Introduction
Laryngeal carcinoma (LC), the second most common
type of head and neck cancer, seriously threatens the health and
longevity of individuals (1).
Among all the subtypes of LC, laryngeal squamous cell carcinoma
(LSCC) accounts for 95–98% of cases (1–3).
Incidence of LSCC ranges from 3–10/100,000 in the USA alone and
LSCC causes 2.1% of all cancer deaths worldwide, demonstrating that
LSCC has become a worldwide public health problem (4,5).
Beneath the complexity of every cancer lies a number of critical
events that occur in the cell cycle that determine growth or
shrinkage of the tumor. Apoptosis, which has an important role in
chemotherapy, is a target of potent and specific therapeutics
(6,7). Carboplatin (CBDCA) is a
platinum-based agent that has been widely used in cancer
chemotherapy. It is characterized by its ability to generate DNA
lesions, thereby inhibiting replication and transcription, and
finally leading to apoptosis (8,9).
Specifically, CBDCA enters the cell by passive diffusion and
undergoes hydrolysis to assume a form that interacts with
nucleophilic purine bases in the DNA strand, resulting in major
intra-strand cross linkages and minor inter-strand cross linkages.
Subsequently, the cross linkages inhibit the process of DNA
replication, causing errors in transcription (10–12).
CBDCA was approved by the US Food and Drug Administration in the
1980s, and since then it has been regularly used in the treatment
of several types of tumors (13).
Despite its extensive clinical application, the
anticancer efficacy and safety of CBDCA remain important issues
(14). The sensitivity of CBDCA
therapy is a major obstacle to its successful clinical application
(15,16). Due to the low incidence of side
effects of CBDCA in clinical treatment, it is widely used in
combination with other strategies (17,18).
What may be inferred, therefore, is that understanding the factors
and underlying mechanisms of cancer cell apoptosis during CBDCA
treatment may provide valuable insight for the rational design of
more efficient therapeutic strategies, as well as development of
novel platinum-based agents.
It has been widely recognized that apoptosis is
initiated via two different routes: The death receptor pathway and
the mitochondrial pathway (19–21).
In the present study, the HN-3 cells were exposed to CBDCA to
demonstrate the order of the occurrence of apoptotic phenomena, as
well as mitochondrial activation, and the mechanism between them
was investigated. In addition, it was reported that increased
levels of glutathione (GSH), which is a reactive oxygen species
(ROS) scavenger, may induce tumor cell resistance to CBDCA
(22,23). Therefore, the effect of GSH on the
mitochondrial activity and apoptosis of HN-3 cells was also
investigated to extensively verify their roles in apoptosis.
Materials and methods
Cell culture
The laryngeal squamous cell carcinoma cell line HN-3
was kindly provided by the Asan Medical Center (Seoul, Korea). The
cell line was cultured in RPMI-1640 medium (Hyclone; GE Healthcare
Life Sciences, Logan, UT, USA) supplemented with 10% fetal bovine
serum (Hyclone; GE Healthcare Life Sciences), penicillin (50 U/ml)
and streptomycin (50 µg/ml) at 37°C in a humidified incubator with
5% CO2 and 95% air. CBDCA was obtained from Qilu
Pharmaceutical Co., Ltd. (Jinan, China). GSH was purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Cell viability assay
An MTT assay was used to measure cell viability. The
cells were treated by CBDCA (0–5.0 mg/ml; 0–48 h) once 90%
confluence was reached. Then the cells were incubated with 50 µl
MTT solution (2 mg/ml) for 2 h. The MTT solution was exchanged with
100 µl dimethyl sulfoxide, shaken for 20 sec and the absorbance at
540 nm was measured. Cell viability was calculated according to the
following equation: Cell viability (%)=(mean absorbance in
treatment group/mean absorbance in control group) ×100.
Hoechst 33342 and propidium iodide
(PI) double staining
The concentration of GSH used was 5 mM. When the
cells (5×105 cells/well) were at 90% confluence, the
samples were treated by carboplatin with or without GSH at 37°C for
12 h. Following cultivation with CBDCA with or without GSH, cells
were stained with Hoechst 33342/PI and observed by confocal
microscopy as previously described (24). CBDCA-induced apoptosis was
quantified according to the percentage of Hoechst 33342 and/or
PI-positive stained cells by guavaSoft version 3.11 (EMD Millipore,
Billerica, MA, USA).
Assessment of mitochondrial membrane
potential (MMP)
In brief, CBDCA-treated cells (the incubation began
when the cell fusion degree was around 60%) cultured for 6 and 12
h, as well as the cells cultured with CBDCA and/or GSH for 12 h,
were stained with rhodamine 123 (1 µM; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) for 30 min at 37°C for observation by
confocal microscopy, and further monitored using a flow cytometer
(BD Biosciences, San Jose, CA, USA) and data were analyzed with the
BD CellQuest Pro software version 2.0 (BD Biosciences) as described
previously (25).
Detection of intracellular
calcium
EGTA (2 mM) was added to chelate any residual
Ca2+ in the RPMI-1640 medium. Cells were incubated with
the Ca2+-free medium and exposed to CBDCA (0.08 mg/ml)
for 12 h at 37°C, following which cells were incubated with 4 µM
calcium Green-1 (Molecular Probes; Thermo Fisher Scientific, Inc.)
for 30 min at 37°C. Images of green calcium were collected with an
LSM-510-META confocal microscope (Zeiss AG, Oberkochen, Germany)
with an excitation wavelength of 488 nm, a 560-nm dichroic mirror,
and a 505–550-nm band pass barrier filter. Quantification of
calcium green-1 fluorescence was made by ImageJ version k1.45 (NIH,
Bethesda, MD, USA). Intracellular Ca2+ was also
monitored by flow cytometry following the same procedures described
for rhodamine 123.
Western blot analysis
Antibodies against caspase-3 (cat. no. MAB4703),
cleaved caspase-3 (cat. no. AB3623), caspase-8 (cat. no. MAB4708),
poly ADP ribose polymerase (PARP; cat. no. AM30), cleaved PARP
(AB3620) were purchased from Merck KGaA (Darmstadt, Germany);
caspase-9 (cat. no. sc-8355) and apoptosis inducing factor (AIF;
cat. no. sc-5586) antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA); Antibodies against
pro-caspase-3 (cat. no. ab32499), pro-caspase-12 (cat. no. ab8117),
caspase-12 and cleaved-caspase-12 (cat. no. ab18766), GAPDH (cat.
no. ab9485) was obtained from Abcam (Cambridge, UK); cytochrome
c antibody (cat. no. 556433) was purchased from BD
Biosciences. Horseradish peroxidase (HRP)-labeled goat anti-mouse
(cat. no. A0216) and goat anti-rabbit immunoglobulin G (cat. no.
A0208) secondary antibodies were purchased from Beyotime Institute
of Biotechnology (Jiangsu, China).
Prior to protein extraction, cells were treated with
CBDCA (0.04 or 0.08 mg/ml) with or without GSH (5 mM) for the
indicated duration. Using RIPA lysis buffers (Sigma-Aldrich; Merck
KGaA), total protein was extracted from HN-3 cells. Equivalent
amounts of protein (100 µg/lane), whose concentration was
determined by a BCA kit (Pierce; Thermo Fisher Scientific, Inc.),
were loaded onto 10% polyacrylamide gels, subjected to
electrophoresis, and transferred onto polyvinylidene fluoride
membranes. Following blocking with 5% skim milk for 1 h at room
temperature, primary antibodies were incubated overnight at 4°C
[cytochrome c (1:200); AIF (1:200); GAPDH (1:2,000); PARP
(1:200); cleaved-PARP (1:200); caspase-3 (1:1,000); cleaved
caspase-3 (1:1,000); pro-caspase-3 (1:1,000); caspase-9 (1:500);
caspase-8 (1:500); caspase-12 (1:500); pro-caspase-12 (1:500)].
Each membrane was probed with horseradish peroxidase conjugated
anti-mouse or anti-rabbit IgG antibody (1:1,000) for 1 h at room
temperature. Labeled protein bands were visualized by an ECL + plus
western blotting system kit (cat. no. RPN3352; GE Healthcare Life
Sciences, Little Chalfont, UK) and were detected by a Kodak in
vivo image analyzer (Kodak, Rochester, NY, USA). The bands were
analyzed by densitometry using ImageJ version k1.45 (NIH, Bethesda,
MD, USA).
Statistical analysis
Data are expressed as the mean ± standard deviation
(n=3). The significance of differences was evaluated by one-way
analysis of variance followed by the Least Significant Difference
procedure by GraphPad Prism 6 software (GraphPad Software, Inc., La
Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
The influence of CBDCA on HN-3
cells
The influence of CBDCA alone or in combination with
GSH on HN-3 cells was initially investigated in terms of
concentration and exposure duration.
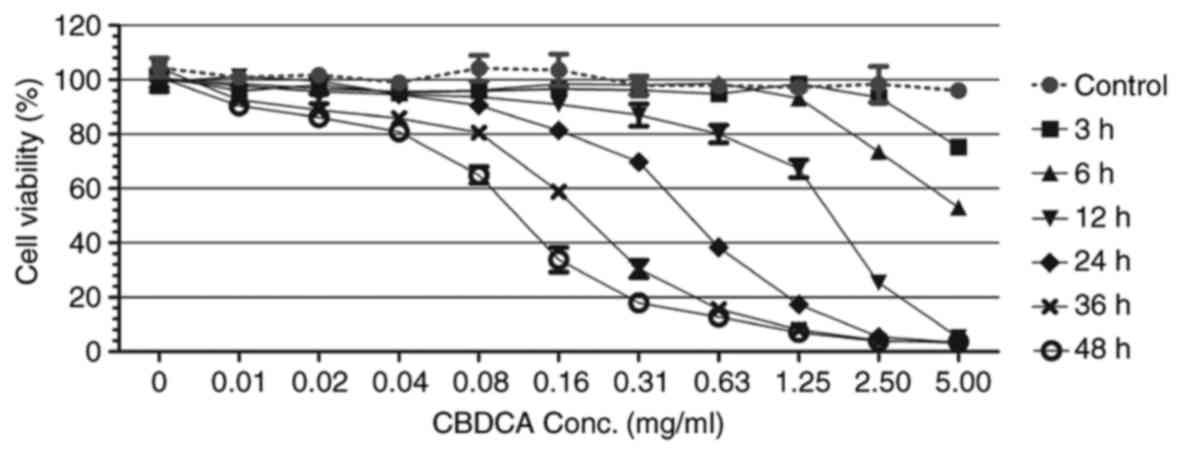
The MTT assay results revealed that HN-3 cells
exhibited various levels of cell viability, dependent on the
concentration of CBDCA as well as the exposure time (Fig. 1). For each exposure duration, there
was a minimal CBDCA concentration that resulted in a cell viability
of ≤80%. Following 3 h exposure, this minimal concentration was
~5.00 mg/ml; following 6, 12, 24, 36 and 48 h, the minimal
concentration was 2.50, 0.63, 0.16, 0.08 and 0.04 mg/ml,
respectively. This revealed a downward trend in the concentration
required as the exposure time increased.
The mode of cell death at each exposure duration,
namely apoptosis and necrosis, was evaluated by Hoechst 33342/PI
double staining following CBDCA and/or GSH treatment (Fig. 2). When exposed to 0.08 mg/ml CBDCA
(Fig. 2A), apoptosis was initiated
in a time-dependent manner; no obvious indication of apoptosis
(<20%) was observed prior to 24 h of exposure. When HN-3 cells
were exposed to various concentrations of CBDCA for 24 h,
apoptosis/necrosis was concentration dependent (Fig. 2B). In the concentration range of 0
to 0.64 mg/ml, apoptosis was significantly increased as the
concentration increased. At higher concentrations, a gradual
increase in the proportion of necrotic cells, as well as a decrease
in the proportion of apoptotic cells was observed. These results
demonstrated that the influence of CBDCA on HN-3 cells was exerted
in a dose and time-dependent manner.
In the confocal microscopy images (Fig. 2C), intact homogeneous blue nuclei
were considered viable; condensed/fragmented blue nuclei were
considered early apoptotic; condensed/fragmented pink nuclei were
considered late apoptotic; and pink intact nuclei were considered
necrotic. In the control group (no CBDCA or GSH treatment), intact
homogeneous blue and round nuclei were observed. This was also
observed in cells cultured with added GSH. Ubiquitous
condensed/fragmented blue nuclei and pink nuclei were observed in
the CBDCA treated group, whereas fewer pink nuclei were observed in
cells exposed to CBDCA and GSH simultaneously. It can be inferred
that CBDCA at a concentration of 0.08 mg/ml induced both early and
late stage apoptosis following 24 h incubation, and the addition of
GSH caused a decrease in the amount of late apoptotic cells.
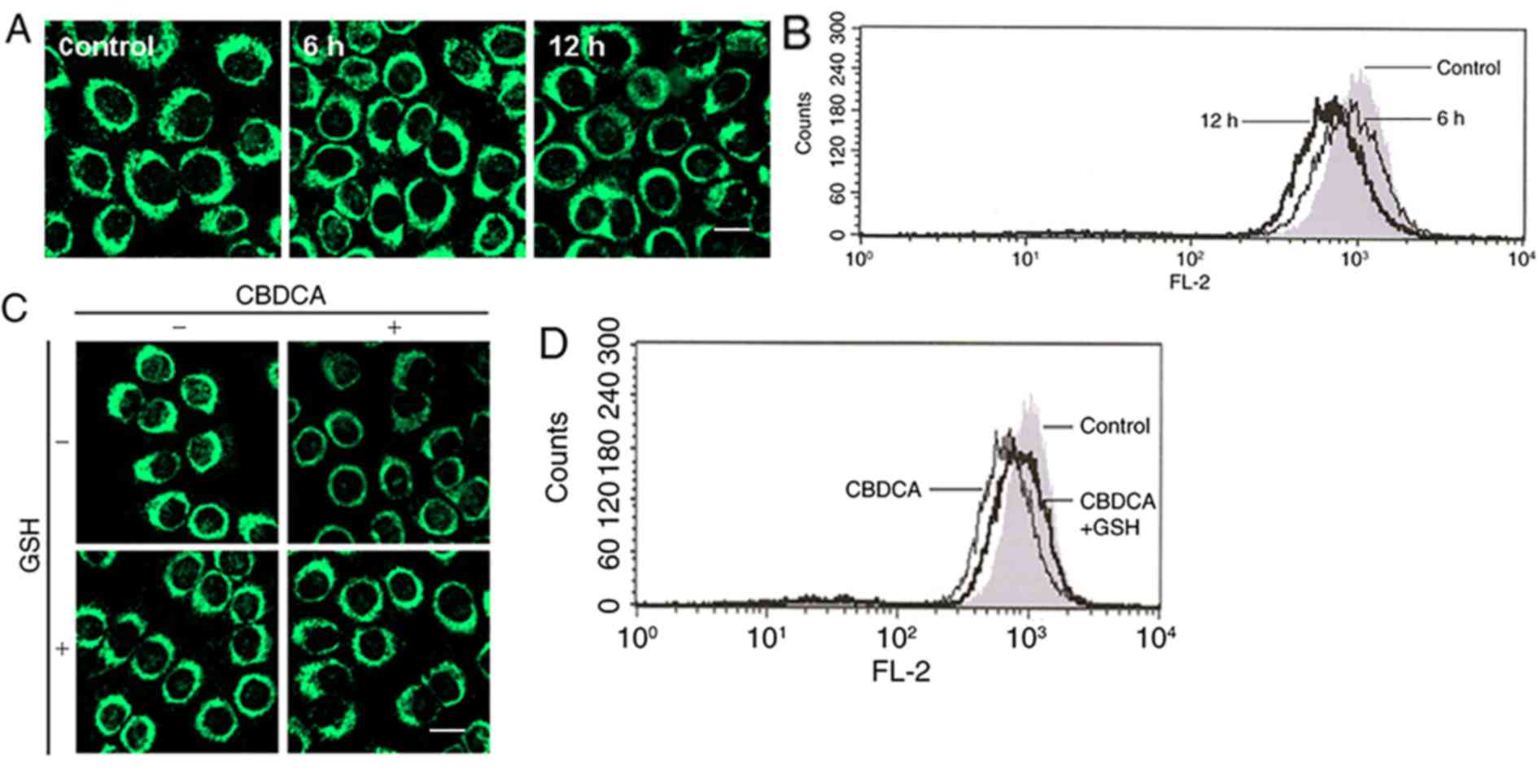
Mitochondrial depolarization
An event now recognized to be important in apoptosis
is the mitochondrial permeability transition (26,27).
In the present study, the response of mitochondria in HN-3 cells
exposed to CBDCA was investigated via staining methods and
monitoring the MMP, as well as the membrane potential (MP), in a
single cell by flow cytometry combined with the fluorescence
detection of rhodamine 123. Under confocal microscopy, it was
observed that the majority of bright fluorescent spheres became
faint following exposure to CBDCA for 12 h (Fig. 3A), and MMP was noticeably shifted
left (Fig. 3B). Based on these
results, it was concluded that mitochondrial depolarization
occurred in HN-3 cells exposed to CBDCA for 12 h. However, when
exposed to the mixture of CBDCA and GSH, the faintness and
intensity of the sphere fluorescence, as well as the MMP value,
were partially restored (Fig. 3C and
D), indicating that mitochondrial depolarization was
alleviated. No marked differences were observed in cells treated
with CBCDA alone in comparison to cells treated with GSH alone
(Fig. 3C).
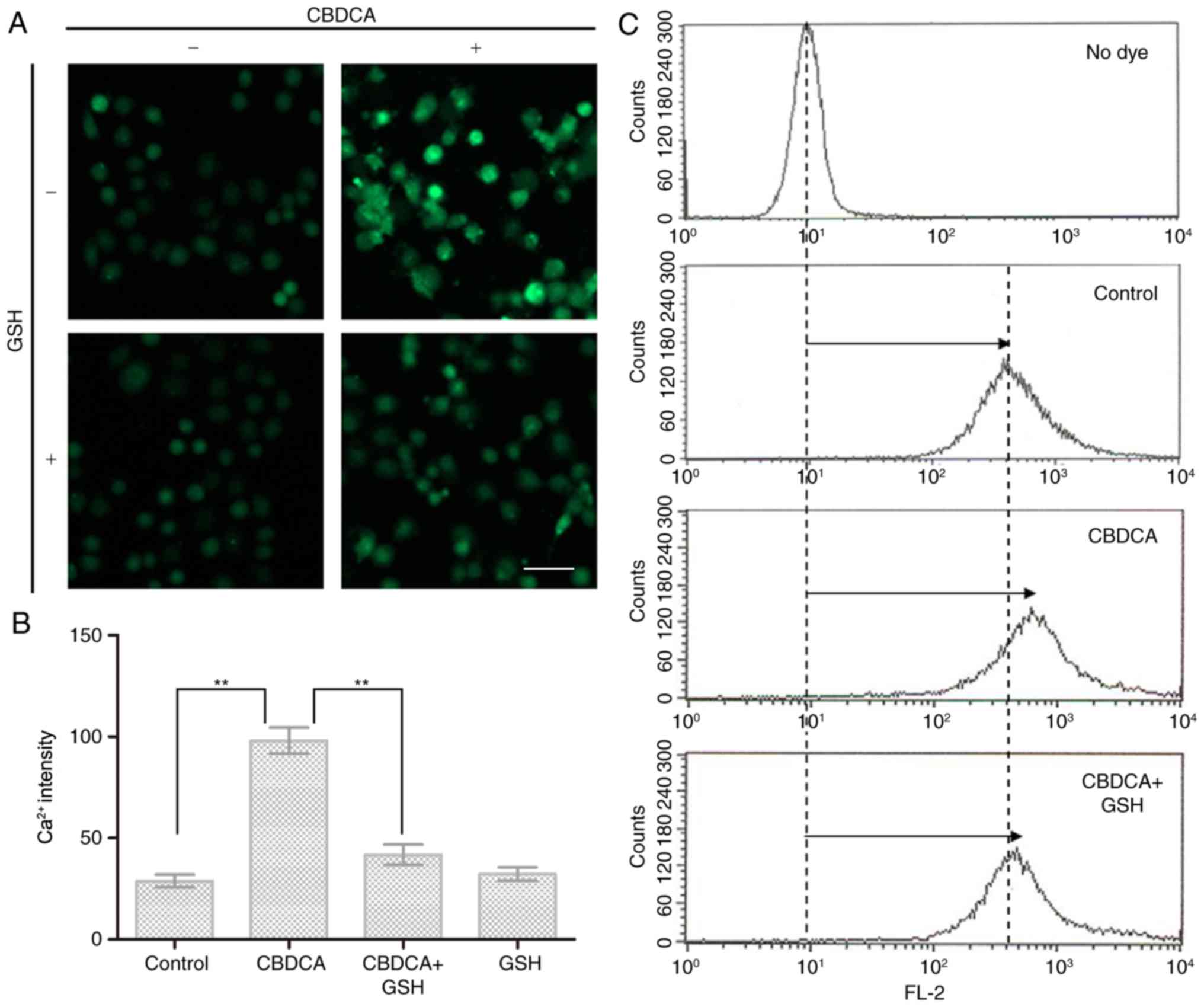
Ca2+ is a universal signaling molecule
that controls a variety of physiological functions, including those
involving the mitochondria (28).
In the present study, exposure to CBDCA alone induced a significant
elevation of Ca2+ concentration in HN-3 cells (Fig. 4A). This increase was effectively
inhibited by the addition of GSH, and no obvious impact on the
intracellular Ca2+ level was observed when cells were
exposed to GSH alone (Fig. 4A and
B). Accordingly, the intracellular level of Ca2+
displayed a distinct rightward shift in the flow cytometry results
(Fig. 4C), upon exposure to CBDCA,
which was restored by the addition of GSH into the cell
culture.
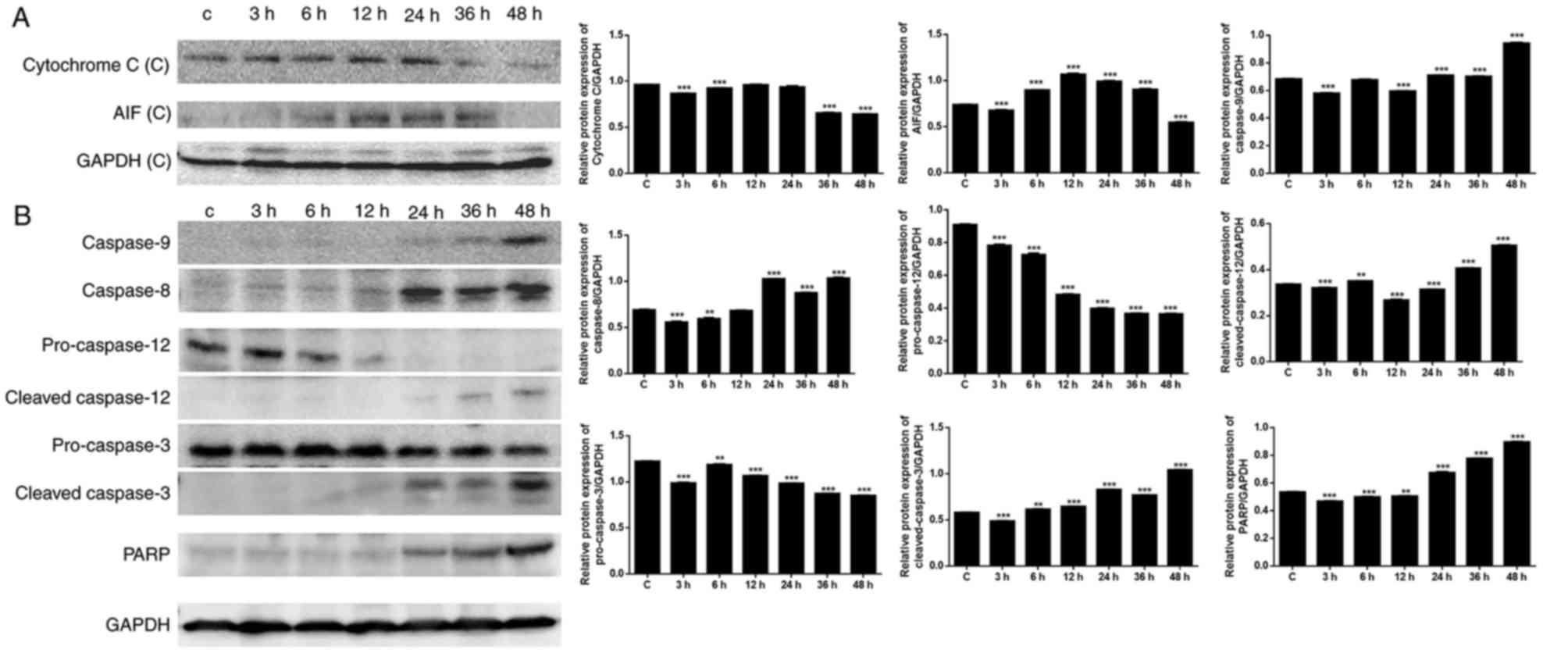
Activation of caspases and PARP
Mitochondria serve an important role in cell death
through the release of pro-apoptotic factors, including cytochrome
c and apoptosis-inducing factor (AIF), which activate
caspase-dependent and caspase-independent cell death, respectively
(29). As presented in Fig. 5A, the expression levels of
cytochrome c and AIF were upregulated following exposure to
CBDCA, particularly in the first 24 h. The decrease in cytochrome
c and AIF expression observed following 24 h may have
resulted from loss of mitochondrial function. To confirm the
effects of CBDCA on apoptosis, apoptosis-associated protein
expression was further evaluated (Fig.
5B). Western blot analysis revealed that there was a
time-dependent increase in the expression of the downstream caspase
cascade, including caspase-8/−9 and PARP. Increased expression of
caspase-8 was observed following 12 h, and increased expression of
caspase-9, cleaved caspase-3 and PARP was observed following ~36 h.
These results were in accordance with the apoptosis results
summarized above, confirming that apoptosis predominantly occurred
when cells were exposed to CBDCA for a duration of >24 h.
Caspase-8 may have been induced when Ca2+ and
mitochondrial depolarization took effect after 12 h, and caspase-9
may have been induced by cytochrome c release after 36
h.
When the exposure time was fixed at 12 h, a
comparison was made among the control and CBDCA and GSH treatment
groups, alone or in combination. A weak dose dependent release of
cytochrome c and AIF was detected (Fig. 6A). By contrast, there was a marked
dose-dependent increase in the expression of caspase-12/−8/−9/−3
and cleaved PARP (Fig. 6B).
Upregulation of apoptosis-associated protein expression was
observed in the group treated with 0.08 mg/ml CBDCA alone, but this
was less evident when the CBDCA concentration was 0.04 mg/ml.
Furthermore, treatment with CBDCA and GSH almost entirely inhibited
the activating effects of CBDCA at 0.08 mg/ml. These data indicated
that the activation of apoptosis-associated proteins was sensitive
to the concentration of CBDCA. Fig.
7 demonstrates that apoptosis predominantly occurred through
two signal-transduction pathways: The death receptor pathway and
the mitochondrial pathway, which are also termed the extrinsic and
intrinsic apoptotic pathways, respectively.
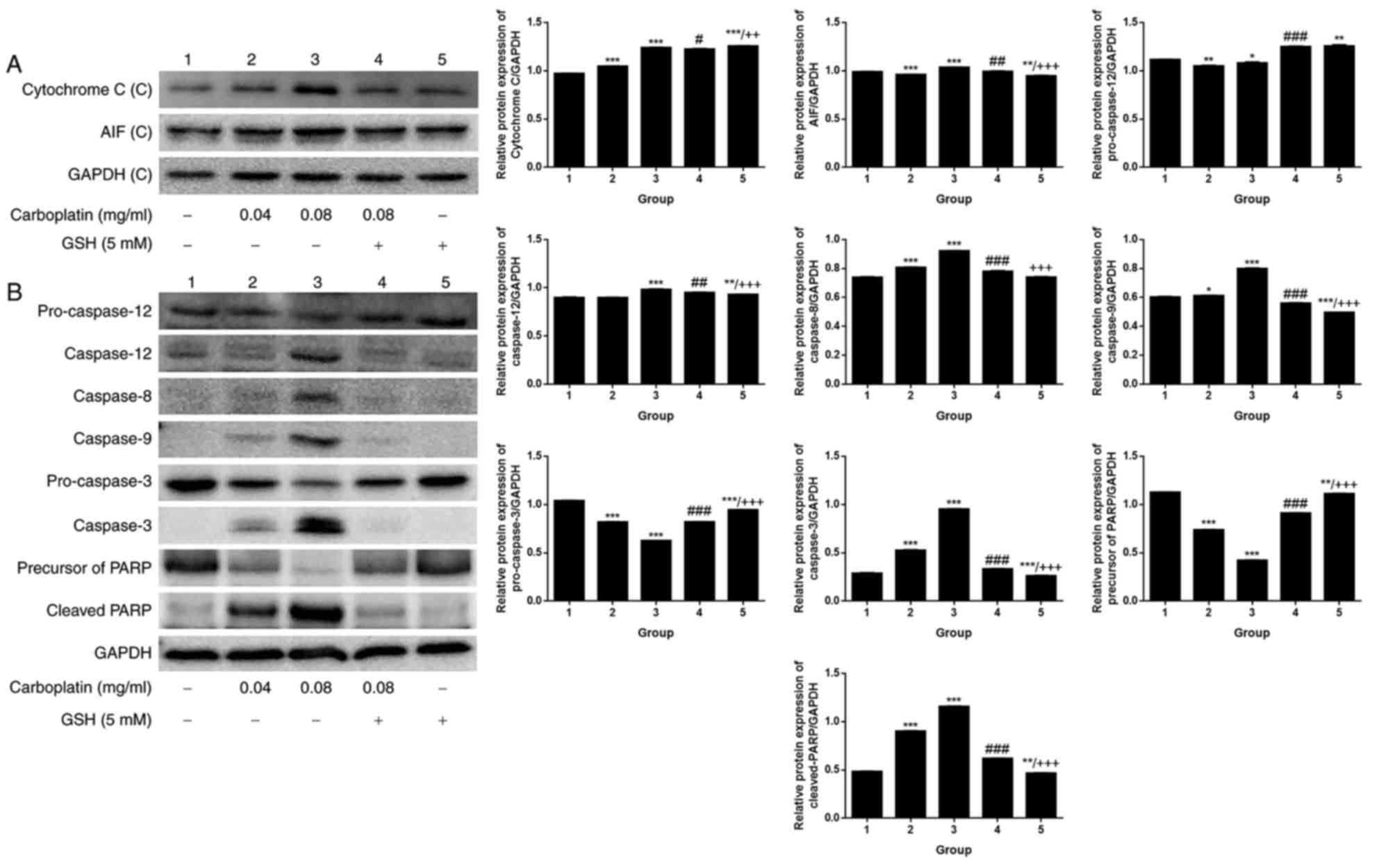 | Figure 6.Expression of apoptosis-associated
proteins following CBDCA treatment with or without GSH. (A)
Cytochrome c and AIF expression, as well the (B) expression
of caspase-3, −8, −9, −12, cleaved PARP, pro-caspase-3 and −12
following treatment with CBDCA, GSH or a combination for 12 h.
GAPDH was included as a loading control. GSH, glutathione; CBDCA,
carboplatin; PARP, poly ADP ribose polymerase; AIF, apoptosis
inducing factor. Data were presented as the mean ± standard
deviation. *P<0.05, **P<0.01, ***P<0.001 vs. group 1.
#P<0.05, ##P<0.01,
###P<0.001 vs. group 3. ++P<0.01,
+++P<0.001 vs. group 4. |
Discussion
As an anti-cancer drug, CBDCA was demonstrated to
inhibit cell viability and have an apoptosis-inducing effect in the
present study. Cell viability was notably suppressed by CBDCA but
was protected by the addition of GSH. These effects were dose- and
time-dependent.
The mitochondrial pathway of apoptosis is initiated
by death-inducing stimuli including oxidative stress and UV
radiation, which leads to the activation of caspase-9 through the
mitochondria (30,31). Extrinsic apoptosis initiated by
death-inducing stimuli involves the complex formation of
death-domain-containing-proteins and consequent activation of
caspase-8. In addition, in the presence of death-inducing stimuli,
the endoplasmic reticulum may also initiate apoptosis via the
activation of caspase-12 (20,21,32).
As for the death-inducing stimuli mentioned above, there is
increasing evidence to support the theory that the major mechanism
of cell apoptosis induced by CBDCA includes direct covalent binding
between CBDCA and DNA, to form DNA adducts, as well as oxidative
stress due to ROS generation (33,34).
These results indicate that CBDCA-induced apoptosis is mediated
through the mitochondrial pathway (8,35).
In the present study, the protective effect of GSH indicated that
ROS served a causative role. Regarding endoplasmic
reticulum-related apoptosis initiation, the death-inducing stimulus
generated may have been excessive Ca2+, as demonstrated
by the significant increase in Ca2+ in response to CBDA
detected in the present study, as well as in previous research
(16).
When HN-3 cells were exposed to CBDCA in sufficient
concentrations, the agent successfully crossed the cell membrane,
underwent hydrolysis and became positively charged. The current
experimental results revealed that when exposed to CBDCA for 12 h,
by which time apoptosis was, although not significant, notably
increased, the HN-3 cells had enhanced MP and increased
intracellular Ca2+ concentration, as well as
mitochondrial depolarization. Upregulated expression of caspase-8
was observed when cells were exposed to CBDCA for 24 h (12 h
following Ca2+ overload and mitochondrial
depolarization). PARP took effect at a later stage, after cells
were exposed to CBDCA for ~36 h, and caspase-9 was activated last,
when cells were exposed to CBDCA for ~48 h.
After 12 h, the activated apoptotic signaling
molecules were released into the cytoplasm, which contains many
pro-apoptotic factors such as cytochrome c, AIF and
pro-caspase-9 in the intermembrane space (28,36,37).
In addition, mitochondrial depolarization was almost fully restored
by the addition of GSH, which is an ROS scavenger. Oxidative stress
during apoptosis is thought to be associated with the malfunction
of the mitochondrial respiratory chain and disengagement of
cytochrome c, as well as alteration in mitochondrial
transmembrane potential and membrane permeability (38,39).
Therefore, it can be inferred that the release of AIF is
responsible for the enhanced ROS and oxidative stress. Oxidative
stress may have induced mitochondrial permeability transition,
which leads to the release of AIF. Furthermore, the mitochondrial
permeability transition may have generated more ROS from the
mitochondria in a positive feedback loop.
In the present study, simultaneous with AIF release
(at ~12 h), an increase in the amount of Ca2+ was
detected. The occurrence of MP excluded the possibility that CBDCA
directly destroyed the membrane function of the cells. Instead,
membrane polarization suggested that HN-3 cells were processing the
excessive intracellular Ca2+ by increasing membrane
polarization to inhibit Ca2+ influx. Oxidative stress
has been reported to be induced by increased intracellular
Ca2+ (40). Excessive
cytosolic Ca2+ induced by exposure to CBDCA in HN-3
cells is transferred to the mitochondria by the endoplasmic
reticulum (28). This may have led
to alterations in ultra-structural integrity and a reduction in the
MMP in the present study. CBDCA may increase Ca2+
release from the endoplasmic reticulum, leading to the production
of nitric oxide (NO). Mitochondrial uptake of Ca2+ and
peroxynitrite, which is generated from the interaction of NO and
superoxide anion, block mitochondrial respiration resulting in the
generation of ROS (29). However,
although oxidative stress is induced by Ca2+, this
excessive release of Ca2+ is effectively inhibited by
the addition of GSH. This is because the high level of GSH
effectively ‘mops up’ the activated platinum in the cytoplasm by
direct binding prior to its interaction with the organelles. This
causes a loss of the effectiveness of CBDCA to induce additional
Ca2+ release (22).
This CBDCA-induced excessive intracellular Ca2+ release
is sufficient to act as a death-inducing stimulus, which results in
early and persistent mitochondrial depolarization with perturbation
or rupture of the outer mitochondrial membrane, as well as induced
oxidative stress and AIF release. In the present study, excessive
Ca2+-induced AIF release occurred at the earliest stage
(the first 12 h) of CBDCA treatment, but did not persist due to
mitochondrial damage, which ceased functioning following 36 h of
CBDCA exposure. Therefore, AIF release was likely not the main
mechanism of apoptosis.
With prolonged exposure to CBDCA, a variety of
interactions took effect in coordination with cytochrome c,
caspase-8 and PARP. The apoptotic signals triggered caspase
activation by either activating initiator caspases, including
caspase-8, or through the release of caspase-activating factors,
including cytochrome c and AIF from the mitochondria. In
addition, cell death occurred not only through the caspases, but
also through a PARP-mediated cell death pathway, causing AIF to
translocate from the mitochondria into the nucleus. Furthermore, it
has been reported that CBDCA molecule could interact with DNA
molecules (33). The linkage
between DNA and CBDCA may be the most cytotoxic effect, through
inhibiting the process of DNA replication, causing errors in
replication and inducing the signaling molecules of apoptosis
(41). These activities triggered
caspase-8 activation or the PARP-mediated cell death pathway.
The existence of different stages of apoptosis and
the fact that apoptosis-associated factors were activated at
different times accounted for the time-dependent response of HN-3
cells treated with CBDCA. For example, the CBDCA required a
synergistic effect of caspase-8 and −9, as well as the PARP pathway
to exert its full effectiveness. The present study also suggested
that CBDCA-induced apoptosis in HN-3 cells is concentration
dependent. The dose-dependence of CBDCA may be due to the DNA
repair machinery, which is activated when mild damage occurs but
under severe insult that results in extensive DNA damage, caspase
induction occurs over the activation of DNA repair (37).
In conclusion, CBDCA exerted a time- and
dose-dependent inhibitory effect in HN-3 cells in the present
study. The results demonstrated the involvement of mitochondria at
the initiation of apoptosis triggered by excessive intracellular
Ca2+. Investigation of the expression of
apoptosis-associated proteins revealed that mitochondria act as an
independent upstream mediator of the CBDCA-induced apoptosis
pathways, cooperating with the nuclear pathways that take effect
earlier than the mitochondrial pathways. The present findings may
have useful implications for the rational design of more efficient
therapeutic strategies as well as the development of novel
platinum-based agents.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81172557), the
Project of Shanghai Municipal Health and Family Planning Commission
(grant no. 201640104), and partially supported by Leading Foreign
Research Institute Recruitment Program through the National
Research Foundation of Korea funded by the Ministry of Education,
Science and Technology (grant no. 2012K1A4A3053142) and Beckman
Laser Institute Korea, Dankook University.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
PH and PC made substantial contributions to the
concept and design of the present study, and the examination of the
manuscript. BS and WM conducted experiments and produced the
manuscript. JA conducted experiments and analyze the experimental
data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CBDCA
|
carboplatin
|
|
GSH
|
glutathione
|
|
ROS
|
reactive oxygen species
|
|
MMP
|
mitochondrial membrane potential
|
|
MP
|
membrane potential
|
|
PARP
|
poly ADP ribose polymerase
|
|
AIF
|
poly ADP ribose polymerase
|
References
|
1
|
Zhang W, Yan Y, Gu M, Wang X, Zhu H, Zhang
S and Wang W: High expression levels of Wnt5a and Ror2 in laryngeal
squamous cell carcinoma are associated with poor prognosis. Oncol
Lett. 14:2232–2238. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Riga M, Chelis L, Danielides V, Vogiatzaki
T, Pantazis TL and Pantazis D: Systematic review on T3 laryngeal
squamous cell carcinoma; still far from a consensus on the optimal
organ preserving treatment. Eur J Surg Oncol. 43:20–31. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shen Z, Cao B, Lin L, Zhou C, Ye D, Qiu S,
Li Q and Cui X: The clinical signification of claudin-11 promoter
hypermethylation for laryngeal squamous cell carcinoma. Med Sci
Monit. 23:3635–3640. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mao Y, Zhang DW, Lin H, Xiong L, Liu Y, Li
QD, Ma J, Cao Q, Chen RJ, Zhu J and Feng ZQ: Alpha B-crystallin is
a new prognostic marker for laryngeal squamous cell carcinoma. J
Exp Clin Cancer Res. 31:1012012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Radogna F, Dicato M and Diederich M:
Cancer-type-specific crosstalk between autophagy, necroptosis and
apoptosis as a pharmacological target. Biochem Pharmacol. 94:1–11.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sousa GFD, Wlodarczyk SR and Monteiro G:
Carboplatin: Molecular mechanisms of action associated with
chemoresistance. Braz J Pharm Sci. 50:693–701. 2014. View Article : Google Scholar
|
|
9
|
Castrellon AB, Pidhorecky I, Valero V and
Raez LE: The role of carboplatin in the neoadjuvant chemotherapy
treatment of triple negative breast cancer. Oncol Rev. 11:3242017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Johnstone TC, Park GY and Lippard SJ:
Understanding and improving platinum anticancer
drugs-phenanthriplatin. Anticancer Res. 34:471–476. 2014.PubMed/NCBI
|
|
11
|
Fong CW: Platinum based
radiochemotherapies: Free radical mechanisms and radiotherapy
sensitizers. Free Radic Biol Med. 99:99–109. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee CK, Jung M, Choi HJ, Kim HR, Kim HS,
Roh MR2, Ahn JB, Chung HC, Heo SJ, Rha SY and Shin SJ: Results of a
phase II study to evaluate the efficacy of docetaxel and
carboplatin in metastatic malignant melanoma patients who failed
first-line therapy containing dacarbazine. Cancer Res Treat.
47:781–789. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sanborn RE: Cisplatin versus carboplatin
in NSCLC: Is there one ‘best’ answer? Curr Treat Option On.
9:326–342. 2008. View Article : Google Scholar
|
|
14
|
Comella P, Gambardella A, Frasci G,
Avallone A and Costanzo R: SICOG investigators: Comparison of the
safety and efficacy of paclitaxel plus gemcitabine combination in
young and elderly patients with locally advanced or metastatic
non-small cell lung cancer. A retrospective analysis of the
Southern Italy Cooperative Oncology Group trials. Crit Rev Oncol
Hematol. 65:164–171. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barghout SH, Zepeda N, Vincent K, Azad AK,
Xu Z, Yang C, Steed H, Postovit LM and Fu YX: RUNX3 contributes to
carboplatin resistance in epithelial ovarian cancer cells. Gynecol
Oncol. 138:647–655. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lum E, Vigliotti M, Banerjee N, Cutter N,
Wrzeszczynski KO, Khan S, Kamalakaran S, Levine DA, Dimitrova N and
Lucito R: Loss of DOK2 induces carboplatin resistance in ovarian
cancer via suppression of apoptosis. Gynecol Oncol. 130:369–376.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
de Castria TB, da Silva EM, Gois AF and
Riera R: Cisplatin versus carboplatin in combination with
third-generation drugs for advanced non-small cell lung cancer.
Cochrane Db Syst Rev. 8:CD0092562013.
|
|
18
|
Zhang Q, Cheng Y, Huang L, Bai Y, Liang J
and Li X: Inhibitory effect of carboplatin in combination with
bevacizumab on human retinoblastoma in an in vitro and in vivo
model. Oncol Lett. 14:5326–5332. 2017.PubMed/NCBI
|
|
19
|
Montero AJ and Jassem J: Cellular redox
pathways as a therapeutic target in the treatment of cancer. Drugs.
71:1385–1396. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Green DR: Apoptotic pathways: Paper wraps
stone blunts scissors. Cell. 102:1–4. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kelland L: The resurgence of
platinum-based cancer chemotherapy. Nat Rev Cancer. 7:573–584.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nicolson MC, Orr RM, O'Neill CF and Harrap
KR: The role of platinum uptake and glutathione levels in L1210
cells sensitive and resistant to cisplatin, tetraplatin or
carboplatin. Neoplasma. 39:189–195. 1992.PubMed/NCBI
|
|
24
|
He P, Ahn JC, Shin JI, Hwang HJ, Kang JW,
Lee SJ and Chung PS: Enhanced apoptotic effect of combined modality
of 9-hydroxypheophorbide alpha-mediated photodynamic therapy and
carboplatin on AMC-HN-3 human head and neck cancer cells. Oncol
Rep. 21:329–334. 2009.PubMed/NCBI
|
|
25
|
He P, Bo S, Chung PS, Ahn JC and Zhou L:
Photosensitizer effect of 9-hydroxypheophorbide α on diode
laser-irradiated laryngeal cancer cells: Oxidative stress-directed
cell death and migration suppression. Oncol Lett. 12:1889–1895.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Evtodienko YuV, Teplova V, Khawaja J and
Saris NE: The Ca(2+)-induced permeability transition pore is
involved in Ca(2+)-induced mitochondrial oscillations: A study on
permeabilised Ehrlich ascites tumour cells. Cell Calcium.
15:143–152. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hunter DR and Haworth RA: The Ca2+-induced
membrane transition in mitochondria. I. The protective mechanisms.
Arch Biochem Biophys. 195:453–459. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Giorgi C, Romagnoli A, Pinton P and
Rizzuto R: Ca2+ signaling, mitochondria and cell death. Curr Mol
Med. 8:119–130. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hong SJ, Dawson TM and Dawson VL: Nuclear
and mitochondrial conversations in cell death: PARP-1 and AIF
signaling. Trends Pharmacol Sci. 25:259–264. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gonzalez D, Bejarano I, Barriga C,
Rodriguez AB and Pariente JA: Oxidative stress-induced caspases are
regulated in human myeloid HL-60 cells by calcium signal. Curr
Signal Transduct Ther. 5:181–186. 2010. View Article : Google Scholar
|
|
31
|
Wajant H: The Fas signaling pathway: More
than a paradigm. Science. 296:1635–1636. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Su SH, Su SJ, Lin SR and Chang KL:
Cardiotoxin-III selectively enhances activation-induced apoptosis
of human CD8+ T lymphocytes. Toxicol Appl Pharmacol. 193:97–105.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hah SS, Stivers KM, de Vere White RW and
Henderson PT: Kinetics of carboplatin-DNA binding in genomic DNA
and bladder cancer cells as determined by accelerator mass
spectrometry. Chem Res Toxicol. 19:622–626. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng CF, Juan SH, Chen JJ, Chao YC, Chen
HH, Lian WS, Lu CY, Chang CI, Chiu TH and Lin H: Pravastatin
attenuates carboplatin-induced cardiotoxicity via inhibition of
oxidative stress associated apoptosis. Apoptosis. 13:883–894. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hwang H, Biswas R, Chung PS and Ahn JC:
Modulation of EGFR and ROS induced cytochrome c release by
combination of photodynamic therapy and carboplatin in human
cultured head and neck cancer cells and tumor xenograft in nude
mice. J Photochem Photobiol B. 128:70–77. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gunter TE and Pfeiffer DR: Mechanisms by
which mitochondria transport calcium. Am J Physiol. 258:C755–C786.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Joza N, Susin SA, Daugas E, Stanford WL,
Cho SK, Li CY, Sasaki T, Elia AJ, Cheng HY, Ravagnan L, et al:
Essential role of the mitochondrial apoptosis-inducing factor in
programmed cell death. Nature. 410:549–554. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zamzami N, Marchetti P, Castedo M,
Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B and
Kroemer G: Sequential reduction of mitochondrial transmembrane
potential and generation of reactive oxygen species in early
programmed cell death. J Exp Med. 182:367–377. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Boya P, Morales MC, Gonzalez-Polo RA,
Andreau K, Gourdier I, Perfettini JL, Larochette N, Deniaud A,
Baran-Marszak F, Fagard R, et al: The chemopreventive agent
N-(4-hydroxyphenyl)retinamide induces apoptosis through a
mitochondrial pathway regulated by proteins from the Bcl-2 family.
Oncogene. 22:6220–6230. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Richter C and Kass GE: Oxidative stress in
mitochondria: Its relationship to cellular Ca2+ homeostasis, cell
death, proliferation, and differentiation. Chem Biol Interact.
77:1–23. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Los G, Smals OA, van Vugt MJ, van der
Vlist M, den Engelse L, McVie JG and Pinedo HM: A rationale for
carboplatin treatment and abdominal hyperthermia in cancers
restricted to the peritoneal cavity. Cancer Res. 52:1252–1258.
1992.PubMed/NCBI
|