Introduction
Myocardial ischemia-reperfusion injury (MIRI)
frequently occurs after the ischemic myocardium is treated. When
MIRI occurs, the degree of myocardial damage is aggravated and the
infarct area expands. The inflammatory response is one of the main
factors causing reperfusion injury following acute myocardial
ischemia (1). Inflammation is
modulated by a number of factors including inflammatory mediators
and inflammatory cells that promote myocardial damage from ischemia
injury to reperfusion injury.
In acute myocardial infarction (AMI), platelets are
activated and can interact with leukocytes, thereby forming
platelet-leukocyte aggregates (PLAs). Increased PLA levels may be
an indicator of thrombus burden and pro-inflammatory response
(2) and has been observed in a
variety of cardiovascular diseases, including unstable angina and
AMI (3,4). Reperfusion therapy is the cornerstone
for salvaging the myocardium, but it can induce damage to the
working myocardium via ischemia-reperfusion (I/R) injury.
Postconditioning, defined as brief periods of reperfusion
alternating with re-occlusion applied during the very early min of
reperfusion, has been demonstrated to be protective against MIRI
(5,6) and therefore may reduce myocardial
infarct size, but its underlying mechanism is incompletely
elucidated.
The c-Jun N-terminal kinase (JNK) is a member of the
mitogen activated protein kinase (MAPK) superfamily. Previous
experiments have demonstrated the role of the JNK signaling pathway
by increasing JNK expression in MIRI (7–9).
However, whether postischemic conditioning exerts its
cardio-protective effects by altering the JNK signalling pathway
and the levels of PLA have not been examined.
In the present study, a rat model of myocardial
ischemia reperfusion in vivo was established. The expression
of phosphorylated P38 MAPK and PLA was observed following
postconditioning treatment, and the effect was investigated using
specific inhibitors of P38 MAPK (SP 600125) and as well as
activators of P38 MAPK (anisomycin). Furthermore, the present study
aimed to investigate the role of P38 MAPK in signal transduction of
postconditioning, as well as its effect on PLA expression.
Materials and methods
Materials
A total of 60 male Sprague Dawley rats (8 weeks old;
weight, 250–280 g) were purchased from Beijing Wei Tong Li Hua
Experimental Animal Co., Ltd. (Beijing, China). The rats were
housed in an environment with a maintained temperature of 22°C, a
relative humidity of 50±15% and a 12-h light/dark cycle. All rats
had free access to standard chow and water. The present study
conformed to the Guide for the Care and Use of Laboratory Animals
published by the US National Institutes of Health (NIH publication
no. 85–23; revised 1996) and was approved by the Research
Commission on Ethics of the Affiliated Yantai Yuhuangding Hospital
of Qingdao University (Yantai, China). For measuring markers of
myocardial damage, creatine kinase-muscle/brain (CK-MB) and
troponin (TnI) kits were purchased from Merck KGaA (Darmstadt,
Germany). The erythrocyte lysate was purchased from BD Biosciences,
Franklin Lakes, (NJ, USA). Cluster of differentiation (CD)45 and
CD41a were purchased from eBioscience; Thermo Fisher Scientific,
Inc. (Waltham, MA, USA). The rabbit anti-mouse phosphorylated JNK
(phospho-JNK MAPK/p-JNK) polyclonal antibody was purchased from
Santa Cruz Biotechnology, Inc., Dallas, TX, USA and the
corresponding secondary antibody was from Jackson ImmunoResearch
Laboratories, Inc. (West Grove, PA, USA).
Myocardial ischemia reperfusion
model
The rats were anesthetized with an initial
intra-peritoneal injection of sodium pentobarbital (0.23 ml/100 g)
and were then intubated and ventilated using a rodent respirator. A
ligature was inserted under the anterior descending branch of the
left coronary artery (LAD), and the model was constructed by
ligating the LAD for 30 min and then loosening the knot of the LAD.
Different drugs, including DMSO (0.8 ml; 10%), SP600125 (6 mg/kg in
0.8 ml of 10% DMSO) and anisomycin (2 mg/kg in 0.8 ml of 10% DMSO)
were injected into the right jugular vein 5 min following
reperfusion. Then 3–4 ml blood was drawn from the carotid artery
cannula and different treatments were given according to the
measured indexes.
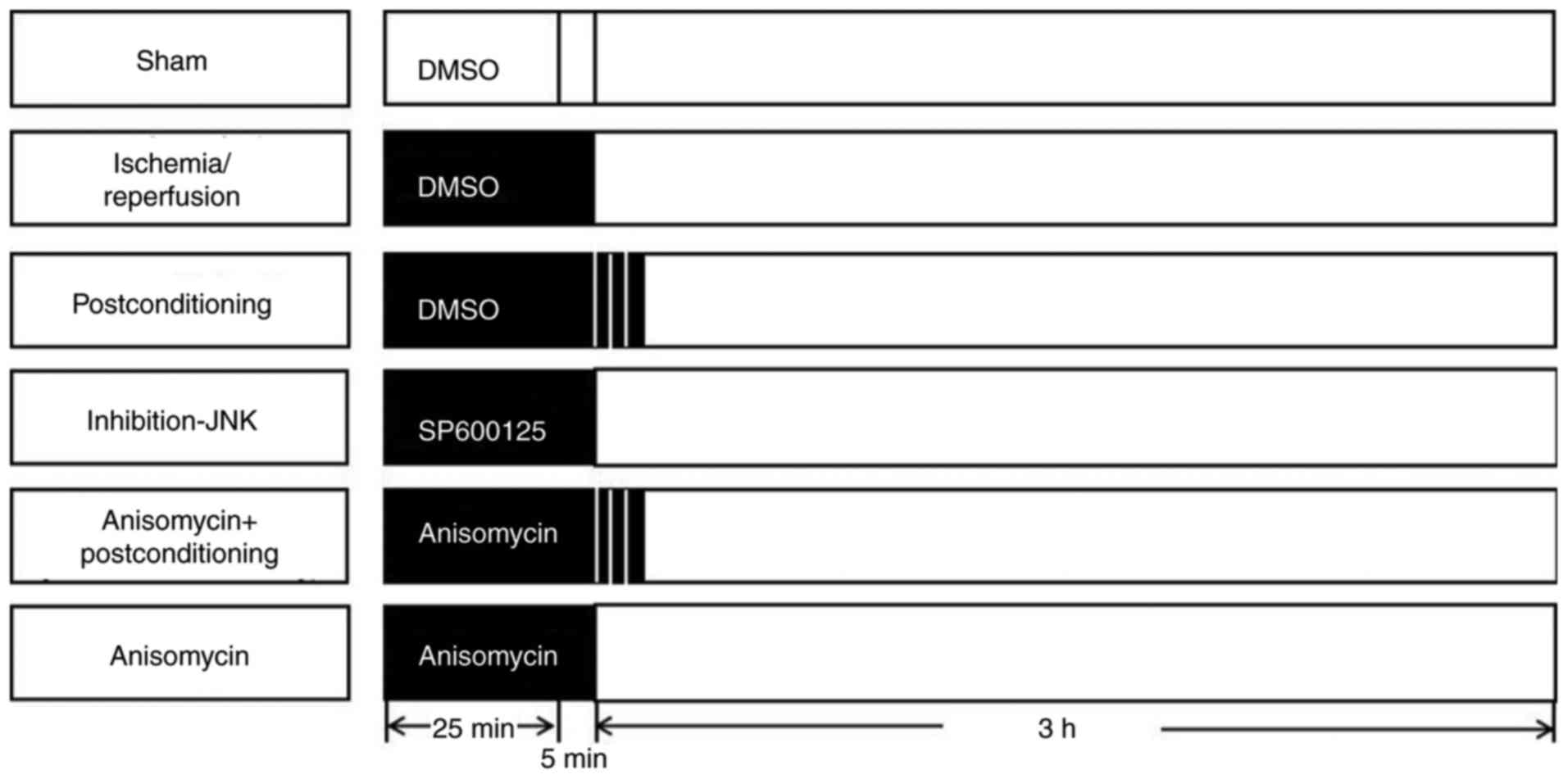
Experimental protocols
A total of 60 rats were randomly allocated to one of
the 6 groups (n=10) as follows: i) The sham group; ii)
ischemia-reperfusion (I/R) group; iii) post-conditioning (PostC)
group; iv) JNK inhibitor SP600125 (I-JNK) group; v) anisomycin/Ani
plus postconditioning (Ani + PostC) group and vi) anisomycin (Ani)
group (Fig. 1).
In the sham group, the left coronary artery was
isolated and threaded but not ligated and left for 30 min. A total
of 0.8 ml 10% DMSO was injected into the right jugular vein 25 min
following ischemia. In the I/R group, 0.8 ml 10% DMSO was injected
into the right jugular vein 25 min following ischemia. Following
the end of ischemia, continuous reperfusion was resumed and
performed for 3 h. In the postconditioning (PostC) group, 0.8 ml of
10% DMSO into the right jugular vein 25 min following ischemia.
After the ischemia was completed, 3 cycles of 1/1 min
reperfusion/reocclusion began immediately at the onset of
reperfusion and then continuous reperfusion was performed for 3 h.
In the I-JNK group, SP600125 (Merck KGaA; 6 mg/kg in 0.8 ml of 10%
DMSO) was injected into the right jugular vein 25 min following
ischemia. Following the end of ischemia, continuous reperfusion was
resumed and performed for 3 h. In the Ani + PostC group, anisomycin
(2 mg/kg in 0.8 ml of 10% DMSO) was injected into the right jugular
vein 25 min following ischemia. At the end of ischemia, 3 cycles of
1/1 min reperfusion/reocclusion began immediately at the onset of
reperfusion and continuous reperfusion was performed for 3 h. In
the Ani group, anisomycin (Merck KGaA; 2 mg/kg in 0.8 ml of 10%
DMSO) was injected into the right jugular vein 25 min following
ischemia. Following the end of ischemia, continuous reperfusion was
resumed and performed for 3 h.
Release of serum markers
The serum levels of TnI and CK-MB were analyzed
using an automatic biochemistry analyzer with the CK-MB kit. All
experiments were carried out according to the manufacturer's
protocol.
Measurement of infarct size
A total of 3 h following reperfusion, the hearts
from the rats were excised and maintained at −20°C for 20 min. The
hearts were sliced into four sections (thickness, 2 mm) from the
base to the apex for staining with triphenyltetrazolium chloride
(1%) at 37°C for 15 min for measuring the area of necrosis. The
myocardium of the infarct area was pale, while the active
myocardium was red in color. The infarct size was expressed as the
ratio of the infarct area to the total left ventricular volume.
Measurement of PLA
Flow cytometry was used to detect the level of PLA
as described previously (10).
Briefly, sodium citrate anticoagulant was added to 200 µl of blood
followed by centrifugation at 1,000 g for 5 min at room
temperature. The supernatant was discarded. A total of 6–10 times
the cell volume of red blood cell lysate was added and then gently
mixed. A total of 1–2 min post-mixing, the cells were lysed using
Red Blood Cell Lysis Buffer (cat. no. 3702; Beyotime Institute of
Biotechnology, Shanghai, China). This step was repeated until
cracking was complete. Then, PBS was added. The precipitate was
resuspended and centrifuged at 1,000 g for 3 min. The supernatant
was discarded. A total of 200 µl paraformaldehyde (2%) was added
and fixed for 60 min at room temperature. PBS (1,600 µl) was added
for 15 min, and centrifuged at 1,000 × g for 15 min at room
temperature followed by resuspension with PBS for 3 times, and
finally dissolved in 200 µl PBS. To each group of two tubes, CD45
(cat. no. 11-0461-82; 0.25 µg/test) and CD41-phycoerythrin (cat.
no. PA5-79526; 1 µg/1×106 cells) were used, maintained
in a dark room for 30 min and centrifuged at 1,000 × g at 4°C for 5
min. PBS suspension was detected using a BD FACSCanto II flow
cytometer and BD FACSDiva™ software (BD Biosciences).
Measurement of p-JNK MAPK
Western blot analysis was performed as previously
described (11). Briefly, the left
ventricular myocardium was homogenized in radioimmunoprecipitation
assay lysis buffer [hepes (20 mmol/l; pH 7.7), MgC12 2.5
mmol/l, EDTA (0.1 mmol/l), Β-glycerophosphate (20 mmol/l),
dithiothreitol (0.5 mmol/l), sodium orthovanadate (0.1 mmol/l),
NaCl (75 mmol/l), leupeptin (4 µg/ml), phenyl-methylsulfonyl
fluoride (20 µg/ml) and Triton X-100 (0.05%; v/v)]. To quantify
protein levels, equal amounts of total protein (10 µg) were loaded
into lanes. Protein concentration was determined using the
bicinchoninic acid method (cat. no. A53225; Pierce; Thermo Fisher
Scientific, Inc.). After the separation of proteins via SDS-PAGE
gel (10%) electrophoresis, the proteins were transferred to
nitrocellulose membranes and incubated with antibodies against
p-JNK MAPK (cat. no. sc-6254; 1:200) and β-actin (cat. no.
sc-47778; 1:200; both Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA) for 4 h at room temperature, which was followed by
incubation with horseradish peroxidase-conjugated secondary
antibodies (cat. no. 016-030-084; 1:1,000; Jackson ImmunoResearch
Laboratories, Inc.) for 1 h at room temperature. The
antigen-antibody complexes were visualized using enhanced
chemiluminescence (Beyotime Institute of Biotechnology) at room
temperature. The integrated optical density (IOD) of the bands were
analysed using Image Pro Plus image analysis software (version 4.1;
Media Cybernetics, LP, USA). The IOD was calculated by multiplying
the value of the average optical density by area. The ratio of IOD
values of the target protein and β-actin was used to reflect the
relative level of the target protein.
Statistical analysis
The data were analysed using the statistical
software package SPSS 20.0 (IBM Corp., Armonk, NY, USA) for
Windows. The values are expressed as the mean ± standard error of
the mean. A one-way analysis of variance followed by the SNK post
hoc test or the Student's t-test was used as appropriate to
determine the differences between groups. P<0.05 was considered
to indicate a statistically significant difference. All experiments
were performed in triplicate.
Results
Infarct size and serum markers of
myocardial damage
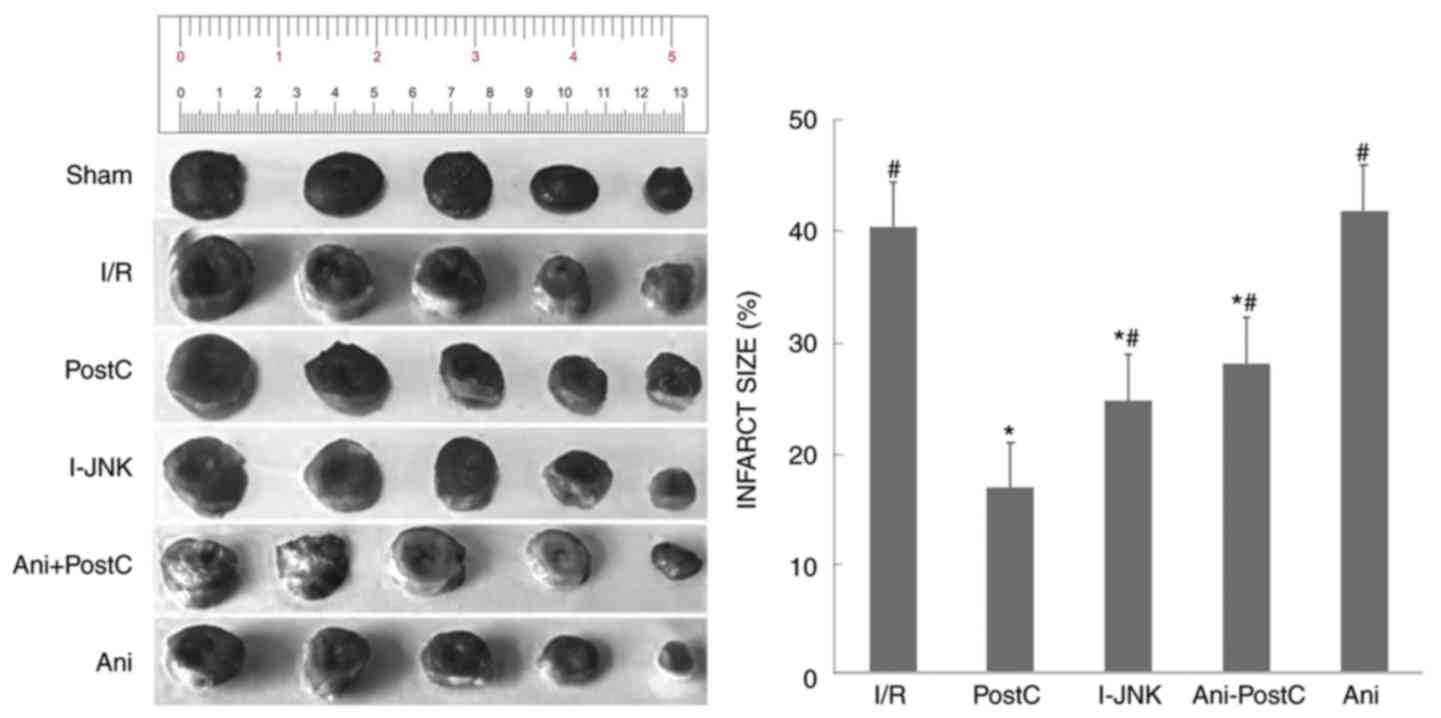
Initial experiments examined infarct size following
AMI with the infarcted myocardium stained white and the viable
myocardium stained red (Fig. 2).
Compared with the sham group, the infarct size was 42.2% in the I/R
injury group. The infarct size was significantly reduced in the
postconditioning group (P<0.05) and also following application
of the JNK inhibitor, SP600125 (I-JNK). Furthermore, this reduction
in infarct size was abolished when the JNK activator, Ani was
applied, but not when the hearts underwent postconditioning
(Table I).
 | Table I.Serum markers and infarct area in all
groups. |
Table I.
Serum markers and infarct area in all
groups.
| Group | CK-MB (pg/ml) | TnI (pg/ml) | Infarct area (%) |
|---|
| Sham |
166.02±33.26a,b |
7.79±0.54a,b |
0.00±0.00a,b |
|
Ischemia-reperfusion |
637.31±43.99b |
19.67±0.66b |
40.17±1.77b |
| Postconditioning |
257.38±53.12a |
10.05±0.81a |
16.68±2.06a |
| JNK inhibitor
SP600125 |
402.10±47.94a,b |
14.26±1.31a,b |
24.62±1.58a,b |
| JNK activator
anisomycin and postconditioning |
500.84±52.93a,b |
17.32±0.77a,b |
27.91±1.79a,b |
| JNK activator
anisomycin |
638.34±25.87b |
19.96±0.72b |
40.62±1.59b |
In the sham group the levels of CK-MB and TnI were
166±33 pg/ml and 7.8±0.5, respectively (Table I). Compared with the Sham group,
the levels of CK-MB and TnI were significantly increased in the I/R
injury group (P<0.05). Compared with the I/R injury group, the
levels of CK-MB and TnI were significantly lower in the
postconditioning group (P<0.05), and the JNK inhibitor SP600125,
exhibited a similar cardioprotective effect as ischemic
postconditioning. By contrast, when the JNK activator, Ani was
applied, both CK-MB and TnI were high and not significantly
different from those of the I/R group. However, ischemic
postconditioning partially protected against myocardial damage even
when anisomycin was applied.
Levels of the PLAs and p-JNK
The level of PLAs in the Sham group was 8.70±0.56,
8.99±0.56, 8.80±0.36 and 10.11±0.87 at 30 min following ischemia,
and at 30, 60 min and 3 h following reperfusion, respectively
(Table II). Ischemia-reperfusion
injury led to a significant increase in PLA level 60 min and 3 h
following reperfusion compared with the post-conditioning group
(P<0.05). By contrast, ischemic postconditioning or application
of the JNK inhibitor, SP600125, prevented the rise of PLA level at
60 min and 3 h post-reperfusion compared with the I/R injury group
(P<0.05). Neither anisomycin treatment nor anisomycin with
postconditioning significantly affected PLA level compared with
ischemia-reperfusion conditioning (Table II and Fig. 3).
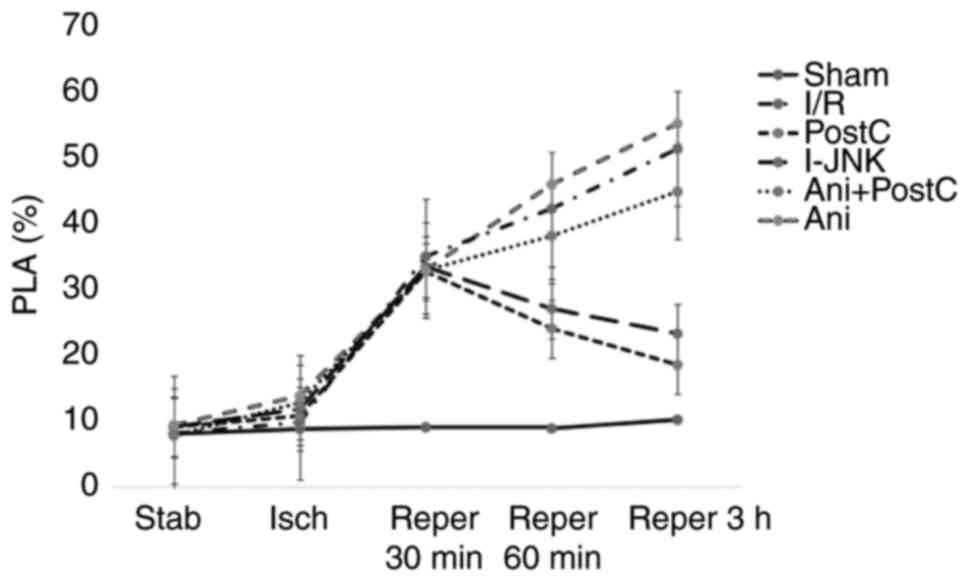 | Figure 3.The level of PLAs at different time
points following ischemia or reper in different groups. CD45 and
CD41-Phycoerythrin were used to detect PLAs levels in each group.
At the time point of baseline, 30 min following ischemia, 30, 60
min and 3 h following reper, the level of PLAs was detected by flow
cytometry. Reper, reperfusion; CD, cluster of differentiation; I/R,
ischemia reperfusion; Ani, anisomycin, postC, postconditioning;
I-JNK, inhibitor of c-Jun N-terminal kinase; PLA,
platelet-lymphocyte aggregates. |
| Table II.Expression of platelet-lymphocyte
aggregates at different time points. |
Table II.
Expression of platelet-lymphocyte
aggregates at different time points.
| Group | Baseline | 30 min following
ischemia | 30 min following
reperfusion | 60 min following
reperfusion | 3 h following
reperfusion |
|---|
| Sham | 7.96±0.30 |
8.70±0.56a,b |
8.99±0.56a,b |
8.80±0.36a,b |
10.11±0.87a,b |
|
Ischemia-reperfusion | 8.06±0.23 | 23.70±2.79 | 34.78±2.79 |
42.00±2.02b |
51.03±2.02b |
| Postconditioning | 9.01±0.26 | 22.76±1.93 | 32.50±1.93 |
23.90±0.89a |
18.40±1.27a |
| JNK inhibitor
SP600125 | 8.43±0.22 | 23.90±2.06 | 32.90±2.06 |
29.57±1.46a,b |
26.64±1.18a,b |
| JNK activator
anisomycin and postconditioning | 9.30±0.29 | 21.81±2.14 | 32.69±2.14 |
45.66±0.94b |
54.35±1.45b |
| JNK activator
anisomycin | 9.23±0.40 | 22.20±2.82 | 32.7±2.82 |
45.70±1.69b |
54.80±1.40b |
Finally, p-JNK expression was measured (Fig. 4). Compared with the I/R group,
postconditioning or SP600125 significantly reduced p-JNK expression
(P<0.05), while treatment with anisomycin alone significantly
increased p-JNK expression (P<0.05). The treatment with
anisomysin with postconditioning partially also reduced the level
of PLA.
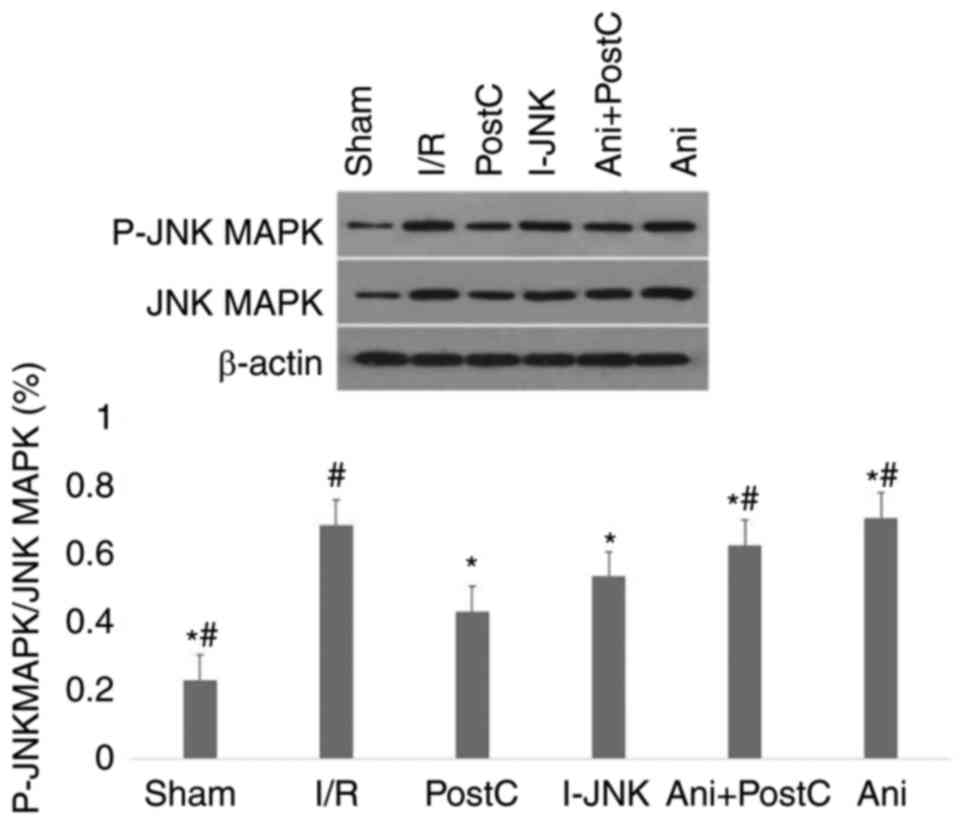 | Figure 4.Expression of P-JNK MAPK in different
groups. Western blot analysis of the expression of P-JNK MAPK in
the sham, ischemia-reperfusion (I/R), ischemic PostC, I-JNK, Ani
and ischemic postconditioning with anisomycin (Ani+PostC) groups.
Statistical analysis of levels of protein expression. *P<0.05
vs. the I/R group; #P<0.05 vs. the post-conditioning
group. I/R, ischemia reperfusion; Ani, anisomycin, postC,
postconditioning; I-JNK, inhibitor of c-Jun N-terminal kinase; p,
phosphorylated. |
Discussion
Using an ischemia-reperfusion model in rat hearts,
ischemic postconditioning reduced infarct size and prevented the
rise in TnI, CK-MB, p-JNK expression and the levels of PLA. These
protective effects were similarly observed using the JNK inhibitor
SP600125, but the effects were abolished with the JNK activator,
anisomycin.
Ischemic postconditioning is an important cardiac
protective mechanism, which reduces myocardial infarct size
(12), the occurrence of
arrhythmias (13), and improves
vascular endothelial function (14) and myocardial systolic function. In
ischemia-reperfusion injury, inflammation has a critical role in
mediating myocardial damage. This involves the recruitment of
leukocytes and the activation of platelets, leading to enhanced
interactions between P-selectin glycoprotein ligand-1, which is
expressed in the leukocytes, with P-selectin, which is expressed in
the platelets to form PLAs. PLA is a sensitive index of thrombus
load and inflammatory burden in vivo (1). Previous studies have demonstrated
that high levels of PLA were not only positively and significantly
associated with the risk of acute coronary syndrome (15) but were also closely associated with
the no-reflow phenomenon (16).
Postconditioning is an important mechanism that
protects against myocardial damage, reducing damage that is
mediated by ischemia-reperfusion and improving cardiac contractile
function. However, the molecular pathways responsible for
protection against myocardial damage have not been entirely
elucidated.
JNK is a member of the MAPK family that modulates
multiple cellular functions, including proliferation,
differentiation and apoptosis (17). JNK can be activated by ischemia and
reperfusion insult. In the absence of JNK, mouse hearts subjected
to ischemia-reperfusion have significantly less necrosis and
apoptosis (18). On the contrary,
the inhibition of JNK reduced the apoptosis of cardiomyocytes and
infarct size following ischemia-reperfusion injury (19).
Nuclear factor (NF)-κB is an important transcription
factor that is involved in ischemia and reperfusion injury. A
recent study demonstrated that intrinsic activation of
AMP-activated protein kinase modulated JNK-NF-κB signaling cascade
during hypoxia and reoxygenation stress conditions. It was critical
to prevent excess mitochondrial reactive oxygen production and
consequent JNK signaling during reperfusion, thereby protecting
against mitochondrial permeability transition pore opening,
irreversible mitochondrial damage and myocardial injury (20). In the present study, at different
time points of MIRI, the level of PLA gradually increased. Compared
with the injury-reperfusion group, the level of PLA in the PostC
and I-JNK groups was significantly reduced at 60 min and 3 h
following reperfusion, which suggested that PLA (a marker of
inflammation and thrombosis) was involved in the pathogenesis of
MIRI. Ischemic postconditioning or inhibition of JNK reduced the
level of PLA. In addition, ischemia-reperfusion injury was able to
increase p-JNK expression, which was prevented by ischemic
postconditioning or JNK inhibition. This result suggested that
myocardial ischemia-reperfusion was able to activate the JNK
signaling pathway. Ischemic postconditioning or the inhibition of
JNK served a critical role in inhibiting the inflammatory response
during the ischemia/reperfusion process. Based on the above
results, it was speculated that the cardioprotective effect of
ischemic postconditioning may be associated with the attenuation of
inflammation, which occurred during ischemia/reperfusion via the
modulation of the JNK-mediated NF-κB signaling pathway. Therefore,
regulating JNK activation may represent an important novel strategy
in the prevention and treatment of MIRI.
Although the current study provided interesting
results, there remain certain weaknesses. Firstly, MIRI is a
complicated pathological process, however, only JNK MAPK signaling
pathway was included in the present study, the whole signaling
network was not investigated. Secondly, the infarct size in I-JNK
group was significantly reduced, but the expression level of
apoptosis associated proteins was not detected. Finally, although
pharmacological inhibition of JNK has cardioprotective effects, it
is not clear whether this JNK inhibitory effect will produce
adverse effects in other tissues.
JNK is a critical mediator of MIRI. Ischemic
postconditioning can reduce the level of PLA during reperfusion by
inhibiting the phosphorylation of JNK MAPK, thereby reducing MIRI.
Pharmacological inhibition and activation of JNK can improve and
reduce the cardioprotective effects, respectively. The results of
the present study help to explain the mechanism of the
cardioprotection against MIRI of postconditioning in rats. In
addition, it provided novel insight and targets for the therapeutic
strategy of MIRI. JNK inhibition may represent a novel therapeutic
strategy for the alleviation of MIRI.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant no.
81500271).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
FR, NM and MG performed experiments and drafted the
manuscript. CZ and XS performed the experiments. LL and TL analyzed
the data. JL and JS performed the animal experiments. MD and GT
made substantial contributions to the design of the present study.
All authors approved the final version of manuscript.
Ethics approval and consent to
participate
The present study was approved by the Research
Commission on Ethics of the Affiliated Yantai Yuhuangding Hospital
of Qingdao University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Bonvini RF, Hendiri T and Camenzind E:
Inflammatory response post-myocardial infarction and reperfusion: A
new therapeutic target? European Heart J Suppl. 7 suppl_I:I27–I36.
2005. View Article : Google Scholar
|
|
2
|
Schächinger V, Britten MB and Zeiher AM:
Prognostic impact of coronary vasodilator dysfunction on adverse
long-term outcome of coronary heart disease. Circulation.
101:1899–1906. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Furman MI, Barnard MR, Krueger LA, Fox ML,
Shilale EA, Lessard DM, Marchese P, Frelinger AL III, Goldberg RJ
and Michelson AD: Circulating monocyte-platelet aggregates are an
early marker of acute myocardial infarction. J Am Coll Cardiol.
38:1002–1006. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Faraday N, Braunstein JB, Heldman AW,
Bolton ED, Chiles KA, Gerstenblith G and Schulman SP: Prospective
evaluation of the relationship between platelet-leukocyte conjugate
formation and recurrent myocardial ischemia in patients with acute
coronary syndromes. Platelets. 15:9–14. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thuny F, Lairez O, Roubille F, Mewton N,
Rioufol G, Sportouch C, Sanchez I, Bergerot C, Thibault H, Cung TT,
et al: Post-conditioning reduces infarct size and edema in patients
with ST-segment elevation myocardial infarction. J Am Coll Cardiol.
59:2175–2181. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xue F, Yang X, Zhang B, Zhao C, Song J,
Jiang T and Jiang W: Postconditioning the human heart in
percutaneous coronary intervention. Clin Cardiol. 33:439–444. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang C, Jacobson K and Schaller MD: A
role for JNK-paxillin signaling in cell migration. Cell Cycle.
3:4–6. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Uehara T, Bennett B, Sakata ST, Satoh Y,
Bilter GK, Westwick JK and Brenner DA: JNK mediates hepatic
ischemia reperfusion injury. J Hepatol. 42:850–859. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Z, Tang B, Tang F, Li Y, Zhang G,
Zhong L, Dong C and He S: Protection of rat intestinal epithelial
cells from ischemia/reperfusion injury by (D-Ala2,
D-Leu5)-enkephalin through inhibition of the MKK7-JNK signaling
pathway. Mol Med Rep. 12:4079–4088. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang DH, Tan N, He PC, Liu Y, Wen JY, Chen
JY, Zhou YL and Huang WH: Increased platelet-leukocyte aggregates
in patients with acute coronary syndrome. Zhonghua Xin Xue Guan
Bing Za Zhi. 40:482–486. 2012.(In Chinese). PubMed/NCBI
|
|
11
|
Li Y, Ge X and Liu X: The cardioprotective
effect of postconditioning is mediated by ARC through inhibiting
mitochondrial apoptotic pathway. Apoptosis. 14:164–172. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Staat P, Rioufo TG, Piot C, Cottin Y, Cung
TT, L'Huillier I, Aupetit JF, Bonnefoy E, Finet G, André-Fouët X
and Ovize M: Postconditioning the human heart. Circulation.
112:2143–2148. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kloner RA, Dow J and Bhandari A:
Postconditioning markedly attenuates ventricular arrhythmias after
ischemia-reperfusion. J Cardiovasc Pharmacol Ther. 11:55–63. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang DH, Tan N, He PC, Liu Y, Wen JY, Chen
JY, Zhou YL and Huang WH: Increased platelet-leukocyte aggregates
in patients with acute coronary syndrome. Zhonghua Xin Xue Guan
Bing Za Zhi. 40:482–486. 2012.(In Chinese). PubMed/NCBI
|
|
15
|
Sivaraman V, Mudalagiri NR, Di Salvo C,
Kolvekar S, Hayward M, Yap J, Keogh B, Hausenloy DJ and Yellon DM:
Postconditioning protects human atrial muscle through the
activation of the RISK pathway. Basic Res Cardiol. 102:453–459.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ren F, Mu N, Zhang X, Tan J, Li L, Zhang C
and Dong M: Increased platelet-leukocyte aggregates are associated
with myocardial no-reflow in patients with ST elevation myocardial
infarction. Am J Med Sci. 352:261–266. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weston CR and Davis RJ: The JNK signal
transduction pathway. Curr Opin Cell Biol. 19:142–149. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kaiser RA, Liang Q, Bueno O, Huang Y,
Lackey T, Klevitsky R, Hewett TE and Molkentin JD: Genetic
inhibition or activation of JNK1/2 protects the myocardium from
ischemia-reperfusion-induced cell death in vivo. J Biol Chem.
280:32602–32608. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ferrandi C, Ballerio R, Gaillard P,
Giachetti C, Carboni S, Vitte PA, Gotteland JP and Cirillo R:
Inhibition of c-Jun N-terminal kinase decreases cardimoyocyte
apoptosis and infarct size after myocardial ischemia and
reperfusion in anaesthetized rats. Br J Pharmacol. 142:953–960.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zaha VG, Qi D, Su KN, Palmeri M, Lee HY,
Hu X, Wu X, Shulman GI, Rabinovitch PS, Russell RR III and Young
LH: AMPK is critical for mitochondrial function during reperfusion
after myocardial ischemia. J Mol Cell Cardiol. 91:104–113. 2016.
View Article : Google Scholar : PubMed/NCBI
|