Introduction
With the growth and aging of the population and the
increasing number of risk factors in the lives of individuals,
including smoking and heavy drinking, the incidence of cancer
continues to increase annually. According to GLOBOCAN estimates,
18.1 million individuals were diagnosed with cancer and 9.6 million
individuals succumbed to cancer in 2018 worldwide (1). Among all cancer-related mortalities,
colorectal cancer (CRC) ranks third in both males and females
(2). Although CRC mortality is
declining overall, its incidence has increased in America among
adults <55 years old, between 1970 and 2014 (3). It is well known that tumor cells
exist in a state of uncontrolled growth and invasion. Such
undisciplined cells may suffer from programmed cell death to
protect organisms. Cancer development is due to the inactivation of
the apoptotic pathway (4),
resulting in drugs resistant of cancer cells. Previous studies have
reported that resistance to drugs is occasionally due to Bax
gene mutations; Bcl-2-like protein 4 (Bax) is an important
regulatory factor of the mitochondrial apoptosis pathway (5–8).
Apoptosis is one mechanism by which cell death
occurs. p53 is a tumor-suppression gene that serves an
important role in apoptosis, cell aging and cell cycle arrest
(9). The inactivation of p53 in
the p53-dependent apoptosis pathway may promote tumor occurrence,
tumor development and resistance to antitumor drugs (10). The Bcl-2 gene family serves
important functions in the intrinsic mitochondrial-mediated
apoptosis pathway. In this family, certain members exhibit
inhibitory effects on apoptosis, whereas others such as Bax promote
apoptosis (11). Bax is normally
located in the cytoplasm; once the apoptotic signal is received,
Bax undergoes oligomerization and is translocated to the
mitochondrial membrane, which subsequently leads to the release of
cytochrome c and other apoptosis factors into the cytoplasm
(12,13). Cytochrome c combines apoptotic
protease activating factor-1 and pro-caspase-9, which form
apoptosomes, which lead to the activation of caspase-9 and
caspase-3. Previous studies have reported that Bax deficiency may
cause cancer cells to become insensitive to certain antitumor drugs
by preventing the translocation of Bax to the mitochondria
(14).
Cordycepin (3′ deoxyadenosine), a derivative of the
nucleoside adenosine, is a metabolic product extracted from
Cordyceps militaris (15)
and is a major bioactive component with important anticancer
potential (16). Previous studies
in several disease models have demonstrated that cordycepin
possesses antitumor and anti-inflammatory effects that occur
through the inhibition of mRNA synthesis (17,18).
Cordycepin possesses anticancer activities, including
antiproliferation, autophagy promotion, anti-migration and
apoptosis induction (19,20). Although the anticancer activity of
cordycepin has been examined in human bladder, brain and lung
cancer cells, the mechanism by which cordycepin affects CRC remains
poorly understood (21–23).
Results from the present study indicated that
cordycepin suppresses colon cancer cell growth in vitro and
demonstrated that cordycepin may accelerate apoptosis in HCT116
cells by inducing the translocation of Bax to the mitochondrial
membrane (24). However,
cordycepin-induced apoptosis and Bax translocation was notably
inhibited in isogenic Bax-null (Bax−/−)
human colon cancer HCT116 cells. Cordycepin-activated Bax
translocation induced apoptosis, which was recovered through the
reintroduction of Bax expression into Bax−/−
cells. Taken together, these results suggested that cordycepin may
establish the foundation of a therapeutic approach for cancers by
targeting the Bax protein.
Materials and methods
Cell lines and plasmids
HCT116 human colorectal carcinoma cells were
purchased from the American Type Culture Collection (Manassas, VA,
USA). p53−/−, Bax−/− and
p21−/− HCT116 cells were obtained from Dr Bert
Vogelstein (Johns Hopkins University, Baltimore, MD, USA). The
pEGFP-C3-Bax expression vectors were provided by Dr Quan
Cheng (Institute of Zoology, Chinese Academy of Sciences, Beijing,
China).
Cell culture
Wild-type (WT) HCT116,
HCT116-p53−/−, HCT116-p21−/−
and HCT116-Bax−/− cells were cultured in McCoy's
5A Medium (cat. no. A1324-9050; AppliChem, Inc., Maryland Heights,
MO, USA) with 10% (v/v) fetal bovine serum (GE Healthcare Life
Sciences, Logan, UT, USA) and 100 U penicillin/streptomycin (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C in a 5%
CO2 incubator.
Reagents and antibodies
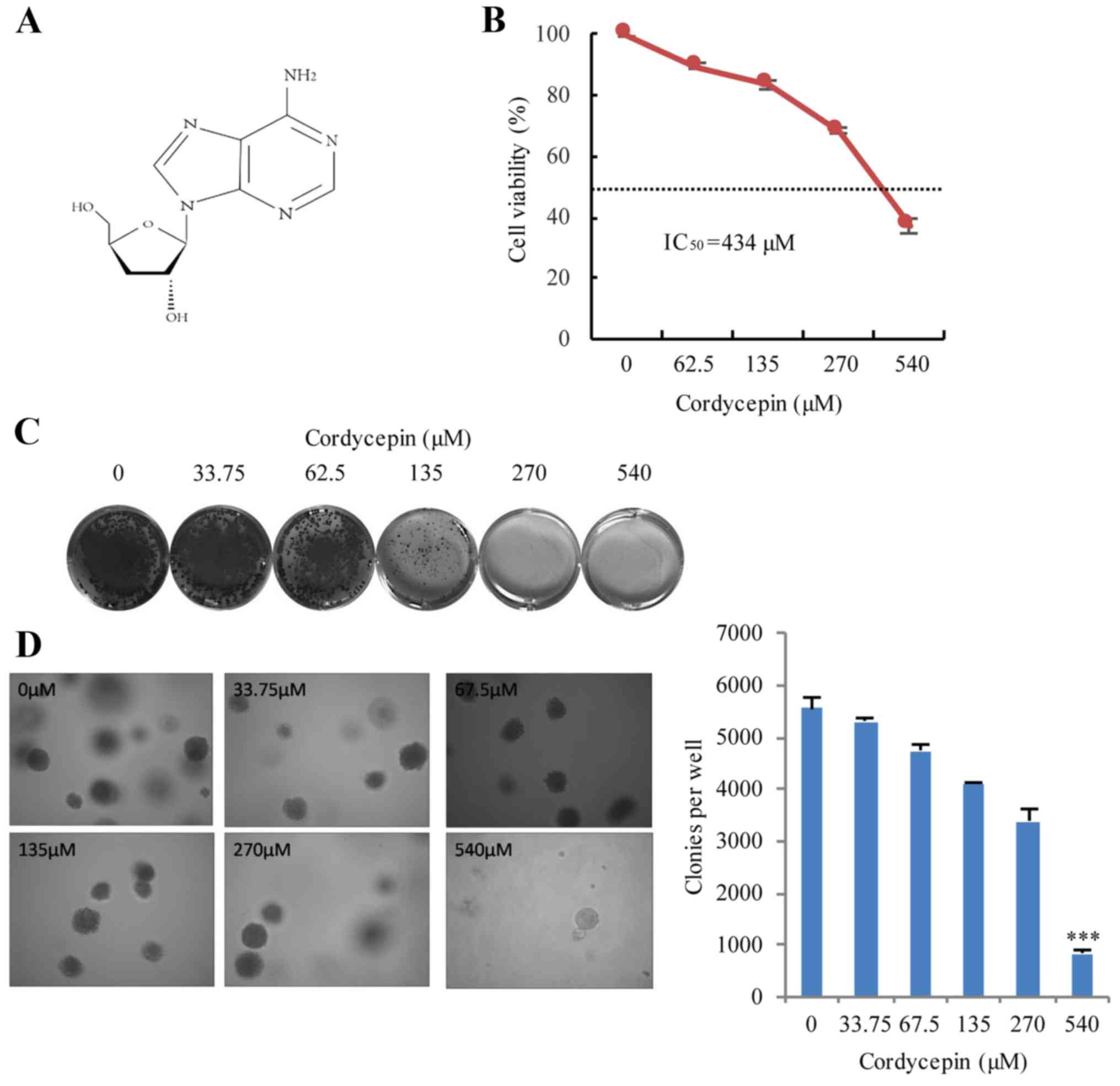
Cordycepin
(C10H13N5O3; 251 Da;
cat. no. C3394; Fig. 1A) and
caspase-3 inhibitor (cat. no. 219007) were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Rabbit monoclonal
antibodies against Bax (cat. no. 5023), pro-caspase-3 (cat. no.
9665), cytochrome c oxidase IV (CoxIV; cat. no. 4850) and
cleaved poly(ADP-ribose) polymerase (PARP; cat. no. 9541) were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
The anti-mouse monoclonal p53 antibody (cat. no. sc-126) and
anti-mouse cytochrome c (cat. no. sc-126) were obtained from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). The anti-mouse
monoclonal β-actin antibody (cat. no. AC004) was purchased from
ABclonal Biotech Co., Ltd. (Woburn, MA, USA). The anti-mouse
monoclonal β-tubulin antibody (cat. no. AbM59005-37B-PU) was
obtained from Beijing Protein Innovation (Beijing, China). The
horseradish peroxidase (HRP)-conjugated secondary antibodies (cat.
nos. 111-035-003 or 115-035-003) were obtained from Jackson
ImmunoResearch Laboratories, Inc. (West Grove, PA, USA).
Transient and stable transfection
Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) was used for transient transfections
according to the manufacturer's protocols. Transfection reagents
and DNA were mixed in Opti-MEM (Invitrogen; Thermo Fisher
Scientific, Inc.); For 6-well plate, 200 µl (10 µg/ml) transfection
DNA complex was added to cells grown to 30–60% confluency, and the
culture was incubated for ~3 h, and the medium was replaced with
fresh full McCoy's 5A medium. For EGFP-Bax stable
transfections, 0.5 mg/ml G418 was added to the medium for 48 h
following transient transfection, and the cells were selected after
2 weeks. Thereafter, stable cells were always maintained in 0.25
mg/ml G418 medium.
Colony formation and soft agar
assay
Soft agar and colony formation assays were used to
examine the viability and tumorigenicity of HCT116 cells following
treatment with cordycepin. Briefly, 3×103 HCT116 cells
were treated with various concentrations of cordycepin (0, 62.5,
135, 270 and 540 µM) for 24 h; the medium and drugs were
subsequently replaced with fresh medium. After 2 weeks incubation,
cell clones were stained with 0.05% crystal violet at room
temperature for 30 min and images were captured by scanner
(MRS-2400U2; Microtek, Shanghai, China).
For the soft agar test, 2 ml of 0.7% lower
agar-McCoy's 5A with cordycepin (0–540 µM, as aforementioned) was
plated onto each well of 6-well plates. Subsequently, 1 ml of
HCT116 cells (1×104) was mixed with 1 ml of 0.7%
agar-McCoy's 5A/cordycepin (0–540 µM) mix and added to the curdled
lower agar; 2 ml of McCoy's 5A medium was added to the upper agar,
and the plates were incubated at 37°C in a 5% CO2
incubator for 3 weeks, and the numbers of clones were counted and
images captured by an inverted microscope (ICX41; SDPTOP, Shanghai,
China).
Cell viability assay
Cell viability was examined using Cell Counting
Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan), following the
manufacturer's protocol. The HCT116 cell lines [wild-type (WT)
HCT116, HCT116-p53−/−, HCT116-p21−/− and
HCT116-Bax−/−] aforementioned were seeded
(1×104 cells/well) into a 96-well plate, cultured to 60%
confluency and exposed to different doses of cordycepin (0, 33.75,
62.5, 135, 270 and 540 µM) for 24 h. The medium was replaced with
100 µl of fresh McCoy's 5A full medium with 10% CCK-8 reagent and
incubated for 1 h. Absorbance was detected at 450 nm using an
ELx800 microplate reader (BioTek Instruments, Inc., Winooski, VT,
USA). The results were expressed as percentages of cell
viability.
Apoptosis assay
The HCT116 cell lines (1×104 cells/well)
were seeded in 6-well plates and cultured to 60% confluency.
Following exposure to the various concentration of cordycepin (0,
135 and 270 µM) for 24 h, cells were collected, washed with
phosphate buffered saline (PBS) twice. Subsequently, the cells were
centrifuged at 120 × g in the room temperature for 5 min and
suspended in apoptosis binding buffer (50 mM HEPES, 700 mM NaCl,
12.5 mM CaCl2, pH 7.4) at a concentration of
1×106 cells/ml. Cells were stained with propidium iodide
(PI) and Annexin V fluorescein isothiocyanate (FITC) for 10–15 min
at room temperature. Samples were subjected to flow cytometry
(Beckman Coulter, Fullerton, CA) and data were analyzed using
FlowJo software (10.07 version). For apoptosis inhibition assay,
the cells were incubated with 25 µM caspase-3 inhibitor for 24 h to
prevent PARP cleavage and cell apoptosis.
Mitochondrial isolation
HCT116 cells (5×106) were exposed to 270
µM cordycepin for 24 h. The Mitochondria/Cytosol Fractionation Kit
(Beyotime Institute of Biotechnology, Beijing, China) was used to
isolate the mitochondrial and cytosolic proteins. Briefly, cells
were resuspended in the mitochondria isolation buffer and
homogenized with a 2 ml glass homogenizer on ice. Cell lysates were
centrifuged at 1,000 × g for 10 min at 4°C, and the supernatant was
centrifuged at 14,000 × g for 15 min at 4°C. The supernatant
included the cytosolic fraction, and the pellet contained the
mitochondrial fraction.
GFP-Bax translocation assay
GFP-Bax plasmids were stably expressed in
Bax−/− HCT116 cells. Cells were exposed to
cordycepin (0 or 270 µM) for 12 h and fixed with 4%
paraformaldehyde for 30 min at room temperature. The fixed cells
were observed using a confocal microscope using an ×60 oil
objective. Images were captured using 2.0b Olympus FluoView
software (Olympus Corporation, Tokyo, Japan), and distribution
pattern of GFP-Bax was examined.
Western blot assay
An immunoblotting assay was performed as described
previously (25). Briefly, cells
were cultured on 6-well plates to 60% confluency and subsequently
treated with cordycepin (0–540 µM) for 24 h. Cells were lysed with
SDS sample buffer (62.5 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol)
at 95°C for 10 min. Protein concentrations were determined using a
BCA protein assay kit (Thermo Fisher Scientific, Inc.). Protein
samples (20 µg) were separated by 10% SDS-PAGE, transferred and
incubated with indicated primary antibodies (1:1,000) overnight at
4°C and HRP-conjugated secondary antibodies (1:10,000) at room
temperature for 1 h. The β-actin was used as loading control.
Protein expressions were visualized with the Immobilon Western
Chemiluminescent HRP Substrate kit (Merck KGaA, Minneapolis, MN,
USA).
Statistical analysis
All data were analyzed using SPSS software version
20.0 (IBM Corp., Armonk, NY, USA). For data with a Gaussian
distribution, parametric statistical analysis was performed using
the two-tailed Student's t-test for two groups; one-way analysis of
variance was used for multiple comparisons followed by Bonferroni's
post hoc analysis for data meeting homogeneity of variance or with
Tamhane's T2 analysis for data of heteroscedasticity. For data sets
with skewed distribution, non-parametric statistical analysis was
performed using the Kruskal-Wallis test followed by the Dunn's test
for multiple comparisons. Only in the cases in which the global
null hypothesis was rejected was pairwise comparison subsequently
performed. For all analyzed variables, a pre-specification approach
was performed. P<0.05 was considered to indicate a statistically
significant difference.
Results
Cordycepin represses HCT116 cell
growth in vitro
To determine the most suitable concentration of
cordycepin to treat colon cancer cells, the half-maximal inhibitory
concentration (IC50) of cordycepin in HCT116 cells was
determined. HCT116 cells were treated with 0–540 µM cordycepin for
24 h and further cultured at 37°C in a 5% CO2 incubator
for ~10 days. The viability of the HCT116 cells clearly decreased
following cordycepin treatment (Fig.
1B and C); and the results demonstrated that the
IC50 of cordycepin was 434 µM for HCT116 cells (Fig. 1B). Soft agar assays were conducted
to examine the effects of cordycepin treatment on tumor formation
in vitro. At 2 weeks post-treatment, anchorage-independent
tumor growth was significantly inhibited in a dose-dependent manner
in cells incubated with cordycepin (Fig. 1D). These results demonstrated that
cordycepin has significant inhibitory effects on human HCT116 cell
viability, proliferation and tumorigenesis in vitro.
Cordycepin induces apoptosis in HCT116
cells
To determine the molecular mechanism underlying the
anticancer activity of cordycepin, cordycepin-activated apoptotic
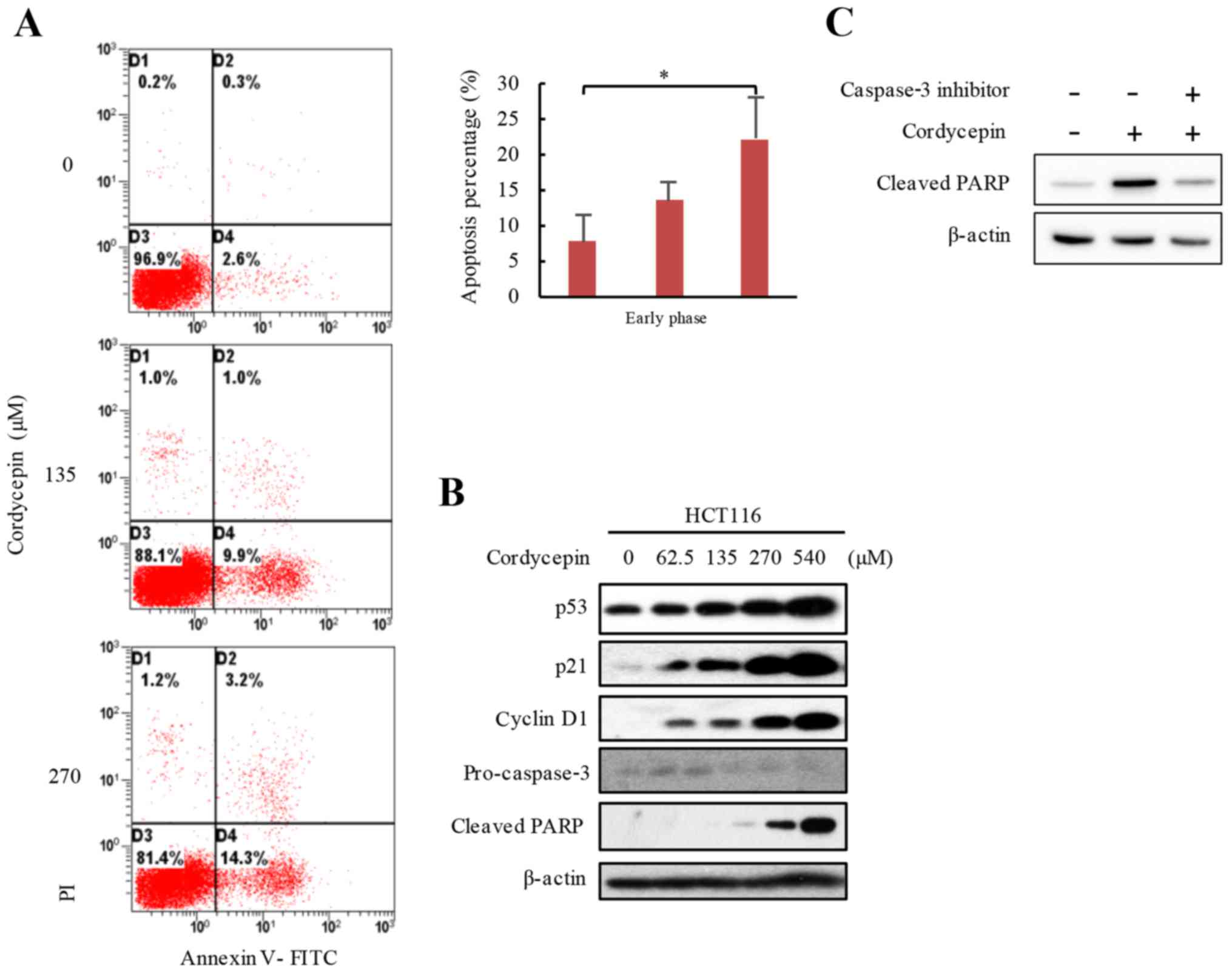
activity was examined in HCT116 cells. The results demonstrated
that cells treated with 0, 135 and 270 µM cordycepin for 24 h
exhibited average 7.8, 13.4 and 22.1% early apoptotic rates,
respectively (Fig. 2A). To confirm
the apoptotic phenotype, the levels of certain proteins that are
closely associated with apoptosis were tested. HCT116 cells were
exposed to 0–540 µM cordycepin for 24 h, and the protein expression
levels of p53 and its target gene p21 were investigated by western
blot analysis, which demonstrated notably higher protein expression
levels in cells treated with cordycepin, and the increased
expressions were in a dose-dependent manner (Fig. 2B). The apoptosis markers,
pro-caspase-3 and cleaved PARP were also analyzed by western
blotting, and the results demonstrated lower pro-caspase-3 and
higher cleaved PARP levels in response to increased cordycepin
concentrations (Fig. 2B). These
data suggested that cordycepin induced apoptosis in HCT116 cells.
To further demonstrate that increased cleaved-PARP occurred through
a caspase-3-dependent pathway, cells were incubated with 25 µM
caspase-3 inhibitor for 24 h to prevent cell apoptosis, and the
results demonstrated that the caspase-3 inhibitor blocked
cordycepin-induced cleaved-PARP expression in HCT116 (Fig. 2C). These results suggested that
cordycepin may induce apoptosis in HCT116 CRC cells.
Cordycepin induces HCT116 cell death
through Bax- dependent apoptosis
Bax, a pro-apoptosis Bcl-2 family member,
translocates from the cytoplasm to the mitochondria in response to
stimulation, which subsequently results in the release of
cytochrome c and cell apoptosis (26). The tumor suppressor protein p53
serves an important role in the maintenance of genomic stability
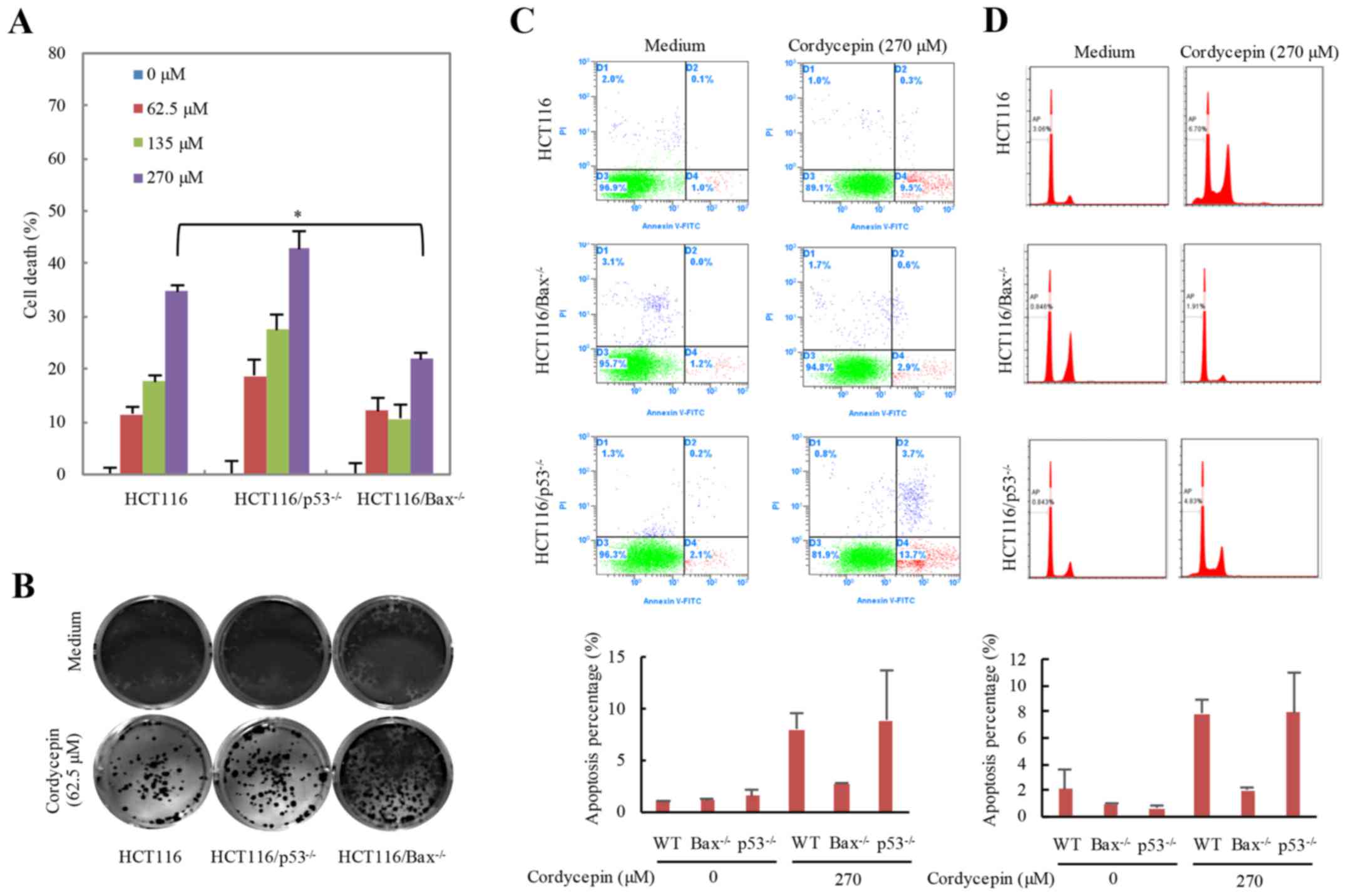
and cell apoptosis (27,28). To understand whether p53 or Bax
serves a role in cordycepin-induced apoptosis, the apoptotic
effects following cordycepin treatment in HCT116-WT,
HCT116-p53−/− and HCT116-Bax−/−
cells were examined (Fig. 3).
HCT116 cells were treated with 0–270 µM cordycepin for 24 h and
cell viability was examined (Fig.
3A). The cell viability of HCT116 and
HCT116-p53−/− cells was significantly decreased
with increasing cordycepin concentration; however,
Bax−/− cells treated with various concentrations
appeared to be insensitive to cordycepin treatment compared with
the HCT116-WT and p53−/− cells. These results
indicated that Bax may serve a more essential part in
cordycepin-induced cell death compared with p53. In addition, the
mutant cell lines were exposed to 62.5 µM cordycepin for 24 h,
cultured for an additional 10 days and the colony number reduced in
HCT116-WT and HCT116-p53−/− cells but not in
HCT116-Bax−/− cells (Fig. 3B). HCT116-WT,
p53−/− and Bax−/− HCT116 cells
were also incubated with 270 µM cordycepin for 24 h, and the
Annexin V/FITC staining results demonstrated that average 2.76% of
the Bax−/− cells were early apoptotic compared
with 8.03% of the HCT116-WT cells, which also suggested that
HCT116-Bax−/− cells were insensitive to cordycepin
treatment (Fig. 3C). The reason
why p53−/− cells are more sensitive to cordycepin
treatment is that p53 is an essential gene for cell life
activities; thus, the amount of cell death in this line is already
substantially higher than that of WT cells. Apoptosis will result
in DNA fraction; therefore, we determined the DNA fraction using PI
staining and the results consistent with the Annexin V/FITC
staining (Fig. 3D). These results
indicated that cordycepin-induced apoptosis may be Bax
dependent.
Cordycepin releases cytochrome c to
induce apoptosis is dependent on Bax
As aforementioned, cordycepin-induced apoptosis may
be Bax-dependent. To assess whether cordycepin induces the
cleavage of PARP in p53−/− or
Bax−/− HCT116 cells, cleaved-PARP protein
expression levels were analyzed. Cells were treated with 0–270 µM
cordycepin for 24 h, and the protein expression levels of p53, Bax
and cleaved-PARP were notably increased with higher concentrations
of cordycepin treatment in HCT116-WT cells. Additionally,
significant levels of cell apoptosis were observed in the
p53−/− HCT116 cells, but apoptosis markers
cleaved-PARP were blocked in the Bax−/− HCT116
cells (Fig. 4A). The inhibition of
cordycepin-induced PARP protein cleavage in the
Bax−/− HCT116 cells was rescued by reintroducing
the Bax gene into Bax−/− HCT116 cells
(Fig. 4B). The translocation of
Bax to the mitochondria may exert an important effect on apoptotic
initiation (29). The stable
EGFP-Bax cell line allowed for the tracking of the location
of Bax protein expression. Untreated control cells exhibited a
diffuse distribution of green fluorescence among whole cells,
whereas almost 70% of the cordycepin-treated cells demonstrated
only punctate fluorescence, which suggested the translocation of
Bax reached 70% in the cordycepin-treated cells (Fig. 4C). In addition, western blot
analysis revealed increased Bax protein expression in the
mitochondria when exposed to cordycepin (Fig. 4D). cytochrome c is located
in the mitochondrial inner membrane; its translocation from the
mitochondria to the cytosol induces cell apoptosis (30). As a downstream marker of Bax
aggregation, the protein levels of cytochrome c in the
cytoplasm of HCT116 cells exposed to cordycepin was examined. The
results demonstrated increased cytochrome c expression the
cytosol in response to cordycepin exposure (Fig. 4E). These data demonstrated that Bax
is employed in cordycepin-induced apoptosis.
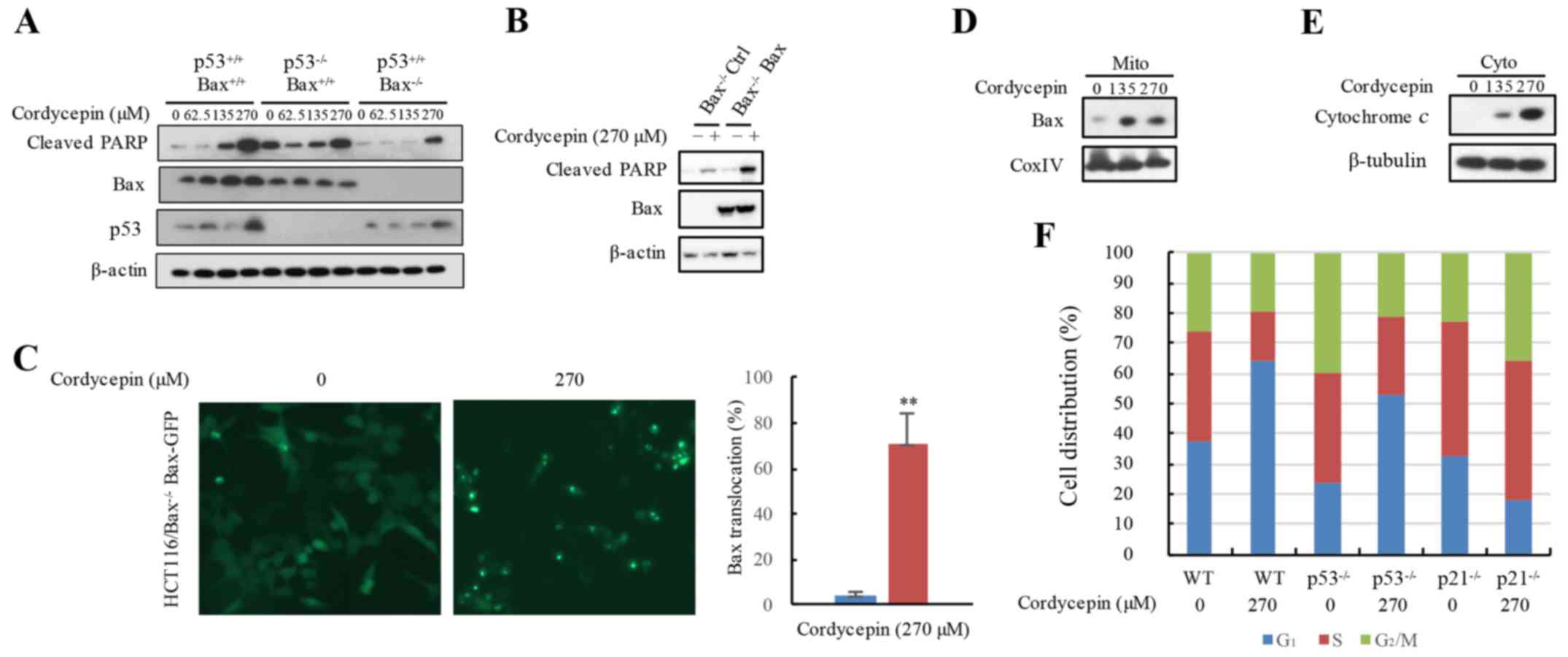 | Figure 4.Cordycepin utilizes Bax releasing
cytochrome c to induce apoptosis. (A) Apoptosis marker
protein expression levels in HCT116,
HCT116-p53−/− and HCT116-Bax−/−
cells treated with 0 or 270 µM cordycepin were measured by western
blotting. (B) Cordycepin-induced apoptosis is hindered in
Bax−/− HCT116 cells, but recovered by
reintroduction of the Bax gene. (C) The EGFP-Bax plasmid is
stably overexpressed in Bax−/− HCT116 cells, and
the cells were exposed to 270 µM cordycepin for 24 h; the location
of the EGFP-Bax protein was determined using a fluorescence
microscope and compared with that of untreated EGFP-Bax
control cells. Magnification, ×20. (D and E) HCT116 cells were
exposed to 270 µM cordycepin for 24 h, and mitochondria and
cytoplasm were isolated; cytochrome c and Bax protein
expressions were detected by western blot analysis; β-tubulin and
CoxIV were used as loading controls. (F) Cordycepin arrested the
cell cycle at the G1/S phase through p21 in the HCT116
cell line. PI staining flow cytometry was used to analyze the cell
cycle profiles in HCT116, HCT116-p53−/− and
HCT116-p21−/− cells treated with or without 270
µM cordycepin. **P<0.01 vs. the controls. Bax, Bcl-2-like
protein 4; CoxIV, cytochrome c oxidase IV; cyto, cytoplasm;
GFP, green fluorescent protein; mito, mitochondria; PARP,
poly(ADP-ribose) polymerase; PI, propidium iodide; WT,
wild-type. |
Cordycepin arrests the cell cycle at
the G1/S phase through p21 in HCT116 cells
In addition to investigating cordycepin-induced
apoptosis, whether cordycepin was involved in cell cycle profile
changes was examined. Flow cytometric assay results demonstrated
that cordycepin treatment resulted in cell cycle arrest in the
HCT116-WT cells in the G1/S phase (Fig. 4F). However, in the
p21−/− HCT116 cells, G1/S phase arrest
was reduced. Whether p53, which is directly controlled by
the expression of p21 (31), is involved in G1/S phase
arrest was also examined. As expected, in
p53−/−deficient HCT116 cells, G1/S
phase arrest was clearly ameliorated when compare to HCT116-WT
cells treated with 270 µM cordycepin (Fig. 4F). As the protein expression levels
of p21 and p53 were increased in response to cordycepin, it was
hypothesized that cordycepin treatment may induce G1/S
phase cell cycle arrest in HCT116 cells by inducing the expression
of p53 and its target gene p21.
Discussion
The antitumor activity of cordycepin on human CRC
cell growth in vitro was examined in the present study, and
it was identified that its molecular mechanism may be associated
with Bax-dependent apoptosis. In addition, it was demonstrated that
cordycepin-induced apoptosis may be reversed by an inhibitor of
caspase. Using the Bax−/− and
p53−/− HCT116 cell lines, it was demonstrated
that apoptosis induced by cordycepin was notably increased in the
Bax-dependent pathway, but not in the p53-dependent
pathway. The localization of Bax to the mitochondria following
cordycepin treatment indicated that the Bax protein may be required
for cordycepin-induced apoptosis. Notably, the reintroduction of
Bax into Bax−/− cells recovered the
cordycepin-induced apoptosis effect.
Apoptosis is a programmed cell death process that
occurs in multicellular organisms during both normal cell growth
and aging. Improper apoptosis results in a variety of diseases,
including CRC (32). The induction
of apoptosis by antitumor drugs is mediated by a variety of
mechanisms, including cell cycle and replication arrest,
transcription regulation, DNA repair, apoptosis and autophagy
(33). The initiation of apoptosis
has extrinsic and intrinsic pathways. Mitochondria serve a pivotal
role in the intrinsic pathway and are involved in drug-induced
apoptosis; in addition, members of the Bcl-2 family take part in
intrinsic pathway regulation, including regulation of the
Bax gene (34).
To identify whether p53 or Bax serves
a pivotal role in cordycepin-induced apoptosis, apoptosis was
examined in WT, p53−/− and
Bax−/− HCT116 cells in the presence or absence of
cordycepin. Increased apoptosis was observed in HCT116 and
p53−/− cells, but not in Bax−/−
cells, and Bax was demonstrated to translocate to the mitochondria
in an EGFP-Bax stable cell line treated with cordycepin. The
data indicated that cordycepin-induced apoptosis may depend on
Bax activity.
Cordycepin is a purine nucleoside antimetabolite and
antibiotic isolated from the fungus Cordyceps militaris that
has certain antineoplastic activities (35). The results of the present study are
consistent with cordycepin induced apoptotic cell death, and
demonstrated that cordycepin-induced apoptosis in human CRCs may be
more dependent on Bax than p53. Although the present study has
clarified the role and apoptotic mechanism of cordycepin in human
CRC, additional studies, including investigations in an animal
model, are required to determine a therapeutic strategy.
In conclusion, the results of the present study
suggested that cordycepin effectively reduced HCT116 cell viability
by activating apoptotic responses through a Bax-dependent and
p53-independent manner. The results revealed the mechanisms of
cordycepin-induced apoptosis and indicated that cordycepin may be a
novel therapeutic approach to cancers with wild-type Bax.
Acknowledgements
Authors would like to thank Dr Feng Zhou for
statistical analysis and Dr Bert Vogelstein and Dr Quan Cheng for
providing cell lines and plasmids.
Funding
The present study was supported by grants from The
National Science and Technology Support Project (grant no.
2014BAI02B00), The National Natural Science Foundation of China
(grant no. 81470375) and The National Science Foundation for Young
Scientists of China (grant no. 31501148).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
SZL, RLD and XDZ designed the research; SZL, JWR and
JF performed the research. SZL and RLD wrote the manuscript. SZL
revised the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kan WL, Yin C, Xu HX, Xu G, To KK, Cho CH,
Rudd JA and Lin G: Antitumor effects of novel compound, guttiferone
K, on colon cancer by p21Waf1/Cip1-mediated
G0/G1 cell cycle arrest and apoptosis. Int J
Cancer. 132:707–716. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel RL, Miller KD and Jemal A:
Colorectal cancer mortality rates in adults aged 20 to 54 years in
the United States, 1970–2014. JAMA. 318:572–574. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rampino N, Yamamoto H, Ionov Y, Li Y,
Sawai H, Reed JC and Perucho M: Somatic frameshift mutations in the
BAX gene in colon cancers of the microsatellite mutator
phenotype. Science. 275:967–969. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang L, Yu J, Park BH, Kinzler KW and
Vogelstein B: Role of BAX in the apoptotic response to
anticancer agents. Science. 290:989–992. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Haneef J, Parvathy M, Thankayyan RS,
Sithul H and Sreeharshan S: Bax translocation mediated
mitochondrial apoptosis and caspase dependent photosensitizing
effect of Ficus religiosa on cancer cells. PLoS One.
7:e400552012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhu S, Li T, Tan J, Yan X, Zhang D, Zheng
C, Chen Y, Xiang Z and Cui H: Bax is essential for death
receptor-mediated apoptosis in human colon cancer cells. Cancer
Biother Radiopharm. 27:577–581. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li T, Liu X, Jiang L, Manfredi J, Zha S
and Gu W: Loss of p53-mediated cell-cycle arrest, senescence and
apoptosis promotes genomic instability and premature aging.
Oncotarget. 7:11838–11849. 2016.PubMed/NCBI
|
|
10
|
McCurrach ME, Connor TM, Knudson CM,
Korsmeyer SJ and Lowe SW: bax-deficiency promotes drug
resistance and oncogenic transformation by attenuating
p53-dependent apoptosis. Proc Natl Acad Sci USA. 94:2345–2349.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chittenden T, Flemington C, Houghton AB,
Ebb RG, Gallo GJ, Elangovan B, Chinnadurai G and Lutz RJ: A
conserved domain in Bak, distinct from BH1 and BH2, mediates cell
death and protein binding functions. EMBO J. 14:5589–5596. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ferrer PE, Frederick P, Gulbis JM, Dewson
G and Kluck RM: Translocation of a Bak C-terminus mutant from
cytosol to mitochondria to mediate cytochrome c release:
Implications for Bak and Bax apoptotic function. PLoS One.
7:e315102012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gahl RF, He Y, Yu S and Tjandra N:
Conformational rearrangements in the pro-apoptotic protein, Bax, as
it inserts into mitochondria: A cellular death switch. J Biol Chem.
289:32871–32882. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chandrika BB, Maney SK, Lekshmi SU, Joseph
J, Seervi M, K S P and T R S: Bax deficiency mediated drug
resistance can be reversed by endoplasmic reticulum stress induced
death signaling. Biochem Pharmacol. 79:1589–1599. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cunningham KG, Manson W, Spring FS and
Hutchinson SA: Cordycepin, a metabolic product isolated from
cultures of Cordyceps militaris (Linn.) link. Nature. 166:9491950.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jagger DV, Kredich NM and Guarino AJ:
Inhibition of Ehrlich mouse ascites tumor growth by cordycepin.
Cancer Res. 21:216–220. 1961.PubMed/NCBI
|
|
17
|
Kondrashov A, Meijer HA, Barthet-Barateig
A, Parker HN, Khurshid A, Tessier S, Sicard M, Knox AJ, Pang L and
De Moor CH: Inhibition of polyadenylation reduces inflammatory gene
induction. RNA. 18:2236–2250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nakamura K, Yoshikawa N, Yamaguchi Y,
Kagota S, Shinozuka K and Kunitomo M: Antitumor effect of
cordycepin (3′-deoxyadenosine) on mouse melanoma and lung carcinoma
cells involves adenosine A3 receptor stimulation. Anticancer Res.
26:43–47. 2006.PubMed/NCBI
|
|
19
|
Choi KS: Autophagy and cancer. Exp Mol
Med. 44:109–120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tao X, Ning Y, Zhao X and Pan T: The
effects of cordycepin on the cell proliferation, migration and
apoptosis in human lung cancer cell lines A549 and NCI-H460. J
Pharm Pharmacol. 68:901–911. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chaicharoenaudomrung N, Jaroonwitchawan T
and Noisa P: Cordycepin induces apoptotic cell death of human brain
cancer through the modulation of autophagy. Toxicol In Vitro.
46:113–121. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cao HL, Liu ZJ and Chang Z: Cordycepin
induces apoptosis in human bladder cancer cells via activation of
A3 adenosine receptors. Tumour Biol. 39:10104283177069152017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hwang JH, Park SJ, Ko WG, Kang SM, Lee DB,
Bang J, Park BJ, Wee CB, Kim DJ, Jang IS, et al: Cordycepin induces
human lung cancer cell apoptosis by inhibiting nitric oxide
mediated ERK/Slug signaling pathway. Am J Cancer Res. 7:417–432.
2017.PubMed/NCBI
|
|
24
|
An WG, Hwang SG, Trepel JB and
Blagosklonny MV: Protease inhibitor-induced apoptosis: Accumulation
of wt p53, p21WAF1/CIP1, and induction of apoptosis are independent
markers of proteasome inhibition. Leukemia. 14:1276–1283. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li SZ, Zeng F, Li J, Shu QP, Zhang HH, Xu
J, Ren JW, Zhang XD, Song XM and Du RL: Nemo-like kinase (NLK)
primes colorectal cancer progression by releasing the E2F1 complex
from HDAC1. Cancer Lett. 431:43–53. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
De Giorgi F, Lartigue L, Bauer MK,
Schubert A, Grimm S, Hanson GT, Remington SJ, Youle RJ and Ichas F:
The permeability transition pore signals apoptosis by directing Bax
translocation and multimerization. FASEB J. 16:607–609. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gotz C and Montenarh M: P53 and its
implication in apoptosis (Review). Int J Oncol. 6:1129–1135.
1995.PubMed/NCBI
|
|
28
|
Choisy-Rossi C, Reisdorf P and
Yonish-Rouach E: Mechanisms of p53-induced apoptosis: In search of
genes which are regulated during p53-mediated cell death. Toxicol
Lett. 102–103. 491–496. 1998.
|
|
29
|
Peña-Blanco A and García-Sáez AJ: Bax, Bak
and beyond-mitochondrial performance in apoptosis. FEBS J.
285:416–431. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yamamoto T, Yamada A, Yoshimura Y, Terada
H and Shinohara Y: The mechanisms of the release of cytochrome
c from mitochondria revealed by proteomics analysis.
Yakugaku Zasshi. 132:1099–1104. 2012.(In Japanese). View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tarangelo A and Dixon S: The p53-p21
pathway inhibits ferroptosis during metabolic stress. Oncotarget.
9:24572–24573. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li J, Liu YY, Yang XF, Shen DF, Sun HZ,
Huang KQ and Zheng HC: Effects and mechanism of STAT3 silencing on
the growth and apoptosis of colorectal cancer cells. Oncol Lett.
16:5575–5582. 2018.PubMed/NCBI
|
|
33
|
Blankenberg FG: Apoptosis imaging:
Anticancer agents in medicinal chemistry. Anticancer Agents Med
Chem. 9:944–951. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hardwick JM and Soane L: Multiple
functions of BCL-2 family proteins. Cold Spring Harb Perspect Biol.
5:a0087222013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zheng ZL, Qiu XH and Han RC:
Identification of the genes involved in the fruiting body
production and cordycepin formation of Cordyceps militaris
fungus. Mycobiology. 43:37–42. 2015. View Article : Google Scholar : PubMed/NCBI
|