Introduction
Non-alcoholic fatty liver disease (NAFLD) is a
clinicopathological syndrome that is characterized by fatty
deposits in the liver cells; it is caused by genetic, environmental
and metabolic stress-associated factors (1). In China, the incidence of NAFLD is
increasing, and NAFLD is one of the major diseases that seriously
threatens public health (2); NAFLD
is able to cause hepatocellular carcinoma (3,4). The
prevalence of NAFLD is 17–33% in the Americas and the prevalence of
nonalcoholic steatohepatitis (NASH), a type of NAFLD, accounts for
~10–20%. Noticeably, the incidence rate of cirrhosis among patients
with NASH, within 10–15 years, reaches as high as 15–25% (5,6). At
present, it is believed that the core factor leading to NASH is
lipotoxicity (7).
A number of studies have demonstrated that free
fatty acid (FFA) produce a toxic effect, which contributed to liver
cell dysfunction and apoptosis (8–10).
When the level of FFA exceeds the oxidative and metabolic capacity
of the liver, lipids accumulate in the liver and inflammatory
factors are released, subsequently initiating apoptotic signals,
increasing the sensitivity of hepatocytes to inflammatory reactions
and various injury factors. This damage will ultimately lead to
hepatocellular dysfunction or death that is called lipotoxicity
(11,12). In liver cells, excessive amounts of
fatty acids are mainly stored in lipid droplets (LD) (13); therefore, the abnormal metabolism
of LD is closely related to lipotoxicity.
LD are a major storage site for neutral lipids in
cells, and are widely found in bacteria, fungi, plants and animals
(14,15). LD is considered a simple structure
of energy storage; however, it has been reported that LD serves a
role in diseases, including obesity and fatty liver (16). Structurally, LD is composed of
neutral lipid and phospholipid, which contains many LD-related
proteins embedded in the phospholipid monolayer; according to
previous studies, these proteins serve a vital role in maintaining
the stability and metabolic processes of LD (17,18).
Lipid storage droplet protein 5 (LSDP5; also known as myocardial
lipid droplet protein or oxidative tissue-enriched PAT protein) is
mainly distributed in tissues with high oxidative metabolism, such
as liver, heart and skeletal muscle (19–21).
In addition, LSDP5 is a member in PAT family of lipid droplet
proteins (19). A number of
previous studies have reported PAT family proteins serve a
regulatory role in lipid droplet metabolism and in maintaining
lipid balance in cells (22,23).
However, the specific function and mechanism of LSDP5 in
hepatocellular lipid metabolism is still not fully understood.
In the present study, it was hypothesized that LSDP5
may produce a protective effect on the liver cells with
lipotoxicity by regulating lipid metabolism. An in vitro
model of lipotoxicity was established by treating LO2 normal human
liver cells with sodium palmitate. The effects of LSDP5 on lipid
metabolism, oxidative stress, apoptosis and mitochondrial function
in sodium palmitate-induced LO2 cells were examined.
Materials and methods
Cell culture
LO2 normal human liver cells were purchased from
Procell Life Technology Co., Ltd. (Wuhan, China). Cells were
cultured in Dulbecco's modified Eagle's medium (DMEM; Huayueyang
Biology, Beijing, China) that contained 10% foetal bovine serum
(HyClone; GE Healthcare Life Sciences, Logan, UT, USA) and 100 U/ml
streptomycin and 100 µg/ml penicillin (Beijing Solarbio Science
& Technology Co., Ltd., Beijing, China) in a Forma
CO2 incubators (Thermo Fisher Scientific, Inc., Waltham,
MA, USA) with 5% CO2 at 37°C.
Preparation of sodium palmitate
solution
Solid sodium palmitate was purchased from Heowns
Biochemical Technology Co., Ltd. (Tianjin, China). The sodium
palmitate was dissolved in BSA solution, which was subsequently
diluted in DMEM; the final concentrations of sodium palmitate were
25, 50, 75, 100, 125 and 150 µmol/l.
Cell transfection and treatment
The pCMV5-LSDP5 and pCMV5-NC plasmids were obtained
from Genewiz Inc. (Suzhou, China). LO2 cells (1×105 ml)
were transfected with 2 µg of pCMV5-LSDP5 or pCMV5-NC plasmid using
Lipofectamine® 2000 transfection reagent (Thermo Fisher
Scientific, Inc.). The experiments comprised four groups: i)
Control group, which was treated with 0.1% PBS; ii) Model group,
which was treated with 100 µmol/l sodium palmitate; iii) Model +
negative control (M + NC) group, which was treated with 100 µmol/l
sodium palmitate transfected with pCMV5-NC plasmid and; and iv)
Model + LSDP5 group (M + LSDP5), which was transfected with
pCMV5-LSDP5 plasmid and treated with 100 µmol/l sodium palmitate at
37°C for 24 h.
Cell Counting Kit-8 (CCK-8) viability
assay
CCK-8 (Beijing Saichi Biological Technology Co.,
Ltd., Beijing, China) was used to detect cell viability, following
the manufacturer's protocol. Briefly, cells (3.5×103
cells/well) were cultured in 96-well plates in a humidified
incubator with 5% CO2 at 37°C. Following a protocol from
a previous study (24), cells were
treated with sodium palmitate at 25, 50, 75, 100, 125 or 150 µmol/l
and incubated for 12, 24 or 48 h. Following incubation, CCK-8
reagent was added into each well and the cells were incubated in
humidified environment with 5% CO2 at 37°C for 2 h.
Absorbance was measured at 450 nm using a SpectraMax iD3 microplate
reader (Molecular Devices, LLC, Sunnyvale, CA, USA).
Non-esterified fatty acid (NEFA)
assay
The content of NEFA was measured using a NEFA
Detection kit (Wako Pure Chemical Industries, Ltd., Osaka, Japan),
following the manufacturer's protocol. Briefly, cells were treated
with drugs and grouped as aforementioned. Following treatments, 22
µM [9,10-3H]oleate was added to cells for 4 h. Cells
were subsequently washed two times with PBS and cultured in DMEM
for 4 h. The cells were digested with 0.25% EDTA-trypsin (Beijing
Solarbio Science & Technology Co., Ltd.), resuspended in DMEM
and centrifuged with 1,000 × g for 10 min. The supernatant was
transferred into a clean centrifuge tube. Lipid was extracted from
supernatant using N-hexane: Isopropanol solution (3:2) and a thin
layer chromatography silica gel plate (25). The silica gel of containing NEFA
fraction was scraped from the silica gel plate and dissolved in
N-hexane: Isopropanol solution (3:2). Scintillation solution was
added and the 3H-labeled NEFA protein content in the
cells was measured using a L6500 liquid scintillation counter
(Beckman Coulter, Inc., Brea, CA, USA). NEFA protein concentration
was detected using a Bradford protein assay kit (Beyotime Institute
of Biotechnology, Haimen, China).
Cell apoptosis assay
An Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) Apoptosis Detection kit
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was used to
determine the rates of cell apoptosis, following the manufacturer's
protocol. Briefly, cells (5×104 cells/well) were
cultured overnight in 6-well plates in a humidified incubator with
5% CO2 at 37°C. Cells were treated with drugs and
grouped as aforementioned. The cells were washed two times with
cold PBS, centrifuged at 300 × g at 4°C for 5 min and resuspended
in 1X Binding Buffer. Subsequently, the cells were stained with
Annexin V-FITC/PI staining solution in the dark at room temperature
for 10–15 min. Apoptotic cells were detected using an A28999 flow
cytometer (Thermo Fisher Scientific, Inc.) and analyzed with BD
CellQuest™ Pro Software version 1.2; BD Biosciences, San Jose, CA,
USA).
Reactive oxygen species (ROS)
assay
ROS activity was determined with a ROS assay kit
(Beyotime Institute of Biotechnology), following the manufacturer's
protocol. Briefly, cells were treated with drugs and grouped, as
aforementioned. Subsequently, the cells were incubated with
2′,7′-dichlorofluorescein diacetate at 37°C for 25 min. The cells
were washed three times with PBS, and ROS was detected using a flow
cytometer and analyzed by Summit Software V4.3 (Dako; Agilent
Technologies, Inc., Santa Clara, CA, USA).
Mitochondrial membrane potential (MMP)
assay
MMP was measured using a JC-1 kit (Beyotime
Institute of Biotechnology), following the manufacturer's protocol.
Briefly, cells were treated with drugs and grouped, as
aforementioned. Following treatments, cells were digested with
0.25% EDTA-trypsin, re-suspended in DMEM and stained with JC-1
solution at 37°C for 20 min. Subsequently, cells were centrifuged
at 600 × g at 4°C for 3 min. The supernatant was discarded and
cells were washed two times with PBS. MMP was detected by a flow
cytometer with analysis software version 1.2 (BD CellQuest™ Pro
Software; BD Biosciences).
ELISA
Levels of oxidative stress-related factors were
respectively measured using an MDA Elisa kit (cat. no. E-EL-0060c,
Elabscience, Wuhan, China) and an SOD ELISA kit (cat. no. S0109;
Beyotime Institute of Biotechnology), following the protocols of
the manufacturer. Briefly, cells were treated with drugs and
grouped, as aforementioned. Following treatments, cells were
digested with 0.25% EDTA-trypsin, centrifuged at 800 × g for 6 min.
The cells (1×106 ml) were re-suspended in DMEM and added
to wells at 37°C for 24 h. A total of 100 µl biotinylated antibody
(included in kits) was added and the cells were incubated at 37°C
for 30 min. Cells were washed three times with PBS, followed by the
addition of the termination solution into wells. Absorbance was
measured at 450 nm using a microplate reader (SkanIt software V3.1,
Thermo Fisher Scientific, Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Cells were treated with drugs and grouped, as
aforementioned. Total RNA was extracted using TRIzol®
Reagent (Takara Biotechnology Co., Ltd., Dalian, China). RNA (1 µg)
was used to synthesize cDNA using an RT Master Mix kit (Takara
Biotechnology Co., Ltd.) and the following parameters: 85°C for 15
min, followed by 4°C for 10 min. SYBR Premix Taq II kit
(Takara Biotechnology Co., Ltd.) was used for amplifying cDNA, and
the 50 µl reactions were set up as follows: 25 µl SYBR-Green Mix, 1
µl forward/reverse primers (Table
I), 19 µl ddH2O and 4 µl cDNA. PCR thermocycling
conditions were: 85°C for 15 min; followed by 30 cycles at 85°C for
20 sec and 65°C for 45 sec; and 85°C for 20 sec, at 37°C for 2 min.
β-actin was used as a loading control for the normalization of
expression. The formula 2−ΔΔCq was used to calculate
relative mRNA expression levels (26).
 | Table I.Sequences of forward and reverse
primers used for reverse transcription-quantitative polymerase
chain reaction. |
Table I.
Sequences of forward and reverse
primers used for reverse transcription-quantitative polymerase
chain reaction.
| Gene | Primer sequence
(5′→3′) |
|---|
| LSDP5 | F:
GTGATCAGACAGCTCAGGACCCT |
|
| R:
CGATTCACCACATTCTGCTG |
|
Active-caspase-3 | F:
TGCCCAAGTGACTGACATCA |
|
| R:
CATCCCCATTGACTGTGCAG |
| Bax | F:
GACCCGGTGCCTCAGGATGC |
|
| R:
AGGTCAGCTCATCATGCTTG |
| Bcl-2 | F:
GTGGAGGAGCTCTTCAGGGA |
|
| R:
GTCTGTGTCCACGGCGGCAA |
| Cytc | F:
CCAAATCTCCACGGTCTGTTC |
|
| R:
ATCAGGGTATCCTCTCCCCAG |
| Cox IV | F:
CGGCGTGACTACCCCTTG |
|
| R:
TGAGGGATGGGGCCATACA |
| CPT1a | F:
TGTCCAAGTATCTGGCAGTCG |
|
| R:
CATAGCCGTCATCAGCAACC |
| ACC1 | F:
TTTGTTTGGTCGTGACTG |
|
| R:
CCCAGCACTCACATAACC |
| ACC2 | F:
GACGCCCGAGGATCTGAAG |
|
| R:
GGGACAGGGACGTACTGATC |
| Fas | F:
ATGACAGGAGATGGAAGG |
|
| R:
CTGACTTCCACCAGCAGC |
| PPARα | F:
CGGCGTGACTACCCCTTG |
|
| R:
TGAGGGATGGGGCCATACA |
| β-actin | F:
GACTCCTATGTGGGTGACGA |
|
| R:
ACGGTTGGCCTTAGGGTTCA |
Western blot assay
Cells were treatment as aforementioned and
subsequently washed twice with PBS, lysed in
Radioimmunoprecipitation Assay Buffer (high) (Beijing Solarbio
Science & Technology Co., Ltd.) to obtain protein extracts.
Protein concentrations were detected by a Bradford protein assay
kit. Proteins (20 µg/lane) were separated by 10% SDS-PAGE. Proteins
were transferred to polyvinylidene fluoride membranes (Hangzhou
Renomem Technology Co., Ltd., Hangzhou, China). Subsequently, the
membranes were blocked in 5% non-fat milk for 2 h at room
temperature. The membranes were incubated with primary antibodies
against LSDP5 (1:1,000; cat. no. ab222811; Abcam, Cambridge, UK),
active-caspase-3 (1:2,000; cat. no. AF-605-NA; R&D Systems,
Inc., Minneapolis, MN, USA), B-cell lymphoma-2 (Bcl-2; 1:800; cat.
no. AF810; R&D Systems, Inc.), Bcl-2-associated X protein (Bax;
1:1,000; cat. no. AF820; R&D Systems, Inc.,), cytochrome
c (Cytc; 1:1,000; cat. no. MAB897; R&D Systems, Inc.),
Cytc oxidase subunits IV (Cox IV; 1:1,200; cat. no. MAB6980;
R&D Systems, Inc.), carnitine palmitoyltransferase 1a (CPT1a;
1:1,200; cat. no. ab83862; Abcam), acetyl-co A carboxylase1 (ACC1;
1:1,000; cat. no. 4190; Cell Signaling Technology, Inc., Danvers,
MA, USA), ACC2 (1:1,000; cat. no. 8578; Cell Signaling Technology,
Inc.), anti-Fas (1:1,000, cat. no. 3180; Cell Signaling Technology,
Inc.), peroxisome proliferator-activated receptors α (PPARα; 1:100;
cat. no. PP-H0723-00) and β-actin (1:2,000; cat. no. MAB8969) on
the rocking table at 4°C overnight. Subsequently, the membranes
were washed 2–3 times with PBS and 0.05% Tween-20 for 6 min,
followed by incubation with the following horseradish
peroxidase-conjugated secondary antibodies at 37°C for 60 min:
Mouse anti-goat immunoglobulin G (1:7,000; cat. no. 31107;
Invitrogen; Thermo Fisher Scientific, Inc.), anti-Mouse IgG
Secondary Antibody (1:1,000; cat. no. HAF007; R&D Systems,
Inc.) and mouse anti-rabbit (1:7,000; cat. no. 31213; Invitrogen;
Thermo Fisher Scientific, Inc.). The membranes were washed 2–3
times with PBST, and the proteins were visualized using Enhanced
Chemiluminescent Detection Reagent (Taixin Biotechnology, Beijing,
China). β-actin was treated as a loading control for normalization.
The blot density was analyzed by Quantity One software version
4.6.2 (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Data were analysed by SPSS 13.0 software (SPSS,
Inc., Chicago, IL, USA); results were expressed as the mean ±
standard deviation. The differences between groups were analysed
using one-way analysis of variance followed by a Dunnett's
post-test. Each experiment was independently repeated in
triplicate. P<0.05 was considered to indicate a statistically
significant difference.
Results
LSDP5 increases the viability of
sodium palmitate-treated LO2
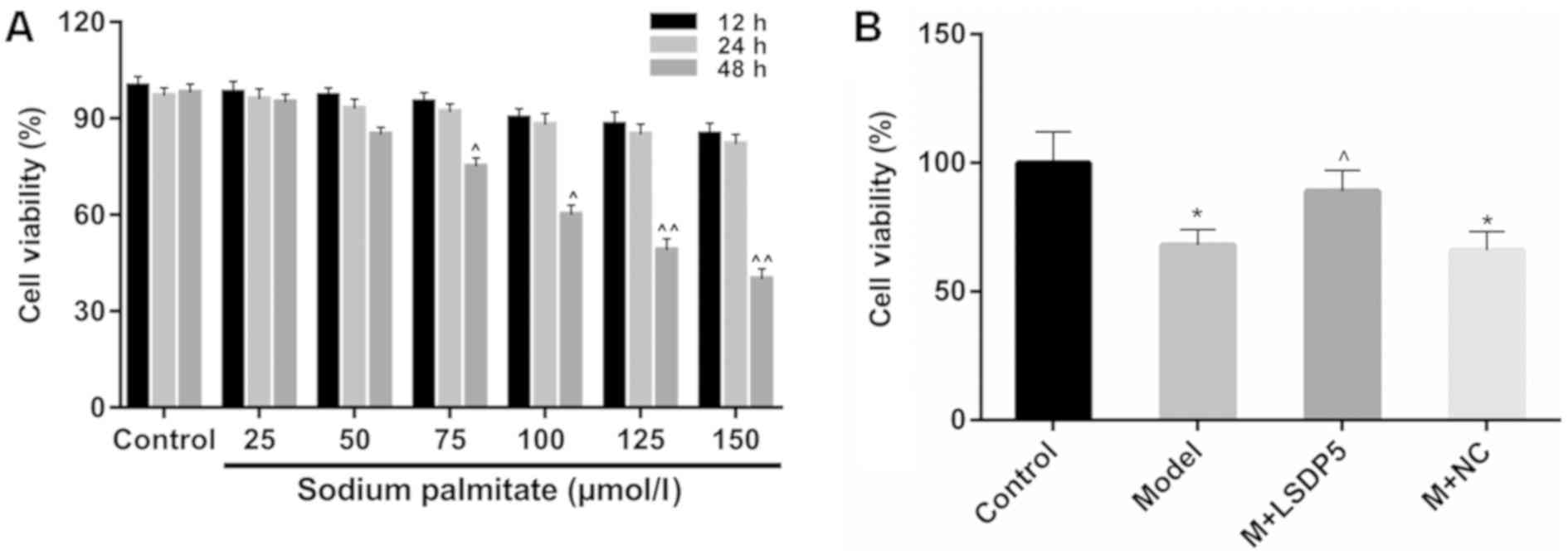
CCK-8 analysis was performed to examine cell
viability to assess the lipotoxic effects of sodium palmitate on
LO2 cells. The data revealed that viability remained relatively
stable when LO2 cells were treated with sodium palmitate at low
doses (25 and 50 µmol/l) for a short period of treatment (12 and 24
h; Fig. 1A). Viability was
significantly inhibited in cells treated with 125 and 150 µmol/l of
sodium palmitate for 48 h, compared with untreated control cells at
the same time (P<0.01; Fig.
1A). Treatment with 100 µmol/l sodium palmitate for 48 h also
significantly inhibited viability, compared with the respected
Control cells (P<0.05; Fig.
1A), and this concentration and incubation time was used in
subsequent experiments as the lipotoxicity Model.
Model cells were transfected with LSDP5
overexpression vectors, which resulted in a significant increase in
viability compared with untransfected Model cells (P<0.05;
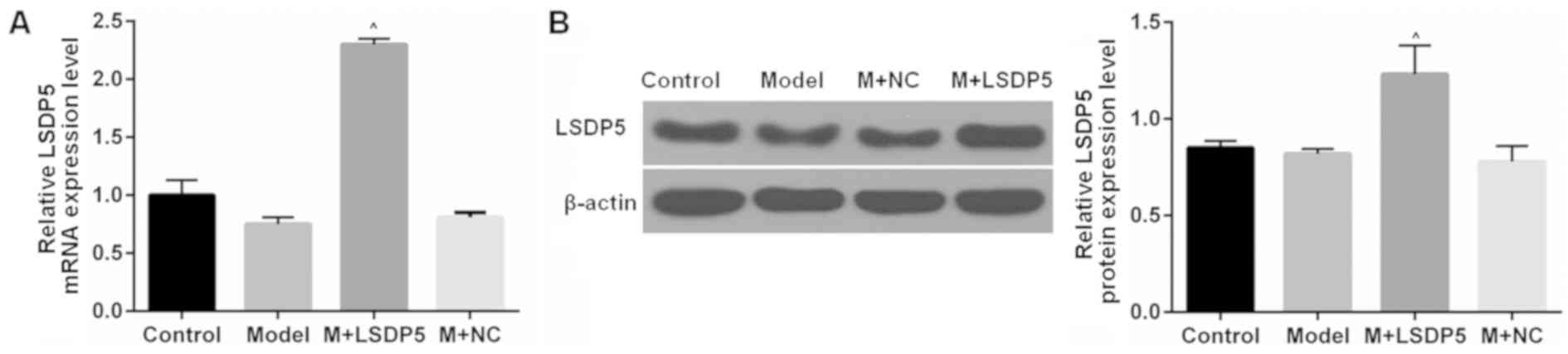
Fig. 1B). RT-qPCR and western blot
assays were conducted to examine the LSDP5 mRNA and protein
expressions levels, respectively, in LO2 Model cells transfected
with pCMV5-LSDP5 plasmid (Fig. 2).
The expression levels of LSDP5 mRNA and protein were significantly
increased in cells transfected with pCMV5-LSDP5 (both P<0.05;
Fig. 2A and B, respectively),
which indicated successful transfection of the LSDP5 overexpression
vector in LO2 cells. Notably, the expression levels of LSDP5 were
slightly reduced in untransfected Model cells, compared with
Control cells (Fig. 2).
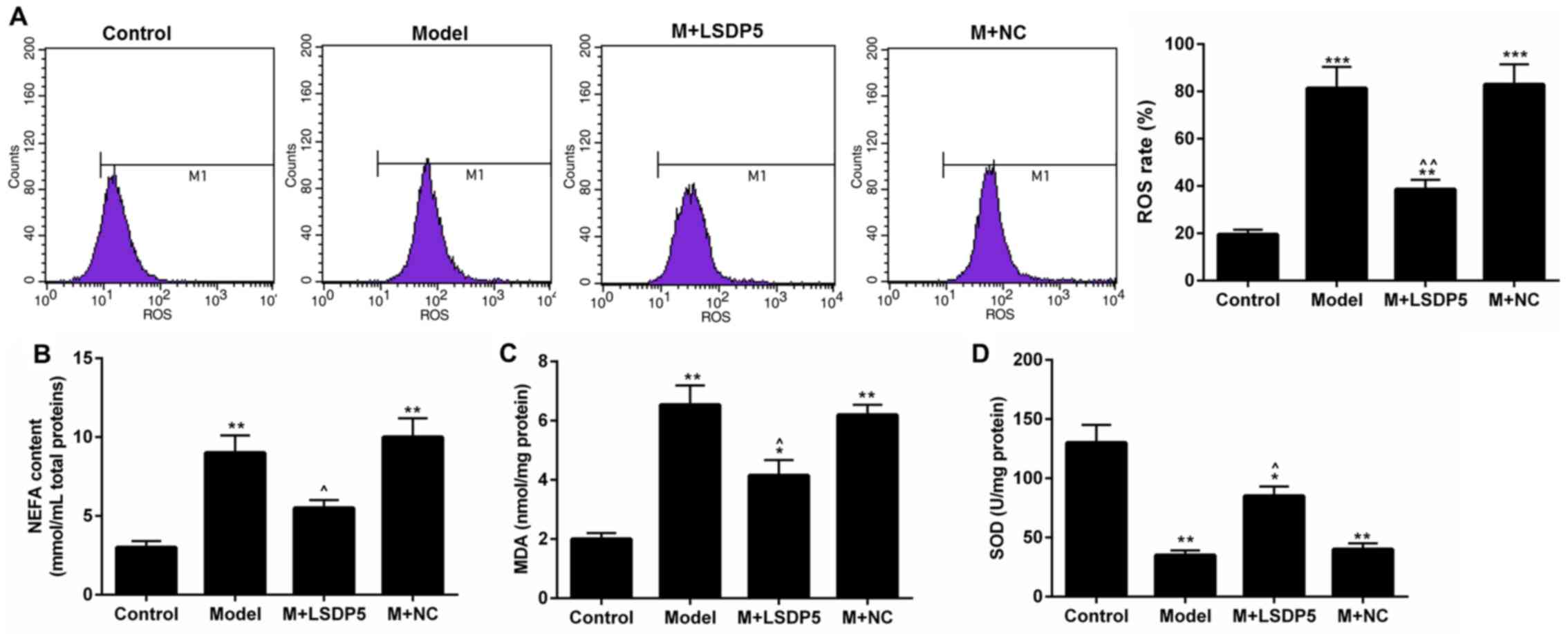
LSDP5 suppresses the activity of
oxidative stress in LO2 lipotoxicity Model cells
Compared with Control cells, Model cells exhibited a
significantly increased rate of ROS production (P<0.001;
Fig. 3A). Although the contents of
NEFA and MDA were increased (P<0.001; Fig. 3B and C, respectively), SOD content
was decreased (P<0.001; Fig.
3D) in Model cells compared with Control. Conversely, LSDP5
overexpression suppressed the oxidative stress of LO2 Model cells;
it was also demonstrated that LSDP5 reduced the ROS level and NEFA
and MDA content, and increased SOD, compared with the untransfected
Model group (P<0.05; Fig.
3).
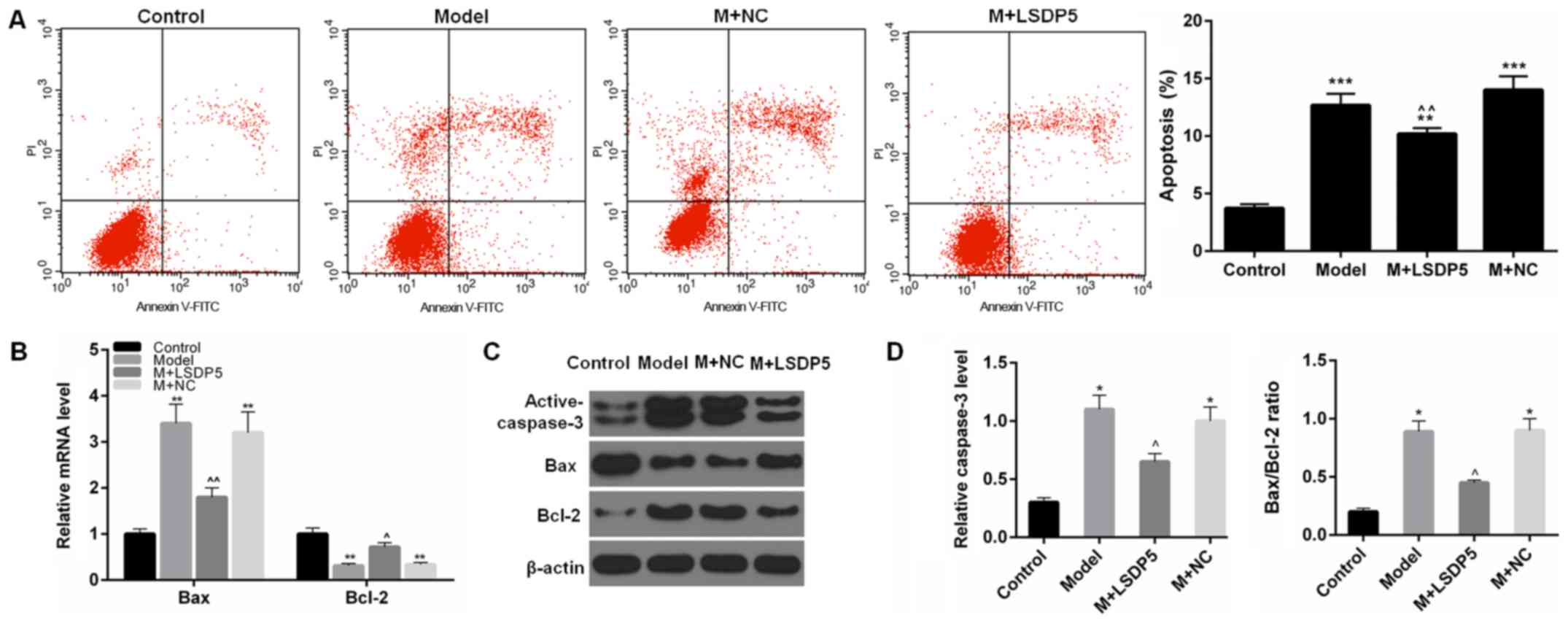
LSDP5 reduces apoptotic rates of LO2
Model cells
The results demonstrated that the rate of apoptosis
was significantly increased (P<0.001; Fig. 4A), and the expression levels of
active-caspase-3 and Bax were also significantly increased in Model
cells compared with Control, whereas Bcl-2 expression levels were
reduced compared with Control (P<0.05 or P<0.01; Fig. 4B-D). In LSDP5-transfected Model
cells, apoptotic rates were significantly decreased, expression
levels of activated-caspase-3 and Bax were reduced, and Bcl-2
expression levels were increased compared with the Model group
(P<0.05 or P<0.01; Fig.
4).
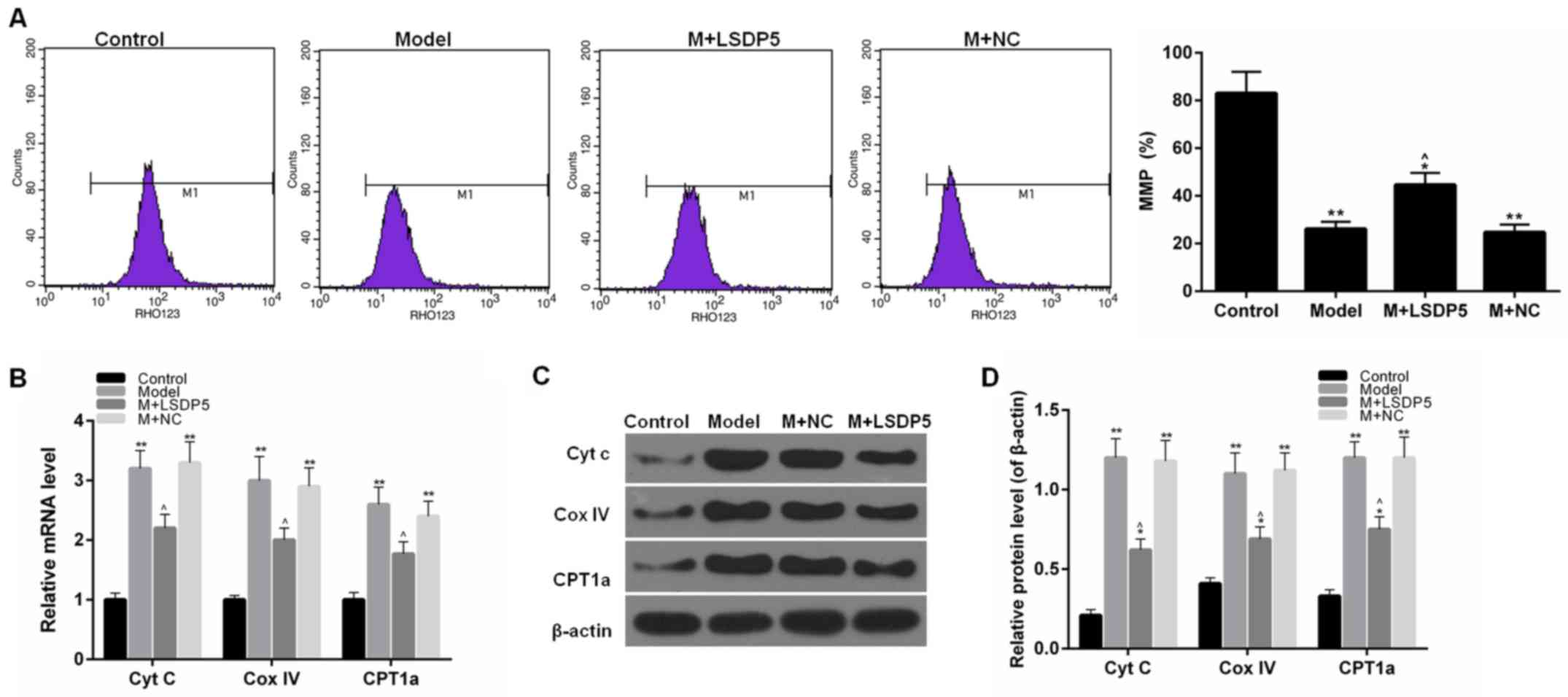
LSDP5 reduces mitochondrial damage in
Model cells
To examine mitochondrial activity, the rate of MMP
and the expression levels of Cytc, Cox IV and CPT1A. The data
indicated that sodium palmitate reduced the MMP rate of LO2 cells
and increased the expression levels of Cytc, Cox IV and CPT1A,
compared with Control. However, LSDP5 overexpression increased the
rate of MMP and reduced Cytc, Cox IV and CPT1A expression levels
compared with the untransfected Model group (P<0.05; Fig. 5).
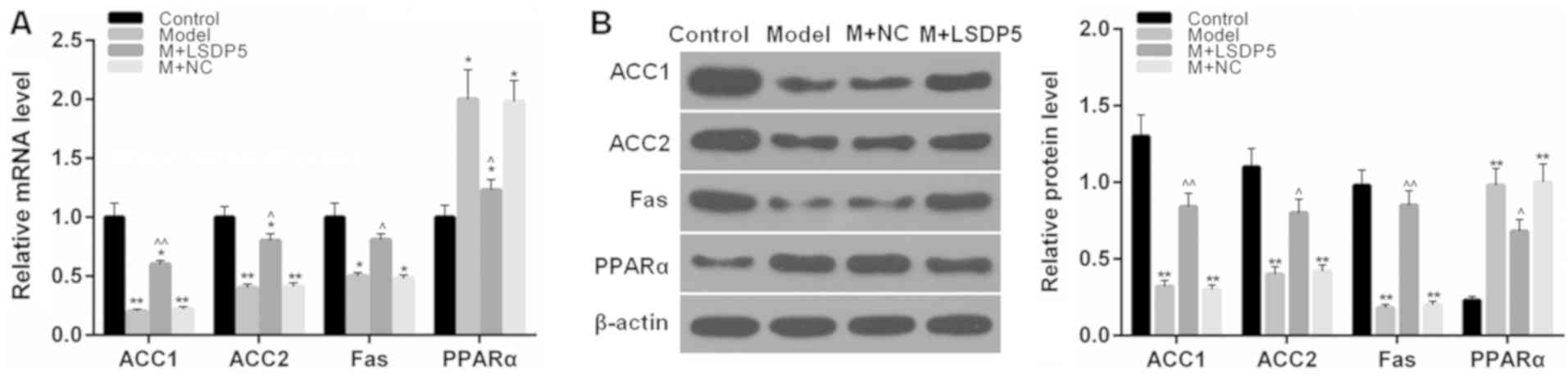
LSDP5 regulates lipid
metabolism-related factors in LO2 Model cells
RT-qPCR and western blotting assays were performed
to determine the mRNA and protein expression levels, respectively,
of lipid metabolism-related factors. The data indicated that the
expression levels of ACC1, ACC2 and Fas were significantly
decreased, whereas PPARα expression levels were increased following
sodium palmitate treatment, compared with Control cells (P<0.05
or P<0.01; Fig. 6). However,
LSDP5 overexpression upregulated the expression levels of ACC1,
ACC2 and Fas, and downregulated PPARα expression levels, compared
with the untransfected Model group (P<0.05 or P<0.01;
Fig. 6).
Discussion
In the clinic, increased FFA levels in patients with
NASH have been associated with the severity of the disease
(27). Previous studies have
demonstrated that sodium palmitate inhibited the viability of cells
and induced lipotoxicity, which led to lipoapoptosis in liver cells
(24,28). In addition, oxidative stress
produced by NEFA oxidation was reported to be the main mechanism of
liver lipotoxicity (29,30). Mitochondrial damage is able to
mediate apoptosis, which serves important roles in the pathogenesis
of NASH (31). Based on these
findings, the present study used sodium palmitate-treated LO2 cells
to establish the lipotoxicity model. In this study, sodium
palmitate was demonstrated to increase the NEFA content, and it
contributed to the accumulation of oxidative stress and apoptosis,
as well as mitochondrial damage in LO2 cells.
LSDP5 is a member of PAT family, which are
specifically expressed in tissues with high oxidative metabolism. A
previous report indicated that the overexpression of LSDP5
increased lipid accumulation, while that siRNA-LSDP5 promoted fatty
acid oxidation in liver cells (32). Though LSDP5 deletion significantly
reduces LD content, it increases ROS content in cardiomyocytes
(33). Therefore, LSDP5 may
produce a certain effect on fatty acid-induced lipotoxicity. LSDP5
was observed to be overexpressed in transfected with pCMV5-LSDP5
and treated with sodium palmitate LO2 cells; LSDP5 overexpression
decreased NEFA content in Model LO2 cells. It was suggested that
overexpression of LSDP5 may inhibit lipolysis and the accompanied
liver injury. A previous study reported that the knockdown of LSDP5
may stimulate lipolysis and increase the level of fatty-acid β
oxidation (32), which may be a
primary cause of liver injury (7).
These data suggested that knockdown of LSDP5 accelerated the
lipolysis and induced liver injury, which was consistent with the
present study results. However, although these previous studies
reported that LSDP5 promoted lipid accumulation, the present study
considered that it was relative to the lipolysis by LSDP5
knockdown. Taken together, the present study results indicated that
LSDP5 reduced the effect of lipotoxicity by regulating lipid
metabolism-related factors.
A previous study reported that ROS may attack
unsaturated fatty acids in a biofilm and enable lipid peroxidation
to form end products such as MDA (34), an important index of cellular
oxidative damage. SOD is also an antioxidant substance (35). The present study results
demonstrated that LSDP5 reduced ROS and MDA content, and increased
SOD content, which indicated that LSDP5 may suppress oxidative
stress in sodium palmitate-treated LO2 cells.
Hepatocellular apoptosis was also reported to serve
a vital role in the development of NASH (36,37).
It is known that apoptosis may cause the change of the apoptotic
protein. Bcl-2 is an indispensable anti-apoptotic gene, and Bax
belongs to the Bcl-2 gene family promoting apoptosis (38). Active-caspase-3 also contributes to
and can stimulate apoptosis (39).
The present data indicated that LSDP5 overexpression inhibited
apoptosis in lipotoxicity Model cells, and upregulated Bcl-2
expression and downregulated the levels of activated-caspase-3 and
Bax. In addition, LSDP5 increased the rate of MMP and increased the
expression levels of CPT1a; however, the expression levels of Cytc
and Cox IV were reduced. These results suggested that LSDP5 may
reduce apoptotic rates and mitochondrial damage in lipotoxicity
Model LO2 cells. The results were in support of a literature that
the number of mitochondria increased in LSDP5-deficient cells
(32).
A previous study reported that exogenous LSDP5
increased the content of lipid in oleic acid-induced COS7 and OP9
liver cells (40). Another study
suggested that LSDP5 may function to regulate the interaction
between lipase and LD, and such a function is similar to perilipin,
which regulated the lipolytic activity of adipocyte triglyceride
lipase (41). In addition, PPARα
is a key regulator of lipid oxidation; LSDP5 is downstream of PPARα
and the upregulation of PPARα may significantly increase LSDP5
expression in hepatocytes (18).
ACC1, ACC2 and Fas are related to fatty acid synthesis (42). Results from the present study
demonstrated that the overexpression of LSDP5 in LO2 Model cells
upregulated the expression levels of ACC1, ACC2 and Fas, and
downregulated PPARα expression levels; these data are consistent
with a previous study, which reported that the PPARα activity was
increased by small interfering (si)RNA-mediated LSDP5 knockdown
(32). In addition, the expression
levels of ACC1 and Fas were not altered by si-LSDP5 in that study.
Considering these findings, it was hypothesized that the
downregulation of ACC1 and Fas may be a stress response when the
cells were stimulated with high concentration of sodium palmitate;
in this condition, the fatty acid synthesis may be inhibited.
However, the present study data were acquired using
only one cell line in vitro, which was a limitation. Thus,
to investigate the role of LADP5 in other cell lines or in
vivo may further confirm the results from this study.
To conclude, sodium palmitate induced lipotoxicity
in LO2 cells. However, overexpression of LSDP5 in lipotoxicity
Model LO2 cells reduced the activity of oxidative stress and
mitochondrial damage, and inhibited apoptosis by regulating lipid
metabolism-related factors. These results demonstrated that LSDP5
may produce a protective effect on sodium palmitate-induced
lipotoxicity in LO2 cells. However, the specific signalling
pathway(s) of LSDP5 regulation of lipid metabolism remains to be
further examined. This study provided a molecular basis for
understanding lipotoxicity in liver, as well as a indicated a
possible candidate target for treating liver lipotoxicity.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The analysed data sets generated during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XM and TZ made substantial contributions to the
design of the present study. Data acquisition and interpretation
was conducted by FC, KY and KJ. TZ and FC critically revised the
manuscript for important intellectual content. All authors approved
the final version of the manuscript. KY and KJ aggree to be
accountable for all aspects of the work in ensuring that questions
related to the accuracy or integrity of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zheng X, Gong L, Luo R, Chen H, Peng B,
Ren W and Wang Y: Serum uric acid and non-alcoholic fatty liver
disease in non-obesity Chinese adults. Lipids Health Dis.
16:2022017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fan JG and Farrell GC: Epidemiology of
non-alcoholic fatty liver disease in China. J Hepatol. 50:204–210.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Angulo P: GI epidemiology: Nonalcoholic
fatty liver disease. Aliment Pharmacol Ther. 25:883–889. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
de Alwis NM and Day CP: Non-alcoholic
fatty liver disease: The mist gradually clears. J Hepatol. 48
(Suppl 1):S104–S112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fan JG, Saibara T, Chitturi S, Kim BI,
Sung JJ and Chutaputti A; Asia-Pacific Working Party for NAFLD, :
What are the risk factors and settings for non-alcoholic fatty
liver disease in Asia-Pacific? J Gastroenterol Hepatol. 22:794–800.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Farrell GC and Larter CZ: Nonalcoholic
fatty liver disease: From steatosis to cirrhosis. Hepatology. 43
(Suppl 1):S99–S112. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Garbarino J and Sturley SL: Saturated with
fat: New perspectives on lipotoxicity. Curr Opin Clin Nutr Metab
Care. 12:110–116. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weiss TS, Lupke M, Ibrahim S, Buechler C,
Lorenz J, Ruemmele P, Hofmann U, Melter M and Dayoub R: Attenuated
lipotoxicity and apoptosis is linked to exogenous and endogenous
augmenter of liver regeneration by different pathways. PLoS One.
12:e01842822017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kapoor A and Sanyal AJ: Endoplasmic
reticulum stress and the unfolded protein response. Clin Liver Dis.
13:581–590. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bellanti F, Mitarotonda D, Tamborra R,
Blonda M, Iannelli G, Petrella A, Sanginario V, Iuliano L,
Vendemiale G and Serviddio G: Oxysterols induce mitochondrial
impairment and hepatocellular toxicity in non-alcoholic fatty liver
disease. Free Radic Biol Med. 75 (Suppl 1):S16–S17. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kusminski CM, Shetty S, Orci L, Unger RH
and Scherer PE: Diabetes and apoptosis: Lipotoxicity. Apoptosis.
14:1484–1495. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Unger RH and Orci L: Lipoapoptosis: Its
mechanism and its diseases. Biochim Biophys Acta. 1585:202–212.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
McCullough AJ: The clinical features,
diagnosis and natural history of nonalcoholic fatty liver disease.
Clin Liver Dis. 8521–533. (viii)2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Murphy DJ: The biogenesis and functions of
lipid bodies in animals, plants and microorganisms. Prog Lipid Res.
40:325–438. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zehmer JK, Huang Y, Peng G, Pu J, Anderson
RG and Liu P: A role for lipid droplets in inter-membrane lipid
traffic. Proteomics. 9:914–921. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Geng F and Guo D: Lipid droplets,
potential biomarker and metabolic target in glioblastoma. Intern
Med Rev (Wash DC). May 3–2017.(Epub ahead of print). doi:
10.18103/imr.v3i5.443.
|
|
17
|
Bartz R, Zehmer JK, Zhu M, Chen Y, Serrero
G, Zhao Y and Liu P: Dynamic activity of lipid droplets: Protein
phosphorylation and GTP-mediated protein translocation. J Proteome
Res. 6:3256–3265. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brasaemle DL: Thematic review series:
Adipocyte biology. The perilipin family of structural lipid droplet
proteins: Stabilization of lipid droplets and control of lipolysis.
J Lipid Res. 48:2547–2559. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dalen KT, Dahl T, Holter E, Arntsen B,
Londos C, Sztalryd C and Nebb HI: LSDP5 is a PAT protein
specifically expressed in fatty acid oxidizing tissues. Biochim
Biophys Acta. 1771:210–227. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wolins NE, Quaynor BK, Skinner JR, Tzekov
A, Croce MA, Gropler MC, Varma V, Yao-Borengasser A, Rasouli N,
Kern PA, et al: OXPAT/PAT-1 is a PPAR-induced lipid droplet protein
that promotes fatty acid utilization. Diabetes. 55:3418–3428. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yamaguchi T, Matsushita S, Motojima K,
Hirose F and Osumi T: MLDP, a novel PAT family protein localized to
lipid droplets and enriched in the heart, is regulated by
peroxisome proliferator-activated receptor alpha. J Biol Chem.
281:14232–14240. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gandotra S, Le Dour C, Bottomley W,
Cervera P, Giral P, Reznik Y, Charpentier G, Auclair M, Delépine M,
Barroso I, et al: Perilipin deficiency and autosomal dominant
partial lipodystrophy. N Engl J Med. 364:740–748. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tansey JT, Sztalryd C, Gruia-Gray J, Roush
DL, Zee JV, Gavrilova O, Reitman ML, Deng CX, Li C, Kimmel AR and
Londos C: Perilipin ablation results in a lean mouse with aberrant
adipocyte lipolysis, enhanced leptin production, and resistance to
diet-induced obesity. Proc Natl Acad Sci USA. 98:6494–6499. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cao J, Feng XX, Yao L, Ning B, Yang ZX,
Fang DL and Shen W: Saturated free fatty acid sodium
palmitate-induced lipoapoptosis by targeting glycogen synthase
kinase-3β activation in human liver cells. Dig Dis Sci. 59:346–357.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li F, Gu Y, Dong W, Li H, Zhang L, Li N,
Li W, Zhang L, Song Y, Jiang L, et al: Cell death-inducing
DFF45-like effector, a lipid droplet-associated protein, might be
involved in the differentiation of human adipocytes. FEBS J.
277:4173–4183. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bhala N, Younes R and Bugianesi E:
Epidemiology and natural history of patients with NAFLD. Curr Pharm
Des. 19:5169–5176. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cao J, Dai DL, Yao L, Yu HH, Ning B, Zhang
Q, Chen J, Cheng WH, Shen W and Yang ZX: Saturated fatty acid
induction of endoplasmic reticulum stress and apoptosis in human
liver cells via the PERK/ATF4/CHOP signaling pathway. Mol Cell
Biochem. 364:115–129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Malhi H and Gores GJ: Molecular mechanisms
of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver
Dis. 28:360–369. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ning B, Bai M and Shen W: Reduced
glutathione protects human hepatocytes from palmitate-mediated
injury by suppressing endoplasmic reticulum stress response.
Hepatogastroenterology. 58:1670–1679. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cao SS and Kaufman RJ: Targeting
endoplasmic reticulum stress in metabolic disease. Expert Opin Ther
Targets. 17:437–448. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li H, Song Y, Zhang LJ, Gu Y, Li FF, Pan
SY, Jiang LN, Liu F, Ye J and Li Q: LSDP5 enhances triglyceride
storage in hepatocytes by influencing lipolysis and fatty acid
β-oxidation of lipid droplets. PLoS One. 7:e367122012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kuramoto K, Okamura T, Yamaguchi T,
Nakamura TY, Wakabayashi S, Morinaga H, Nomura M, Yanase T, Otsu K,
Usuda N, et al: Perilipin 5, a lipid droplet-binding protein,
protects heart from oxidative burden by sequestering fatty acid
from excessive oxidation. J Biol Chem. 287:23852–23863. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Engin A: Non-alcoholic fatty liver
disease. Adv Exp Med Biol. 960:443–467. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kishikawa N and Kuroda N:
Chemiluminescence assay for the investigation of reactive oxygen
species generator. Yakugaku Zasshi. 135:191–196. 2015.(In
Japanese). View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Feldstein AE, Canbay A, Angulo P, Taniai
M, Burgart LJ, Lindor KD and Gores GJ: Hepatocyte apoptosis and fas
expression are prominent features of human nonalcoholic
steatohepatitis. Gastroenterology. 125:437–443. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schattenberg JM, Galle PR and Schuchmann
M: Apoptosis in liver disease. Liver Int. 26:904–911. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cao Z, Zhang H, Cai X, Fang W, Chai D, Wen
Y, Chen H, Chu F and Zhang Y: Luteolin promotes cell apoptosis by
inducing autophagy in hepatocellular carcinoma. Cell Physiol
Biochem. 43:1803–1812. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Diamantis A, Magiorkinis E, Sakorafas GH
and Androutsos G: A brief history of apoptosis: From ancient to
modern times. Onkologie. 31:702–706. 2008.PubMed/NCBI
|
|
40
|
Granneman JG, Moore HP, Mottillo EP and
Zhu Z: Functional interactions between Mldp (LSDP5) and Abhd5 in
the control of intracellular lipid accumulation. J Biol Chem.
284:3049–3057. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang H, Bell M, Sreenivasan U, Hu H, Liu
J, Dalen K, Londos C, Yamaguchi T, Rizzo MA, Coleman R, et al:
Unique regulation of adipose triglyceride lipase (ATGL) by
perilipin 5, a lipid droplet-associated protein. J Biol Chem.
286:15707–15715. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mansour M, Schwartz D, Judd R, Akingbemi
B, Braden T, Morrison E, Dennis J, Bartol F, Hazi A, Napier I and
Abdel-Mageed AB: Thiazolidinediones/PPARγ agonists and fatty acid
synthase inhibitors as an experimental combination therapy for
prostate cancer. Int J Oncol. 38:537–546. 2011. View Article : Google Scholar : PubMed/NCBI
|