted triangle denotes a downregulated gene. The top
five genes with higher degrees compared with the other genes in the
network are represented by five extra-large green inverted
triangles, separately. PPI, protein-protein interaction; BioGRID,
Biological General Repository for Interaction Datasets; HPRD, Human
Protein Reference Database; LCK, LCK proto-oncogene, Src family
tyrosine kinase; EHMT2, euchromatic histone lysine
methyltransferase 2; IMPDH2, inosine monophosphate dehydrogenase 2;
PPP4C, protein phosphatase 4 catalytic subunit; ZAP70, ζ-chain of
T-cell receptor associated protein kinase 70. |
Functional annotation
The enrichment analysis indicated that genes in the
PPI network were significantly associated with numerous GO BP
terms, including the ‘transmembrane receptor protein tyrosine
kinase signaling pathway’ and ‘peptidyl-tyrosine
autophosphorylation’ (Table
III). With regard to KEGG pathways, there were five significant
pathways, including ‘metabolic pathways’, ‘natural killer cell
mediated cytotoxicity’, ‘fructose and mannose metabolism’, ‘primary
immunodeficiency’, and ‘Fc gamma R-mediated phagocytosis’ (Table IV). Notably, ‘natural killer cell
mediated cytotoxicity’ and ‘primary immunodeficiency’ pathways were
significantly enriched with LCK and ZAP70.
| Table III.Significant GO terms. |
Table III.
Significant GO terms.
| Category | Term | Count of genes | P-value |
|---|
| Biology
process | Transmembrane
receptor protein tyrosine kinase signaling pathway | 9 | <0.001 |
|
| Peptidyl-tyrosine
autophosphorylation | 5 | <0.001 |
|
| Oxidation-reduction
process | 13 | <0.001 |
|
| Inflammatory
response | 10 | <0.001 |
|
| Mesoderm
development | 4 | <0.001 |
|
| Innate immune
response | 9 | 0.003 |
|
| Carbohydrate
phosphorylation | 3 | 0.007 |
|
| Regulation of
defense response to virus by virus | 3 | 0.011 |
|
| Chromatin
remodeling | 4 | 0.013 |
|
| Response to
lipopolysaccharide | 5 | 0.014 |
|
| Protein
autophosphorylation | 5 | 0.017 |
|
| Peptidyl-lysine
modification to peptidyl-hypusine | 2 | 0.022 |
|
| Lymphocyte
proliferation | 2 | 0.028 |
|
| Fc-gamma receptor
signaling pathway involved in phagocytosis | 4 | 0.035 |
|
| Response to
fungicide | 2 | 0.039 |
|
| Negative regulation
of B cell activation | 2 | 0.039 |
|
| Apoptotic
process | 8 | 0.042 |
| Cellular
component | Extrinsic component
of cytoplasmic side of plasma membrane | 5 | 0.000 |
|
| Ruffle | 4 | 0.012 |
|
| Protein phosphatase
4 complex | 2 | 0.021 |
|
| Neuron
projection | 5 | 0.038 |
| Molecular
function | Protein tyrosine
kinase activity | 5 | 0.007 |
|
| Kinase
activity | 6 | 0.013 |
|
| Insulin receptor
binding | 3 | 0.014 |
|
| Electron carrier
activity | 4 | 0.015 |
|
| Cholestenone
5-alpha-reductase activity | 2 | 0.017 |
|
| Catalytic
activity | 5 | 0.023 |
|
| Poly(A) RNA
binding | 13 | 0.028 |
|
| Oxidoreductase
activity | 5 | 0.028 |
|
| mRNA
3′-untranslated region binding | 3 | 0.032 |
|
| Receptor
activity | 5 | 0.037 |
|
| Enzyme binding | 6 | 0.043 |
| Table IV.Significant KEGG signaling
pathways. |
Table IV.
Significant KEGG signaling
pathways.
| Term | Count | P-value | Genes |
|---|
| Metabolic
pathways | 22 | 0.001 | GCDH, CES1, GNE,
AMACR, SPHK1, PISD, CKB, GCH1, ALDH1A1, ADI1, HK3, NT5C2, AKR1B1,
SMPD1, MBOAT2, CDA, CYP4F3, ACSL4, PCYT2, IMPDH2, CBS, AOC3 |
| Natural killer cell
mediated cytotoxicity | 5 | 0.022 | PRF1, LCK, ZAP70,
PAK1, FAS |
| Fructose and
mannose metabolism | 3 | 0.032 | PFKFB4, HK3,
AKR1B1 |
| Primary
immunodeficiency | 3 | 0.036 | CD8B, LCK,
ZAP70 |
| Fc gamma R-mediated
phagocytosis | 4 | 0.038 | HCK, SPHK1, PAK1,
CRK |
Discussion
CAD remains a primary public health concern
(32). The present study attempted
to dissect the underlying pathogenic mechanisms of CAD via a
combined analysis of four GEO datasets, which contained CAD samples
and healthy control samples. A total of 11 modules were detected in
the WGCNA network, five of which were highly preserved across all
datasets. Using the metaDE method, 836 common DEGs in the four
datasets were identified. Furthermore, a PPI network was
constructed with the 177 overlapping genes of the DEGs with
significant consistency and the five preserved WGCNA modules.
According to degree, the top five genes of the PPI network were
LCK, EHMT2, IMPDH2, PPP4C and ZAP70. Notably,
multiple significant pathways for genes in the PPI network were
identified, including ‘natural killer cell mediated cytotoxicity’,
‘primary immunodeficiency’, and ‘Fc gamma R-mediated phagocytosis’
pathways.
The LCK protein encoded by the gene LCK,
additionally termed lymphocyte-specific protein tyrosine kinase, is
a member of the Src family of tyrosine kinases, which are involved
in T cell signaling (33).
Insufficient deactivation of LCK has been demonstrated to render
patients with acute coronary syndrome (ACS) vulnerable to abnormal
T cell responses (34). The
ZAP-70 gene encodes the ZAP-70 enzyme, which belongs to the
protein-tyrosine kinase family and is a T cell receptor. In T cell
signaling, ZAP-70 binds to the CD3 complex that is phosphorylated
by the LCK protein (35). It has
been established that the ZAP-70 protein expression is able to act
as a marker for chronic lymphocytic leukemia or small lymphocytic
lymphoma (36).
In the present study, LCK (degree=15) and
ZAP70 (degree=11) were highlighted in the PPI network.
Furthermore, based on the KEGG pathway enrichment analysis,
LCK and ZAP70 were significantly associated with
‘natural killer cell mediated cytotoxicity’ and ‘primary
immunodeficiency’ pathways, which were associated with immune
processes. Similarly, there is evidence that dysregulated adaptive
immunity serves a causative role in ACS (37). Therefore, it may be inferred that
LCK and ZAP70 serve a role in CAD, which may partly
be by regulating natural killer cell mediated cytotoxicity and
primary immunodeficiency pathways. Additionally, a previous study
with a novel knowledge-based approach revealed that the Fc gamma
R-mediated phagocytosis pathway is a pathogenic mechanism for CAD
(38). Likewise, the present study
suggests that ‘Fc gamma R-mediated phagocytosis’ may have a
function in CAD.
EHMT2, encoded by the gene EHMT2, termed G9a,
is a histone methyltransferase that serves a critical role in
epigenetic regulation within the nucleus accumbens (39). Histone methylation has emerged as a
crucial epigenetic mechanism for cardiovascular development and
homeostasis (40). Papait et
al (41) demonstrated that
EHMT2 orchestrates important epigenetic alterations in
cardiomyocyte homeostasis and hypertrophy. Furthermore, EHMT1/2 has
been suggested to be a therapeutic target against pathological
cardiac hypertrophy (42). These
results are in accordance with the present results, which
demonstrated that EHMT2 was a predominant gene in the PPI network
(degree=14). These collectively suggest a potentially critical role
of EHMT2 in CAD.
IMPDH2, encoded by gene IMPDH2, termed IMP
dehydrogenase 2, acts as a rate-limiting enzyme in guanine
nucleotide biosynthesis (43). It
has been reported to be involved in a number of cancer types,
including primary nasopharyngeal carcinoma (44) and prostate cancer (45). Nonetheless, the role of IMPDH2 in
CAD has been poorly defined. The PPP4C gene encodes the PP4C
protein, which is a member of the type 2 serine/threonine protein
phosphatase family (46), and
partly regulates the phosphorylation of numerous proteins
implicated in cardiac physiology and hypertrophy (47,48).
The present study indicated that IMPDH2 and PPP4C may
be important genes in CAD.
Although a comprehensive bioinformatics analysis
using four datasets was performed, there were a number of
limitations. The potentially important genes associated with CAD
lacked gene expression and functional validation. Further studies
are required with substantial experiments in vivo and in
vitro. Furthermore, the four datasets were not from the same
platform, which may cause numerous deviations. Nevertheless, the
present study provides novel insight into CAD pathogenesis.
In conclusion, the present study revealed that
LCK, EHMT2, IMPDH2, PPP4C and ZAP70 may be proposed
as genetic biomarkers for CAD. They may function via the
involvement of the ‘natural killer cell mediated cytotoxicity’,
‘primary immunodeficiency’, and ‘Fc gamma R-mediated phagocytosis
pathways’ in CAD. These results require further validation with
substantial experiments.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SY performed data analyses, wrote the manuscript,
conceived and designed the study.
Ethics approval and consent to
participate
In the original article of the datasets, the trials
were approved by the local institutional review boards of all
participating centers and informed consent was obtained from all
patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ohira T and Iso H: Cardiovascular disease
epidemiology in Asia: An overview. Circ J. 77:1646–1652. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sayols-Baixeras S, Lluís-Ganella C, Lucas
G and Elosua R: Pathogenesis of coronary artery disease: Focus on
genetic risk factors and identification of genetic variants. Appl
Clin Genet. 7:15–32. 2014.PubMed/NCBI
|
|
3
|
Torpy JM, Burke AE and Glass RM: Coronary
heart disease risk factors. JAMA. 302:23882009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hanson MA, Fareed MT, Argenio SL,
Agunwamba AO and Hanson TR: Coronary artery disease. Prim Care.
40:1–16. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Callaway JL, Shaffer LG, Chitty LS,
Rosenfeld JA and Crolla JA: The clinical utility of microarray
technologies applied to prenatal cytogenetics in the presence of a
normal conventional karyotype: A review of the literature. Prenat
Diagn. 33:1119–1133. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abulwerdi FA and Schneekloth JS Jr:
Microarray-based technologies for the discovery of selective,
RNA-binding molecules. Methods. 103:188–195. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ren J, Zhang J, Xu N, Han G, Geng Q, Song
J, Li S, Zhao J and Chen H: Signature of circulating microRNAs as
potential biomarkers in vulnerable coronary artery disease. PLoS
One. 8:e807382013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sharma P, Garg G, Kumar A, Mohammad F,
Kumar SR, Tanwar VS, Sati S, Sharma A, Karthikeyan G, Brahmachari V
and Sengupta S: Genome wide DNA methylation profiling for
epigenetic alteration in coronary artery disease patients. Gene.
541:31–40. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu J, Jing L and Tu X: Weighted gene
co-expression network analysis identifies specific modules and hub
genes related to coronary artery disease. BMC Cardiovasc Disord.
16:542016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang X, Kang DD, Shen K, Song C, Lu S,
Chang LC, Liao SG, Huo Z, Tang S, Ding Y, et al: An R package suite
for microarray meta-analysis in quality control, differentially
expressed gene analysis and pathway enrichment detection.
Bioinformatics. 28:2534–2536. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cai H, Xu J, Han Y, Lu Z, Han T, Ding Y
and Ma L: Integrated miRNA-risk gene-pathway pair network analysis
provides prognostic biomarkers for gastric cancer. Onco Targets
Ther. 9:2975–2986. 2016.PubMed/NCBI
|
|
12
|
Qi C, Hong L, Cheng Z and Yin Q:
Identification of metastasis-associated genes in colorectal cancer
using metaDE and survival analysis. Oncol Lett. 11:568–574. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sinnaeve PR, Donahue MP, Grass P, Seo D,
Vonderscher J, Chibout SD, Kraus WE, Sketch M Jr, Nelson C,
Ginsburg GS, et al: Gene expression patterns in peripheral blood
correlate with the extent of coronary artery disease. PLoS One.
4:e70372009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Elashoff MR, Wingrove JA, Beineke P,
Daniels SE, Tingley WG, Rosenberg S, Voros S, Kraus WE, Ginsburg
GS, Schwartz RS, et al: Development of a blood-based gene
expression algorithm for assessment of obstructive coronary artery
disease in non-diabetic patients. BMC Med Genomics. 4:262011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Beineke P, Fitch K, Tao H, Elashoff MR,
Rosenberg S, Kraus WE and Wingrove JA: PREDICT Investigators: A
whole blood gene expression-based signature for smoking status. BMC
Med Genomics. 5:582012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Parrish RS and Spencer HJ III: Effect of
normalization on significance testing for oligonucleotide
microarrays. J Biopharm Stat. 14:575–589. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: Limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang B and Horvath S: A general framework
for weighted gene co-expression network analysis. Stat Appl Genet
Mol Biol. 4:Article17. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhai X, Xue Q, Liu Q, Guo Y and Chen Z:
Colon cancer recurrence-associated genes revealed by WGCNA
co-expression network analysis. Mol Med Rep. 16:6499–6505. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vilne B, Skogsberg J, Asl Foroughi H,
Talukdar HA, Kessler T, Björkegren JLM and Schunkert H: Network
analysis reveals a causal role of mitochondrial gene activity in
atherosclerotic lesion formation. Atherosclerosis. 267:39–48. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Min S, Sun T, He Z and Xiong B:
Identification of two novel biomarkers of rectal carcinoma
progression and prognosis via co-expression network analysis.
Oncotarget. 8:69594–69609. 2017.PubMed/NCBI
|
|
23
|
Yip AM and Horvath S: Gene network
interconnectedness and the generalized topological overlap measure.
BMC Bioinformatics. 8:222007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hui L, Zhang J, Ding X, Guo X and Jang X:
Identification of potentially critical differentially methylated
genes in nasopharyngeal carcinoma: A comprehensive analysis of
methylation profiling and gene expression profiling. Oncol Lett.
14:7171–7178. 2017.PubMed/NCBI
|
|
25
|
Chatraryamontri A, Breitkreutz BJ,
Oughtred R, Boucher L, Heinicke S, Chen D, Stark C, Breitkreutz A,
Kolas N, O'Donnell L, et al: The BioGRID interaction database: 2015
update. Nucleic Acids Res. 43:(Database Issue). D470–D478. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Prasad Keshava TS, Goel R, Kandasamy K,
Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R,
Shafreen B, Venugopal A, et al: Human protein reference
database-2009 update. Nucleic Acids Res. 37:(Database Issue).
D767–D772. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Szklarczyk D, Franceschini A, Kuhn M,
Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork
P, et al: The STRING database in 2011: Functional interaction
networks of proteins, globally integrated and scored. Nucleic Acids
Res. 39:(Database Issue). D561–D568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lopes CT, Franz M, Kazi F, Donaldson SL,
Morris Q and Bader GD: Cytoscape Web: An interactive web-based
network browser. Bioinformatics. 26:2347–2348. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gene Ontology Consortium: Gene ontology
consortium: Going forward. Nucleic Acids Res. 43:(Database Issue).
D1049–D1056. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kanehisa M, Sato Y, Kawashima M, Furumichi
M and Tanabe M: KEGG as a reference resource for gene and protein
annotation. Nucleic Acids Res. 44(D1): D457–D462. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
da Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hanson MA, Fareed MT, Argenio SL,
Agunwamba AO and Hanson TR: Coronary artery disease. Prim Care.
40:1–16. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Patil DP and Kundu GC: LCK
(lymphocyte-specific protein tyrosine kinase). Atlas Genet
Cytogenet Oncol Haematol. 9:229–230. 2005.
|
|
34
|
Pryshchep S, Goronzy JJ, Parashar S and
Weyand CM: Insufficient deactivation of the protein tyrosine kinase
lck amplifies T-cell responsiveness in acute coronary syndrome.
Circ Res. 106:769–778. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pelosi M, Di Bartolo V, Mounier V, Mège D,
Pascussi JM, Dufour E, Blondel A and Acuto O: Tyrosine 319 in the
interdomain B of ZAP-70 is a binding site for the Src homology 2
domain of Lck. J Biol Chem. 274:14229–14237. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Staudt LM, Rosenwald A, Wilson W, Barry TS
and Wiestner A: ZAP-70 expression as a marker for chronic
lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). US
Patent 7,981,610 B2. Filed December 12, 2007; issued July 19.
2011.
|
|
37
|
Flego D, Liuzzo G, Weyand CM and Crea F:
Adaptive immunity dysregulation in acute coronary syndromes: From
cellular and molecular basis to clinical implications. J Am Coll
Cardiol. 68:2107–2117. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li H, Zuo X, Ouyang P, Lin M, Zhao Z,
Liang Y, Zhong S and Rao S: Identifying functional modules for
coronary artery disease by a prior knowledge-based approach. Gene.
537:260–268. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tachibana M, Sugimoto K, Fukushima T and
Shinkai Y: Set domain-containing protein, G9a, is a novel
lysine-preferring mammalian histone methyltransferase with
hyperactivity nd specific selectivity to lysines 9 and 27 of
histone H3. J Biol Chem. 276:25309–25317. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang QJ and Liu ZP: Histone methylations
in heart development, congenital and adult heart diseases.
Epigenomics. 7:321–330. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Papait R, Serio S, Pagiatakis C, Rusconi
F, Carullo P, Mazzola M, Salvarani N, Miragoli M and Condorelli G:
Histone methyltransferase G9a is required for cardiomyocyte
homeostasis and hypertrophy. Circulation. 136:1233–1246. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Thienpont B, Aronsen JM, Robinson EL,
Okkenhaug H, Loche E, Ferrini A, Brien P, Alkass K, Tomasso A,
Agrawal A, et al: The H3K9 dimethyltransferases EHMT1/2 protect
against pathological cardiac hypertrophy. J Clin Invest.
127:335–348. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bremer S, Rootwelt H and Bergan S:
Real-time PCR determination of IMPDH1 and IMPDH2 expression in
blood cells. Clin Chem. 53:1023–1029. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xu Y, Zheng Z, Gao Y, Duan S, Chen C, Rong
J, Wang K, Yun M, Weng H, Ye S and Zhang J: High expression of
IMPDH2 is associated with aggressive features and poor prognosis of
primary nasopharyngeal carcinoma. Sci Rep. 7:7452017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhou L, Xia D, Zhu J, Chen Y, Chen G, Mo
R, Zeng Y, Dai Q, He H, Liang Y, et al: Enhanced expression of
IMPDH2 promotes metastasis and advanced tumor progression in
patients with prostate cancer. Clin Transl Oncol. 16:906–913. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gingras AC, Caballero M, Zarske M, Sanchez
A, Hazbun TR, Fields S, Sonenberg N, Hafen E, Raught B and
Aebersold R: A novel, evolutionarily conserved protein phosphatase
complex involved in cisplatin sensitivity. Mol Cell Proteomics.
4:1725–1740. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Eleftheriadou O, Longman MR, Boguslavskyi
A, Ryan A, Wadzinski BE, Shattock MJ and Snabaitis AK: Expression
of type 2a protein phosphatases in cardiac health and disease.
Heart. 100:A162014. View Article : Google Scholar
|
|
48
|
Eleftheriadou O, Boguslavskyi A, Longman
MR, Cowan J, Francois A, Heads RJ, Wadzinski BE, Ryan A, Shattock
MJ and Snabaitis AK: Expression and regulation of type 2A protein
phosphatases and alpha4 signalling in cardiac health and
hypertrophy. Basic Res Cardiol. 112:372017. View Article : Google Scholar : PubMed/NCBI
|