Introduction
Congenital obstructive nephropathy (CON) is a main
cause of kidney insufficiency in child and infant (1). It is associated with various
diseases, such as prune belly syndrome (2). As a result, CON generates a huge
social burden especially in the morbidity and mortality (2). The different challenges associated
with CON in areas like diagnose and therapy highlight the
importance of the molecular mechanism of CON (3). Thus, the emerging theories of the
biology of CON suggest new targets for therapeutic interventions
(4).
Identification of novel biomarkers provides
prognostic value for this process remains a major goal in the study
of kidney disease (5). In renal
and urinary tract, previous study showed that disruption of
angiotensin type 2 receptor (AGTR2) induced a huge
scale of anomaly (6). Meanwhile,
the Monocyte chemotactic protein 1 (MCP-1) as well as
the Epidermal growth factor (EGF) seem to be involved
in the pathogenesis of tubulointerstitial damage of CON, and their
urine excretion may serve as a powerful prognostic marker for this
form renal disease (7). Not only
gene, but also pathways enriched by differentially expressed genes
(DEGs) are closed related with CON. Previous study demonstrated
that TGF-β signaling pathway plays an important role in regulating
kidney injury process following by obstruction (8). In a rat model, Hermens et al
(9) show that there is an
age-dependent role of Wnt signaling pathway in the pathophysiology
of CON. Despite of revealing the potential genes or pathways,
understanding the molecular mechanism of CON also brings a
breakthrough for the novel treatment of CON (10). The development of biomarkers
contribute to molecular therapies of CON (11). However, the molecular mechanisms
underlying these histologic changes are still poorly defined.
To better understand the pathophysiology of CON,
Brian Becknell and his colleagues evaluated the global
transcription in kidneys with graded hydronephrosis in the
megabladder (mgb2/2) mouse (12).
Based on normal, mild, moderate, and severe mouse models, they
prove that the development of progressive hydronephrosis can result
in renal adaptation, including significant changes in the
morphology and potential functionality of the renal urothelium.
However, the co-regulated gene in all models, transcription factor
(TF) associated with DEGs, gene expression in severe disease
status, as well as the potential chemicals for CON treatment are
still unclear. In the current study, a bioinformatics research was
designed based on the gene expression profile provided by Becknell
et al (12). Then,
investigations of DEGs, function and pathway enrichment,
protein-protein interaction network (PPI) analyses, as well as
TF-target gene regulatory network were performed in all disease
status. Furthermore, the chemical-gene interaction network in
severe models were also investigated. We aimed to explore the
potential pathoenesis of CON and provided information about novel
gene targets, as well as chemicals for CON treatment.
Materials and methods
Microarray data
Expression data of GSE48041 (12), sequenced on the platform of
GPL10787 Agilent-028005 SurePrint G3 Mouse GE 8×60K Microarray,
were obtained from the Gene Expression Omnibus (GEO) database
(www.ncbi.nlm.nih.gov/geo/). A total of
24 kidney tissue samples from control and megabladder (mgb−/−)
mouse aged 23–30 days were included in this dataset, and
hydronephrosis was induced by renal ultrasound as previously
reported (13). According to the
hydronephrosis degrees (www.sfu-urology.org/sfu_hydrone_grading.cfm),
animals were divided into 4 groups: Control (n=6), mild (n=7),
moderate (n=5) and severe (n=6) according to their disease
stages.
Data preprocessing
The preprocessing were performed based on robust
multi-array average (RMA) method (14) in Affy package (15) in R (v.3.10.3, bioconductor.org/biocLite.R). Contents of
processing included background correct microarray expression
intensities, normalize the expression within each array, and
expression calculation. The probe ID was convert to the gene symbol
based on the chip platform annotation files.
DEGs analysis
Classical Bayes method in Linear Models for
Microarray Data (limma) package (16) of R was used to reveal DEGs by
comparing expression value among samples from three comparisons:
Mild vs. control, moderate vs. control, and severe vs. control.
P-value <0.05 and |log2 log fold change (FC)|≥1 were
selected as the cut-off criteria for DEG screening.
VennPlot analysis of DEGs
VENNY (v.2.1) (17)
is an online tool used for Venn diagram analysis based on gene
expression value. The number of up-regulated, down-regulated, and
contra-regulated genes can be marked by VennPlex. In the present
study, VennPlex software was used to investigate the difference of
DEGs among 3 comparative groups. Then, the co-regulated DEGs (Set
1) and severe group DEGs (Set 2) were selected for further
investigation.
Enrichment analysis of DEGs
The Database for Annotation, Visualization, and
Integrated Discovery (DAVID; v.6.8) (18) provides a synthetic set of
functional annotation tools for users for revealing the biological
significances among huge scale of genes. Gene Ontology (GO)
function enrichment analysis and Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathways enrichment analysis were performed using
DAVID online tool (19). GO
functional categories including molecular function (MF), biological
process (BP), and cellular component (CC) (20). In the present study, GO functions
assembled by DEGs in severe group, as well as KEGG pathways
enriched by the co-regulated DEGs were investigated. P-value
<0.05 and enriched gene numbers (count) ≥2 were considered as
cut-off values.
PPI network investigation
Search Tool for the Retrieval of Interacting
Genes/Proteins (STRING; v.10.0) is an online tool for the
predication of PPI based on a large scale of biological database
(21). In the present study,
STRING was used to predict the interaction relations between DEGs
corresponding coding proteins. PPI were revealed based on the
STRING database with score (median confidence)=0.4, and nodes
represent DEGs in the PPI interaction. The degree was defined as
the number of the connections for the target proteins. Cytoscape
software (v.3.2.0) (22,23) was used for the visualization of PPI
network obtained above. Then, the CytoNCA software (v.2.1.6,
apps.cytoscape.org/apps/cytonca) (24) was used for the nodes topological
analysis (Parameter: Without weight). Furthermore, the sub-networks
with score >4.5 were identified by MOCDE (v.1.4.2; apps.cytoscape.org/apps/MCODE) (23) plugin in Cytoscape software.
TF-target gene regulatory network
construction
TF is an important class of participators in the
regulation of gene expression. Analysis of TF binding sites is of
great significance for the study of gene regulation system. In the
current research, the TFs-co-regulated DEGs network was constructed
with iRegulon (25) (v.1.3,
apps.cytoscape.org/apps/iRegulon). The Parameters of
this analysis were set as: Minimum identity between orthologous
genes=0.05, and maximum false discovery rate (FDR) on motif
similarity=0.001. TF-target gene relations with normalized
enrichment score (NES) >4 were considered as the meaningful
results.
Chemical-DEGs interaction network
construction
Comparative Toxicogenomics Database (CTD) provides
relationships, such as chemical-gene/protein interactions,
chemical-disease as well as gene-disease (26). Relationship between disease
(Nephrosis, congenital) associated chemical-gene or interactions
was revealed by CDT. Then Chemical-DEGs interaction network was
constructed by Cytoscape software.
Results
DEGs investigation in mild, moderate
and severe groups
Considering a huge amount of calculation in gene
expression profile, the original data was analyzed and filtered.
The results revealed that totally 220 up-regulated DEGs and 225
down-regulated DEGs were obtained in the mild group. In the
moderate group, there were 375 up-regulated DEGs and 142
down-regulated DEGs screened out. Furthermore, a total of 484
up-regulated DEGs and 280 down-regulated DEGs uncovered in the
severe group.
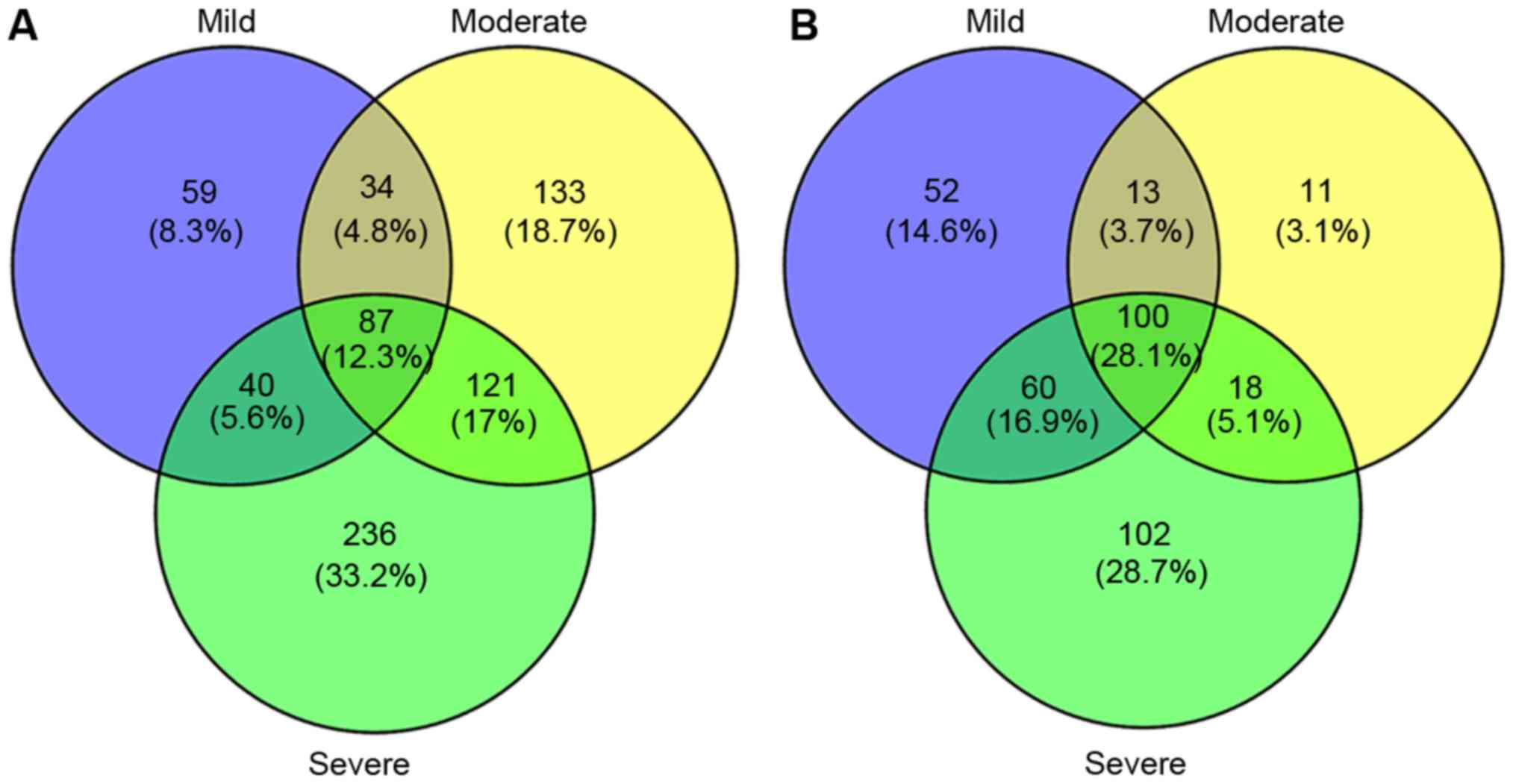
VennPlot analysis
VennPlot for DEGs in the mild, moderate, and severe
groups were investigated in the present study (Fig. 1). The results revealed 187
co-regulated DEGs in Set 1, including 87 up-regulated DEGs and 100
down-regulated DEGs. In Set 1, Serpina6, Hpd, and
Nr4a1 were the 3 most outstanding up-regulated genes, while
Proz, 4122401K19Rik, and Nudt19 were the 3 most
outstanding down-regulated genes. Meanwhile, there were 139 DEGs in
Set 2, including 121 up-regulated DEGs and 18 down-regulated DEGs.
In Set 2, Sprr2f, Lcn2 and Saa2 were the 3 most
outstanding up-regulated gene, while Symbol, Vps8 and
Ang2 were the 3 most down-regulated gene.
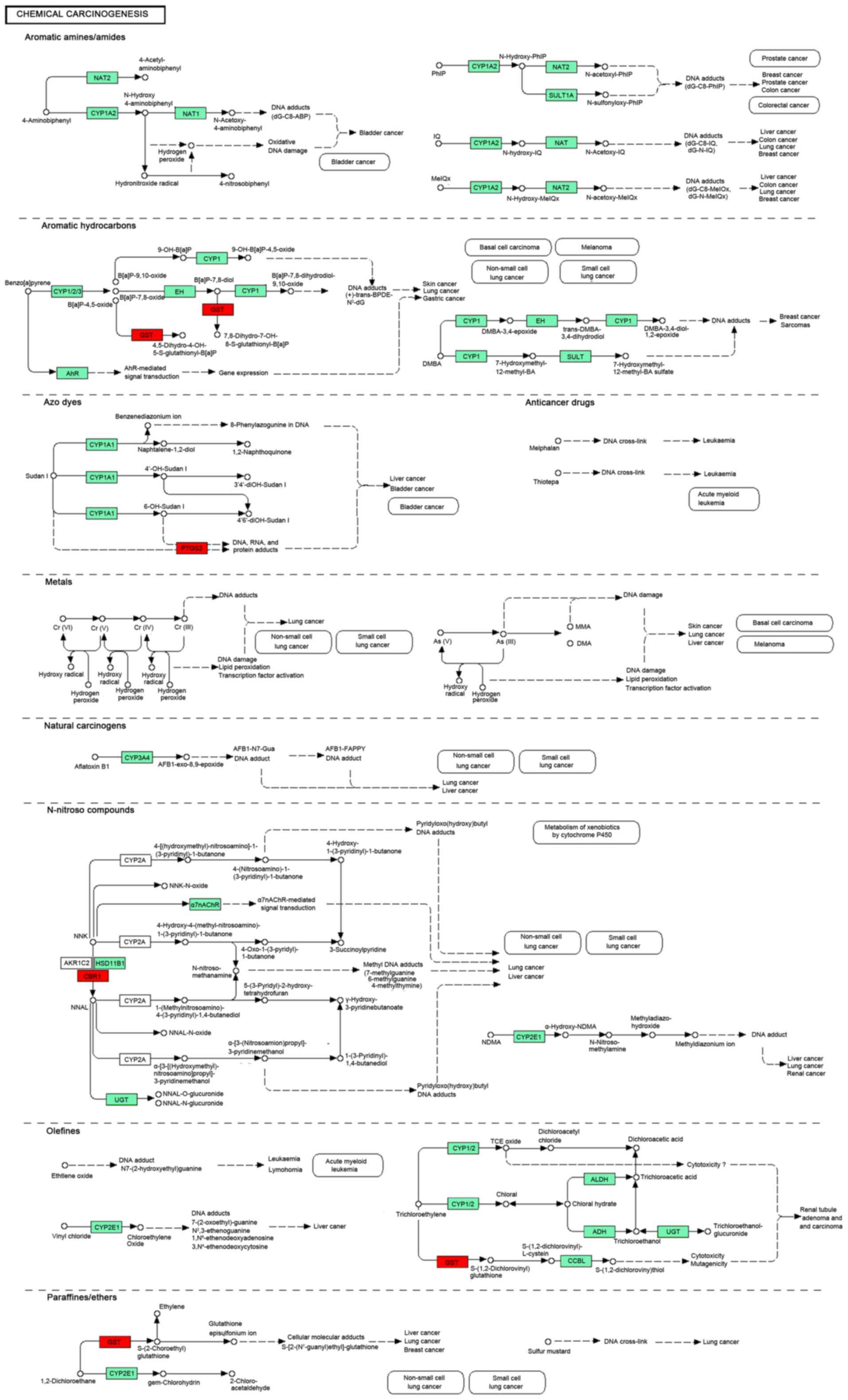
KEGG and GO enrichment analysis
The KEGG analysis of DEGs in Set 1 showed that a
total of 16 pathways were significantly enriched (Table I). The up-regulated DEGs were
mostly enriched in TNF signaling pathway (mmu04668,
P=3.59×10−4), while the down-regulated DEGs were mainly
assembled in Retinol metabolism (mmu00830, P=1.36×10−7).
Among these pathways, Chemical carcinogenesis (mmu05204, Gene:
CBR1, UGT2B38, GSTA2, UGT2B37 and UGT2B36) was the
common pathway in all 3 group of Set 1. The detail pathway map of
Chemical carcinogenesis was showed in Fig. 2.
 | Table I.Kyoto Encyclopedia of Genes and
Genomes pathway enrichment analysis for common DEGs in the mild,
moderate and severe groups. |
Table I.
Kyoto Encyclopedia of Genes and
Genomes pathway enrichment analysis for common DEGs in the mild,
moderate and severe groups.
| Regulation | Pathway ID | Pathway name | Count | P-value |
|---|
| ALL | mmu00980 | Metabolism of
xenobiotics by cytochrome P450 | 9 | 2.84×10-7 |
|
| mmu00830 | Retinol
metabolism | 10 | 3.16×10-7 |
|
| mmu05204 | Chemical
carcinogenesis | 10 | 4.21×10-7 |
|
| mmu00982 | Drug
metabolism-cytochrome P450 | 8 | 5.09×10-6 |
|
| mmu00053 | Ascorbate and
aldarate metabolism | 5 | 1.64×10-4 |
|
| mmu01100 | Metabolic
pathways | 27 | 4.04×10-4 |
|
| mmu00040 | Pentose and
glucuronate interconversions | 5 | 5.13×10-4 |
|
| mmu00860 | Porphyrin and
chlorophyll metabolism | 5 | 8.48×10-4 |
|
| mmu00983 | Drug
metabolism-other enzymes | 5 | 1.93×10-3 |
|
| mmu00140 | Steroid hormone
biosynthesis | 6 | 2.12×10-3 |
|
| mmu00590 | Arachidonic acid
metabolism | 6 | 2.34×10-3 |
|
| mmu04668 | TNF signaling
pathway | 6 | 5.60×10-3 |
|
| mmu04976 | Bile secretion | 5 | 6.42×10-3 |
|
| mmu00071 | Fatty acid
degradation | 4 | 1.45×10-2 |
|
| mmu05140 | Leishmaniasis | 4 | 2.93×10-2 |
|
| mmu04610 | Complement and
coagulation cascades | 4 | 4.53×10-2 |
| UP | mmu04668 | TNF signaling
pathway | 6 | 3.59×10-4 |
|
| mmu05140 | Leishmaniasis | 4 | 5.65×10-3 |
|
| mmu04010 | MAPK signaling
pathway | 6 | 1.39×10-2 |
|
| mmu05204 | Chemical
carcinogenesis | 4 | 1.52×10-2 |
|
| mmu05152 | Tuberculosis | 5 | 1.74×10-2 |
|
| mmu05166 | HTLV–I
infection | 6 | 2.01×10-2 |
|
| mmu05142 | Chagas disease
(American trypanosomiasis) | 4 | 2.06×10-2 |
|
| mmu04978 | Mineral
absorption | 3 | 2.26×10-2 |
| DOWN | mmu00830 | Retinol
metabolism | 8 | 1.36×10-7 |
|
| mmu00053 | Ascorbate and
aldarate metabolism | 5 | 6.48×10-6 |
|
| mmu00980 | Metabolism of
xenobiotics by cytochrome P450 | 6 | 1.03×10-5 |
|
| mmu00982 | Drug
metabolism-cytochrome P450 | 6 | 1.20×10-5 |
|
| mmu01100 | Metabolic
pathways | 18 | 1.73×10-5 |
|
| mmu00040 | Pentose and
glucuronate interconversions | 5 | 2.11×10-5 |
|
| mmu00860 | Porphyrin and
chlorophyll metabolism | 5 | 3.57×10-5 |
|
| mmu00140 | Steroid hormone
biosynthesis | 6 | 4.64×10-5 |
|
| mmu05204 | Chemical
carcinogenesis | 6 | 6.07×10-5 |
|
| mmu00983 | Drug
metabolism-other enzymes | 5 | 8.52×10-5 |
|
| mmu04146 | Peroxisome | 4 | 6.63×10-3 |
|
| mmu00071 | Fatty acid
degradation | 3 | 2.17×10-2 |
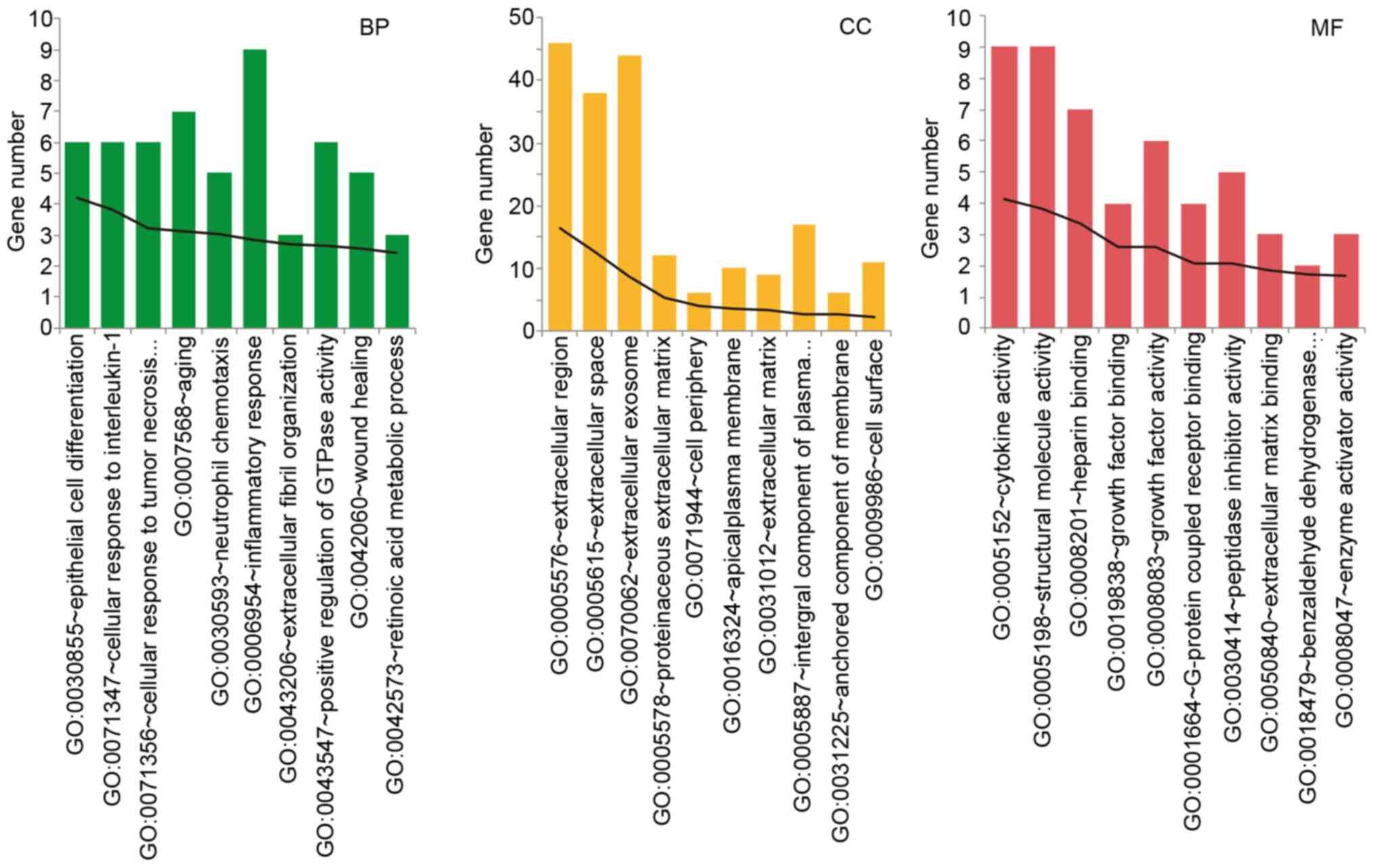
GO functional analysis was performed in the severe
group DEGs. With count number >10, the top 10 of BP, CC, and MF
functions were revealed, respectively. The DEGs in the severe group
was mainly assembled in functions like inflammatory response (BP,
GO: 0006954, P=1.43×10−3), cellular response to
interleukin-1 (BP, GO: 0071347, P=8.87×10−3), cellular
response to tumor necrosis (BP, GO: 0071356,
P=6.34×10−4), extracellular region (CC, GO: 0005576,
P=3.53×10−7), and cytokine activity (MF, GO: 0005125,
P=7.63×10−5). The detail information was showed in
Fig. 3.
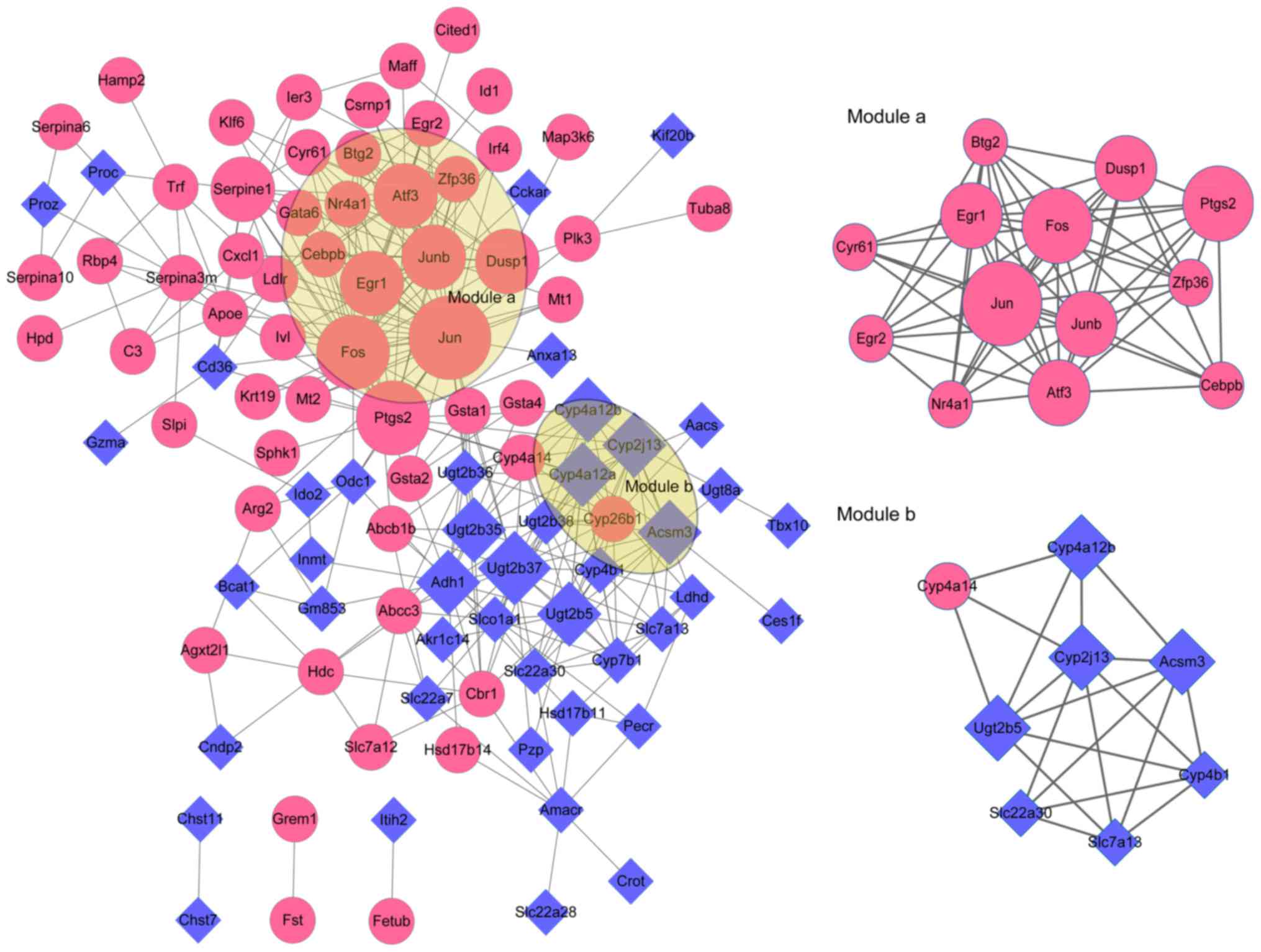
PPI network and modules analysis
To dig out the potential interactions of the DEGs,
PPI network and related modules were constructed based on the
protein interactions of DEGs. A total of 102 nodes (Jun, Fos,
Ptgs2, Ugt2b37, Junb, Atf3, Cyp4a12a, Adh1, Ugt2b5, Egr1, etc.)
and 320 interactions were identified in the PPI network of Set 1.
With score >4.5, 2 modules were further obtained from the Set 1
PPI network. There were 13 nodes (Jun, Fos, Ptgs2, Junb, Atf3,
Egr1, Dusp1, Nr4a1, Zfp36, Btg2, etc.) and 60 interactions in
module a (score=10), while 8 nodes (Ugt2b5, Cyp2j13, Acsm3,
Cyp4a12b, Cyp4a14, Cyp4b1 Slc22a30 and Slc7a13) and 20
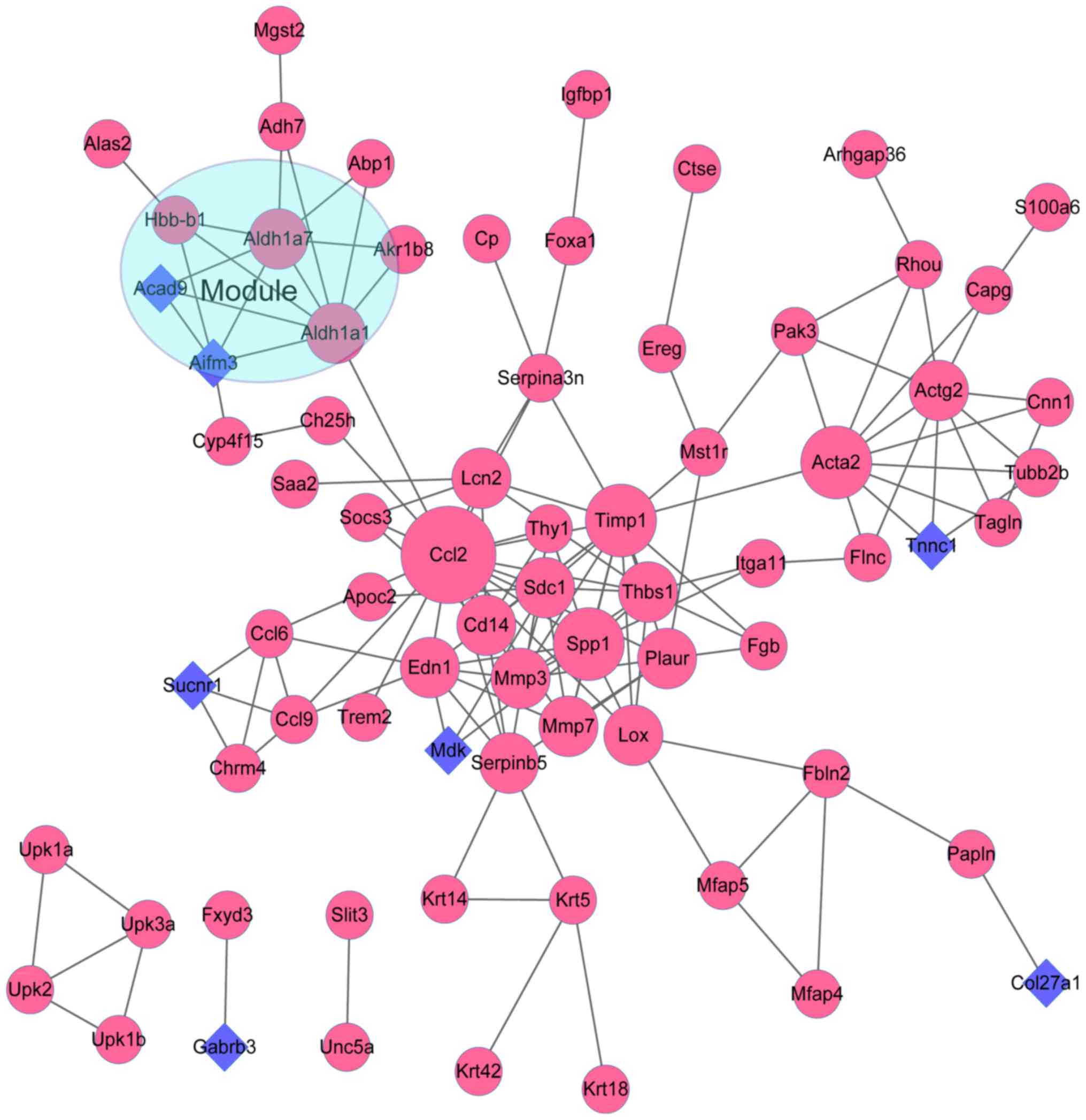
interactions contained in module b (score=5.714) (Fig. 4). Furthermore, there were a total
of 73 nodes (Ccl2, Timp1, Acta2, Spp1, Actg2, Thbs1, Edn1,
Aldh1a1, Mmp3, Aldh1a7, etc.) and 143 interactions included in
the PPI network of Set 2. With score >4.5, one module was
further obtained from the Set 2 PPI network. A total of 5 nodes
(Aldh1a1, Aldh1a7, Aifm3, Hbb-b1 and Acad9) and 9
interactions were revealed in the module of Set 2 (Fig. 5).
TF-target gene regulatory network
analysis
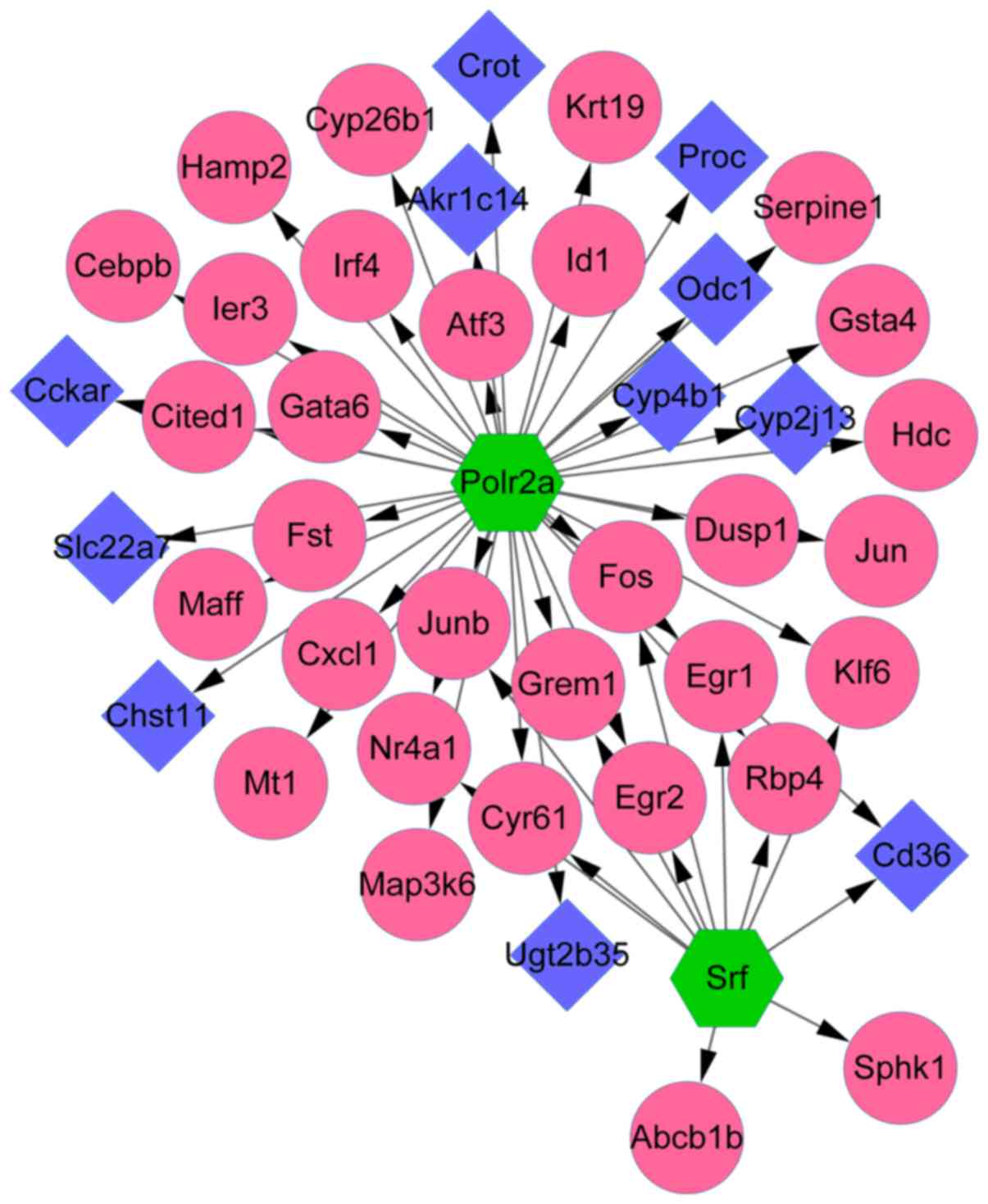
By using the iRegulon software, 2 TFs (Polr2a
and Srf) were obtained from the Set 1 PPI network. To
further investigate the relationship between TFs and their target
genes, the TF-target gene network was visualized by cytoscape
software. With NES >4, the results showed that there were
totally 52 regulatory relations contained in this network. Some
genes like FOS were co-regulated by both Polr2a and
Srf. The detail information was showed in Fig. 6.
Chemical-gene interaction network
analysis
The present study revealed totally 12012
chemical-gene interactions related to Nephrosis or congenital.
Among these interactions, totally 60 interactions were matched with
the 139 DEGs in Set 2. The results showed that Cyclosporine was the
most important chemical that target with either the most number of
up-regulated or down-regulated DEGs in Set 2. Detailed information
was listed in Table II.
| Table II.Chemical-target gene interaction
network. |
Table II.
Chemical-target gene interaction
network.
| Gene | Chemical name | Description | Gene | Chemical name | Description | Gene | Chemical name | Description |
|---|
| ACTA2 | Cyclosporine | UP | EREG | Cyclosporine | UP | RGS16 | Cyclosporine | UP |
| ACTG2 | Cyclosporine | UP | FGB | Cyclosporine | UP | RHOU | Cyclosporine | UP |
| AIFM3 | Cyclosporine | DOWN | FOXA1 | Cyclosporine | UP | S100A6 | Cyclosporine | UP |
| ALDH1A1 | Cyclosporine | UP | FOXQ1 | Cyclosporine | UP | SAA2 | Cyclosporine | UP |
| APOC2 | Cyclosporine | UP | GDF15 | Cyclosporine | UP | SAMD5 | Cyclosporine | UP |
| BCAS1 | Cyclosporine | UP | GPR87 | Cyclosporine | UP | SDC1 | Cyclosporine | UP |
| BCL2L14 | Cyclosporine | UP | IGFBP1 | Cyclosporine | UP | SERPINA3N | Cyclosporine | UP |
| CAPG | Cyclosporine | UP | IGFBP6 | Cyclosporine | UP | SLC16A3 | Cyclosporine | UP |
| CCL2 | Cyclosporine | UP | ITGA11 | Cyclosporine | UP | SLC17A3 | Cyclosporine | DOWN |
| CCL6 | Cyclosporine | UP | LCN2 | Cyclosporine | UP | SLC39A4 | Cyclosporine | UP |
| CD14 | Cyclosporine | UP | LOX | Cyclosporine | UP | SPP1 | Cyclosporine | UP |
| CES1E | Cyclosporine | DOWN | LTBP2 | Cyclosporine | UP | TACSTD2 | Cyclosporine | UP |
| CLDN4 | Cyclosporine | UP | MDK | Cyclosporine | DOWN | TAGLN | Cyclosporine | UP |
| CNN1 | Cyclosporine | UP | MFAP5 | Cyclosporine | UP | TGFB3 | Cyclosporine | UP |
| COL27A1 | Cyclosporine | DOWN | MGST2 | Cyclosporine | UP | THBS1 | Cyclosporine | UP |
| CP | Cyclosporine | UP | MMP3 | Cyclosporine | UP | TIMP1 | Cyclosporine | UP |
| CPE | Cyclosporine | UP | MMP7 | Cyclosporine | UP | TRPV6 | Cyclosporine | UP |
| CTSE | Cyclosporine | UP | OSMR | Cyclosporine | UP | TUBB2B | Cyclosporine | UP |
| EDN1 | Cyclosporine | UP | PAK3 | Cyclosporine | UP | UCHL1 | Cyclosporine | UP |
| ENTPD3 | Azathioprine | UP | PLAUR | Cyclosporine | UP | UPK3A | Cyclosporine | UP |
Discussion
CON is the leading cause of chronic kidney disease
in child. However, the molecular mechanisms underlying disease
progression are still poorly defined. To reveal the potential genes
or pathways associated with CON, the present bioinformatics study
was performed on mild, moderate, and severe CONs. The results
showed that totally 187 and 139 co-regulated DEGs were obtained in
Set 1 and Set 2, respectively. KEGG analysis for Set 1 showed that
DEGs were mainly enriched in pathways like chemical carcinogenesis.
Meanwhile, the GO analysis for Set 2 showed that DEGs were mainly
assembled in functions like cellular response to interleukin-1 and
cellular response to tumor necrosis. Two modules in Set 1 and one
module in Set 2 were revealed based on PPI network analysis.
Furthermore, the genes like FOS were co-regulated by TFs
like Polr2a and Srf. Cyclosporine was the most
important chemical that target with DEGs in Set 2.
Based on the Set 1 (including all disease status
like mild, moderate and severe), KEGG pathway analysis, PPI network
analysis, and TF-target investigation were carried out in this
study to provide a potential mechanism understanding for the
process of CON. Chemical carcinogenesis is a process that the cells
need genetic and epigenetic variations based on elements including
oncogenes and tumor suppressors (27,28).
Carbonyl reductase 1 (CBR1) metabolized by the body
many toxic environmental quinones and pharmacological relevant
substrates, such as the anticancer doxorubicin (27). Previous study has shown that
CBR1 is vital for the cell protection in functions like
oxidative stress and apoptosis (29). Pathway analysis showed that the
chemical carcinogenesis was the common pathway enriched by both up-
and down-regulated DEGs (especially CBR1 in Set 1). Thus, we
speculated that DEGs like CBR1 in chemical carcinogenesis pathway
might be the potential pathogenesis of CON. However, the related
study associated with chemical carcinogenesis pathway about
CBR1 and CON is very rare. Furthermore, c-Fos
(FOS) gene has been considered as the regulators of cell
proliferation, differentiation, transformation, and apoptotic cell
death (30,31). Otsuka et al (32) indicates that the expression of
FOS is potentially an early indicator of late radiation
damage to the kidney. In rats model, previous study indicated that
the renoprotective effects of drugs partly through inhibiting the
upregulation of FOS mRNA and protein in renal cortices
(33). In the present study, the
PPI network analysis showed that FOS was one of the most
outstanding DEGs in Set 1. Interestingly, the TF-target gene
network in this study showed that FOS was the co-target gene
of Polr2a and Srf. Thus, we speculated that DEGs like
FOS might play an important role in the process of CON via
TFs like Polr2a and Srf. However, the related study
associated with FOS and CON is very rare. Thus, an
additional research with a wide scale of gene expression analysis
is necessary to confirm this speculation.
Based on Set 2 (only severe group), GO function
analysis and chemical-gene interaction network investigation in
this study were performed and intended to provide a potential
therapy strategy for CON. Interleukin-1 family plays a vital role
in the immune and inflammatory responses regulation (34). Previous study showed that the
protective effect of interleukin-1 in renal injury might be induced
by reducing the induction of the nuclear factors (35). Actually, Pindjakova et al
(36) indicate that the production
of interleukin-1 based on the resident dendritic cells enhances the
situation of acute kidney injury. Moreover, tumor necrosis factor
(TNF) is a superfamily of cytokines that can lead to cell death,
and it is a potent pyrogen that can be caused by stimulation of
interleukin-1 secretion (37).
Meldrum et al (38) prove
that TNF-α mediates renal tubular cell apoptosis in the process of
obstructive uropathy. Recent study indicates that the production of
TNF-α by macrophages take part in the progression of renal injury
(39). Actually, interleukin-1 and
TNF-α gene polymorphism are associated with the susceptibility,
disease activity and long-term outcome in human nephropathy
(40). In the current research,
the results of GO analysis showed that cellular response to
interleukin-1 and cellular response to tumor necrosis is two of
most outstanding function assembled by DEGs. Thus, we speculated
that these two functions might be vital for the process of severe
CON. The deteriorated or even canceration might take place in
severe CON, which needed an early therapy intervention.
Furthermore, cyclosporine, also spelled ciclosporin or cyclosporin,
is an immunosuppressant medication and natural product (41). As an immunosuppressant with
therapeutic indications in various immunological diseases, the use
of cyclosporine is reported to be associated with chronic
nephropathy (42). In the present
study, cyclosporine was the most outstanding chemical associated
with DEGs identified in severe CON. Thus, cyclosporine might still
be the preferences for the therapy of CON. However, due to the
negative effects of cyclosporine in CON clinical treatment, a more
effective drug with less adverse reaction is needed.
However, there were still some limitations in this
study. First, findings in the present study were all analyzed by
bioinformatics methods, and experimental validations were
deficient. Second, parameters used in in silico analyses
were set manually, and some results might be missed or
overemphasized. Therefore, further experimental validations in both
in vivo and in vitro were required in the future.
In conclusion, the DEGs like CBR1 and
FOS, TFs like Polr2a and Srf, as well as
pathways like chemical carcinogenesis might play important roles in
the process of CON. Interleukin-1 and tumor necrosis function were
associated with the deteriorate of CON, and might be novel targets
for CON treatment. Furthermore, a more effective drug with less
adverse reaction for severe CON is needed.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GX contributed to the conceptualization of the
research. GX and RC acquired the data and performed the analysis.
RC and XZ interpreted the data. GX wrote the manuscript, and XZ
revised the manuscript for important intellectual content.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Piepsz A, Ham H and Josephson S: RE:
Biomarkers of congenital obstructive nephropathy: Past, present and
future. J Urol. 173:2207–2208. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ingraham SE and Mchugh KM: Current
perspectives on congenital obstructive nephropathy. Pediatr
Nephrol. 26:1453–1461. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chevalier RL, Thornhill BA, Forbes MS and
Kiley SC: Mechanisms of renal injury and progression of renal
disease in congenital obstructive nephropathy. Pediatr Nephrol.
25:687–697. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liapis H: Biology of congenital
obstructive nephropathy. Nephron Exp Nephrol. 93:e87–e91. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chevalier RL: Prognostic factors and
biomarkers of congenital obstructive nephropathy. Pediatr Nephrol.
31:1411–1420. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hahn H, Ku SE, Kim KS, Park YS, Yoon CH
and Cheong HI: Implication of genetic variations in congenital
obstructive nephropathy. Pediatr Nephrol. 20:1541–1544. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grandaliano G, Gesualdo L, Bartoli F,
Ranieri E, Monno R, Leggio A, Paradies G, Caldarulo E, Infante B
and Schena FP: MCP-1 and EGF renal expression and urine excretion
in human congenital obstructive nephropathy. Kidney Int.
58:182–192. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Inazaki K, Kanamaru Y, Kojima Y, Sueyoshi
N, Okumura K, Kaneko K, Yamashiro Y, Ogawa H and Nakao A: Smad3
deficiency attenuates renal fibrosis, inflammation, and apoptosis
after unilateral ureteral obstruction. Kidney Int. 66:597–604.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hermens JS, Thelen P, Ringert RH and
Seseke F: Alterations of selected genes of the Wnt signal chain in
rat kidneys with spontaneous congenital obstructive uropathy. J
Pediatr Urol. 3:86–95. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chevalier RL: Promise for gene therapy in
obstructive nephropathy. Kidney Int. 66:1709–1710. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chevalier RL: Obstructive nephropathy:
Towards biomarker discovery and gene therapy. Nat Clin Pract
Nephrol. 2:157–168. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Becknell B, Carpenter AR, Allen JL,
Wilhide ME, Ingraham SE, Hains DS and McHugh KM: Molecular basis of
renal adaptation in a murine model of congenital obstructive
nephropathy. PLoS One. 8:e727622013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Carpenter AR, Becknell B, Ingraham SE and
McHugh KM: Ultrasound imaging of the murine kidney. Methods Mol
Biol. 886:403–410. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Smyth GK: Limma: Linear models for
microarray data. In: Bioinformatics and Computational Biology
Solutions Using R and BioconductorStatistics for Biology and
Health. Gentleman R, Carey VJ, Huber W, Irizarry RA and Dudoit S:
Springer; New York, NY: pp. 397–420. 2005, View Article : Google Scholar
|
|
17
|
Oliveros JC: Venny. An interactive tool
for comparing lists with Venn Diagrams. BioinfoGP of CNB-CSIC.
http://bioinfogp.cnnb.csic.es/tools/venny/index.htJuly
1–2017
|
|
18
|
da Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gene Ontology Consortium: The gene
ontology in 2010: Extensions and refinements. Nucleic Acids Res.
38:(Database Issue). D331–D335. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43:(Database Issue). D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bandettini WP, Kellman P, Mancini C,
Booker OJ, Vasu S, Leung SW, Wilson JR, Shanbhag SM, Chen MY and
Arai AE: MultiContrast Delayed Enhancement (MCODE) improves
detection of subendocardial myocardial infarction by late
gadolinium enhancement cardiovascular magnetic resonance: A
clinical validation study. J Cardiovasc Magn Reson. 14:832012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang Y, Li M, Wang J, Pan Y and Wu FX:
CytoNCA: A cytoscape plugin for centrality analysis and evaluation
of protein interaction networks. Biosystems. 127:67–72. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Janky R, Verfaillie A, Imrichová H, Van de
Sande B, Standaert L, Christiaens V, Hulselmans G, Herten K,
Sanchez Naval M, Potier D, et al: iRegulon: From a gene list to a
gene regulatory network using large motif and track collections.
PLoS Comput Biol. 10:e10037312014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Davis AP, Grondin CJ, Lennon-Hopkins K,
Saraceni-Richards C, Sciaky D, King BL, Wiegers TC and Mattingly
CJ: The comparative toxicogenomics database's 10th year
anniversary: Update 2015. Nucleic Acids Res. 43:(Database Issue).
D914–D920. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Arlt VM, Zuo J, Trenz K, Roufosse CA, Lord
GM, Nortier JL, Schmeiser HH, Hollstein M and Phillips DH: Gene
expression changes induced by the human carcinogen aristolochic
acid I in renal and hepatic tissue of mice. Int J Cancer.
128:21–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jin K, Su KK, Li T, Zhu XQ, Wang Q, Ge RS,
Pan ZF, Wu BW, Ge LJ, Zhang YH, et al: Hepatic premalignant
alterations triggered by human nephrotoxin aristolochic acid I in
canines. Cancer Prev Res (Phila). 9:324–334. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ismail E, Al-Mulla F, Tsuchida S, Suto K,
Motley P, Harrison PR and Birnie GD: Carbonyl reductase: A novel
metastasis-modulating function. Cancer Res. 60:1173–1176.
2000.PubMed/NCBI
|
|
30
|
Gao S, Tang K, Zhang J, Yang Z, Cui H, Li
P, Tang H and Zhou M: Department of Orthopedics, Orthopedic Center
of Chinese PLA, Southwest Hospital, Third Military Medical
University; Department of Neurobiology, Third Military Medical
University: Effect of cyclic stretch on expression of c-fos gene in
rat Achilles-derived tendon stem cells. Chin J Rep Reconstr Surg.
2017.
|
|
31
|
Ge R, Wang Z, Zeng Q, Xu X and Olumi AF:
F-box protein 10, an NF-κB-dependent anti-apoptotic protein,
regulates TRAIL-induced apoptosis through modulating c-Fos/c-FLIP
pathway. Cell Death Differ. 18:1184–1195. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Otsuka M, Hatakenaka M, Ishigami K and
Masuda K: Expression of the c-myc and c-fos genes as a potential
indicator of late radiation damage to the kidney. Int J Radiat
Oncol Biol Phys. 49:169–173. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou F, Zhang L, Su Y, Zhang J and An Z:
Inhibitory effect of artemisinin on the upregulation of c-fos and
c-jun gene expression in kidney tissue of diabetic rats. Modern
Journal of Integrated Traditional Chinese & Western Medicine.
23:2294–2295. 2014.
|
|
34
|
Yazdi AS and Ghoreschi K: The
interleukin-1 family. Adv Exp Med Biol. 941:21–29. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rusai K, Huang H, Sayed N, Strobl M, Roos
M, Schmaderer C, Heemann U and Lutz J: Administration of
interleukin-1 receptor antagonist ameliorates renal
ischemia-reperfusion injury. Transpl Int. 21:572–580. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pindjakova J, Hanley SA, Duffy MM, Sutton
CE, Weidhofer GA, Miller MN, Nath KA, Mills KH, Ceredig R and
Griffin MD: Interleukin-1 accounts for intrarenal Th17 cell
activation during ureteral obstruction. Kidney Int. 81:379–390.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wajant H, Pfizenmaier K and Scheurich P:
Tumor necrosis factor signaling. Cell Death Differ. 10:45–65. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Meldrum K, Misseri R, Rink R and Meldrum
D: Tumor necrosis factor alpha (TNF) mediates renal tubular cell
apoptosis during obstructive uropathy. J Am Coll Surg.
197:S912003.
|
|
39
|
Awad AS, You H, Gao T, Cooper TK,
Nedospasov SA, Vacher J, Wilkinson PF, Farrell FX and Reeves Brian
W: Macrophage-derived tumor necrosis factor-α mediates diabetic
renal injury. Kidney Int. 88:722–733. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shu KH, Lee SH, Cheng CH, Wu MJ and Lian
JD: Impact of interleukin-1 receptor antagonist and tumor necrosis
factor-alpha gene polymorphism on IgA nephropathy. Kidney Int.
58:783–789. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
World Health Organization, . Stuart MC,
Kouimtzi M and Hill S: WHO model formulary 2008. World Health
Organization; Geneva: 2009
|
|
42
|
Sattarinezhad E, Panjehshahin MR,
Torabinezhad S, Kamali-Sarvestani E, Farjadian S, Pirsalami F and
Moezi L: Protective effect of edaravone against
cyclosporine-induced chronic nephropathy through antioxidant and
nitric oxide modulating pathways in rats. Iran J Med Sci.
42:170–178. 2017.PubMed/NCBI
|