Introduction
CaMKII is a multi-polymer serine threonine kinase
consisting of 12 subunits. Each subunit contains three conserved
domains: i) A catalytic domain in an amino terminal, which supplies
the binding site for adenosine triphosphate (ATP) and multiple
substrate enzymes; ii) a central autoregulatory domain, which has
different models of post-translational modifications and exhibits a
suppressive effect by the mimic sequence of substrate enzyme; iii)
a carboxy-terminal association domain, which has the subunits of
oligomerization to produce holoenzyme and the variable sites of a
variety of splice variants. Typically, the pseudosubstrate sequence
of the autoregulatory domain maintains the basal kinase activity
levels at 100–1,000-fold lower than the activity level stimulated
by calcified calmodulin (Ca2+/CaM) (1).
The autoinhibitory region has residues that mimic a
protein or nucleotide substrate. They interact with the catalytic
domain and block the ATP- and substrate-binding pockets (1,2). The
binding of an allosteric activator, such as Ca2+/CaM,
alters the configuration of the autoinhibitory region, allowing
access to the catalytic site (1).
The sustained activation of the enzyme results in
autophosphorylation at Thr287 (3).
This leads to a 1,000-fold increase in the affinity of CaM, and
generates Ca2+/CaM-independent enzyme activity (3). The activation of CaMKII further
triggers the exchange of subunits between the holoenzymes,
including the inactive ones, thus enabling the calcium-independent
activation of new holoenzymes (4).
The activation of CaMKII is improved by reactive
oxygen species (ROS), leading to cardiovascular disease,
inflammation and cancer. The present review summarizes the current
findings associated with the function of oxidized CaMKII
(ox-CaMKII) in inflammatory diseases, including asthma, identifies
important gaps in current knowledge and suggests novel approaches
for future research (Table I).
 | Table I.Ox-CamKII promotes the development of
multiple diseases. |
Table I.
Ox-CamKII promotes the development of
multiple diseases.
| Author, year | Disease | Relevant
functions | (Refs.) |
|---|
| Gu et al,
2016 | Acute ischemic
stroke | Ox-CaMKII can
induce nuclear factor-κB to exacerbate neurovascular inflammation
and cerebral tissue damage in ischemic stroke. | (32) |
| Erickson et
al, 2008; Singh et al, 2012; He et al, 2011 | MI | Ox-CaMKII is
increased by Ang II and aldosterone and induces MI. | (5,12,14) |
| Ho et al,
2014; Swaminathan et al, 2011; Purohit et al, 2013;
Wagner et al, 2011 | Arrhythmias | Ox-CaMKII is
increased in atrial fibrillation in the presence of Ang II and a
key mediator of the cardiac glycoside reactive oxygen
species-induced arrhythmogenic effects on myocyte Ca2+
handling. | (11,13,37,38) |
| He et al,
2011; Zhu et al, 2014; Scott et al, 2012 | Vascular
disease | Ox-CaMKII is
involved in vascular smooth muscle responses to injury and may
provide feedback information for cellular redox balance. | (14,39,40) |
| Rajtik et
al, 2016 | Acute myocardial
ischemia/reperfusion injury | Ox-CaMKII induces
the development of acute myocardial ischemia/reperfusion
injury. | (41) |
| Luo et al,
2013 | Diabetic
mellitus | Ox-CaMKII causes
higher mortality rates in diabetic patients with MI. | (8) |
| Hart et al,
2015 | Breast cancer | Ox-CaMKII as a key
signal for breast cancer cells and CaMKII inhibition may have
therapeutic benefit in types of cancer relying on glycolysis for
selective advantage over non-tumor cells. | (51) |
| Qu et al,
2017 | Asthma | Ox-CaMKII is
predominantly present in the epithelium (9) Ox-CaMKII significantly promotes asthma
through mast cell activation. | (64) |
| Wang et al,
2018 | CRSwNP | ox-CaMKII is
involved in KYN/AhR signaling-mediated mast cell activation in
CRSwNP. | (69) |
CaMKII is activated by ROS
Erickson et al (5) was the first to observe that the
activity and oxidation of CaMKII are enhanced in the myocardium
under conditions of pressure, such as hypertrophy and infarction.
Furthermore, the inhibition of CaMKII has been implicated in a
reduction in the death rate of cardiomyocytes and an improvement of
poor left ventricular remodeling. Of note, the level of ROS is
decreased, suggesting that the inhibition of CaMKII hinders its
oxidative role (5). ROS are
produced by NADPH oxidase and mitochondria-oxidized methionine at
281 and 282, promoting autonomic enzyme activity (6). However, the suppressive role of
pseudosubstrates requires the initial binding of
Ca2+/CaM (7). The
removal of ROS by inhibiting any of the pathways results in a
marked decrease in the expression of oxidative CaMKII (ox-CaMKII)
(8,9). Several other factors, including
hyperglycemia (8), excess
intracellular Na+ load (10), cardiac glycoside toxicity (11), and a variety of cytokines,
including endotoxin (12),
angiotensin II (Ang II) (13) and
aldosterone (14), are able to
enhance the expression of ox-CaMKII in the myocardium.
Methionine sulfoxide is the initial oxidation status
of methionine, and methionine sulfoxide reductase A (MsrA)
dynamically decreases the expression of ox-CaMKII by reducing the
methionine residues, thus portraying the reversibility of the
oxidative activation. Another novel mechanism underlying the
autonomous activation of CaMKII can be observed in diabetes.
Hyperglycemia causes the O-linked N-acetylglucosamine modification
of CaMKII at Ser279/280, activating it even at a low
Ca2+ levels (15).
β-adrenergic receptor signaling in cardiomyocytes results in the
activation of CaMKII via a direct pathway, which involves the role
of nitrosylation at C290. This NO-induced independent activation of
CaMKII downstream to β-adrenergic signaling is responsible for
increased sarcoplasmic reticulum-mediated Ca2+ leak and
arrhythmogenesis. In fact, NO can also suppress the activation of
CaMKII through the nitrosylation of C273, thus revealing the dual
effects of NO on the activity of CaMKII (16–19).
The phosphorylation of Ser26, a residue located within the ATP
binding site of CaMKIIγ, has been shown to shut off the activity of
CaMKII in vascular tissues, thus terminating its sustained
activation (20). Of note,
methionine pairs are the most recent activators in vertebrate
evolution, whereas S280, T287, C273, Ser26 and C290 are earlier
activators, which also exist in invertebrate organisms. This
indicates that M281/282 has physiological advantages in connection
to the redox signal, but may have potential disadvantages in
connection to excessive ROS caused by ox-CaMKII in several
diseases.
Four isoforms (α, β, γ and δ) of CaMKII have been
identified in Homo sapiens (21). The alternative splicing of these
isoforms results in the further expansion of CaMKII types by
generating splice variants (22).
These enzymes are differentially expressed in tissues. While
CaMKIIα and β are predominantly present in the brain, the γ and δ
isoforms are expressed in various tissues (21,23,24).
The γ isoform has been investigated most in the vasculature
(25–27) and the δ isoform in cardiac tissues
(12,14,28–30).
All isoforms share 89–93% sequence similarity in their catalytic
and autoregulatory domains (21).
All isoforms have autophosphorylation sites, although their
location differs marginally (Thr286 in α and Thr287 in β, γ and δ)
in the core regulatory and CaM-binding domains (Thr305/306 in α and
Thr306/307 in β, γ and δ). Whereas all other isoforms have paired
methionine residues within the self-inhibiting areas of the
regulatory regions, the α isoform has cysteine-methionine residues,
all of which are able to autonomously activate Ca2+/CaM
during oxidation (5). In its
resting state, without Ca2+/CaM binding, CaMKII is
almost inactive, as the self-inhibitory region of the regulatory
and catalytic domains combine and inhibit the release of ATP.
However, M281/282 oxidation transforms the hydrophobic residues
into hydrophilic species, which tend to bind to become involved in
the substrate binding site, even without Ca2+/CaM
binding (6). The following
summarizes the role of ox-CaMKII in inflammatory diseases.
Role of ox-CaMKII in inflammatory
diseases
Ox-CaMKII promotes acute ischemic
stroke
Acute ischemic stroke is the process of rapid loss
of neurological function as a result of insufficient blood flow to
affected brain areas. According to the model of transient cerebral
ischemia, ROS aggravate the severity of stroke, resulting in
neurological disorders (31). It
has been reported that MsrA, an antioxidant enzyme that can reverse
methionine oxidation, decreased the ROS-induced activation of
nuclear factor (NF)-κB in endothelial cells by inhibiting the
oxidation of methionine residues in the regulatory region of CaMKII
(32). CaMKII is the upstream
protein kinase of NF-κB, which has been shown to modulate the
activation of NF-κB in myocardial ischemia/reperfusion injury
(5,12). The treatment of human umbilical
vein endothelial cells (HUVECs) with H2O2
leads to CaMKII Met281/282 oxidation. This means that ROS can lead
to the oxidation and activation of CaMKII. To confirm that the
stimulatory effect of CaMKII Met281/282 oxidation can induce the
activation of NF-κB in endothelial cells, HUVECs were infected with
oxidation-resistant CaMKII tandem mutants with methionine at
Met281/282. The results showed that the overexpression of CaMKII at
Met281/282 almost inhibited the H2O2-induced
activation of NF-κB to inhibit cerebral ischemia/reperfusion
injury. The administration of the CaMKII inhibitor KN-93 also
protected MsrA−/− mice from exacerbated neurological
deficits following cerebral ischemia/reperfusion injury. In
conclusion, with the assistance of ROS, ox-CaMKII can cause NF-κB
to aggravate neurovascular inflammation and brain tissue injury in
ischemic stroke (32).
Ox-CaMKII promotes cardiovascular
disease
The sources of ROS within cardiac myocytes include
mitochondria, NADPH oxidase, xanthine oxidase and uncoupled nitric
oxide synthases (33). The
production of ROS increases in myocardial infarction (MI) and heart
failure (34). Enhanced oxidative
stress leads to NF-κB-mediated cytokine release and contributes to
inflammation (35). It also causes
myocardial contractile dysfunction and remodeling, leading to heart
failure (36). Ox-CaMKII levels
are also elevated in conditions of increased oxidative stress,
including MI (5,12,14),
arrhythmia (11,14,37,38),
vascular smooth muscle migration and cell apoptosis (14,39,40),
and acute myocardial ischemia (41). The inhibition of ox-CaMKII blunts
the expression of inflammatory genes (6).
The expression of ox-CaMKII was found to be enhanced
following Ang II exposure in a model of MI, in addition to
myocardial death, dysfunction and unfavorable left ventricular
function (5). The level of
ox-CaMKII is particularly enhanced during the initial 24 h
following MI, depending on the generation of ROS by regulating
innate immune adaptors, including Toll-like receptor 4 and myeloid
differentiation factor-88 (MyD88) (12). However, MyD88-deficient mice
exhibited reduced expression of ox-CaMKII, myocardial hypertrophy
and increased mortality due to MI, and reduced transcription levels
(12). Furthermore, infusion
aldosterone enhances myocardial ROS and ox-CaMKII and leads to MI,
and ox-CaMKII and matrix metalloproteinase 9 mediate the activation
of myocyte enhancer factor 2, which causes myocardial rupture
(14). These results demonstrate
that the expression of ox-CaMKII is enhanced following Ang II and
aldosterone injections, and induces MI.
Ox-CaMKII is a vital modulator of ROS cardiac
glycosides, which can trigger an arrhythmogenic response to myocyte
Ca2+ signaling. It has been reported that digitoxin
(DGT) treatment enhances the expression of ox-CaMKII in wide-type
(WT) mice (11). Accordingly, an
increasing expression of ox-CaMKII is observed with the
phosphorylation of RyR2 at Ser2814. Of note, the arrhythmogenic
responses of DGT are significantly reduced in myocytes from mice
without this site (RyR2-S2814A mice), suggesting that ox-CaMKII
serves a vital role in RyR2 CaMKII phosphorylation in
arrhythmogenic responses of DGT. Sinoatrial node (SAN) dysfunction
causes bradycardia and leads to damaged conduction speed or
conduction block. Hypertension and heart failure can induce SAN
dysfunction accompanied by increased myocardial ROS. Ox-CaMKII has
been shown to be increased in the SAN myocardium by performing
patient gene detection, indicating that ox-CaMKII can serve as a
potential marker for those with heart failure and SAN dysfunction
(14). Furthermore, ox-CaMKII is
enhanced in patients with atrial fibrillation (AF), compared with
those with a sinus rhythm, which is consistent with the results
obtained from mice injected with Ang II. Knock-in mice lacking the
critical oxidation M281/282 sites in CaMKIIδ (MM-VV) showed reduced
AF following Ang II injection (37,38).
Vascular smooth muscle (VSMC) proliferation and
migration are important in the response to vascular injury.
Previous reports have shown ox-CaMKII to be critical to VSMC
proliferation and migration, as MM-VVδ mice lacked this vascular
remodeling response (14,40). Preventing the oxidative activation
of CaMKII decreased VSMC migration and apoptosis. Furthermore, the
mRNA levels of CaMKIIγ and -δ were clearly enhanced in MM-VVδ mice,
when compared with WT VSMCs, suggesting that the expression of
CaMKII is regulated by ROS (14,39).
These findings show that ox-CaMKII is involved in the response of
damaged vascular smooth muscle and suggests that ox-CaMKII is able
to supply the feedback signaling pathway to balance the
intercellular redox response.
Ox-CaMKII is also critical in the development of
acute myocardial ischemia/reperfusion injury. Ox-CaMKIIδ is
downregulated at the end of reperfusion compared with levels in
ischemia. The NADPH oxidase 2-ox-CAMKIIδ signaling pathway is
unlikely to be involved in the cardioprotective obstruction of Ang
II receptor type 1 activation, which can be inhibited with CaMKII
(41).
Ox-CaMKII promotes diabetes
mellitus
Diabetes mellitus is a major public health concern
that influences >8% of the US population (42,43).
MI is one of the most important causes of mortality in patients
with diabetes, with a study reporting that MI-associated mortality
rates in patients with diabetes are twice those of patients without
(42). It has been reported that
diabetic patients with MI exhibit higher ox-CaMKII levels in their
right atrium, compared with non-diabetic patients with MI. In
models of diabetic mice with MI, increased SAN cell death and
fibrosis were present, which was prevented by CaMKII inhibition and
the oxidation-resistant CaMKII mutant (MM-VV), highlighting the
importance of ox-CaMKII as a cause of higher mortality rates in
diabetic patients with MI (8). The
elevated expression of ox-CaMKII in diabetes is due to increased
mitochondrial superoxide production and hyperglycemia-induced ROS
production (8). In MM-VV and WT
mice treated with streptozotocin and injected with ROS-targeted
inhibitor, the expression of ox-CaMKII was decreased, the survival
rate of pacemaker cells was enhanced, no significant difference was
observed in the heart rate, and diabetes-attributable mortality was
reduced following MI (8),
suggesting that the ROS-ox-CaMKII signaling pathway can induce an
increase in sudden mortality in diabetic patients following MI.
Ox-CaMKII promotes cancer
The overactivation of CaMKII has been shown to
promote several types of cancer. CAMKII serves an important role in
cancer cell proliferation, differentiation and survival, and has
been implicated in various types of cancer, including lung
(44), breast (45,46),
prostate (47) and colon cancer
(48,49). Cancer cells favor glycolysis in
low-oxygen environments to ensure survival, a phenomenon commonly
known as the Warburg effect (50).
Evidence shows that CaMKII is a key ROS sensor and that ox-CaMKII
facilitates the activation of AMPK phosphorylation at Thr172
(51). The use of a CaMKII
inhibitor resulted in the dephosphorylation of AMPK, decreased
glycolysis and reduced steady-state ATP levels. It also markedly
reduced the survivability of cells with a high expression of MnSOD
(51). A comparison between KN-93,
a CaMKII inhibitor and chemotherapeutic agent, paclitaxel and
fluorouracil in aggressive breast cancer revealed a greater
efficacy of KN-93 (51).
Ox-CaMKII promotes asthma
Asthma is a chronic airway inflammatory disease,
typically featuring airway hyperreactivity and reversible airflow
obstruction, influx and activation of inflammatory cells, and
release of inflammatory cytokines ultimately leading to airway
remodeling (52,53). It is a major public health burden
and is now considered one of the most common chronic disorders
worldwide (54). The total number
of individuals with asthma may be as high as 334,000,000. Asthma
affects ~8.5% of the total US population and, in 2007, the
estimated cost related to medical expenses and lost work due to
asthma was $56,000,000,000 annually (55). High-oxygen environments, large
airway surface area and rich blood predispose lungs to oxidative
stress. The two primary sources of ROS in the lungs are
environmental, including gaseous and particulate air pollution, and
cellular. The main sources of cellular ROS are mitochondrial
respiration, NADPH oxidase and the xanthine/xanthine oxidase system
in epithelial and inflammatory cells (52). Increased levels of ROS have been
detected in the gases exhaled by patients with asthma, compared
with healthy controls (56). ROS
induce goblet cell proliferation and metaplasia, and mucus
hypersecretion by increasing the gene and protein expression of
MUC5AC (57). They are also
implicated in eosinophil recruitment and airway hyperresponsiveness
(58). However, the exact
molecular mechanisms linking the ability of ROS to cause several
phenotypic features of asthma remain to be fully elucidated. The
role of ox-CaMKII has been widely examined in heart diseases, and
the presence of both increased ROS and ox-CaMKII levels in the
diseased myocardia has prompted investigations med at identifying a
potential link between the two in airways.
It is well-known that the level of ROS is elevated
in asthma. CaMKII is a critical ROS sensor. A study showed that
smooth muscle CaMKIIδ facilitates allergen-induced airway
hyperresponsiveness and inflammation (59). However, detection of ox-CaMKII in
the bronchial epithelium and smooth muscle revealed that it mainly
exists in the epithelium rather than in the smooth muscle, and that
ox-CaMKII is enhanced in patients with severe asthma compared to
total CaMKII (9). Increased
ox-CaMKII levels were also detected in ovalbumin (OVA)-treated mice
compared with vehicle-treated mice; there was no marked difference
in total CaMKII. In addition, the expression of ox-CaMKII was found
to be elevated further in patients with severe asthma compared with
either patients with mild asthma or healthy individuals, suggesting
that ox-CaMKII can be used to not only diagnose asthma, but also
determine its severity (9). The
P47 gene encodes NADPH oxidase, which is required for the
production of ROS (60).
P47−/− OVA mice exhibited a lower expression of ROS and
ox-CaMKII compared with WT OVA mice. These findings indicated the
crucial role of P47-induced NADPH oxidase in ROS generation and the
production of ox-CaMKII. Compared with WT OVA mice,
P47−/− mice exhibited a significant decline in goblet
cell proliferation, bronchial epithelium thickness, more positive
MUC5AC airway cells, mRNA expression of MUC5AC, airway eosinophilia
and one of the eosinophil chemoattractant molecules (Ccl-11), when
compared with WT OVA mice. These changes were similar to those
observed following corticosteroid use (61). These results indicated that NADPH
oxidase-derived ROS serve a vital role in the phenotypic features
of asthma and CaMKII oxidation. The antioxidant enzyme MsrA reduces
CaMKII by reversing the paired oxidation of Met281/282 (32,62).
Compared with WT OVA mice, MsrA−/− OVA mice exhibited a
marked enhancement in goblet cell proliferation, bronchial
epithelium thickness, positive MUC5AC airway cells, mRNA expression
of MUC5AC, airway eosinophilia and Ccl-11, when compared with WT
OVA mice (9). NF-κB is a major
modulator of asthma progression, inflammatory signaling and
expression of MUC5AC (63). OVA
mice expressing NF-κB reporter exhibited a notable increase in
NF-κB levels compared with saline-treated mice. However, the
genetic manipulation of pathways for ROS generation and CaMKII
oxidation affected the levels of NF-κB. MsrA−/−
exhibited an increase in NF-κB, indicating that the effects of
NF-κB signaling pathways lie downstream of ROS and ox-CaMKII in the
pathogenesis of asthma (9).
The γ-aminobutyric acid type A (GABA) receptors
present in the pulmonary epithelial cells exhibit a markedly
increased expression when sensitized and challenged with OVA.
Chloride currents (ICl) activity in the bronchial
epithelium is mostly due to these GABA receptors; an increase in
ICl activity was observed in OVA mice compared with that
in the tracheal epithelial cells of saline-treated mice. The
addition of GABA receptor antagonist picrotoxin and CaMKII
inhibitor KN-93 (8) significantly
reduced ICl activity in OVA mice but did not further
reduce activity if both were introduced in the mice simultaneously,
suggesting the existence of a pathway for ICl-mediated
goblet cell hyperplasia. ICl activity was also enhanced
considerably more in cells overexpressing ox-CaMKII than in cells
expressing MM-VV CaMKII mutant or those expressing CaMKII when
H2O2 was added. OVA-induced ICl
activity was decreased in P47−/− OVA mice. This
indicates the importance of ox-CaMKII derived from NADPH
oxidase-derived ROS in ICl activity. WT OVA mice
exhibited a marked enhancement in airway hyper-reactivity compared
with saline-treated controls. Transgenic CaMKII with inhibitory
peptide in OVA epi-AC3-I mice reduced goblet cell proliferation and
bronchial epithelium thickness, and increased the mRNA expression
of MUC5AC, airway eosinophilia, airway hyperreactivity and
ICl activity, compared with the same features in WT OVA
mice (9).
Ox-CaMKII promotes asthma through the
activation of mast cells
It has been shown that ox-CaMKII can be used to not
only diagnose asthma, but also determine its severity. In the
present review, the function of ox-CaMKII in asthma and its
underlying mechanism were further examined. For the first time, the
oxidant-resistant CaMKII MM-VV was used to construct asthma using
M281/282 valines (MM-VVδ) in the isotype genetic background of
isoform δ mutation in knock-in mice (64). Cockroach allergen (CRE)-treated
MM-VVδ mice exhibited considerably decreased airway inflammation,
lower airway resistance and lower concentrations of Th2 cytokines,
compared with CRE-treated WT mice. The role of ROS and/or ox-CaMKII
in the activation of mast cells remains to be elucidated. It was
demonstrated that ox-CaMKII modulates the migration and activation
of mast cells in the lung tissues of CRE-exposed mice, thus
promoting the occurrence of asthma.
In vitro, the enhanced expression of
mitochondrial ROS was identified in OVA-treated bone marrow mast
cells (BMMCs) compared with that in controls. Similar to
mitochondrial ROS, the expression of intracellular ROS was markedly
decreased in MM-VVδ BMMCs, compared with that in the WT group,
which is consistent with the previous observations of increased ROS
production in OVA-activated mast cells (65). It is noteworthy that MM-VVδ mast
cells exhibited a decline in mast cell degranulation following OVA
stimulation, decreased histamine release, and decreased expression
of leukotriene C4 and interleukin (IL)-13, indicating that the
promotion of mast cell activation by ox-CaMKII relies on
intracellular Ca2+ generation.
KN-93 inhibited OVA-induced asthma in a previous
study (9), therefore, KN-93 was
used to further investigate the mechanism underlying the effect of
CaMKII in the activation of mast cells. The results showed that the
dose-dependent suppression of IgE regulates mast cell granules and
the secretion of IL-13 in BMMCs and human mast cells (HMC-1),
suggesting that reducing the expression of CaMKII protects against
allergies caused by mast cell activation. The present review is the
first time, to the best of our knowledge, that the
CRE-ROS-ox-CaMKII axis has been linked to the modulation of mast
cell activation following the progress of asthma and allergies
(Fig. 1).
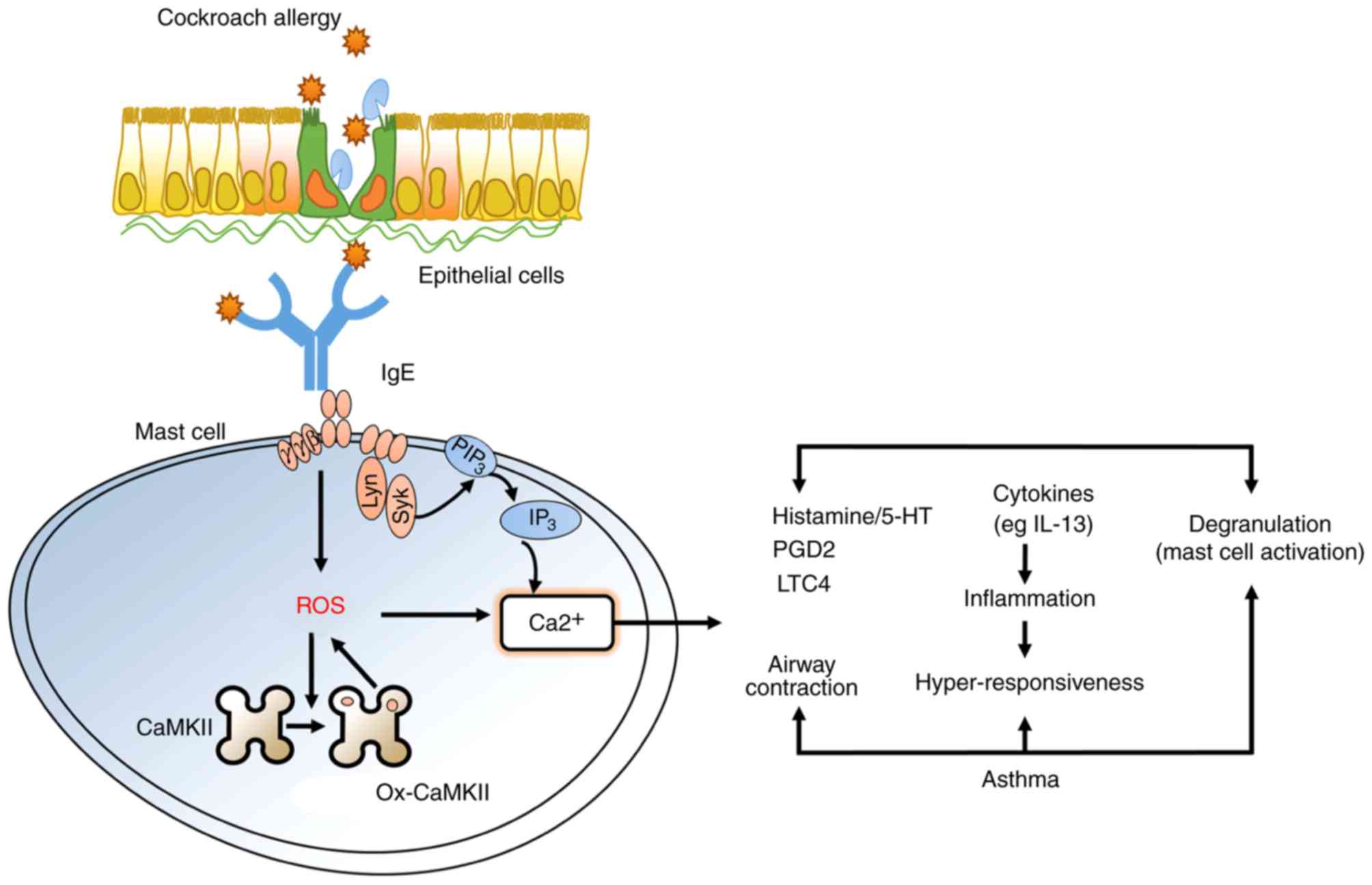 | Figure 1.Ox-CaMKII promotes asthma through the
activation of mast cells. CRE induces the generation of ROS to
modulate the ox-CaMKII activation of mast cells, which is dependent
on intracellular Ca2+ generation. In particular,
ox-CaMKII regulates mast cell degranulation, histamine release, and
the expression of LTC4 and IL-13. Ox-CaMKII, oxidized CaMKII; CRE,
cockroach allergen; ROS, reactive oxygen species; PGD4,
prostaglandin 4; LTC4, leukotriene C4; IL-13, interleukin 13. |
Role of ox-CaMKII in chronic
rhinosinusitis with nasal polyps (CRSwNP)
CRSwNP is an inflammatory sinonasal disease with
Th2-skewed eosinophilic inflammation in mast cells, which is vital
in regulating the environmental antigen-induced pathogenesis of
CRSwNP (66). It is known that
aryl hydrocarbon receptor (AhR), a ligand-induced transcriptional
factor, triggers the production of ROS and the
Ca2+-reliant activation of mast cells (65). AhR can also detect endogenous
tryptophan metabolites produced by kynurenine (KYN) or indoleamine
2,3-dioxygenase and tryptophan 2,3-dioxygenas, which modulate the
activation of mast cells (67,68).
Our previous study examined whether ROS, serving as the upstream of
ox-CaMKII, aggravated CRE-mediated lung inflammation through
activating mast cells (64).
However, the role of ox-CaMKII associated with the KYN/AhR
signaling pathway induced by mast cell activation in CRSwNP remains
unknown. Another study reported that KYN activated AhR signaling in
CRSwNP, and that AhR regulated the generation of ROS and activation
of mast cells. Furthermore, the expression of ox-CaMKII was
considerably increased in the airway epithelial cells and cells
infiltrating the sinonasal mucosa. In addition, ox-CaMKII was shown
to be involved in the KYN/AhR signaling pathway and modulate the
activation of mast cells in CRSwNP. These studies suggested an
important function of the KYN/AhR axis in mediating the activation
of mast cells through ox-CaMKII in the pathogenesis of CRSwNP,
although further elucidation is required (69).
Conclusions
Ox-CaMKII is a vital sensor of ROS in different
diseases, which regulates calcium signals and the ROS signaling
pathway in several subcellular loci. In recent years, ox-CaMKII has
been shown to affect immune regulation, particularly through
regulating the activation of mast cells in asthma and CRSwNP.
However, global antioxidant treatments are mostly unsuccessful. It
is imperative to obtain an understanding of the mechanisms through
which the ROS-modulated signaling pathway leads to the development
of related diseases. Our recent studies and those of others have
supported the hypothesis that CaMKII can mediate an effective
antioxidant treatment for asthma and CRSwNP (64,69).
Studies involving MM-VV mice may assist in assessing the
therapeutic potential of ox-CaMKII.
Acknowledgements
Not applicable.
Funding
This work was supported in part by the National
Youth Science Foundation of China (grant no. 81802278 to QJJ).
Availability of data and materials
Data sharing is not applicable to this article, as
no datasets were generated or analyzed during the current
study.
Authors' contributions
JJQ and RCN wrote the manuscript; JJQ and QHM
collected the references and modified the manuscript; QHM analyzed
the data; RCN designed the manuscript and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hudmon A and Schulman H:
Structure-function of the multifunctional
Ca2+/calmodulin-dependent protein kinase II. Biochem J.
364:593–611. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosenberg OS, Deindl S, Sung RJ, Nairn AC
and Kuriyan J: Structure of the autoinhibited kinase domain of
CaMKII and SAXS analysis of the holoenzyme. Cell. 123:849–860.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Erickson JR, He BJ, Grumbach IM and
Anderson ME: CaMKII in the cardiovascular system: Sensing redox
states. Physiol Rev. 91:889–915. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stratton M, Lee IH, Bhattacharyya M,
Christensen SM, Chao LH, Schulman H, Groves JT and Kuriyan J:
Activation-triggered subunit exchange between CaMKII holoenzymes
facilitates the spread of kinase activity. Elife. 3:e016102013.
View Article : Google Scholar
|
|
5
|
Erickson JR, Joiner ML, Guan X, Kutschke
W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE,
Aykin-Burns N, et al: A dynamic pathway for calcium-independent
activation of CaMKII by methionine oxidation. Cell. 133:462–474.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brookes PS, Yoon Y, Robotham JL, Anders MW
and Sheu SS: Calcium, ATP, and ROS: A mitochondrial love-hate
triangle. Am J Physiol Cell Physiol. 287:C817–C833. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nickel AG, Kohlhaas M, Bertero E, Wilhelm
D, Wagner M, Sequeira V, Kreusser MM, Dewenter M, Kappl R, Hoth M,
et al: CaMKII does not control mitochondrial Ca2+ uptake
in cardiac myocytes. J Physiol. Feb 16–2019.(Epub ahead of print).
doi: 10.1113/JP276766. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Luo M, Guan X, Luczak ED, Lang D, Kutschke
W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, et al:
Diabetes increases mortality after myocardial infarction by
oxidizing CaMKII. J Clin Invest. 123:1262–1274. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sanders PN, Koval OM, Jaffer OA, Prasad
AM, Businga TR, Scott JA, Hayden PJ, Luczak ED, Dickey DD,
Allamargot C, et al: CaMKII is essential for the proasthmatic
effects of oxidation. Sci Transl Med. 5:195ra972013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Viatchenko-Karpinski S Kornyeyev D,
El-Bizri N, Budas G, Fan P, Jiang Z, Yang J, Anderson ME, Shryock
JC, Chang CP, et al: Intracellular Na+ overload causes
oxidation of CaMKII and leads to Ca2+ mishandling in
isolated ventricular myocytes. J Mol Cell Cardiol. 76:247–256.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ho HT, Liu B, Snyder JS, Lou Q, Brundage
EA, Velez-Cortes F, Wang H, Ziolo MT, Anderson ME, Sen CK, et al:
Ryanodine receptor phosphorylation by oxidized CaMKII contributes
to the cardiotoxic effects of cardiac glycosides. Cardiovasc Res.
101:165–174. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Singh MV, Swaminathan PD, Luczak ED,
Kutschke W, Weiss RM and Anderson ME: MyD88 mediated inflammatory
signaling leads to CaMKII oxidation, cardiac hypertrophy and death
after myocardial infarction. J Mol Cell Cardiol. 52:1135–1144.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Swaminathan PD, Purohit A, Soni S, Voigt
N, Singh MV, Glukhov AV, Gao Z, He BJ, Luczak ED, Joiner ML, et al:
Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J
Clin Invest. 121:3277–3288. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
He BJ, Joiner ML, Singh MV, Luczak ED,
Swaminathan PD, Koval OM, Kutschke W, Allamargot C, Yang J, Guan X,
et al: Oxidation of CaMKII determines the cardiotoxic effects of
aldosterone. Nat Med. 17:1610–1618. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Erickson JR, Pereira L, Wang L, Han G,
Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM and
Bers DM: Diabetic hyperglycaemia activates CaMKII and arrhythmias
by O-linked glycosylation. Nature. 502:372–376. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gutierrez DA, Fernandez-Tenorio M,
Ogrodnik J and Niggli E: NO-dependent CaMKII activation during
β-adrenergic stimulation of cardiac muscle. Cardiovasc Res.
100(392-401): 12013
|
|
17
|
Coultrap SJ and Bayer KU: Nitric oxide
induces Ca2+-independent activity of the
Ca2+/calmodulin-dependent protein kinase II (CaMKII). J
Biol Chem. 289:19458–19465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Curran J, Tang L, Roof SR, Velmurugan S,
Millard A, Shonts S, Wang H, Santiago D, Ahmad U, Perryman M, et
al: Nitric oxide-dependent activation of CaMKII increases diastolic
sarcoplasmic reticulum calcium release in cardiac myocytes in
response to adrenergic stimulation. PLoS One. 9:e874952014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Erickson JR, Nichols CB, Uchinoumi H,
Stein ML, Bossuyt J and Bers DM: S-Nitrosylation induces both
autonomous activation and inhibition of
Calcium/Calmodulin-dependent protein kinase II δ. J Biol Chem.
290:25646–25656. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yilmaz M, Gangopadhyay SS, Leavis P,
Grabarek Z and Morgan KG: Phosphorylation at Ser26 in
the ATP-binding site of Ca2+/calmodulin-dependent kinase
II as a mechanism for switching off the kinase activity. Biosci
Rep. 33:e000242013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tobimatsu T and Fujisawa H:
Tissue-specific expression of four types of rat
calmodulin-dependent protein kinase II mRNAs. J Biol Chem.
264:17907–17912. 1989.PubMed/NCBI
|
|
22
|
Tombes RM and Krystal GW: Identification
of novel human tumor cell-specific CaMK-II variants. Biochim
Biophys Acta. 1355:281–292. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takaishi T, Saito N and Tanaka C: Evidence
for distinct neuronal localization of gamma and delta subunits of
Ca2+/calmodulin-dependent protein kinase II in the rat
brain. J Neurochem. 58:1971–1974. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bayer KU, Löhler J, Schulman H and Harbers
K: Developmental expression of the CaM kinase II isoforms:
Ubiquitous gamma- and delta-CaM kinase II are the early isoforms
and most abundant in the developing nervous system. Brain Res Mol
Brain Res. 70:147–154. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim I, Je HD, Gallant C, Zhan Q, Riper DV,
Badwey JA, Singer HA and Morgan KG:
Ca2+-calmodulin-dependent protein kinase II-dependent
activation of contractility in ferret aorta. J Physiol.
526:367–374. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gangopadhyay SS, Barber AL, Gallant C,
Grabarek Z, Smith JL and Morgan KG: Differential functional
properties of calmodulin-dependent protein kinase IIgamma variants
isolated from smooth muscle. Biochem J. 372:347–357. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Marganski WA, Gangopadhyay SS, Je HD,
Gallant C and Morgan KG: Targeting of a novel
Ca+2/calmodulin-dependent protein kinase II is essential
for extracellular signal-regulated kinase-mediated signaling in
differentiated smooth muscle cells. Circ Res. 97:541–549. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guo T, Zhang T, Ginsburg KS, Mishra S,
Brown JH and Bers DM: CaMKIIδC slows Ca]i decline in cardiac
myocytes by promoting Ca sparks. Biophys J. 102:2461–2470. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mishra S, Ling H, Grimm M, Zhang T, Bers
DM and Brown JH: Cardiac hypertrophy and heart failure development
through Gq and CaM kinase II signaling. J Cardiovasc Pharmacol.
56:598–603. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Singh MV, Kapoun A, Higgins L, Kutschke W,
Thurman JM, Zhang R, Singh M, Yang J, Guan X, Lowe JS, et al:
Ca2+/calmodulin-dependent kinase II triggers cell
membrane injury by inducing complement factor B gene expression in
the mouse heart. J Clin Invest. 119:986–996. 2009.PubMed/NCBI
|
|
31
|
Crack PJ, Taylor JM, Ali U, Mansell A and
Hertzog PJ: Potential contribution of NF-kappaB in neuronal cell
death in the glutathione peroxidase-1 knockout mouse in response to
ischemia-reperfusion injury. Stroke. 37:1533–1538. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gu SX, Blokhin IO, Wilson KM, Dhanesha N,
Doddapattar P, Grumbach IM, Chauhan AK and Lentz SR: Protein
methionine oxidation augments reperfusion injury in acute ischemic
stroke. JCI Insight. 1(pii): e864602016.PubMed/NCBI
|
|
33
|
Kimura W, Muralidhar S, Canseco DC, Puente
B, Zhang CC, Xiao F, Abderrahman YH and Sadek HA: Redox signaling
in cardiac renewal. Antioxid Redox Signal. 21:1660–1673. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fraccarollo D, Galuppo P, Neuser J,
Bauersachs J and Widder JD: Pentaerythritol tetranitrate targeting
myocardial reactive oxygen species production improves left
ventricular remodeling and function in rats with ischemic heart
failure. Hypertension. 66:978–987. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Frantz S, Brandes RP, Hu K, Rammelt K,
Wolf J, Scheuermann H, Ertl G and Bauersachs J: Left ventricular
remodeling after myocardial infarction in mice with targeted
deletion of the NADPH oxidase subunit gp91PHOX. Basic Res Cardiol.
101:127–132. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Murdoch CE, Zhang M, Cave AC and Shah AM:
NADPH oxidase-dependent redox signalling in cardiac hypertrophy,
remodelling and failure. Cardiovasc Res. 71:208–215. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Purohit A, Rokita AG, Guan X, Chen B,
Koval OM, Voigt N, Neef S, Sowa T, Gao Z, Luczak ED, et al:
Oxidized Ca(2+)/calmodulin-dependent protein kinase II triggers
atrial fibrillation. Circulation. 128:1748–1757. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wagner S, Ruff HM, Weber SL, Bellmann S,
Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J, et al:
Reactive oxygen species-activated Ca/calmodulin kinase IIδ is
required for late I(Na) augmentation leading to cellular Na and Ca
overload. Circ Res. 108:555–565. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhu LJ, Klutho PJ, Scott JA, Xie L, Luczak
ED, Dibbern ME, Prasad AM, Jaffer OA, Venema AN, Nguyen EK, et al:
Oxidative activation of the Ca(2+)/calmodulin-dependent protein
kinase II (CaMKII) regulates vascular smooth muscle migration and
apoptosis. Vascul Pharmacol. 60:75–83. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Scott JA, Xie L, Li H, Li W, He JB,
Sanders PN, Carter AB, Backs J, Anderson ME and Grumbach IM: The
multifunctional Ca2+/calmodulin-dependent kinase II
regulates vascular smooth muscle migration through matrix
metalloproteinase 9. Am J Physiol Heart Circ Physiol.
302:H1953–H1964. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rajtik T, Carnicka S, Szobi A, Giricz Z,
O-Uchi J, Hassova V, Svec P, Ferdinandy P, Ravingerova T and
Adameova A: Oxidative activation of CaMKIIδ in acute myocardial
ischemia/reperfusion injury: A role of angiotensin AT1
receptor-NOX2 signaling axis. Eur J Pharmacol. 771:114–122. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Knowler WC, Barrett-Connor E, Fowler SE,
Hamman RF, Lachin JM, Walker EA and Nathan DM: Reduction in the
incidence of type 2 diabetes with lifestyle intervention or
met-formin. N Engl J Med. 346:393–403. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Donahoe SM, Stewart GC, McCabe CH,
Mohanavelu S, Murphy SA, Cannon CP and Antman EM: Diabetes and
mortality following acute coronary syndromes. JAMA. 298:765–775.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chai S, Qian Y, Tang J, Liang Z, Zhang M,
Si J, Li X, Huang W, Xu R and Wang K: Retracted:
Ca(2+)/calmodulin-dependent protein kinase IIγ, a critical mediator
of the NF-κB network, is a novel therapeutic target in non-small
cell lung cancer. Cancer Lett. 344:119–128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Britschgi A, Bill A, Brinkhaus H, Rothwell
C, Clay I, Duss S, Rebhan M, Raman P, Guy CT, Wetzel K, et al:
Calcium-activated chloride channel ANO1 promotes breast cancer
progression by activating EGFR and CAMK signaling. Proc Natl Acad
Sci USA. 110:E1026–E1034. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kim JH, Kim TW and Kim SJ: Downregulation
of ARFGEF1 and CAMK2B by promoter hypermethylation in breast cancer
cells. BMB Rep. 44:523–528. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang T, Guo S, Liu Z, Wu L, Li M, Yang J,
Chen R, Liu X, Xu H, Cai S, et al: CAMK2N1 inhibits prostate cancer
progression through androgen receptor-dependent signaling.
Oncotarget. 5:10293–10306. 2014.PubMed/NCBI
|
|
48
|
Wang C, Li N, Liu X, Zheng Y and Cao X: A
novel endogenous human CaMKII inhibitory protein suppresses tumor
growth by inducing cell cycle arrest via p27 stabilization. J Biol
Chem. 283:11565–11574. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jing Z, Sui X, Yao J, Xie J, Jiang L, Zhou
Y, Pan H and Han W: SKF-96365 activates cytoprotective autophagy to
delay apoptosis in colorectal cancer cells through inhibition of
the calcium/CaMKIIγ/AKT-mediated pathway. Cancer Lett. 372:226–238.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bhat PJ, Darunte L, Kareenhalli V,
Dandekar J and Kumar A: Can metabolic plasticity be a cause for
cancer? Warburg-Waddington legacy revisited. Clin Epigenetics.
2:113–122. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hart PC, Mao M, de Abreu AL,
Ansenberger-Fricano K, Ekoue DN, Ganini D, Kajdacsy-Balla A,
Diamond AM, Minshall RD, Consolaro ME, et al: MnSOD upregulation
sustains the Warburg effect via mitochondrial ROS and
AMPK-dependent signalling in cancer. Nat Commun. 6:60532015.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kirkham P and Rahman I: Oxidative stress
in asthma and COPD: Antioxidants as a therapeutic strategy.
Pharmacol Ther. 111:476–494. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Jaffer OA, Carter AB, Sanders PN, Dibbern
ME, Winters CJ, Murthy S, Ryan AJ, Rokita AG, Prasad AM, Zabner J,
et al: Mitochondrial-targeted antioxidant therapy decreases
transforming growth factor-β-mediated collagen production in a
murine asthma model. Am J Respir Cell Mol Biol. 52:106–115. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Anandan C, Nurmatov U, van Schayck OC and
Sheikh A: Is the prevalence of asthma declining? Systematic review
of epidemiological studies. Allergy. 65:152–167. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lambrecht BN and Hammad H: The immunology
of asthma. Nat Immunol. 16:45–56. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Huang SK, Zhang Q, Qiu Z and Chung KF:
Mechanistic impact of outdoor air pollution on asthma and allergic
diseases. J Thorac Dis. 7:23–33. 2015.PubMed/NCBI
|
|
57
|
Casalino-Matsuda SM, Monzón ME and Forteza
RM: Epidermal growth factor receptor activation by epidermal growth
factor mediates oxidant-induced goblet cell metaplasia in human
airway epithelium. Am J Respir Cell Mol Biol. 34:581–591. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Abdala-Valencia H, Earwood J, Bansal S,
Jansen M, Babcock G, Garvy B, Wills-Karp M and Cook-Mills JM:
Nonhematopoietic NADPH oxidase regulation of lung eosinophilia and
airway hyperresponsiveness in experimentally induced asthma. Am J
Physiol Lung Cell Mol Physiol. 292:L1111–L1125. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Spinelli AM, Liu Y, Sun LY, González-Cobos
JC, Backs J, Trebak M and Singer HA: Smooth muscle CaMKIIδ promotes
allergen-induced airway hyper-responsiveness and inflammation.
Pflugers Arch. 467:2541–2554. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Li JM, Mullen AM, Yun S, Wientjes F,
Brouns GY, Thrasher AJ and Shah AM: Essential role of the NADPH
oxidase subunit p47(phox) in endothelial cell superoxide production
in response to phorbol ester and tumor necrosis factor-alpha. Circ
Res. 90:143–150. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ikeda RK, Nayar J, Cho JY, Miller M,
Rodriguez M, Raz E and Broide DH: Resolution of airway inflammation
following ovalbumin inhalation: Comparison of ISS DNA and
corticosteroids. Am J Respir Cell Mol Biol. 28:655–663. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Anderson ME: Oxidant stress promotes
disease by activating CaMKII. J Mol Cell Cardiol. 89:160–167. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Fujisawa T, Velichko S, Thai P, Hung LY,
Huang F and Wu R: Regulation of airway MUC5AC expression by
IL-1beta and IL-17A; the NF-kappaB paradigm. J Immunol.
183:6236–6243. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Qu J, Do DC, Zhou Y, Luczak E, Mitzner W,
Anderson ME and Gao P: Oxidized CaMKII promotes asthma through the
activation of mast cells. JCI Insight. 2:e901392017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhou Y, Tung HY, Tsai YM, Hsu SC, Chang
HW, Kawasaki H, Tseng HC, Plunkett B, Gao P, Hung CH, et al: Aryl
hydrocarbon receptor controls murine mast cell homeostasis. Blood.
121:3195–3204. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Mahdavinia M, Suh LA, Carter RG, Stevens
WW, Norton JE, Kato A, Tan BK, Kern RC, Conley DB, Chandra R, et
al: Increased noneosinophilic nasal polyps in chronic
rhinosinusitis in US second-generation Asians suggest genetic
regulation of eosinophilia. J Allergy Clin Immunol. 135:576–579.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Totlandsdal AI, Cassee FR, Schwarze P,
Refsnes M and Låg M: Diesel exhaust particles induce CYP1A1 and
pro-inflammatory responses via differential pathways in human
bronchial epithelial cells. Part Fibre Toxicol. 7:412010.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Manners S, Alam R, Schwartz DA and Gorska
MM: A mouse model links asthma susceptibility to prenatal exposure
to diesel exhaust. J Allergy Clin Immunol. 134:63–72. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wang H, Do DC, Liu J, Wang B, Qu J, Ke X,
Luo X, Tang HM, Tang HL, Hu C, et al: Functional role of kynurenine
and aryl hydrocarbon receptor axis in chronic rhinosinusitis with
nasal polyps. J Allergy Clin Immunol. 141:586–600.e6. 2018.
View Article : Google Scholar : PubMed/NCBI
|














