Introduction
Diabetic retinopathy (DR), a microvascular
complication of diabetes mellitus, occurs in almost all patients
with diabetes worldwide, leading to a severe consequence of
blindness (1). A total of two
types of DR have been identified: Non-proliferative DR, which is
characterized by microaneurysms and intraretinal hemorrhage, and
proliferative DR, which has been identified by vitreous hemorrhage
and neovascularization of the eye fundus and iris (1). Previous studies have demonstrated
that chronic inflammatory and oxidative stress is associated with
the development and progression of DR (2–4).
Elevated levels of reactive oxygen species (ROS) and
reduced antioxidant enzyme activity have been observed in DR
(5). The levels of methylglyoxal,
a reactive α-dicarbonyl compound of glucose metabolism, were
elevated in patients with diabetes (6). In cultured bovine retinal pericytes,
the application of methylglyoxal induced apoptosis via increases in
ROS levels (6); however, the
potential molecular mechanisms underlying increased ROS production
in the pathogenesis of DR remain unclear. Mitochondria are the
major source of superoxide production and are subject to direct
attack by ROS (7). In diabetes,
the dysfunction of retinal mitochondria has been reported, and
overexpression of mitochondrial superoxide dismutase was observed
to protect retinal and vascular cells against high glucose
(HG)-induced increases in oxidative stress and DNA damage (8). Additionally, it was reported that
hypoxic and hyperglycemic conditions increased the levels of NADPH
oxidase (Nox) 2 and Nox 4 isoforms, which led to the overproduction
of ROS (9). These findings suggest
that increased Nox activity may serve a critical role in the
development of DR.
Notch signaling, an evolutionarily conserved
pathway, has been demonstrated to serve pivotal roles in retinal
development and vascular homeostasis (10,11).
Notch signaling involves five ligands, including δ-like 1, 3, 4;
Jagged 1, 2 and four receptors, including Notch-1, 2, 3, 4 and the
transcription factor, recombination signal-binding protein J
(RBPj). Upon activation, Notch intracellular domain (NICD) is
released and translocates to the nucleus where NICD binds RBPj
(12). The resulting complex
contributes to the regulation of the expression of downstream
target genes (13). In diabetic
mice and HG-treated human retinal vascular endothelial cells, it
was reported that reduced expression levels of Notch1 were
associated with the induction of apoptosis (14). The present study aimed to
investigate the role and association of Notch signaling and
Nox4-associated ROS in HG-induced injury of human retinal
endothelial cells (HRECs). The results indicated that Nox4
expression levels were significantly increased in HG-treated HRECs,
and that Nox4-mediated ROS may serve a key role in HG-induced HREC
death. In addition, the present study proposed that Notch1
signaling activation mediated increases in Nox4 expression.
Therefore, the inhibition of Notch signaling or Nox4 expression may
be considered as potential therapeutic targets in patients with
DR.
Materials and methods
Cell culture and treatment
HRECs (ACBRI 181; Cell Systems, Kirkland, WA, USA)
were cultured at 37°C in 5% CO2 incubator in a
fibronectin-coated plate with Dulbecco's modified Eagle's medium
(cat. no. 11885-084) supplemented with 10% fetal calf serum (cat.
no. 16000-044) and 100 U/ml of penicillin/streptomycin (cat. no.
15140-122), all obtained from Gibco (Thermo Fisher Scientific,
Inc., Waltham, MA USA). HRECs of passages 3–6 were used for
analysis in the present study. Cells were cultured for the
indicated durations (0, 6, 12, and 24 h) in the presence of 30 mM
glucose (HG; cat. no. G8270; Sigma-Aldrich, Merck KGaA, Darmstadt,
Germany) or osmotic control mannitol (MN; cat. no. PHR1007;
Sigma-Aldrich; Merck KGaA). In addition, cells were treated with 50
µM γ-secretase inhibitor IX (GSI; cat. no. 565770; Merck KGaA), 5
µM diphenyleneiodonium chloride (DPI; cat. no. D2926;
Sigma-Aldrich; Merck KGaA), or 5 µM GKT137831 (cat. no. 17764;
Cayman Chemical Company, Ann Arbor, MI, USA) at 37°C for 24 h,
which were added to the medium alone or in appropriate combinations
(HG plus DPI, GKT or GSI).
Overexpression of NICD
In the present study, overexpression of NICD was
generated by transfection of pCAGGS-NICD (a gift from Professor
Nicholas Gaiano, cat. no. 26891, Addgene, Inc., Cambridge, MA, USA)
using Lipofectamine® 2000 (cat. no. 11668-027;
Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. Transfection with a blank vector pCAGGS
(a gift from Professor Phil Sharp; cat. no. 41583; Addgene, Inc.)
was used as the control. Briefly, cells were grown in 6-well plate
and transfected with pCAGGS-NICD or blank vector pCAGGS using
Lipofectamine 2000 (2.5 µg plasmid DNA and 5.0 µl Lipofectamine
2000 per well) when at 80% confluence. A total of 6 h following
transfection, medium was changed and cells were cultured for 24 h
in the absence or presence of HG.
Cell death detection
Apoptotic cell death was determined by detection of
cytoplasmic histone-associated DNA fragments using the Cell Death
Detection ELISA kit (cat. no. 11 774 425 001; Roche Diagnostics
GmbH, Mannheim, Germany) according to the manufacturer's protocols.
Experiments were conducted in triplicate in each condition as
described above. The absorbance at 405 and 490 nm (reference
wavelength) was determined with a microplate reader (Bio-Tec
Instruments, Inc., Winooski, VT, USA). Signals of the wells
containing the substrate alone were used as the background and the
experimental values obtained from analysis were subtracted.
Caspase 3 activity detection
The levels of activated caspase-3 were measured
using the Caspase-3 Colorimetric Protease Assay kit (cat. no.
KHZ0022; Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocols. The fold change relative to the
control was presented.
Knockdown assay
To downregulate the expression levels of Nox4 and
RBPj, small interfering (si)RNA-Nox4 (siNox4; sc-41586; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and siRNA-RBPj (sc-38214;
Santa Cruz Biotechnology, Inc.) were transfected (final
concentrations, 10 and 20 nM) into cells with RNAiMAX (cat. no.
13778-075; Invitrogen; Thermo Fisher Scientific, Inc.),
respectively. Cells were then collected after 72 h following
transfection. Non-targeted siRNA (siCTL; sc-37007, Santa Cruz
Biotechnology, Inc.) and nontransfected cells were used as the
control.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from nontreated and HG or MN
alone as well as HG plus siRNA treated cells using an RNeasy Mini
kit (cat. no. 74106; Qiagen GmbH, Hilden, Germany) and the
concentration was determined with an ND-1000 spectrophotometer
(NanoDrop Technologies; Thermo Fisher Scientific, Inc., Wilmington,
DE, USA). A total of three independent experiments were performed.
A total of 2 µg RNA was reverse transcribed into cDNA for use with
the SuperScript™ III First-Strand Synthesis kit (cat.
no. 11752-050; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. qPCR was then performed using a 7500 fast
Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The SYBR Green PCR Master Mix (cat. no. 1725270; Bio-Rad
Laboratories, Inc., Hercules, CA, USA) was used, in which 1.5 µl
cDNA and 0.2 µM of specific primer pairs (Table I) were included. A two-step PCR
program was performed: Initial denaturation at 95°C for 10 min; 40
cycles of 95°C for 15 sec and 60°C for 45 sec. The mRNA expression
levels of Nox1-5 and Notch1-3 as well as RBPj were normalized to
the housekeeping gene GAPDH. The relative expression levels of the
target genes were calculated using the 2−ΔΔCq method
(15). The fold-change relative to
the control was presented in the present study.
 | Table I.Primer sequences employed for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primer sequences employed for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Forward primer | Reverse primer |
|---|
| Nox1 |
5′-CCACTGTAGGCGCCCTAAGTT-3′ |
5′-ATGACCGGTGCAAGGATCC-3′ |
| Nox2 |
5′-GCCCAAAGGTGTCCAAGCT-3′ |
5′-TCCCCAACGATGCGGATAT-3′ |
| Nox3 |
5′-CCTTCTGTAGAGACCGCTATGCA-3′ |
5′-GACCACAGGGCCTAAAATCCA-3′ |
| Nox4 |
5′-GACTTTACAGGTATATCCGGAGCAA-3′ |
5′-TGCAGATACACTGGGACAATGTAGA-3′ |
| Nox5 |
5′-CAGGCACCAGAAAAGAAAGCAT-3′ |
5′-TGTTGATCCAGATAAAGTCCACCTT-3′ |
| Notch1 |
5′-CAATGTGGATGCCGCAGTTGTG-3′ |
5′-CAGCACCTTGGCGGTCTCGTA-3′ |
| Notch2 |
5′-AAAAATGGGGCCAACCGAGAC-3′ |
5′-TTCATCCAGAAGGCGCACAA-3′ |
| Notch3 |
5′-AGATTCTCATCCGAAACCGCTCTA-3′ |
5′-GGGGTCTCCTCCTTGCTATCCTGTG-3′ |
| RBPj |
5′-GCTGACTTATGCATTGCCTCAGGA-3′ |
5′-CCACTGCTGTGAACTGGCATGAAA-3′ |
| GAPDH |
5′-TGTGTCCGTCGTGGATCTGA-3′ |
5′-CCTGCTTCACCACCTTCTTGA-3′ |
ROS detection
In nontreated and HG or MN alone as well as HG plus
DPI or GKT treated cells, the levels of cellular ROS were measured
using a DCFDA Cellular ROS Detection Assay kit (cat. no. ab113851;
Abcam, Cambridge, MA, USA) according to the manufacturer's
protocols; the fold change relative to the controls was
compared.
Western blotting
Total cellular protein was extracted using
radioimmunoprecipitation assay buffer (20 mM Tris-HCl pH 7.5, 150
mM NaCl, 1 mM EGTA, 1% NP-40, 1% SDS, 2.5 mM sodium pyrophosphate,
1 mM β-glycerophosphate, 1 mM Na3VO4, 1 µg/ml
leupeptin and 1 mM PMSF). Nuclear protein was isolated using the
Nuclear Extraction kit (ab113474; Abcam). A total of three
independent experiments were performed. Protein concentration was
quantified using a Pierce™ bicinchoninic acid Protein
Assay kit (cat. no. 23225; Thermo Fisher Scientific, Inc.). A total
of 50 µg protein was separated using 7.5 or 12.5% SDS-PAGE, and
then transferred to a nitrocellulose membrane (Abcam). The membrane
was blocked at room temperature for 1 h in 5% low-fat milk prepared
in Tris-buffered saline containing 0.1% Tween-20 (TBST), and then
incubated for overnight at 4°C with the following primary
antibodies: Rabbit anti-B-cell lymphoma 2 (Bcl-2; 1:500; cat. no.
ab32124; Abcam), mouse anti-Bcl-2-associated X (Bax; 1:1,000; cat.
no. ab77566; Abcam), rabbit anti-Nox4 (1:200; cat. no. ABC459;
Merck KGaA), mouse anti-histone H3 (1:11,000; cat. no. 14269S; Cell
Signaling Technology, Inc., Danvers, MA, USA), rabbit anti-RBPj
(1:500; cat. no. SAB1410700; Sigma-Aldrich; Merck KGaA), rabbit
anti-Notch1 (1:400; cat. no. 3608S; Cell Signaling Technology,
Inc.), rabbit anti-Notch 1 Antibody N-terminus (1:500; cat. no.
07-1232; Merck KGaA), and mouse anti-β-actin (1: 10,000; cat. no.
A2228; Sigma-Aldrich; Merck KGaA). Histone H3 and β-actin was used
as an internal reference for the expression level of nuclear
protein and total cellular protein, respectively. Following five
washes with TBST, the membranes were incubated with horseradish
peroxidase-conjugated goat anti-rabbit or goat anti-mouse IgG
antibodies (1:5,000; cat. nos. G-21234 and G-21040; Invitrogen;
Thermo Fisher Scientific, Inc.) for 1 h at room temperature.
Following five washes with TBST, the blots were developed using an
Enhanced Chemiluminescence Western Blotting Substrate (cat. no.
32109; Pierce; Thermo Fisher Scientific, Inc.), and the intensity
of the bands was quantified using ImageJ software (version 1.51s;
National Institute of Health, Bethesda, MD, USA).
Statistical analysis
A total of three independent experiments were
performed in the present study. Data are presented as the mean ±
standard deviation. One-way analysis of variance with the Turkey's
post-hoc test was used to conduct statistical analysis (GraphPad
Prism 6.0, GraphPad Software, Inc., La Jolla, CA, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
Increased ROS production is associated
with HG-induced cell death in HRECs
It has been well reported that oxidative stress
serves a key role in the induction of cellular apoptosis (16). Thus, the present study investigated
the levels of ROS in HG-treated HRECs; MN-treated cells were used
as the osmotic controls. A significant time-dependent increase in
cellular ROS levels was detected in HG-treated cells compared with
in cells at 0 h and the MN group (Fig.
1A). To reveal the role of ROS overproduction in HG-treated
HRECs, the generation of ROS was inhibited by the administration of
DPI. Treatment with DPI has been applied to eliminate the
production of ROS mediated by flavoenzymes, particularly by Nox
(17). The results of the present
study demonstrated that HG-mediated increases in ROS levels were
significantly inhibited by DPI treatment in a dose-dependent manner
compared with in the control and HG groups; however, 10 µM DPI did
not notably affect ROS production compared with in the control
(Fig. 1B). As previously reported,
increased apoptotic cell death was detected in HG-treated HRECs
(18), which may be associated
with the DPI-mediated depletion of ROS; however, 5 and 10 µM DPI
exhibited a significant effect on cell apoptosis compared with in
the MN and control groups (Fig.
1C). Caspase 3, a critical cellular apoptosis-inducing protease
(19), demonstrated significantly
elevated activity levels within HG-treated cells compared with in
the MN and control groups, which was inhibited by DPI in
dose-dependent manner; however 5 and 10 µM DPI did not
significantly affect caspase 3 activity compared with in the MN and
control groups (Fig. 1D).
Consistently, the results from western blot analysis revealed
significant reductions in the expression levels of cellular
survival protein, Bcl-2 and upregulation of proapoptotic protein
Bax in HG-treated cells compared with in the MN and control groups,
which was inhibited by the addition of DPI (Fig. 1E). These data suggested that
activation of oxidative stress signaling-associated proteins may
serve a role in HG-induced HREC injury.
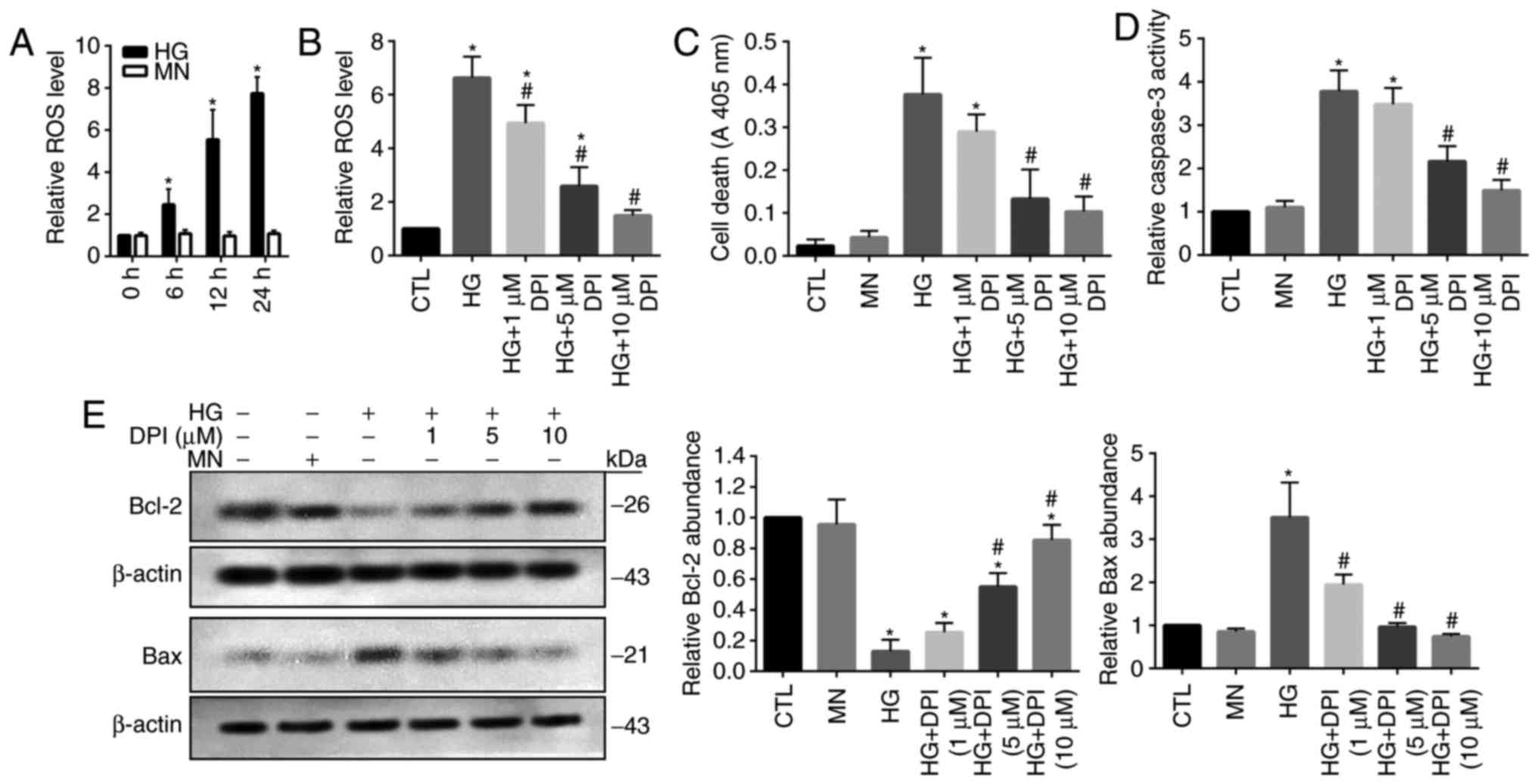 | Figure 1.Increased ROS is associated with
HG-induced cell death in human retinal endothelial cells. (A) Cells
were cultured in the presence of HG or MN, and the ROS levels was
evaluated. (B) Cells were cultured for 24 h in the presence of HG
and treated with DPI. Cells cultured in standard glucose media were
used as the CTL. The ROS levels were then evaluated. Cells were
cultured for 24 h in the presence of HG or MN, and treated with
DPI. (C) Cell death, (D) caspase 3 activity, and (E) protein
expression levels of Bcl-2 and Bax were evaluated. Data are
presented as the mean ± standard deviation. n=3. *P<0.05 vs. 0
h, CTL or MN, #P<0.05 vs. HG. Bcl-2, B-cell lymphoma
2; Bax, Bcl-2-associated X; CTL, control; DPI, diphenyleneiodonium;
HG, high-glucose; MN, mannitol; ROS, reactive oxygen species. |
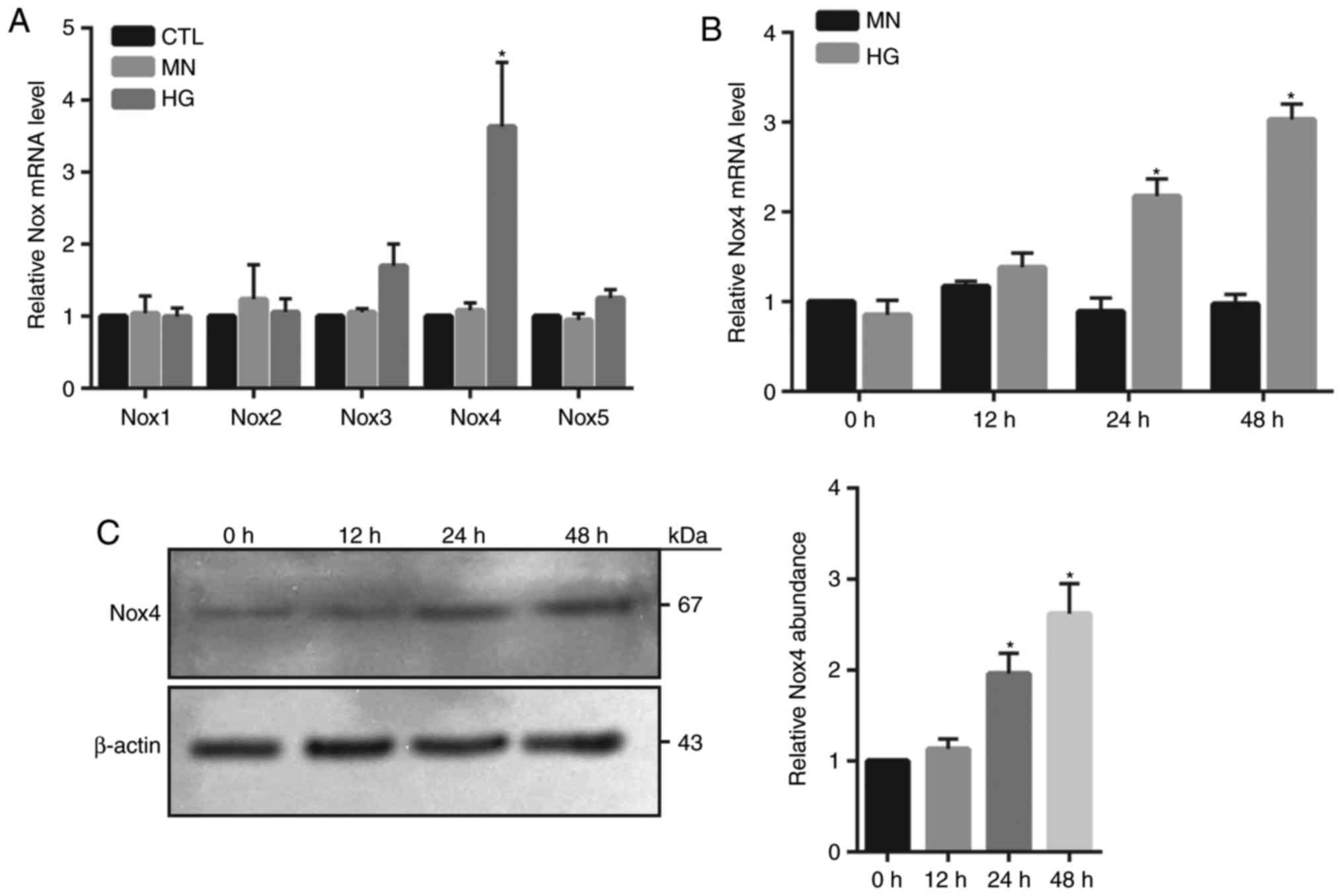
Nox4 upregulation is associated with
HG-induced HRECs injury
NADPH oxidase generates superoxide by transferring
electrons from NADPH within the cell across the membrane and is
coupled to molecular oxygen to produce a superoxide anion, a major
source of ROS (9). In the present
study, the mRNA expression levels of various isoforms of Nox were
investigated, including Nox1, 2, 3, 4 and 5 within HG-treated
HRECs. RT-qPCR demonstrated that only Nox4 mRNA expression levels
were significantly increased at 24 h in HG-treated cells compared
with in the MN and control groups (Fig. 2A). Furthermore, a time-dependent
increase in Nox4 mRNA and protein abundance was observed in
HG-treated cells; however, the expression levels at 24 and 48 h
were significantly elevated compared with 0 h (Fig. 2B and C). To investigate whether
Nox4 serves a major role in the induction of ROS and cellular death
within HG-treated HRECs, Nox4 activity was inhibited by using
GKT137831. The results revealed that treatment with GKT137831
significantly inhibited ROS levels and cell death, as well as the
activity of caspase 3 compared with in the HG group (Fig. 3A-C). To specifically reveal the
role of Nox4, knockdown of Nox4 expression was conducted by using
siNox4. The results revealed that siNox4 (10 and 20 µM)
significantly reduced the protein and mRNA expression levels of
Nox4 compared with in the control groups (Fig. 3D and E). As expected, depletion of
Nox4 expression significantly decreased apoptotic cell death and
caspase 3 activity in HG-treated cells compared with in the control
group (Fig. 3F and G). These
findings indicated that Nox4-mediated oxidative stress may serve an
important role in HG-induced HREC injury.
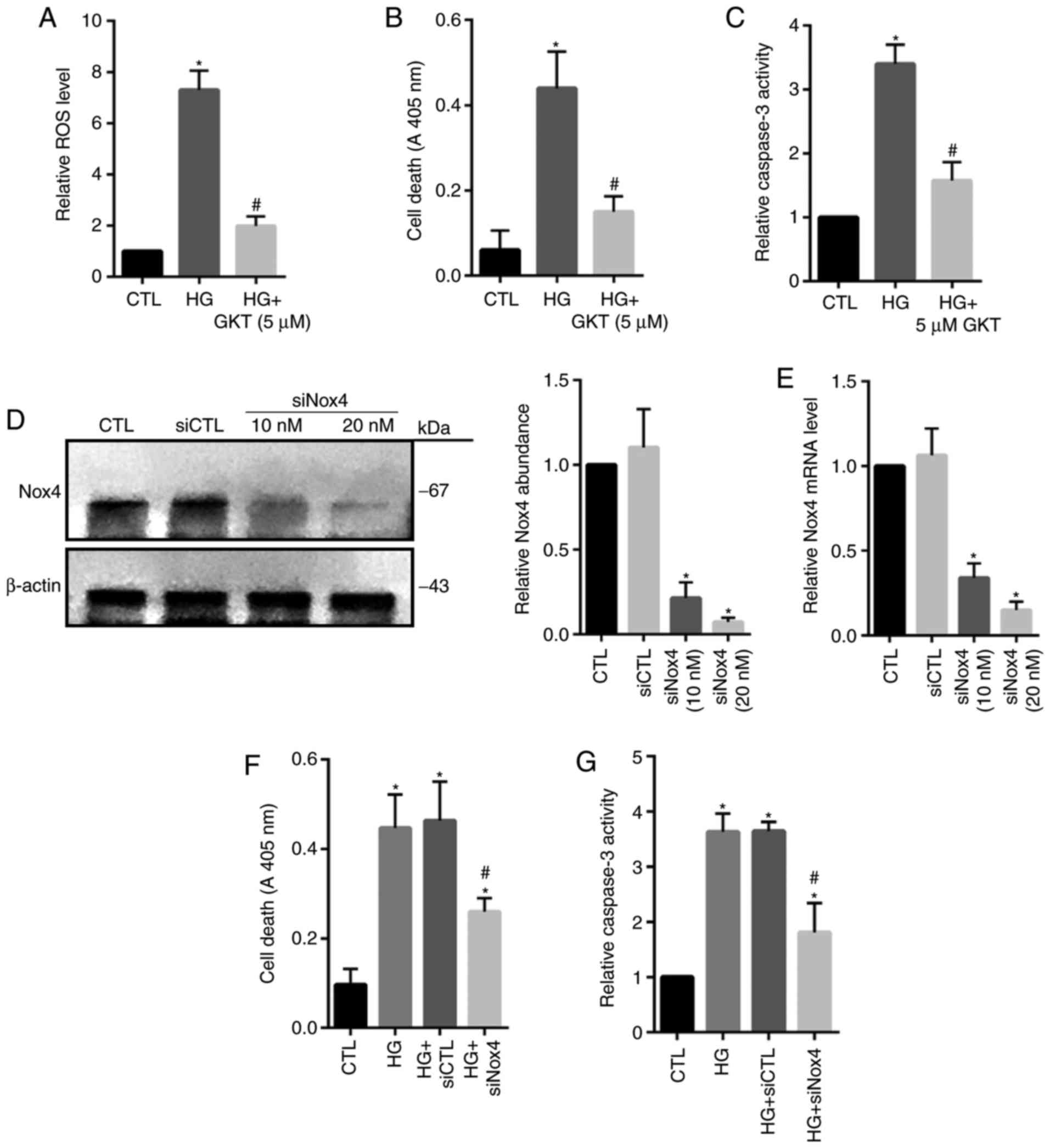 | Figure 3.Inhibition of Nox4 suppresses
HG-induced cell death in human retinal endothelial cells. Cells
were cultured for 24 h in the presence of: Standard glucose as the
CTL, HG, or HG and GKT (5 µM). (A) ROS level, (B) cell death and
(C) caspase 3 activity were assessed, respectively. Cells were
cultured and transfected with siCTL (20 nM) or siNox4 (10 or 20
nM). (D) Protein and (E) mRNA expression levels of Nox4 were
measured via an immunoblot assay and reverse
transcription-quantitative polymerase chain reaction. Cells were
cultured for 24 h in the presence of HG and treated with 20 nM of
siNox4 or siCTL. (F) Cell death and (G) caspase 3 activity were
assessed, respectively. Data are presented as the mean ± standard
deviation. n=3. *P<0.05 vs. CTL or siCTL, #P<0.05
vs. HG or HG + siCTL. CTL, control; Nox4, NADPH oxidase 4; GKT,
GKT137831, Nox4 inhibitor; HG, high-glucose; siRNA, small
interfering RNA; siCTL, control siRNA; siNox4, Nox4 siRNA. |
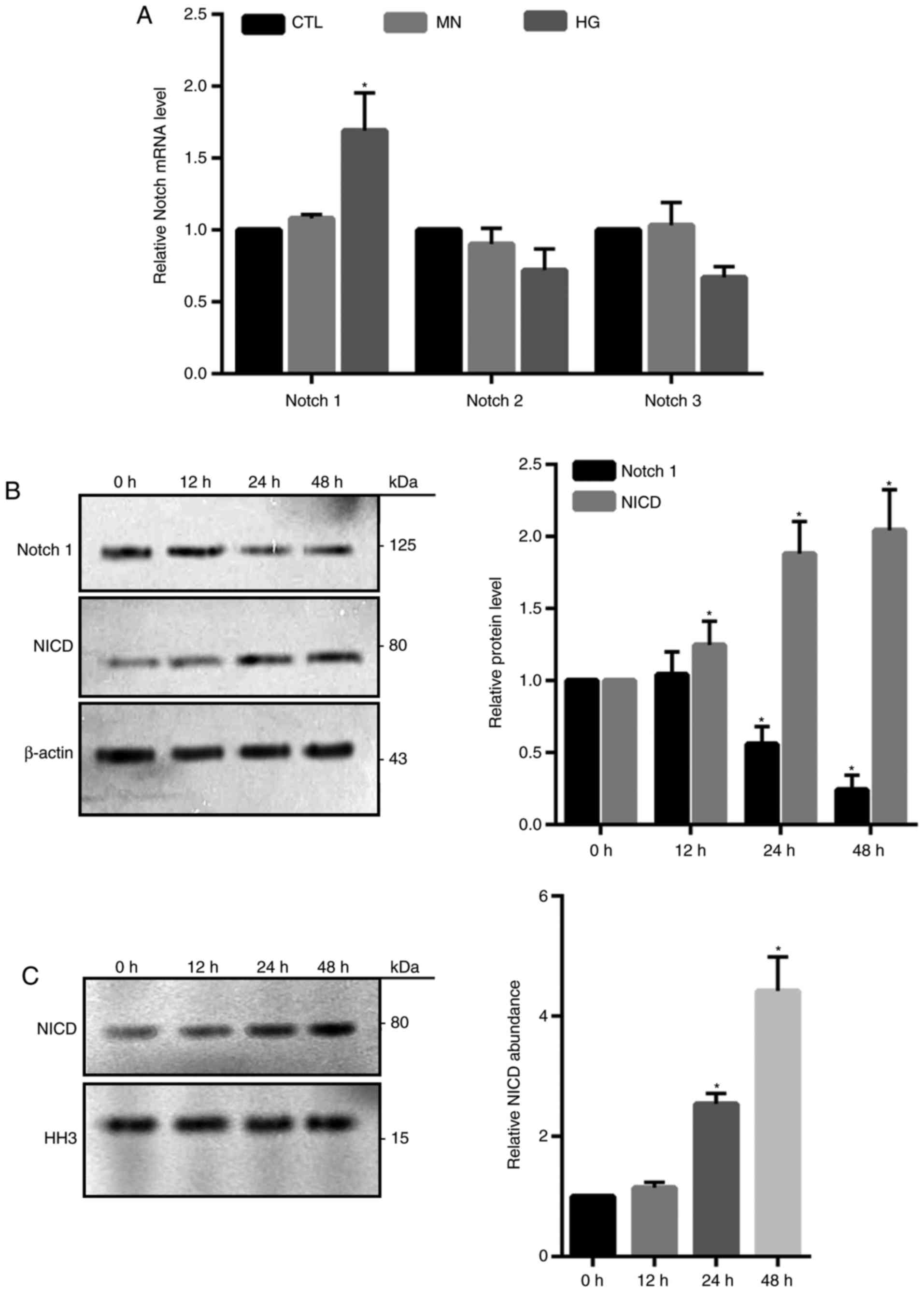
Notch1 signaling is involved in
HG-induced cell death in HRECs
Notch signaling pathway is highly conserved and
involves numerous different Notch receptors (12). It was reported that Notch signaling
serves a crucial role in the regulation of apoptotic cell death
(20). Compared with in the
control groups, Notch1 mRNA expression levels were significantly
increased in HG-treated cells as determined by RT-qPCR; however,
Notch2 and Notch3 exhibited no notable alterations in expression
(Fig. 4A). Western blotting
demonstrated a significant time-dependent increase in the abundance
of NICD and reductions in total Notch1 following HG treatment
compared with 0 h treatment (Fig.
4B). Additionally, significantly increased NICD expression
levels were also detected in the isolated nuclear fractions from
HG-treated cells at 24 and 48 h compared with 0 h (Fig. 4C), indicating that NICD may act as
a transcriptional factor.
To investigate the function of the Notch signaling
pathway, in HG-treated HRECs, Notch activity was suppressed via the
administration of GSI, which inhibits the cleavage of Notch and
therefore NICD production. Western blotting revealed that the
expression levels of Notch1 were significantly increased in
GSI-treated cells compared with in the control, while that of NICD
were significantly reduced (Fig.
5A). HG-induced apoptosis was also significantly inhibited in
response to treatment with GSI (Fig.
5B). In addition, NICD was successfully overexpressed in HRECs
following transfection with pCAGGS-NICD (Fig. 5C); apoptosis was significantly
increased in cells overexpressing NICD compared with in the control
groups (Fig. 5D). Consistently,
HG-induced cell death was significantly enhanced by NICD
overexpression compared with the HG groups and the control
(Fig. 5E). These findings
indicated that overactivation of the Notch signaling pathway may be
associated with HG-mediated HREC injury.
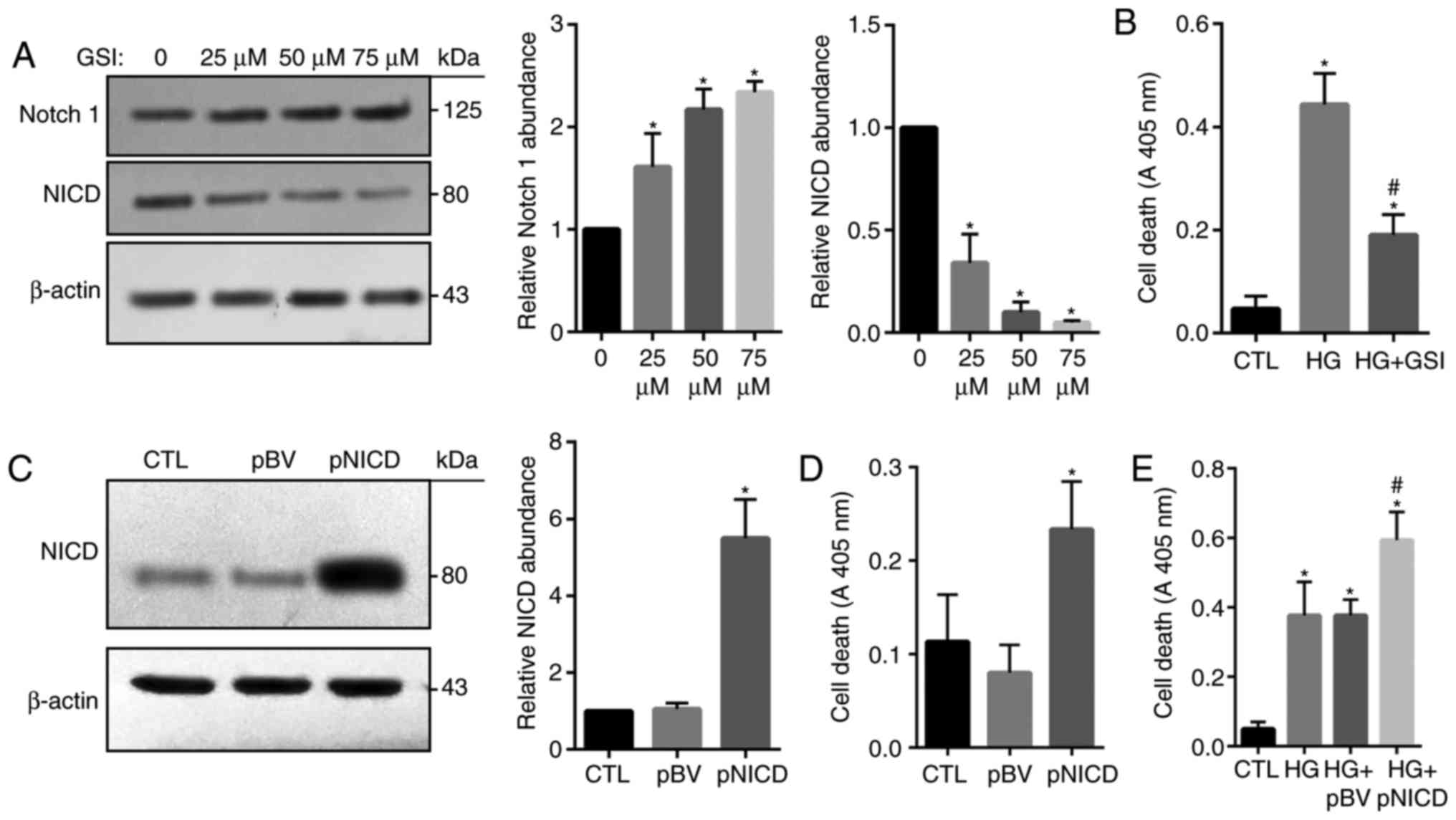 | Figure 5.Notch1 signaling is involved in
HG-induced cell death in human retinal endothelial cells. (A) Cells
were cultured and treated with GSI to inhibit Notch signaling. The
expression levels of NICD and Notch1 were evaluated via
immunoblotting. (B) Cells were cultured for 24 h in the presence
of: Standard glucose as the CTL, HG, or HG and GSI (50 µM). Cell
death was assessed. (C) Cells were transfected with pBV or the
pNICD. Overexpressed NICD was detected via an immunoblot assay; (D)
the effect of NICD on cell death was assessed. (E) Cells were
cultured for 24 h in the presence of HG and pBV or pNICD. Cell
death was then evaluated. Data are presented as the mean ± standard
deviation. n=3. *P<0.05 vs. 0 µM, CTL or pBV,
#P<0.05 vs. HG or HG + pBV. CTL, control; GSI,
γ-secretase inhibitor; HG, high-glucose; NICD, Notch1 intracellular
domain; pBV, black vector; pNICD, overexpressing NICD vector. |
Inhibition of Notch signaling
suppresses Nox4 upregulation in HG-treated HRECs
To determine the potential association between
Notch1 and Nox4, the effects of Notch signaling on Nox4 expression
were investigated in the present study. HG-induced increases in
Nox4 mRNA and protein expression were significantly inhibited via
the application of GSI compared with the HG group (Fig. 6A and B), suggesting that the Notch
signaling pathway may be required for the induction of Nox4 in
HG-treated HRECs. Following Notch activation, NICD is produced by
the cleavage of γ-secretase and is then translocated to nucleus, in
which nuclear NICD associates with RBPj, leading to regulation of
target gene expression (21). In
the present study, RBPj expression was significantly reduced via
siRNA-RBPj (10 and 20 µM) compared with in the control groups
(Fig. 6C and D). Knockdown of RBPj
significantly decreased Nox4 expression levels within HG-treated
cells compared with in the HG groups (Fig. 6E). These data demonstrated that HG
may enhance Nox4 expression potentially by increasing Notch
activity. Over all, the results of the present study suggest that
under the conditions of HG, nuclear NICD is produced and then
induces upregulation of Nox4 expression, thus increasing ROS level
and cell death (Fig. 6F).
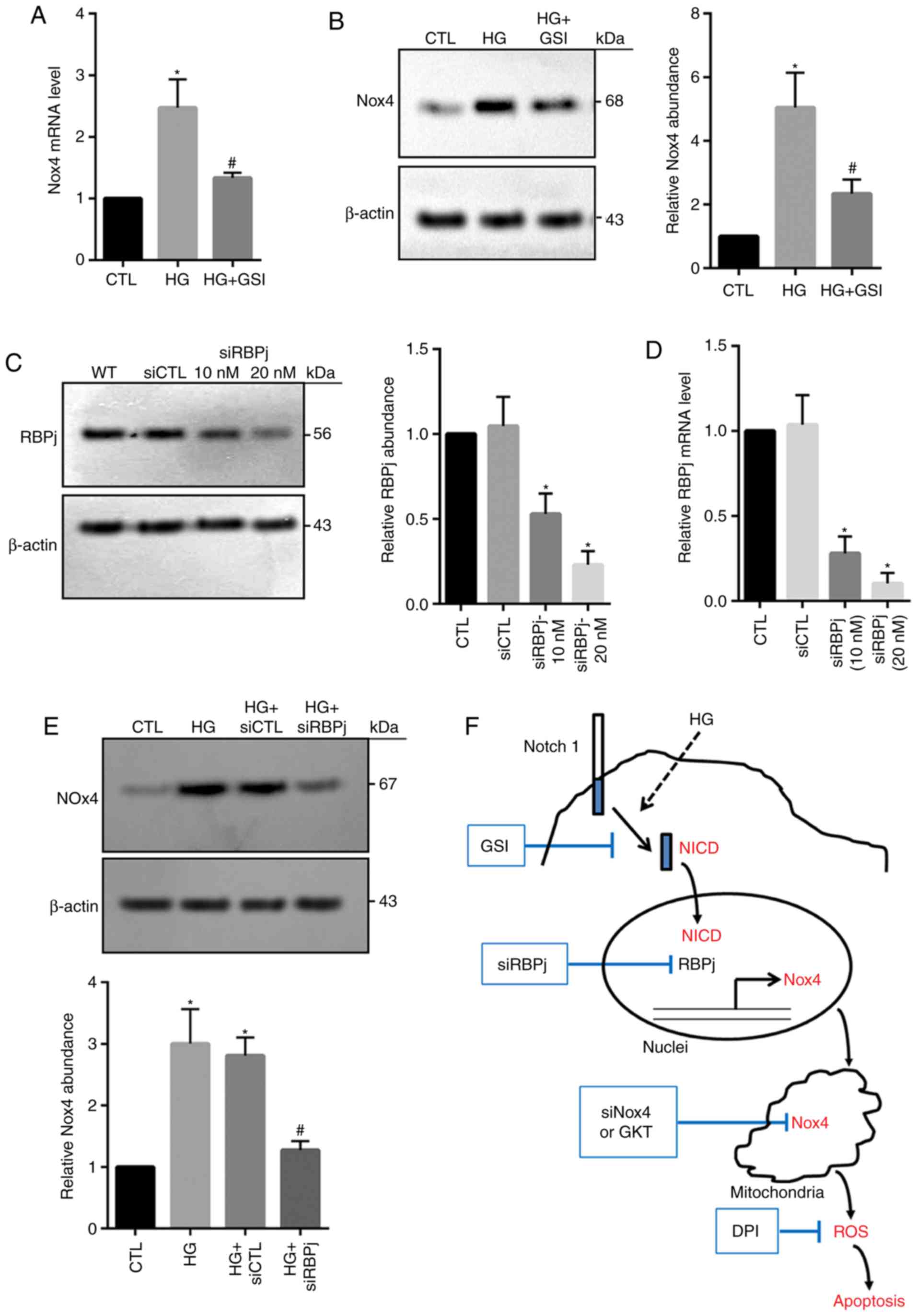 | Figure 6.Inhibition of Notch signaling
suppresses Nox4 expression in HRECs. Cells were cultured for 24 h
in the presence of: Standard glucose as the CTL, HG, or HG and GSI
(50 µM). The expression of Nox4 at the (A) mRNA and (B) protein
levels was evaluated. Cells were cultured and transfected with
siCTL or siRBPj (10 or 20 nM). (C) Protein and (D) mRNA expression
levels of RBPj were measured via immunoblotting and reverse
transcription-quantitative polymerase chain reaction, respectively.
(E) Cells were cultured for 24 h in presence of HG alone, or HG and
20 nM siRBPj. Nox4 protein expression levels were evaluated. Data
are presented as the mean ± standard deviation. n=3. *P<0.05 vs.
CTL or siCTL, #P<0.05 vs. HG or HG+siCTL. (F)
Schematic of the role of the Notch-Nox4-ROS signaling pathway in
HG-induced HRECs injury. Under the conditions of HG, NICD is
expressed and is then translocated to the nucleus; NICD induces the
upregulation of Nox4 via complex formation of NICD and RBPj.
Increased Nox4 upregulates ROS production that is responsible for
the induction of cell death. CTL, control; GSI, γ-secretase
inhibitor; HG, high-glucose; HRECs, human retinal endothelial
cells; NICD, Notch1 intracellular domain; RBPj, recombination
signal-binding protein J; Nox4, NADPH oxidase 4; ROS, reactive
oxygen species; siRNA, small interfering RNA; siCTL, control-siRNA;
siRBPj, RBPj-siRNA. |
Discussion
In the present study, a novel signaling pathway
underlying increased ROS production and retinal vascular cell death
was investigated under HG conditions. The findings of the present
study demonstrated that activation of the Notch signaling pathway
may promote Nox4 expression, which in turn lead to increased ROS
production and cell death in HG-treated HRECs.
In patients with diabetes, increased oxidative
stress has been recognized as a major cause of retinal inflammation
(22). Providing that the retina
requires high levels of oxygen (22), the retina and its vasculature may
be susceptible to oxidative stress. It has been reported that
oxidative stress and ROS serve critical roles in the pathogenesis
of DR (23–25). Excessive ROS can cause endothelial
dysfunction and apoptosis, leading to the loss of vascular cells
(26). In the present study,
exposure to HG significantly increased the apoptosis of cultured
HRECs. Following the addition of HG, a time-dependent increase in
intracellular ROS levels was also detected, whereas depletion of
ROS production by the administration of DPI significantly decreased
HG-induced cell death. In HG-treated cells, increased caspase 3
activity and the proapoptotic Bax protein expression levels were
decreased in response to DPI. These findings suggested that
enhanced oxidative stress serves an important role in HG-induced
HRECs injury, and that the use of specific antioxidant may be a
potential therapeutic target to obtain optimal levels of ROS in DR.
A major source of ROS is the NADPH oxidase system in endothelial
cells (27,28). NADPH oxidase mainly comprises five
isoforms, Nox1-5 (29). The
results of RT-qPCR in the present study revealed that Nox4 mRNA
expression levels had increased significantly, whereas that of
Nox1, 2, and 3 exhibited no notable change within HG-treated HRECs;
however, within cultured bovine retinal endothelial cells (BRECs)
stimulated with 20 mM HG, upregulated Nox2 mRNA expression levels
and activity were reported (30).
In the retinal microvasculature isolated from the retina of
patients with DR, the expression of Nox2 was also increased
(30). Additionally, it was
reported that Nox2 protein expression levels were increased in
retinas isolated from rats with DR, and that inhibition of Nox2
mediated by simvastatin may be associated with normalization of ROS
levels and reductions in retinal vascular injury (31). In the present study, 30 mM HG was
applied to cultured HRECs, which increased the expression of Nox4
at the mRNA and protein levels; however, Nox2 expression levels did
not exhibit significant alterations. Various concentrations of HG
and cellular types may explain these controversial results. Of
note, Nox4 expression levels were not investigated in HG-treated
BRECs, and the retina of patients or rats with DR. In addition, as
a limitation of the present study, in vivo investigations
were not conducted. The varying expression levels of Nox isoforms
require further investigation in animal models of DR. Previous
studies have also reported that Nox4 is a major isoform of NADPH
oxidase in retinal microvascular endothelial cells (32,33).
To investigate the role of Nox4 in HG-treated HRECs, a Nox4
inhibitor GKT137831 and an siRNA designed to target human Nox4 were
employed in the present study. Inhibition of Nox4 prevented
HG-induced increases in ROS production in HRECs. The induction of
caspase 3 activity and apoptosis was also prevented by Nox4
inhibition and knockdown. Therefore, enhanced Nox4 expression may
serve a critical role in HG-induced HREC apoptosis, and Nox4 may be
considered as a therapeutic target to ameliorate vascular injury in
DR. In addition, regarding the activation of Nox proteins, numerous
signaling pathways have been identified to be associated with the
progression of DR, including the activation of protein kinase C,
the formation of advanced glycation end products, the
peroxynitrite, hexosamine and polyol signaling pathways (9).
In the present study, the mechanisms by which HG
induces the upregulation of Nox4 in HRECs were investigated. In
renal tubular epithelial cells, Nox4 was reported to be involved
HG-induced cell death via Notch signaling (34). Additionally, within primary human
umbilical vein endothelial cells, inhibition of Notch signaling led
to increases in intracellular ROS via Nox4 upregulation (35). These findings suggest a potential
association between Nox4 and Notch signaling. The Notch signaling
pathway is a highly conserved cell signaling system that is present
in the majority of multicellular organisms (36). In mammals, there are four different
Notch receptors, referred to as Notch1, Notch2, Notch3 and Notch4.
In the angiogenic process, alternative and distinct roles for
different Notch ligands have been identified in the retina
(37). The present study reported
that Notch1 mRNA expression levels were significantly increased in
HG-treated HRECs, whereas Notch2 and Notch3 exhibited no notable
change. The abundance of NICD was observed to increase in a
time-dependent manner in whole cellular lysates and in the nuclear
fractions from HG-treated cells. Notch is a transmembrane protein;
NICD is produced from the cleavage of Notch by γ-secretase
(37,38). The released NICD then enters the
cell nucleus to regulate the expression of specific genes
associated with the control of cell fate (38).
In retinal pigment epithelium cells, Notch2 was
demonstrated to be the major Notch receptor, and inhibition of
Notch2 markedly attenuated intracellular ROS production and
cellular apoptosis in ultraviolet B-induced damage (39). To reveal the role of Notch
signaling in HG-induced HRECs apoptosis or cell death, GSI was used
to prevent the cleavage of Notch and NICD production in the present
study. GSI was observed to significantly decrease NICD abundance
and HG-induced cell death. Apoptosis was significantly increased in
HG-treated HRECs overexpressing NICD. The results of the present
study suggested that the activation of the Notch signaling pathway
serves a pivotal role in HG-mediated HRECs injury. In retinal
ganglion cells, hypoxia-induced Notch1 expression and signaling
activation, and inhibition of Notch signaling significantly
aggravated hypoxia-induced cell apoptosis (40). In a co-culture system of
ligand-dependent Notch activation using primary cultured retinal
pericytes and a mesenchymal cell line derived from an inducible
mouse model expressing δ-like 1 Notch ligand, ligand-mediated Notch
activity was observed to protect retinal pericytes from
light-induced cell death (41). It
has been reported that Notch signaling is highly pleiotropic and
can affect differentiation, proliferation and/or apoptotic events
in numerous ways that depend on their integration with other
signaling pathways (42). The
present study investigated the effects of inhibiting Notch activity
on Nox4 expression levels in HG-treated HRECs. The results study
revealed that treatment with GSI significantly suppressed
HG-induced upregulation of Nox4 at mRNA and protein levels,
suggesting that HG may enhance Nox4 expression via the activation
of the Notch signaling pathway in HRECs. To further confirm the
role of Notch activation in HG-induced Nox4 expression, the
expression of RBPj was downregulated with a specific siRNA. RBPj is
a co-activator that can promote gene expression by associating with
Notch (43). The constitutive
activation of Notch enhanced retinal pigment epithelium cell
proliferation, which was dependent on the presence of the
transcription factor RBPj (44).
In the present study, knockdown of RBPj prevented HG-induced
increases in the expression levels of Nox4.
Collectively, the results of the present study
demonstrated that HG upregulates Nox4 expression via the activation
of Notch signaling, resulting in increased ROS production and cell
death in HRECs. Inhibition of Notch signaling or Nox4 expression
may be potential therapeutic strategies for the treatment of DR.
However, to further confirm the role of the Notch-Nox4-ROS
signaling pathway in the pathogenesis of DR, future investigations
with primary cultured HRECs, or conditional Notch or Nox4 knockout
mice may be conducted.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of China (grant no. 61572300, 81871508 and
61773246), the Taishan Scholar Program of Shandong Province (grant
no. TSHW201502038), and the Key Research and Development Program of
Shandong Province (grant no. 2017GSF18178).
Availability of data and materials
Not applicable.
Authors' contributions
WJ, JJ and WX conceived designed the experiments. WJ
performed the experiments. WJ, WX, FL and JG analyzed the data. WJ,
YZ and SL performed the statistical analysis. WJ wrote and
submitted the manuscript. WJ and JJ and WX revised the manuscript.
All authors reviewed and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent to publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DR
|
diabetic retinopathy
|
|
HG
|
high glucose
|
|
HRECs
|
human retinal endothelial cells
|
|
NICD
|
Notch intracellular domain
|
|
MN
|
mannitol
|
|
ROS
|
reactive oxygen species
|
|
siRNA
|
small interference RNA
|
|
DPI
|
diphenyleneiodonium
|
|
GSI
|
γ-secretase inhibitor IX
|
|
Nox
|
NADPH oxidase
|
|
RBPj
|
recombination signal-binding protein
J
|
References
|
1
|
Yau JW, Rogers SL, Kawasaki R, Lamoureux
EL, Kowalski JW, Bek T, Chen SJ, Dekker JM, Fletcher A, Grauslund
J, et al: Global prevalence and major risk factors of diabetic
retinopathy. Diabetes Care. 35:556–564. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kanwar M, Chan PS, Kern TS and Kowluru RA:
Oxidative damage in the retinal mitochondria of diabetic mice:
Possible protection by superoxide dismutase. Invest Ophthalmol Vis
Sci. 48:3805–3811. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Adamiec-Mroczek J, Oficjalska-Młyńczak J
and Misiuk-Hojło M: Roles of endothelin-1 and selected
proinflammatory cytokines in the pathogenesis of proliferative
diabetic retinopathy: Analysis of vitreous samples. Cytokine.
49:269–274. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Behl Y, Krothapalli P, Desta T, DiPiazza
A, Roy S and Graves DT: Diabetes-enhanced tumor necrosis
factor-alpha production promotes apoptosis and the loss of retinal
microvascular cells in type 1 and type 2 models of diabetic
retinopathy. Am J Pathol. 172:1411–1418. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kowluru RA, Tang J and Kern TS:
Abnormalities of retinal metabolism in diabetes and experimental
galactosemia. VII. Effect of long-term administration of
antioxidants on the development of retinopathy. Diabetes.
50:1938–1942. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim J, Son JW, Lee JA, Oh YS and Shinn SH:
Methylglyoxal induces apoptosis mediated by reactive oxygen species
in bovine retinal pericytes. J Korean Med Sci. 19:95–100. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Saccà SC, Cutolo CA, Ferrari D, Corazza P
and Traverso CE: The eye, oxidative damage and polyunsaturated
fatty acids. Nutrients. 10(pii): E6682018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kowluru RA, Atasi L and Ho YS: Role of
mitochondrial superoxide dismutase in the development of diabetic
retinopathy. Invest Ophthalmol Vis Sci. 47:1594–1599. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Coucha M, Elshaer SL, Eldahshan WS, Mysona
BA and El-Remessy AB: Molecular mechanisms of diabetic retinopathy:
Potential therapeutic targets. Middle East Afr J Ophthalmol.
22:135–144. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hatakeyama J and Kageyama R: Retinal cell
fate determination and bHLH factors. Semin Cell Dev Biol. 15:83–89.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hellström M, Phng LK, Hofmann JJ, Wallgard
E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N,
et al: Dll4 signalling through Notch1 regulates formation of tip
cells during angiogenesis. Nature. 445:776–780. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Borggrefe T and Oswald F: The Notch
signaling pathway: Transcriptional regulation at Notch target
genes. Cell Mol Life Sci. 66:1631–1646. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zheng M, Zhang Z, Zhao X, Ding Y and Han
H: The Notch signaling pathway in retinal dysplasia and retina
vascular homeostasis. J Genet Genomics. 37:573–582. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qin X, Zhang Z, Xu H and Wu Y: Notch
signaling protects retina from nuclear factor-κB- and
poly-ADP-ribose-polymerase-mediated apoptosis under high-glucose
stimulation. Acta Biochim Biophys Sin (Shanghai). 43:703–711. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dehdashtian E, Mehrzadi S, Yousefi B,
Hosseinzadeh A, Reiter RJ, Safa M, Ghaznavi H and Naseripour M:
Diabetic retinopathy pathogenesis and the ameliorating effects of
melatonin; involvement of autophagy, inflammation and oxidative
stress. Life Sci. 193:20–33. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee JE, Cho KE, Lee KE, Kim J and Bae YS:
Nox4-mediated cell signaling regulates differentiation and survival
of neural crest stem cells. Mol Cells. 37:907–911. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
El-Remessy AB, Rajesh M, Mukhopadhyay P,
Horváth B, Patel V, Al-Gayyar MM, Pillai BA and Pacher P:
Cannabinoid 1 receptor activation contributes to vascular
inflammation and cell death in a mouse model of diabetic
retinopathy and a human retinal cell line. Diabetologia.
54:1567–1578. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shalini S, Dorstyn L, Dawar S and Kumar S:
Old, new and emerging functions of caspases. Cell Death Differ.
22:526–539. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tian DY, Jin XR, Zeng X and Wang Y: Notch
signaling in endothelial cells: Is it the therapeutic target for
vascular neointimal hyperplasia? Int J Mol Sci. 18(pii): E16152017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tanigaki K and Honjo T: Two opposing roles
of RBP-J in Notch signaling. Curr Top Dev Biol. 92:231–252. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang W, Liu H, Al-Shabrawey M, Caldwell
RW and Caldwell RB: Inflammation and diabetic retinal microvascular
complications. J Cardiovasc Dis Res. 2:96–103. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tarr JM, Kaul K, Chopra M, Kohner EM and
Chibber R: Pathophysiology of diabetic retinopathy. ISRN
Ophthalmol. 2013:3435602013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Caldwell RB, Bartoli M, Behzadian MA,
El-Remessy AE, Al-Shabrawey M, Platt DH, Liou GI and Caldwell RW:
Vascular endothelial growth factor and diabetic retinopathy: Role
of oxidative stress. Curr Drug Targets. 6:511–524. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ali TK and El-Remessy AB: Diabetic
retinopathy: Current management and experimental therapeutic
targets. Pharmacotherapy. 29:182–192. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Radak D, Resanovic I and Isenovic ER: Link
between oxidative stress and acute brain ischemia. Angiology.
65:667–676. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Babior BM: The NADPH oxidase of
endothelial cells. IUBMB Life. 50:267–269. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Frey RS, Ushio-Fukai M and Malik AB: NADPH
oxidase-dependent signaling in endothelial cells: Role in
physiology and pathophysiology. Antioxid Redox Signal. 11:791–810.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wingler K, Hermans JJ, Schiffers P, Moens
A, Paul M and Schmidt HH: NOX1, 2, 4, 5: Counting out oxidative
stress. Br J Pharmacol. 164:866–883. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kowluru RA, Kowluru A, Veluthakal R,
Mohammad G, Syed I, Santos JM and Mishra M: TIAM1-RAC1 signalling
axis-mediated activation of NADPH oxidase-2 initiates mitochondrial
damage in the development of diabetic retinopathy. Diabetologia.
57:1047–1056. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Al-Shabrawey M, Bartoli M, El-Remessy AB,
Ma G, Matragoon S, Lemtalsi T, Caldwell RW and Caldwell RB: Role of
NADPH oxidase and Stat3 in statin-mediated protection against
diabetic retinopathy. Invest Ophthalmol Vis Sci. 49:3231–3238.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li J, Wang JJ and Zhang SX: NADPH oxidase
4-derived H2O2 promotes aberrant retinal neovascularization via
activation of VEGF receptor 2 pathway in oxygen-induced
retinopathy. J Diabetes Res. 2015:9632892015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang H, Yang Z, Jiang Y and Hartnett ME:
Endothelial NADPH oxidase 4 mediates vascular endothelial growth
factor receptor 2-induced intravitreal neovascularization in a rat
model of retinopathy of prematurity. Mol Vis. 20:231–241.
2014.PubMed/NCBI
|
|
34
|
Yao M, Gao F, Wang X, Shi Y, Liu S and
Duan H: Nox4 is involved in high glucose-induced apoptosis in renal
tubular epithelial cells via Notch pathway. Mol Med Rep.
15:4319–4325. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cai WX, Liang L, Wang L, Han JT, Zhu XX,
Han H, Hu DH and Zhang P: Inhibition of Notch signaling leads to
increased intracellular ROS by up-regulating Nox4 expression in
primary HUVECs. Cell Immunol. 287:129–135. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Artavanis-Tsakonas S, Rand MD and Lake RJ:
Notch signaling: Cell fate control and signal integration in
development. Science. 284:770–776. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hofmann JJ and Luisa Iruela-Arispe M:
Notch expression patterns in the retina: An eye on receptor-ligand
distribution during angiogenesis. Gene Expr Patterns. 7:461–470.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Oswald F, Täuber B, Dobner T, Bourteele S,
Kostezka U, Adler G, Liptay S and Schmid RM: p300 acts as a
transcriptional coactivator for mammalian Notch-1. Mol Cell Biol.
21:7761–7774. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu L, Zhou X, Kuang X, Long C, Liu W,
Tang Y, Liu H, He J, Huang Z, Fan Y, Zhang Q and Shen H: The
inhibition of NOTCH2 reduces UVB-induced damage in retinal pigment
epithelium cells. Mol Med Rep. 16:730–736. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li H, Zhu Z, Liu J, Wang J and Qu C:
MicroRNA-137 regulates hypoxia-induced retinal ganglion cell
apoptosis through Notch1. Int J Mol Med. 41:1774–1782.
2018.PubMed/NCBI
|
|
41
|
Arboleda-Velasquez JF, Primo V, Graham M,
James A, Manent J and D'Amore PA: Notch signaling functions in
retinal pericyte survival. Invest Ophthalmol Vis Sci. 55:5191–5199.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hori K, Sen A and Artavanis-Tsakonas S:
Notch signaling at a glance. J Cell Sci. 126:2135–2140. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang H, Zang C, Taing L, Arnett KL, Wong
YJ, Pear WS, Blacklow SC, Liu XS and Aster JC: NOTCH1-RBPJ
complexes drive target gene expression through dynamic interactions
with superenhancers. Proc Natl Acad Sci USA. 111:705–710. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schouwey K, Aydin IT, Radtke F and
Beermann F: RBP-Jκ-dependent Notch signaling enhances retinal
pigment epithelial cell proliferation in transgenic mice. Oncogene.
30:313–322. 2011. View Article : Google Scholar : PubMed/NCBI
|