Introduction
Platelets, first identified over 130 years ago, are
produced by megakaryocytes in bone marrow and lungs, and are
critical for maintaining hemostasis and thrombosis balance
(1). In physiological status, the
coagulation system is strictly regulated, but under pathological
status, platelet activation leads to the formation of occlusive
thrombus and causes myocardial infarction and stoke, especially in
patients with hyperlipidemia (2).
When the integrity of blood vessels is destroyed by injury,
atherosclerotic plaque formation or inflammatory reaction,
platelets clot together and form a thrombus at the site of injury.
The flow rate of platelets is reduced in pathological status by the
complex of glycoprotein and von Willebrand factor (vWF), and the
release of thromboxane 2 (TXA2), adenosine diphosphate (ADP) and
5-hydroxytryptamine (5-HT) accelerates the aggregation of platelets
and formation of thrombus (3).
Anti-platelet drugs are commonly administered to patients with a
high risk of thrombogenesis, which can increase the risk of
bleeding and the emergence of a resistance effect to drugs
(4), and the application of
anti-platelet drugs decreases the risk of thrombogenesis.
Anti-platelet drugs are divided into different
groups according to their pharmacological mechanisms, including
TXA2 receptor antagonists, P2Y12 antagonists, GP IIb/IIIa receptor
antagonists and protease-activated receptor (PAR) inhibitors. PAR
is expressed in most cell types in the vasculature. PARs contain
four members, including PAR-1, PAR-2, PAR-3 and PAR-4. Among them,
PAR-1 is a major effector in the thrombin signaling pathway and
negatively regulates the permeability of the endothelial barrier
(5). Antagonists to PARs are a
newly found group of anti-platelet drugs and could specifically
bind with PARs and inhibit the activation of platelets.
Multi-center clinical trial results have shown that patients are
able to tolerate PARs well, and the risk of bleeding does not
increase even when combined with aspirin and clopidogrel (6). The endothelial barrier is an
important component of blood vessels, and the permeability of the
endothelial barrier is transiently increased in the process of
thrombogenesis. The aggregation of platelets induces the release of
thrombin, further activating PAR1 and increasing the activity of
phospholipase C, resulting in activation of the protein kinase C
(PKC) signaling pathway (7). In
addition, a previous study found that multiple factors could
inhibit the expression level of endothelial nitric oxide synthase
(eNOS), such as aging, obesity and disease, including cholesterol,
resulting in the reduction of nitric oxide (NO) (8). This effect is mediated by the
phosphatidylinositide 3-kinase (PI3K)/AKT signaling pathway in
endothelial cells and leads to increased permeability of the
endothelial barrier (9). However,
the detailed mechanism of vorapaxar in sustaining homeostasis of
the endothelial barrier under high lipid stimulation is not fully
understood.
In the present study, we found that high lipid
stimulation reduced the proliferation of endothelial cells, and
vorapaxar alleviated this effect via increasing the intracellular
Ca2+ concentration, activation of the AKT/JNK signaling
pathway and inhibition of the inflammatory response, resulting in
maintenance of permeability of the endothelial barrier and
presenting a protective effect on endothelial cells.
Materials and methods
Materials
Gibco™ high glucose Dulbecco's modified Eagle's
medium (H-DMEM) (10569044) and fetal bovine serum (FBS) (10099141)
were obtained from Thermo Fisher Scientific, Inc. (Waltham, MA,
USA). Cholesterol (C8667) was purchased from Sigma-Aldrich (Merck
KGaA, Darmstadt, Germany). Thrombin receptor activator peptide 6
(TRAP-6, HY-P0078) and vorapaxar (HY-10119) were obtained from
MedChemExpress (MCE, Monmouth Junction, NJ, USA). MTT (IM0280) was
purchased from Beijing Solarbio Science and Technology Co., Ltd.
(Beijing, China). Anti-JNK (ab179461), p-JNK (ab124956), AKT
(ab8805), p-AKT (ab38449), eNOS (ab76198), NF-κB (ab16502), ATM
(ab78), p-ATM (ab81292), ATR (ab101900), p-ATR (ab178407), GSK3β
(ab32391), p-GSK3β (ab68476), Connexin 43/GJA1 (ab11370) and GAPDH
(ab8245) antibodies were purchased from Abcam (Cambridge, UK). Cell
membrane permeable calcium fluorescent probe (40704ES50), Total RNA
extraction reagent (10606ES60), First Strand cDNA Synthesis
SuperMix (11141ES10), and qPCR SYBR−Green Master Mix
(11203ES03) were purchased from Shanghai Yeasen Biotechnology
(Shanghai, China). Protease inhibitor cocktail (CW2200) and
phosphatase inhibitor cocktail (CW2383) were purchased from CWBio
(Beijing, China). Anti-rabbit and anti-mouse HRP-labeling secondary
antibodies (10147552 and 10145703) were obtained from KPL, Inc.
(Gaithersburg, MD, USA).
Cell culture
EA.hy926 cells (GNHu39) were purchased from the Cell
Bank of the Type Culture Preservation Committee of the Chinese
Academy of Sciences (Shanghai, China) and cultured in a humidified
5% CO2 atmosphere at 37°C in H-DMEM containing 10% FBS.
The cells were divided into four groups when cell confluence
reached 60–70%, including a control (NC) group, cholesterol
stimulation (CH) group, cholesterol stimulation with TRAP-6
treatment (CH+T) group and cholesterol stimulation with vorapaxar
treatment (CH+V) group. Cholesterol was dissolved in dimethyl
sulfoxide (DMSO) at a concentration of 100 mmol/l (10), and cells were treated with TRAP-6
for 5 min and vorapaxar for 1 h before cholesterol stimulation.
After being treated with cholesterol for 24 h, the cells were
collected for the further experiments.
MTT assays
Cells were first washed with sterile
phosphate-buffered saline (PBS) to remove redundant cholesterol,
and an MTT assay was performed according to a previous study
(10). Briefly, cells were grouped
as previously described and seeded into each well of 96-well
plates. Each group of cells was first treated with different
concentrations of cholesterol for 24 h, different concentration of
TRAP-6 for 5 min and different concentration of vorapaxar for 1 h
(11), followed by incubation with
5 mg/ml MTT for 3 h. After incubation, the optical density at 490
nm was measured with a SpectraMax Series microplate reader
(Molecular Devices, Sunnyvale, CA, USA). The viability rate was
calculated as:
(ODTreatment-ODBlank)/(ODControl-ODBlank).
Extraction of nuclear proteins
Nuclear protein extraction was performed according
to a previous study (12).
Briefly, cells were divided into four groups when cell confluence
reached 60–70%, including a control (NC) group, a cholesterol
stimulation (CH) group, a cholesterol stimulation with TRAP-6
treatment (CH+T) group and a cholesterol stimulation with vorapaxar
treatment (CH+V) group. Cells were treated with TRAP-6 for 5 min
and vorapaxar for 1 h before cholesterol stimulation. After being
treated with cholesterol for 24 h, cells were washed with sterile
PBS to remove extra cholesterol. Then, cells were lysed with lysis
buffer A (10 mmol/l HEPES pH 7.9, 10 mmol/l KCl, 0.1 mmol/l EDTA,
0.1 mmol/l EGTA, 0.5% NP-40, 1 mmol/l DTT and protease inhibitor),
and the supernatants were collected after centrifugation at 13,523
× g for 10 min. Then, the pellets were lysed with lysis buffer B
[20 mmol/l HEPES (pH 7.9), 0.4 mol/l NaCl, 1 mmol/l EDTA, 1 mmol/l
EGTA, 1 mmol/l DTT and protease inhibitor] for 30 min. The
supernatants were collected after centrifugation at 20,238 × g for
15 min. The concentration of proteins was measured with a BCA
assay.
Western blotting
Cells were divided into four groups when cell
confluence reached 60–70%, including a control (NC) group, a
cholesterol stimulation (CH) group, a cholesterol stimulation with
TRAP-6 treatment (CH+T) group and a cholesterol stimulation with
vorapaxar treatment (CH+V) group. Cells were treated with TRAP-6
for 5 min and vorapaxar for 1 h before cholesterol stimulation.
After being treated with cholesterol for 24 h, cells were washed
with sterile PBS to remove extra cholesterol. Then, the cells in
each group were collected and lysed with urea lysis buffer (8 M
urea, 10 mM DTT, 50 mM IAA, cocktail protease inhibitors and
phosphatase inhibitor cocktail) at 4°C, followed by ultrasonic
lysis. The supernatant in each group was collected after
centrifugation at 13,523 × g for 10 min (4°C). The concentration of
protein was determined using a BCA assay. Protein samples were
firstly balanced with GAPDH, and 60 µg of each protein sample was
loaded and separated using 10% SDS-PAGE. After electrophoresis, the
samples were transferred onto 0.22-µm nitrocellulose membranes
using a semi-dry electro blotter. The membranes were first
incubated with a primary antibody (1:1,000) overnight at 4°C and
then incubated with the corresponding secondary antibody (1:5,000)
for 1 h at room temperature. After being balanced with GAPDH,
protein samples in each group were homogeneously mixed and
sub-packaged in each individual tube as each sample was used in
performing the western blot analysis of GAPDH and stored at −80°C
until performing the following analysis. Then, western blot
analysis of 12 target proteins were performed individually in 12
individual SDS-PAGE electrophoresis. After electrophoresis,
proteins were transferred onto nitrocellulose membranes and
incubated with antibodies as described above. Proteins were
detected using ECL immunoblotting reagent and normalized with
GAPDH. Gray values of each band were quantified using Scion Image
software (version 4.0.3.2; Scion Corp., Frederick, MD, USA).
Experiments were repeated for three times.
Extraction of cellular total RNA and
quantitative real-time PCR analysis for target gene expression
Extraction of total RNA was performed according to
the protocol. Briefly, cells were lysed with total RNA extraction
reagent and centrifuged at 12,000 × g for 15 min after chloroform
was added. Then, isopropanol was added to the water phase, and the
reaction was incubated at room temperature for 10 min. After
ethanol was added and centrifugation at 7,500 × g for 5 min, total
RNA was in the pellet. Reverse transcription was performed
according to the protocol. Quantitative real-time PCR was performed
after the cDNA was synthesized. The primer sequences are listed as
follows: IL-1β: Forward, 5′-CAGAGAGTCCTGTGCTGAAT-3′ and reverse,
5′-GTAGGAGAGGTCAGAGAGGC-3′; TNF-α: Forward,
5′-TCTCGAACCCCGAGTGACAA-3′ and reverse, 5′-TATCTCTCAGCTCCACACCA-3′;
IL-8: Forward, 5′-ATGACTTCCAAGCTGGCCGTG-3′ and reverse,
5′-CTCTTCAAAAACTTCTCCCGACTCTTAAGTATT-3′; IL-13: Forward,
5′-CACCATGCATCCGCTCCTCAATCCT-3′ and reverse,
5′-GTTGAACTGTCCCTCGCGAAA-3′. The annealing temperature was set at
55°C and the reaction was repeated for 40 cycles. The threshold
cycle (CT) was calculated with normalized fluorescence signal after
generation of real-time amplification. The CT value was
proportional with the initial number in the sample of target copies
and was used to perform the kinetic analysis. The quantity of the
target gene in the starting sample was measured after comparison
with the CTs of the positive control in a serial dilution. Relative
analysis on expression of each gene was performed using
2−ΔΔCq method according to previous study (13). The transcripts of GAPDH were used
as an internal reference, and the quantification of clustering mRNA
was normalized with relative GAPDH mRNA.
Detection of
[Ca2+]i using immunofluorescence
The concentration of intercellular Ca2+
([Ca2+]i) was evaluated according to the
protocol of the cell membrane permeable calcium fluorescent probe.
Briefly, 1×104 cells in each groups were seeded into the
confocal plate, cells were cultured as previously described in the
cell culture method section above, and no more specific endothelial
cell growth factors were added. And then, cells were grouped and
treated as previously described in cell culture section above.
Following treatment, the medium was removed and the cells were
incubated with 4 µmol/l Fluo-4 at 37°C for 30 min. Then, the cells
were washed with PBS three times. Images were acquired using
confocal microscopy (objective, 20×; Leica TCS SP8; Leica
Microsystems GmbH, Wetzlar, Germany) and analyzed using IPP 6.0
software (Media Cybernetics, Inc., Rockville, MD, USA).
Statistical analysis
The data are displayed as means ± SEM. Each
experiment was repeated three times independently. One-way analysis
of variance (ANOVA) was used to analyze the differences between
groups using the GraphPad Prism 7.0 software (GraphPad Software,
Inc., La Jolla, CA, USA) followed with Tukey's post-hoc test.
P-value <0.05 was set as a statistically significant
difference.
Results
Effect of cholesterol on the
proliferation of endothelial cells
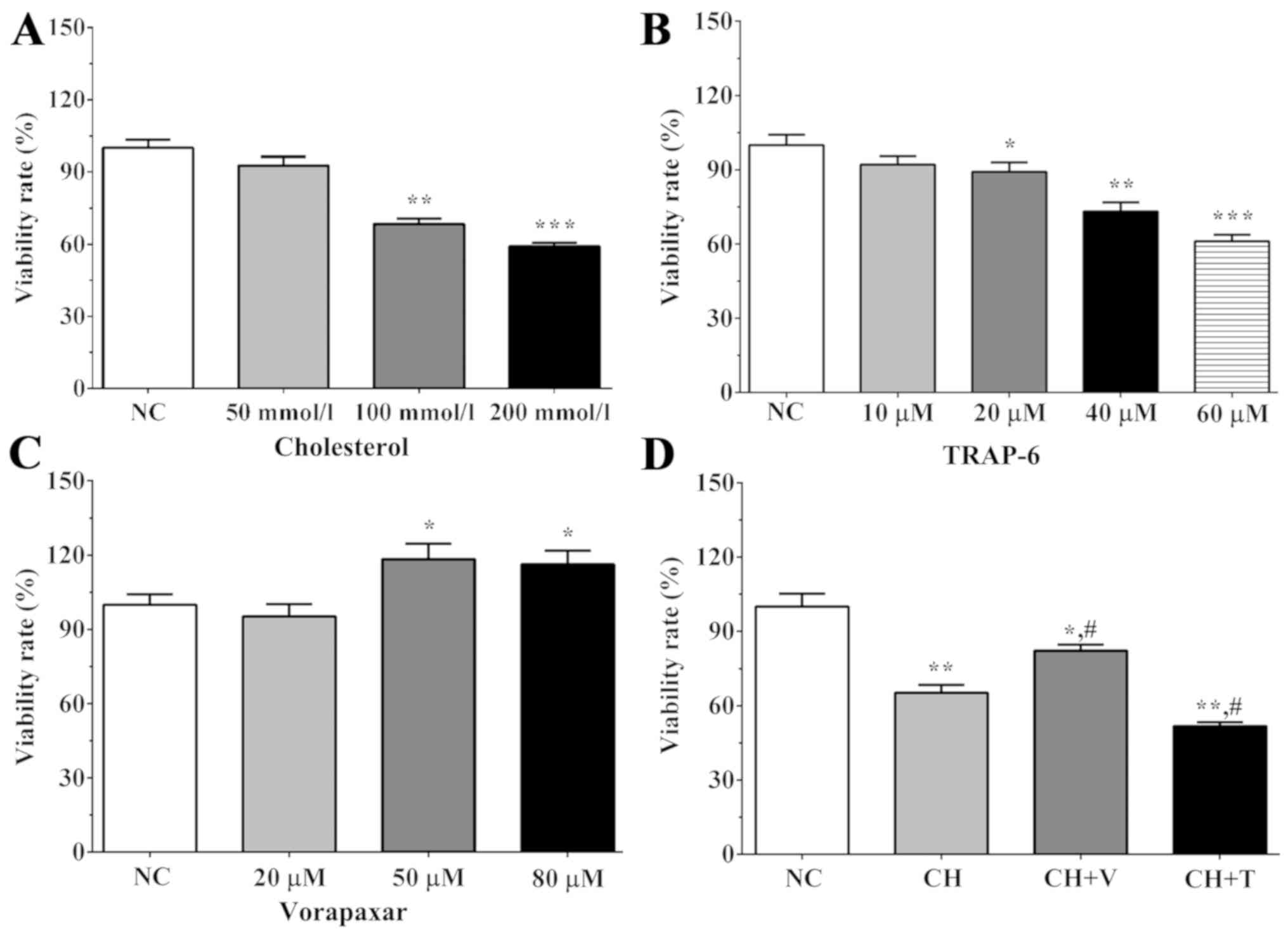
As shown in Fig.
1A, the viability rates following treatment with different
concentrations of cholesterol were 92.6±3.8, 68.3±2.4 and 59.1±1.4
in the 50, 100 and 200 mmol/l cholesterol treatment groups,
respectively. The viability rate was significantly decreased in the
100 and 200 mmol/l cholesterol treatment groups compared with the
NC group (P=0.001 and P<0.001). As shown in Fig. 1B, the viability rates were
92.1±3.4, 89.2±3.8, 73.2±3.6 and 61.1±2.7 for the 10, 20, 40 and 60
µM TRAP-6 treatment groups after 5 min. The viability rate was
significantly increased in the 20, 40 and 60 µM TRAP-6 treatment
groups compared with the NC group (P=0.044, P=0.002 and
P<0.001). As shown in Fig. 1C,
the viability rates were 95.3±4.9, 118.4±6.2 and 116.3±5.5 in the
20, 50 and 80 µM vorapaxar treatment groups. The viability rate was
significantly increased in the 50 and 80 µM vorapaxar treatment
groups compared with the NC group (P=0.018 and 0.020). After
treatment with 100 mmol/l cholesterol for 24 h, we detected the
viability rate using an MTT assay (Fig. 1D). The viability rate was 65.2±3.2
in the CH group, 82.3±2.3 in the CH+V group and 51.7±1.6 in the
CH+T group. Cholesterol treatment significantly decreased the
proliferation of endothelial cells compared with the NC group
(P=0.002 in the CH group), and vorapaxar treatment and TRAP-6
treatment significantly increased or decreased the proliferation
rate of the endothelial cells (P=0.0228 in the CH+V group and 0.001
in the CH+T group). This result indicated that vorapaxar may serve
an important role in the proliferation of endothelial cells.
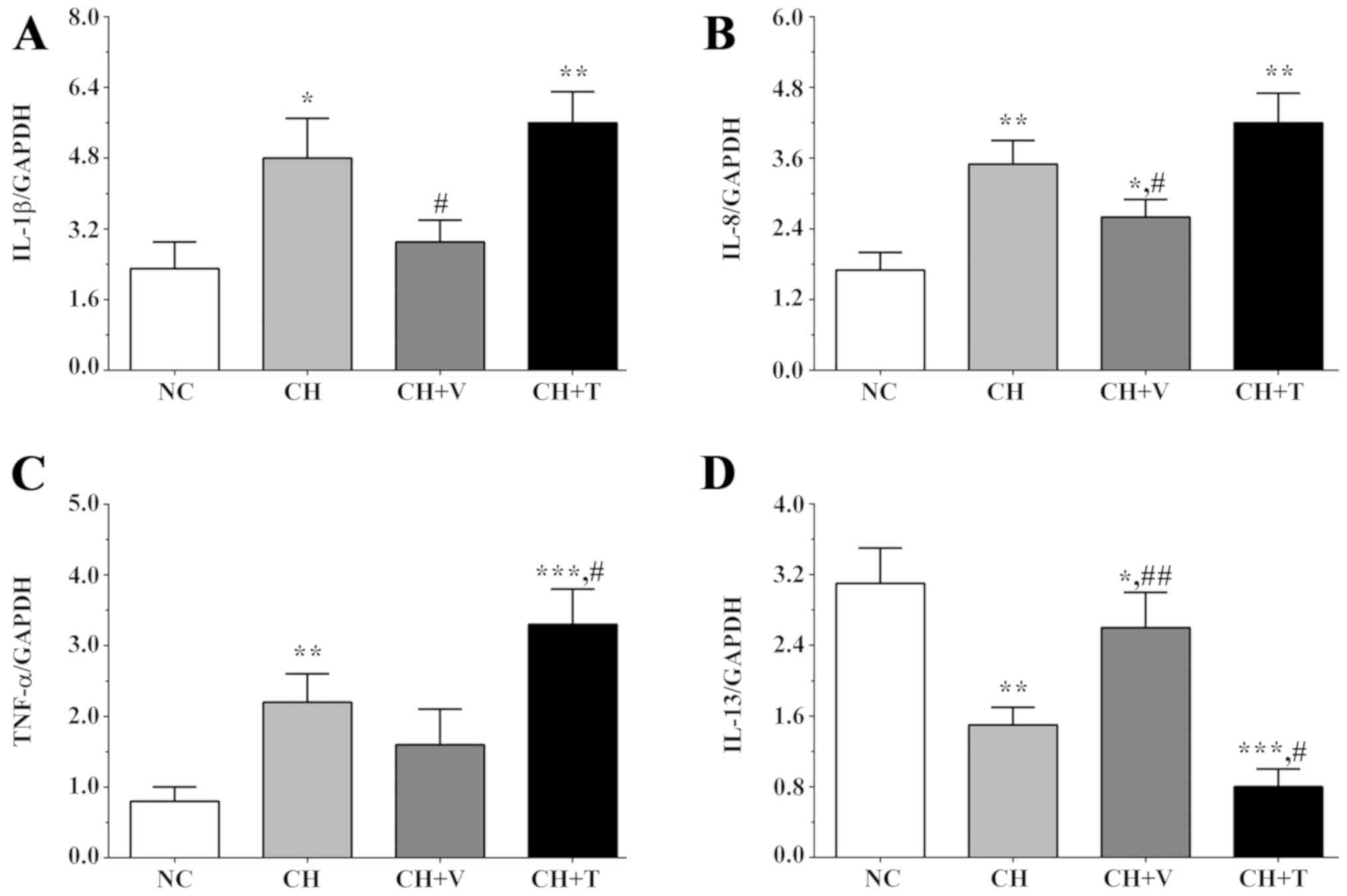
Real-time quantification PCR was used
to detect the expression levels of inflammation-related genes
Cholesterol is able to induce inflammation in
endothelial cells, increasing the expression levels of
inflammation-related genes. Herein, the expression levels of IL-1β,
TNF-α, IL-8 and IL-13 were detected. As shown in Fig. 2A and C, the expression levels of
IL-1β were 2.3±0.6, 4.8±0.9, 2.9±0.5 and 5.6±0.7 in the NC group,
the CH group, the CH+V group and the CH+T group and expression
levels of TNF-α were 0.8±0.2, 2.2±0.4, 1.6±0.5 and 3.3±0.5 in these
groups. As shown in Fig. 2B and C,
the expression levels of IL-8 were 1.7±0.3, 3.5±0.4, 2.6±0.3 and
4.2±0.5 in the NC group, the CH group, the CH+V group and the CH+T
group, and IL-13 levels were 3.1±0.4, 1.5±0.2, 2.6±0.4 and 0.8±0.2
in these groups. The expression level of IL-1β was significantly
increased in the CH and the CH+T group compared with the NC group
(P=0.016 in the CH group and 0.003 in the CH+T group), and
vorapaxar significantly decreased the expression level of IL-1β in
the CH+V group compared with the CH group (P=0.033). The expression
levels of IL-8 were significantly increased in all
cholesterol-treated groups compared with the NC group (P=0.003 in
the CH group, 0.021 in the CH+V group and 0.002 in the CH+T group),
and vorapaxar significantly decreased the expression level of IL-8
in the CH+V group compared with the CH group (P=0.036). The
expression levels of TNF-α were significantly increased in the CH
and the CH+T groups compared with the NC group (P=0.006 in the CH
group and 0.001 in the CH+T group), and TRAP-6 treatment
significantly increased this trend compared with the CH group
(P=0.041). However, the expression level of IL-13, an
anti-inflammatory factor, presented a different trend. The
expression level of IL-13 was significantly decreased after
cholesterol treatment (P=0.003 in the CH group, 0.046 in the CH+V
group and 0.001 in the CH+T group), and vorapaxar or TRAP-6
significantly alleviated or increased this trend, respectively,
compared with the CH group (P=0.008 in the CH+V group and 0.013 in
the CH+T group). These results indicated that cholesterol induced
an inflammatory response in endothelial cells while vorapaxar
inhibited this trend serving a protective role in endothelial
cells.
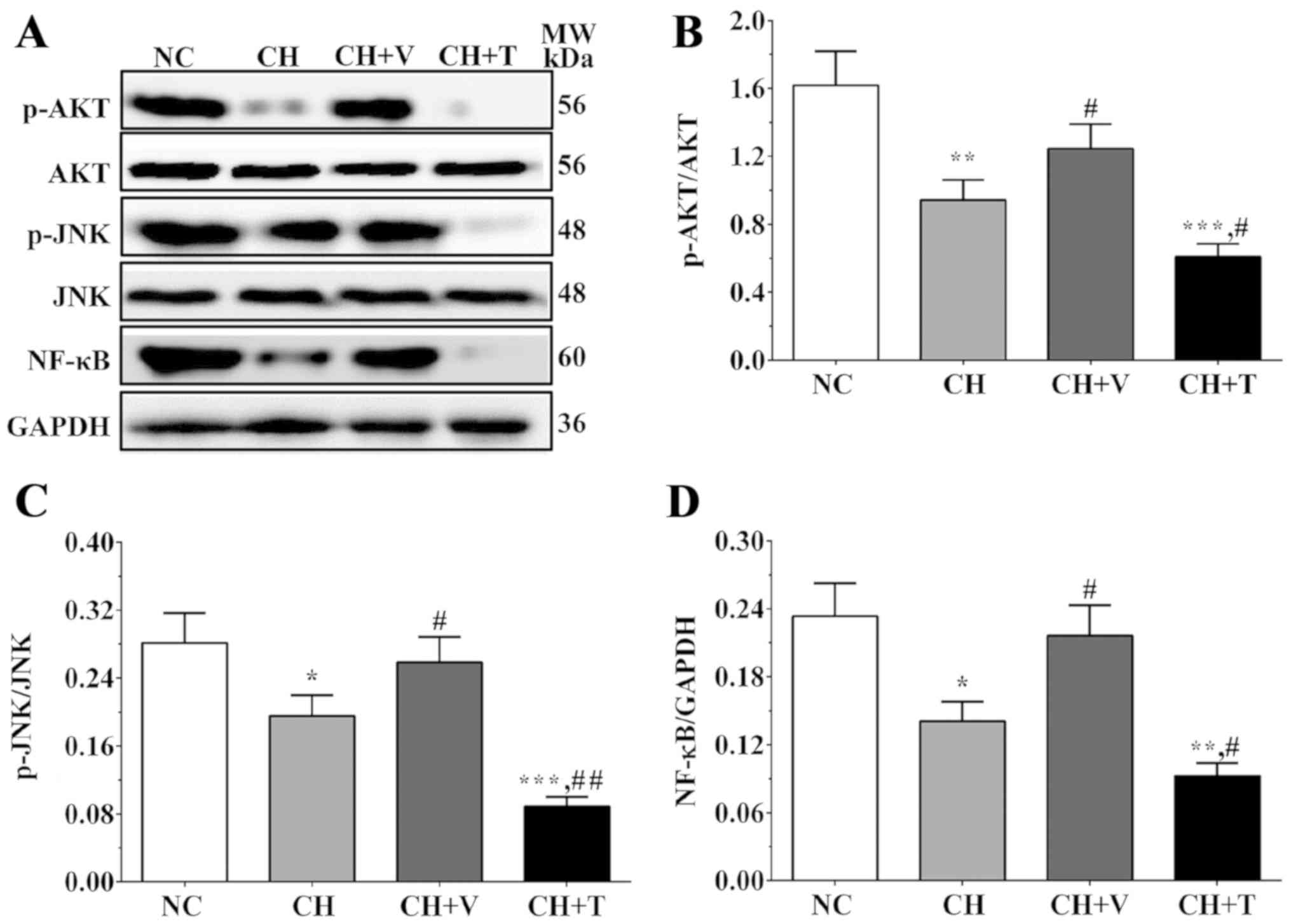
Western blot analysis was used to
detect the expression levels of molecules in the AKT/JNK signaling
pathway
The expression levels of molecules in the AKT/JNK
signaling pathway were evaluated using western blot analysis. As
shown in Fig. 3A and C, the ratios
of p-JNK/JNK were 0.28±0.04, 0.19±0.02, 0.24±0.03 and 0.09±0.01 in
the NC group, the CH group, the CH+V group and the CH+T group. As
shown in Fig. 3A and B, the ratios
of p-AKT/AKT were 1.62±0.20, 0.94±0.12, 1.25±0.14 and 0.61±0.08 in
these groups. As shown in Fig. 3A and
D, the expression levels of NF-κB were 0.23±0.03, 0.14±0.02,
0.22±0.03 and 0.09±0.01 in these groups. The ratios of p-AKT/AKT,
p-JNK/JNK and NF-κB were significantly decreased in the CH group
and the CH+T group compared with the NC group (P=0.007, 0.025 and
0.012 in the CH group and 0.001, 0.001, 0.002 in the CH+T group)
and were significantly increased in the CH+V group compared with
the CH group (P=0.044, 0.046 and 0.018). The expression levels of
these molecules were significantly decreased in the CH+T group
compared with the CH group (P=0.017, 0.002 and 0.018). These
results showed that the AKT/JNK signaling pathway and expression of
NF-κB were inhibited in endothelial cells after cholesterol
stimulation and that vorapaxar could reduce these inhibitory
effects on activation of the AKT/JNK signaling pathway and
expression of NF-κB, whereas TRAP-6 enhanced the inhibitory effects
on activation of the AKT/JNK signaling pathway and expression of
NF-κB.
Western blot analysis was used to
detect the expression levels of DNA damage- and cell cycle-related
proteins
As shown in Fig.
4A-C, the ratios of p-ATM/ATM were 0.24±0.03, 0.74±0.10,
0.50±0.06, 1.22±0.15 in the NC group, the CH group, the CH+V group
and the CH+T group, and the ratios of p-ATR/ATR were 0.27±0.03,
0.62±0.08, 0.54±0.07, 1.13±0.14 in these groups. The expression
levels of cell cycle-related proteins were also detected in
endothelial cells and the effect of cholesterol on the cell cycle
was evaluated. As shown in Fig. 4A, D
and F, the ratios of p-GSK3α/GSK3α were 0.84±0.10, 0.32±0.04,
0.47±0.06 and 0.21±0.03 in the NC group, the CH group, the CH+V
group and the CH+T group, and the expression levels of cyclin D1
were 1.22±0.15, 0.49±0.06, 0.78±0.10 and 0.35±0.04 in these groups.
As a downstream molecule of the AKT/JNK signaling pathway, eNOS
serves an important role in NO synthesis and has an
anti-atherosclerotic effect. As shown in Fig. 4E, the expression levels of eNOS
were 1.38±0.17, 0.61±0.08, 0.93±0.12 and 0.46±0.06 in these groups.
The ratios of p-ATM/ATM and p-ATR/ATR were significantly increased
in the cholesterol treatment groups compared with the NC group
(P=0.001 and 0.002 in the CH group, 0.003 and 0.004 in the CH+V
group, 0.001 and 0.001 in the CH+T group) and were significantly
decreased after vorapaxar treatment and significantly increased
after TRAP-6 treatment in endothelial cells compared with the CH
group (P=0.024 and 0.046 in the CH+V group, 0.001 and 0.005 in the
CH+T group). The ratios of p-GSK/GSK and the expression of cyclin
D1 were significantly decreased after cholesterol treatment
compared with the NC group (P=0.001 and 0.001 in the CH group,
0.005 and 0.013 in the CH+V group, 0.001 and 0.001 in the CH+T
group) and were significantly increased after vorapaxar treatment
and significantly decreased after TRAP-6 treatment in endothelial
cells compared with the CH group (P=0.022 and 0.013 in the CH+V
group, 0.019 and 0.028 in the CH+T group). The expression level of
eNOS was significantly decreased after cholesterol treatment
compared with the NC group (P=0.002 in the CH group, 0.020 in the
CH+V group and 0.001 in the CH+T group), and vorapaxar
significantly increased eNOS expression compared with the CH group
(P=0.018). These results indicate that cholesterol directly induced
DNA damage in the endothelial cells and inhibited cell
proliferation and that vorapaxar could alleviate these effects,
serving a protective role in endothelial cells.
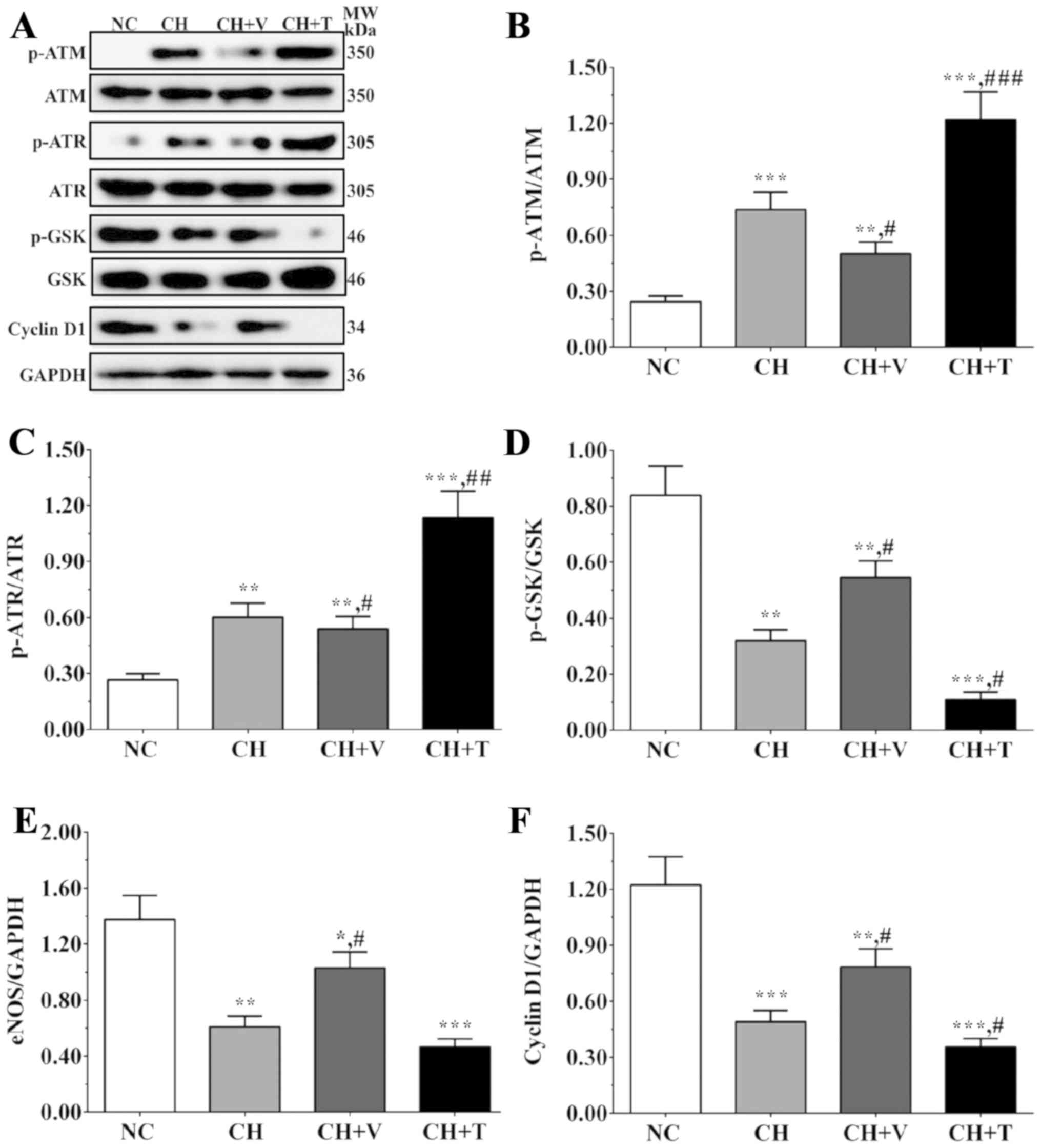 | Figure 4.Expression levels of DNA
damage-related molecules and cell cycle-related molecules. (A)
Detection of expression levels of p-ATM, ATM, p-ATR, ATR, p-GSK,
GSK and cyclin D1 using western blotting. (B-F) Quantitative
analysis of the western blot results. Experiments were repeated
three times independently. Data are represented as the mean ± SEM.
*P<0.05, **P<0.01 and ***P<0.001 vs. the NC group;
#P<0.05, ##P<0.01 and ###P<0.001 vs. the CH group. GAPDH
was used as an internal control. Each image of western blotting
band was separated using a black rectangle. Groups: NC, negative
control group; CH, a cholesterol stimulation group; CH+T,
cholesterol stimulation with TRAP-6 treatment group; CH+V,
cholesterol stimulation with vorapaxar treatment group. |
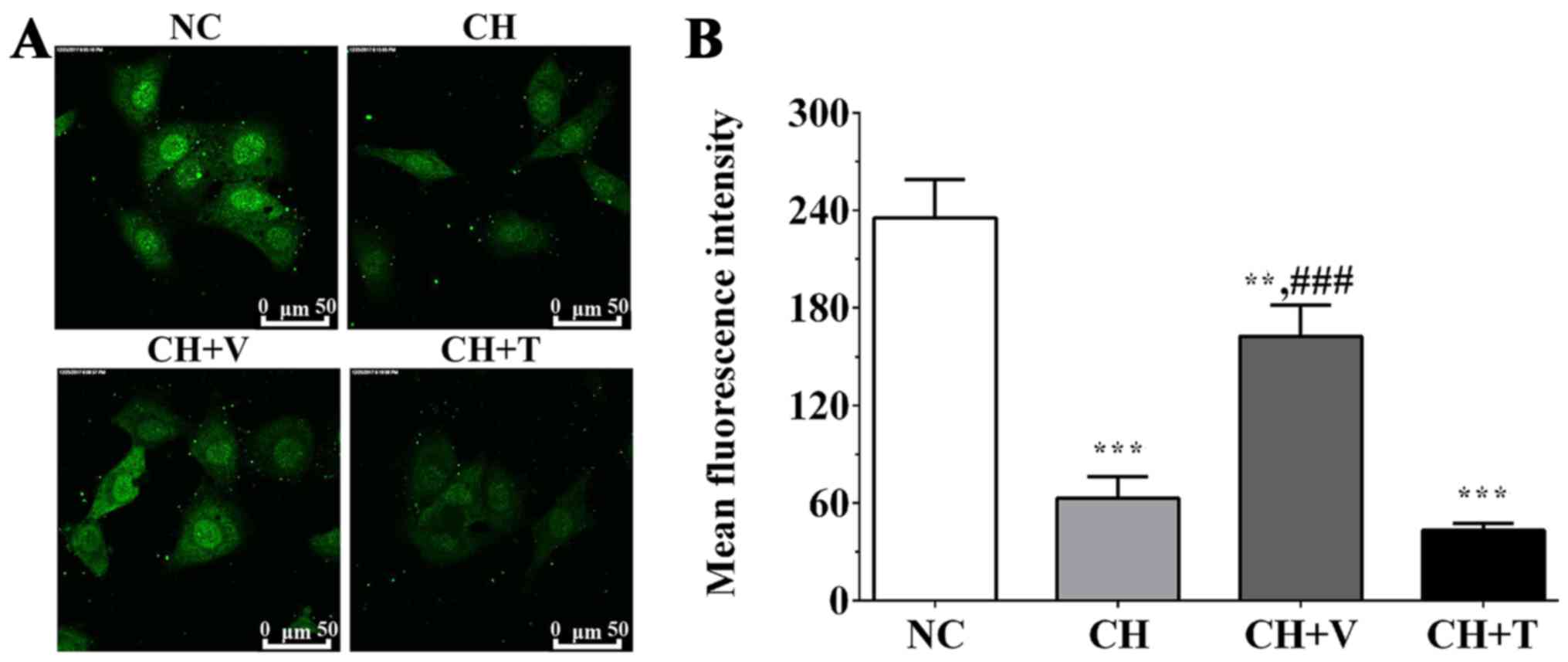
Immunofluorescence was used to measure
the concentration of [Ca2+]i
As shown in Fig. 5,
we detected the concentration of intracellular Ca2+
([Ca2+]i). The mean fluorescence intensity of
the concentration of [Ca2+]i was 235.38±23.56
in the NC group and was 62.92±9.23, 162.42±19.33 and 43.19±4.26 in
the CH group, the CH+V group and the CH+T group, respectively.
These results indicated that cholesterol stimulation significantly
decreased [Ca2+]i compared with the NC group
(P=0.001 in the CH group, 0.014 in the CH+V group and 0.001 in the
CH+T group), and vorapaxar could significantly alleviate this trend
compared with the CH group (P=0.001), whereas there was no
significant change between the CH group and the CH+T group.
Discussion
In the present study, cholesterol stimulation was
found to activate the AKT/JNK signaling pathway, induce an
inflammatory response and decrease the
[Ca2+]i in endothelial cells, and inhibit
proliferation of endothelial cells. The inhibition of PAR-1 via
vorapaxar was able to alleviate the effect of cholesterol on
endothelial cells, whereas the activation of PAR-1 via TRAP-6
enhanced these effects. Therefore, we speculated that vorapaxar
maintains the viability of endothelial cells and the permeability
of the endothelial barrier via activation of the AKT/JNK signaling
pathway, inhibition of the inflammatory response and a decrease in
[Ca2+]i, resulting in a protective role in
endothelial cells.
Cardiovascular disease (CVD) is a serious threat to
public health and the economy worldwide. According to data acquired
from the World Health Organization (WHO) in 2012, cardiovascular
disease caused more than 17 million deaths worldwide, accounting
for over a 30% mortality rate, and cost more than $193 billion for
the treatment of these patients (14). Atherosclerosis (AS) is the leading
cause of CVD and is an inflammatory-related disease characterized
by the progressive accumulation of lipids and inflammatory cells in
the intima of arteries, especially in high lipid states (15). High lipids induce injury of the
endothelial barrier, further causing an inflammatory response and
the formation of thrombus and recruitment of circulating monocytes
attached to the injury site of the endothelial barrier (16). These processes induce the
transformation of monocytes into macrophages and the adhesion of
platelets, resulting in the proliferation of vascular smooth muscle
cells (VSMCs) and leukocytes, promoting the formation of plaque
(17). Disruption of the plaque
may cause exposure of the thrombogenic substrates and initiating
the coagulation cascade, resulting in the formation of thrombus and
manifestations of atherosclerotic disease, even MI or sudden
death.
According to our results, a high concentration of
cholesterol inhibited the proliferation of endothelial cells, and
using TRAP-6 or vorapaxar to activate or inhibit PAR-1
significantly decreased or increased the viability rate of the
endothelial cells compared with the cholesterol treatment and
control groups, indicating that cholesterol inhibits the growth of
endothelial cells, while inhibition or activation of PAR-1 reduces
or increases these trends. We also detected the expression levels
of inflammation-related genes to evaluate the inflammation response
level in endothelial cells after cholesterol stimulation. The
response of endothelial cells to lipoproteins could initiate an
inflammatory response at the atherosclerosis site, resulting in the
activation and release of cytokines including IL-1 and TNF-α
(18). These processes would
attract monocytes to migrate to the injury site and take up
lipoproteins, forming foam cells. IL-13 is a Th2 cytokine that is
involved in regulating inflammatory responses and serves an
anti-inflammatory effect via promoting monocyte/macrophage
transformation into a M2a phenotype and alleviating inflammatory
responses (19). As shown in
Fig. 2, the expression levels of
IL-1β, TNF-α and IL-8 were significantly increased after
cholesterol stimulation and were alleviated or increased after
PAR-1 was inhibited or activated. However, the expression level of
IL-13 presented an opposite trend. These results indicated that
cholesterol stimulation could activate the inflammatory responses
in endothelial cells and PAR-1 serves an important regulatory role
in the maintenance of inflammatory balance, and vorapaxar exhibits
an endothelial cell protective effect via the inhibition of
inflammatory responses.
Multiple signaling pathways are involved in
inflammatory response, and nuclear factor (NF)-κB serves an
important role in the regulation of many genes in inflammatory
responses at the transcription level (20). Other factors such as oxidative
stress, hypoxia, and genotoxic stress could also activate NF-κB via
mitogen-activated protein kinases (MAPKs) and phosphatidylinositol
3-kinase (PI3K)/AKT (21). DNA
damage is closely related to the development of atherosclerosis;
nuclear DNA (nDNA) damage and a lack of mitochondrial DNA (mtDNA)
are commonly observed in circulating blood cells and plaque cells
of coronary heart disease patients (22). Oxidative stress induced by high
cholesterol is one of the main factors leading to DNA damage, and
long-term exposure to risk factors of atherosclerosis including
smoking, diabetes and especially hyperlipidemia, may lead to the
production of reactive oxygen species (ROS) and further cause DNA
damage in endothelial cells (23).
DNA damage initiates the DNA damage response (DDR) to guarantee
proliferation of normal cells through the induction of cell cycle
arrest mechanisms. DNA damage, especially double-stranded DNA
breaks (DSB), activates ataxia-telangiectasia mutated (ATM) and
ataxia telangiectasia and rad3 related (ATR) (24). A previous study found that
cholesterol overload induced DNA damage and activated ATM/ATR, and
ATM/ATR may serve a protective role in this process (25). A previous study found that NEMO
undergoes post-translational modifications including
phosphorylation and ubiquitination regulated by ATM after DNA
damage, and this process may be linked with activation of NF-κB
(26). Activation of NF-κB is one
of the cellular responses to maintain homeostasis after DNA damage.
In this study, we found that vorapaxar may perform a protective
role in endothelial cells under cholesterol stimulation via
increasing the expression of NF-κB.
Endothelial nitric oxide synthase (eNOS) is
constitutively expressed in endothelial cells and is activated by
flowing blood pressure or agonists. Nitric oxide (NO) is regulated
mainly by endothelial NO synthase (eNOS) in endothelial cells and
serves a protective role (27). NO
synthesized by endothelial eNOS could be released into vascular
smooth muscle cells (VSMCs) and induce vasodilatation of the
vascular. The release of endothelial NO could also inhibit the
aggregation and adhesion of platelets and further inhibit the
formation of plaque. Endothelial function serves an important role
in the maintenance of vascular integrity by regulating the
production and activation of NO. The development of atherosclerosis
is closely related to the dysfunction of NO synthesis in
endothelial cells. Previous studies have found that the activity of
endothelial NO is reduced in ApoE-KO mice, which may be due to the
inhibition of eNOS production or eNOS bioactivity, resulting in
accelerated formation of atherosclerosis in ApoE-KO mice (28). A previous study found that genetic
depletion of AKT may lead to dysfunction of endothelial cells,
reducing migration and survival of VSMCs, leading to
atherosclerosis and coronary artery obstruction, resulting in the
formation of plaque (29). The AKT
signaling pathway serves an important role in the regulation of
eNOS. AKT1 is a main subtype of the AKT family expressed in
endothelial cells, and mediates the survival and migration of
endothelial cells and serves an important role in angiogenesis and
the regulation of vascular structure (30,31).
Multiple stimuli including vascular endothelial growth factor
(VEGF) and cholesterol activate AKT in endothelial cells, resulting
in phosphorylation of eNOS and promoting NO release. AKT could
directly phosphorylate eNOS or interact with Hsp90, increasing the
activity of eNOS or its sensitivity to
[Ca2+]i (32). Increasing
[Ca2+]i could induce the activation of eNOS
and the production of NO via a Ca2+/calmodulin
independent kinase II (CaMKII) dependent mechanism (33). Cholesterol stimulation inhibited
the AKT/mTOR signaling pathway, further decreasing the expression
levels of eNOS and [Ca2+]i, leading to a
reduction of NO and reduced integrity of the endothelial cell
barrier, promoting the formation of thrombi and atherosclerosis. We
found that cholesterol could induce DNA damage and decrease the
expression of eNOS via the AKT signaling pathway and
[Ca2+]i, resulting in inhibition of
endothelial cell proliferation, further causing destruction of the
endothelial barrier. However, vorapaxar could alleviate these
changes via inhibition of PAR-1 and serves a protective role in
regulation of endothelial function.
AKT also participates in the regulation of the cell
cycle via the JNK signaling pathway. AKT interacts with
mitogen-activated protein kinase kinase kinase (MAPKKK) and
inhibits c-jun N-terminal kinase (JNK), resulting in maintenance of
cell survival. A previous study found that overexpression or
inhibition of AKT function could increase or decrease the survival
of cardiomyocytes (34). Cell
survival and migration are important process in the maintenance of
vascular structure, and the AKT signaling pathway serves a critical
role in these biological activities. GSK3 is a substrate of AKT,
and the inhibition of GSK leads to proliferation and survival of
smooth muscle cells (SMCs) and sustained homeostasis of blood
vascular walls (35). A previous
study found that this process is related to the development of
atherosclerosis, and an increase in SMC proliferation may lead to
thickening of the vascular adventitia. Arterial injury could also
increase the activity of the AKT signaling pathway, resulting in
inactivation of GSK3β and the activation of multiple transcription
factors. These factors increase glucose metabolism, glycogen, lipid
and protein synthesis, and expression of other specific genes
(36). The AKT signaling pathway
also participates in the process of myocardial remodeling via the
phosphorylation of GSK-3β. The phosphorylation of AKT is the active
form of AKT, whereas the phosphorylation of GSK-3β would inhibit
its activity. A previous study found that activation of AKT is
important in remote conditioning of myocardial cells and serves a
protective role. This effect was displayed via inhibition of GSK-3β
and protects myocardial cells from ischemia-reperfusion injury
(37). A previous study also found
that inhibition of GSK-3β increased the concentration of glucose in
skeletal muscles, which may increase the survival of cardiac muscle
(38). Connexin 43, also known as
gap junction α-1 (GJA-1), is critical for gap junctions between
cells, allowing intercellular communication and physiological
impulses to occur via junctions between cells. A previous study
found that phosphorylation of connexin 43 was parallel to
increasing gap junctions in a rat hypoxic model and serves a
protective role in myocardial cells (39). The results of this study indicated
that downstream molecules of the AKT signaling pathway, such as
GSK-3β and connexin 43, also participate in the cardio-protective
function of AKT via maintaining endothelial cell survival and
cellular junctions and protect endothelial cells from injury
induced by cholesterol stimulation.
The present study indicated that a high
concentration of cholesterol causes an inflammatory response and
DNA damage in endothelial cells, inhibits the survival of
endothelial cells and the release of NO, leading to destruction of
the endothelial barrier and formation of thrombi, and vorapaxar
reverses these processes and serves a protective role in
endothelial cells. However, more clinical experiments are needed,
and we are collecting clinical samples to perform more experiments
to support the findings of the present study.
Acknowledgements
Not applicable.
Funding
This research was supported by Zhejiang medical and
health science and technology project (Grant no.2016KYA197).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JLP and PYH collected the samples and performed the
experiments. JWW and JSJ collected and analysed the data. JLP and
JFL designed the study and wrote the manuscript. All authors read
and approved the manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Institutional Review Board of Tiantai People's Hospital of Zhejiang
Province (Zhejiang, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AS
|
atherosclerosis
|
|
VSMCs
|
vascular smooth muscle cells
|
|
AKT
|
protein kinase B
|
|
GSK-3β
|
glycogen synthase kinase 3β
|
|
[Ca2+]i
|
intracellular concentration of
Ca2+
|
|
PARs
|
protease-activated receptors
|
|
eNOS
|
endothelial nitric oxide synthase
|
|
CVD
|
cardiovascular disease
|
References
|
1
|
Badimón L, Vilahur G and Padró T:
Lipoproteins, platelets and atherothrombosis. Rev Esp Cardiol.
62:1161–1178. 2009.(In English, Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mehta D and Malik AB: Signaling mechanisms
regulating endothelial permeability. Physiol Rev. 86:279–367. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jennings LK: Mechanisms of platelet
activation: Need for new strategies to protect against
platelet-mediated atherothrombosis. Thromb Haemost. 102:248–257.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schaff M, Gachet C and Mangin PH:
Anti-platelets without a bleeding risk: Novel targets and
strategies. Biol Aujourdhui. 209:211–228. 2015.(In French).
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alberelli MA and De Candia E: Functional
role of protease activated receptors in vascular biology. Vascul
Pharmacol. 62:72–81. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gryka RJ, Buckley LF and Anderson SM:
Vorapaxar: The current role and future directions of a novel
protease-activated receptor antagonist for risk reduction in
atherosclerotic disease. Drugs R D. 17:65–72. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Machida T, Dohgu S, Takata F, Matsumoto J,
Kimura I, Koga M, Nakamoto K, Yamauchi A and Kataoka Y: Role of
thrombin-PAR1-PKCθ/δ axis in brain pericytes in thrombin-induced
MMP-9 production and blood-brain barrier dysfunction in vitro.
Neuroscience. 350:146–157. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Suvorava T, Nagy N, Pick S, Lieven O,
Rüther U, Dao VT, Fischer JW, Weber M and Kojda G: Impact of
eNOS-dependent oxidative stress on endothelial function and
neointima formation. Antioxid Redox Signal. 23:711–723. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Abeyrathna P and Su Y: The critical role
of Akt in cardiovascular function. Vascul Pharmacol. 74:38–48.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jin X, Xu Z, Fan R, Wang C, Ji W, Ma Y,
Cai W, Zhang Y, Yang N, Zou S, et al: HO-1 alleviates
cholesterol-induced oxidative stress through activation of Nrf2/ERK
and inhibition of PI3K/AKT pathways in endothelial cells. Mol Med
Rep. 16:3519–3527. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Khoufache K, Berri F, Nacken W, Vogel AB,
Delenne M, Camerer E, Coughlin SR, Carmeliet P, Lina B, Rimmelzwaan
GF, et al: PAR1 contributes to influenza A virus pathogenicity in
mice. J Clin Invest. 123:206–214. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pandey A, Chakraborty S and Chakraborty N:
Nuclear proteome: Isolation of intact nuclei, extraction of nuclear
proteins, and 2-DE analysis. Methods Mol Biol. 1696:41–55. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi Z, Bielecka-Dabrowa AM, Mynbaev OA and
Wei S: Find the essence through the phenomena: Cardiovascular
diseases and biomarkers. Dis Markers. 2018:19291062018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Raggi P, Genest J, Giles JT, Rayner KJ,
Dwivedi G, Beanlands RS and Gupta M: Role of inflammation in the
pathogenesis of atherosclerosis and therapeutic interventions.
Atherosclerosis. 276:98–108. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu H, Yu X, Yu S and Kou J: Molecular
mechanisms in lipopolysaccharide-induced pulmonary endothelial
barrier dysfunction. Int Immunopharmacol. 29:937–946. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yi X, Lin J, Luo H, Wang C and Liu Y:
Genetic variants of PTGS2, TXA2R and TXAS1 are associated with
carotid plaque vulnerability, platelet activation and TXA2 levels
in ischemic stroke patients. PLoS One. 12:e01807042017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li K, Ching D, Luk FS and Raffai RL:
Apolipoprotein E enhances microRNA-146a in monocytes and
macrophages to suppress nuclear factor-κB-driven inflammation and
atherosclerosis. Circ Res. 117:e1–e11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Solís-Martínez R, Cancino-Marentes M,
Hernández-Flores G, Ortiz-Lazareno P, Mandujano-Álvarez G,
Cruz-Gálvez C, Sierra-Díaz E, Rodríguez-Padilla C, Jave-Suárez LF,
Aguilar-Lemarroy A and Bravo-Cuellar A: Regulation of
immunophenotype modulation of monocytes-macrophages from M1 into M2
by prostate cancer cell-culture supernatant via transcription
factor STAT3. Immunol Lett. 196:140–148. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen X, Zong C, Gao Y, Cai R, Fang L, Lu
J, Liu F and Qi Y: Curcumol exhibits anti-inflammatory properties
by interfering with the JNK-mediated AP-1 pathway in
lipopolysaccharide-activated RAW264.7 cells. Eur J Pharmacol.
723:339–345. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moniruzzaman M, Ghosal I, Das D and
Chakraborty SB: Melatonin ameliorates H2O2-induced oxidative stress
through modulation of Erk/Akt/NFkB pathway. Biol Res. 51:172018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gray K and Bennett M: Role of DNA damage
in atherosclerosis-bystander or participant? Biochem Pharmacol.
82:693–700. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Janssens S and Tschopp J: Signals from
within: The DNA-damage-induced NF-κB response. Cell Death Differ.
13:773–784. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maréchal A and Zou L: DNA damage sensing
by the ATM and ATR kinases. Cold Spring Harb Perspect Biol.
5:a0127162013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Enríquez-Cortina C, Bello-Monroy O,
Rosales-Cruz P, Souza V, Miranda RU, Toledo-Pérez R, Luna-López A,
Simoni-Nieves A, Hernández-Pando R, Gutiérrez-Ruiz MC, et al:
Cholesterol overload in the liver aggravates oxidative
stress-mediated DNA damage and accelerates hepatocarcinogenesis.
Oncotarget. 8:104136–104148. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Miyamoto S: Nuclear initiated NF-κB
signaling: NEMO and ATM take center stage. Cell Res. 21:116–130.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sakata K, Kondo T, Mizuno N, Shoji M,
Yasui H, Yamamori T, Inanami O, Yokoo H, Yoshimura N and Hattori Y:
Roles of ROS and PKC-βII in ionizing radiation-induced eNOS
activation in human vascular endothelial cells. Vascul Pharmacol.
70:55–65. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vechoropoulos M, Ish-Shalom M, Shaklai S,
Sack J, Stern N and Tordjman KM: The proatherogenic effect of
chronic nitric oxide synthesis inhibition in ApoE-Null mice is
dependent on the presence of PPAR α. PPAR Res. 2014:1245832014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fernández-Hernando C, Ackah E, Yu J,
Suárez Y, Murata T, Iwakiri Y, Prendergast J, Miao RQ, Birnbaum MJ
and Sessa WC: Loss of Akt1 leads to severe atherosclerosis and
occlusive coronary artery disease. Cell Metab. 6:446–457. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Somanath PR, Razorenova OV, Chen J and
Byzova TV: Akt1 in endothelial cell and angiogenesis. Cell Cycle.
5:512–518. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fulton D, Gratton JP, McCabe TJ, Fontana
J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A and Sessa WC:
Regulation of endothelium-derived nitric oxide production by the
protein kinase Akt. Nature. 399:597–601. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shin MK, Jeong KH, Choi H, Ahn HJ and Lee
MH: Heat shock protein 90 inhibitor enhances apoptosis by
inhibiting the AKT pathway in thermal-stimulated SK-MEL-2 human
melanoma cell line. J Dermatol Sci. 90:357–360. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang B, Zhao J, Yu M, Meng X, Cui X, Zhao
Y, Zhu Y, Xing W and Guan Y: Disturbance of intracellular calcium
homeostasis and CaMKII/CREB signaling is associated with learning
and memory impairments induced by chronic aluminum exposure.
Neurotox Res. 26:52–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sugishita Y, Leifer DW, Agani F, Watanabe
M and Fisher SA: Hypoxia-responsive signaling regulates the
apoptosis-dependent remodeling of the embryonic avian cardiac
outflow tract. Dev Biol. 273:285–296. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Park KW, Yang HM, Youn SW, Yang HJ, Chae
IH, Oh BH, Lee MM, Park YB, Choi YS, Kim HS and Walsh K:
Constitutively active glycogen synthase kinase-3β (GSK-3β) gene
transfer sustains apoptosis, inhibits proliferation of vascular
smooth muscle cells, and reduces neointima. Arterioscler Thromb
Vasc Biol. 23:1364–1369. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nakae J, Park BC and Accili D: Insulin
stimulates phosphorylation of the forkhead transcription factor
FKHR on serine 253 through a wortmannin-sensitive pathway. J Biol
Chem. 274:15982–15985. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rahman S, Li J, Bopassa JC, Umar S, Iorga
A, Partownavid P and Eghbali M: Phosphorylation of GSK-3β mediates
intralipid-induced cardio-protection against ischemia/reperfusion
injury. Anesthesiology. 115:242–253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Forde JE and Dale TC: Glycogen synthase
kinase 3: A key regulator of cellular fate. Cell Mol. Life Sci.
64:1930–1944. 2007. View Article : Google Scholar
|
|
39
|
Brandenburger T, Huhn R, Galas A, Pannen
BH, Keitel V, Barthel F, Bauer I and Heinen A: Remote ischemic
preconditioning preserves Connexin 43 phosphorylation in the rat
heart in vivo. J Transl Med. 12:2282014. View Article : Google Scholar : PubMed/NCBI
|