Introduction
As the leading cause of dementia among the elderly,
Alzheimer's disease (AD) usually induces cognitive deficits in
patients, who require full-time care, burdening their families and
health care systems (1).
Therefore, developing viable anti-AD therapies is urgently
required. As currently approved treatments only partially alleviate
specific symptoms as opposed to effectively treating the complete
AD pathology (2), improved
understanding of the pathogenesis of AD is required for the
development of more effective and potentially curative
treatments.
It is well documented that the accumulation and
aggregation of neurotoxic amyloid β (Aβ) peptides is a principal
pathological feature of the AD brain that is proposed to be
involved in the emergence of clinical manifestations in patients
with AD (3). Aβ is generated from
its precursor amyloid precursor protein (APP) by sequential
cleavage (4). According to the
amyloid hypothesis, the disequilibrium between Aβ production and Aβ
degradation leads to Aβ accumulation, and contributes to the
primary neuropathogenesis of AD (5). In the amyloidogenic pathway, APP is
first hydrolyzed by β-site APP-cleaving enzyme 1 (BACE1) to produce
soluble (s)APPβ and β-C-terminal fragment (β-CTF). The β-CTF is
sequentially cleaved by γ-secretase to release Aβ and the
intracellular domain fragment (AICD) (4,5).
Accumulated Aβ aggregates to generate Aβ oligomers and amyloid
plaques, potentially leading to the dysfunction of synapses and
neuronal injury (6–8). Therefore, inhibition of Aβ production
and/or peptide oligomerization is considered a promising potential
therapeutic strategy for AD (8).
Additionally, there have been increasing efforts to identify
disease-associated biomarkers of AD for diagnosis at early stages
(9–11). This may aid the development of more
effective and potentially curative treatments.
Voltage-gated sodium channel β2 [Navβ2 or Nav2.2,
encoded by the sodium voltage-gated channel β subunit 2
(SCN2B) gene], a voltage-gated sodium channel (VGSC)
auxiliary subunit, is extensively distributed in the central
nervous and cardiac systems (12).
Generally, Navβ2 regulates the activation and inactivation of
Nav1.1 and Nav1.6 in a voltage-dependent manner (12,13).
The upregulation and diffuse distribution of Navβ2 along
demyelinated axons induces conduction block recovery and clinical
remission (14,15). As neuronal channel cell surface
expression is regulated by Navβ2, Navβ2 has also been demonstrated
to be involved in the pathogenic processes underlying multiple
sclerosis and experimental acute encephalitis (EAE) (16). It was reported that Navβ2 regulates
the cell surface expression of Nav1, which modulates propagation of
the action potential and neuronal activity (17–19).
Previous studies have also identified potential roles for Navβ2 in
APP cleavage and AD pathology. Navβ2 was identified as a novel
substrate of BACE1 and γ-secretase in model systems both in
vivo and in vivo (20,21).
Navβ2 cleaved by BACE1 induced a leftward shift in the remodeling
current of Nav1.2 (22). In a
model of AD, BACE1 upregulation was associated with increased
cleavage of Navβ2 cleavage, upregulated expression of Nav1.1 in
cortical neurons and abnormal EEG activity (19,23).
A previous study demonstrated that increased
SCN2B expression in the hippocampus was associated with
cognitive deficits in senescence-accelerated P8 mice (24). It was also reported that Navβ2
knockdown (Navβ2-kd) protected neurons and improved spatial
cognition in aged APP/presenilin 1 (APP/PS1) transgenic (Tg) mice
by partially decreasing pathological amyloidogenic processing of
APP (25). These results suggested
that knockdown of Navβ2 may be a promising therapeutic strategy for
AD; however, the underlying mechanisms remain unclear. The present
study investigated whether Navβ2 knockdown-induced neuroprotection
and inhibition of APP amyloidogenic processing were associated with
the activity and/or expressional regulation of the Aβ-degrading
enzyme neprilysin (NEP).
Materials and methods
Ethics approval
All experimental procedures and animal care was in
compliance with the Guide for the Care and Use of Laboratory
Animals published by the US National Institutes of Health (26). The present study was performed in
accordance with the Care and Use Guidelines of Experimental Animals
established by the Research Ethics Committee of Kunming University
of China that approved the study (permit no. kmu-eac-2017008).
Surgical procedures were performed under anesthesia induced with
diethyl ether. The purchase, storage and use of diethyl ether were
approved by the associated departments and Kunming Medical
University (license no. kmykdx-D-A00258). All efforts were made to
minimize animal suffering during the experiments.
Animal model
In the present study, 16 APPswe/PS1ΔE9 (APP/PS1
transgene with a C57BL/6J genetic background; weight, 20–25 g; age,
3–4 months; female to male ratio, 1:1) mice and the 16 wild type
(WT) littermates were purchased from the Institute of Laboratory
Animal Science (Chinese Academy of Medical Sciences). Navβ2-kd
(60.68% SCN2B transcription reduction) and APP/PS1/Navβ2-kd
Tg mice were generated and bred at Kunming Medical University as
previously described (24,25). Animals were maintained under
standard conditions at 22°C with 55–60% humidity and a 12-h
light/dark cycle. They were housed with free access to food and
water. Mouse genomic DNA extracted from tail tissues was prepared
for PCR, which was employed to screen the potential Tg founders.
The presence of bands with target molecular weights was identified
by PCR for the detection of positive transgenes as previously
described [608 bp for APP/PS1 transgene; 453 bp for Navβ2-kd
transgene; 608 and 453 bp for APP/PS1/Navβ2-kd transgene; and 350
bp for wild-type (WT)] (25).
Cell preparation
Mice were anesthetized via inhalation of diethyl
ether (2–3% volume fraction; Shanghai Wulian Chemical Reagent
Procurement and Supply Chemical Work) for 3–4 min. Anesthesia was
induced using an animal inhalation anesthesia machine (AM Easy-Flex
Auto Flow system; United Well Technologies Ltd.). The hippocampi
were extracted from APP/PS1, Navβ2-kd, APP/PS1/Navβ2-kd, and WT
mice following fast craniotomy, then the animals were sacrificed by
dislocation of the cervical vertebrae. All efforts were made to
minimize animal suffering during all surgical procedures, and the
protocols were approved (permit no. kmu-eac-2017008). Primary
hippocampus cell culture from the WT, APP/PS1/Navβ2-kd, APP/PS1 and
Navβ2-kd mice was conducted as previously described (25). Primary cells were resuspended by
pipetting for ≥10 times in 1 ml Neurobasal medium (Invitrogen;
Thermo Fisher Scientific, Inc.) supplemented with B27 (2%; Gibco;
Thermo Fisher Scientific, Inc.), N2 (1%; Gibco; Thermo Fisher
Scientific, Inc.), penicillin/streptomycin (100 U; Gibco; Thermo
Fisher Scientific, Inc.) and L-glutamine (2 mM; Gibco; Thermo
Fisher Scientific, Inc.) at 37°C. In total, 1×106
neurons were seeded on six-well plates and incubated at 37°C with
5% CO2. The medium was changed every 2 days until use.
Using LEICA DMI6000B (LAS AF system; Leica Microsystems GmbH),
morphological analysis was performed to evaluate the
neuroprotection induced by Navβ2-kd. The axon lengths and areas of
neurons were measured (magnification, ×200).
To evaluate the effects of Navβ2-kd on the
amyloidogenic processing of APP, cell culture medium was collected
from the different groups for the detection of sAPPα, sAPPβ, Aβ40
and Aβ42 expression levels, whereas cell lysates were prepared to
measure Navβ2 and NEP expression, total Aβ degradation detection,
and to perform chromatin immunoprecipitation (ChIP) assays.
Reverse transcription-quantitative PCR
(RT-qPCR)
Cells were prepared for the detection of the gene
expression levels of Navβ2 and NEP by RT-qPCR as previously
described (27,28). TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) was used to isolate
total RNA from cultured cells. A RevertAid First Strand cDNA
Synthesis kit (Fermentas; Thermo Fisher Scientific, Inc.) was used
for RT of RNA. The first-strand cDNA was synthesized from 2 µg
total RNA that was quantified using a NanoDrop spectrophotometer
(NanoDrop 1000; Thermo Fisher Scientific, Inc.). In total, 2 µg
total RNA was incubated with 1 µl oligo (dT), 18 µl random hexamers
primers and 10 µl diethyl pyrocarbonate-treated water at 70°C for 5
min. The reaction was then incubated on ice for 10 sec. This
mixture was subsequently supplied with 4 µl 5X reaction buffer, 1
µl Riboblock™ Ribonuclease inhibitor and 10 mM dNTP (all reagents
were included in the kit), and incubated at 37°C for 5 min. For
cDNA synthesis, 1 µl Revert Aid Moloney murine leukemia virus
transcriptase was added to this mixture for an incubation at 42°C
for 1 h, followed by termination at 70°C for 10 min. Fast SYBR
Green Master Mix (Roche Diagnostics) was used for qPCR, and the
results were analyzed using a Real-Time PCR System (Thermo Fisher
Scientific, Inc.). qPCR was performed in 20 µl reaction volume,
including 2X SYBR Green Master Mix (Roche Diagnostics) 10 µl,
primer (0.25 µl; 10 pmol/l), template DNA (1 µl) and sterile water
(8.5 µl). Then, qPCR amplification was performed at 95°C for 3 min,
followed by 34 thermocycling steps of 94°C for 35 sec, 56.5°C for
30 sec and at 72°C for 30 sec. To analyze the differences between
samples, the relative Cq method was used. Following normalization
to the housekeeping gene (β-actin), fold changes (decrease or
increase) were determined relative to a blank control using the
2−ΔΔCq method (29).
Each reaction was repeated for three times. The following primer
sequences were used (Takara Biotechnology Co., Ltd.): β-actin,
forward 5′-TGGCACCCAGCACAATGAA-3′, reverse
5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′ (30); Navβ2, forward
5′-CTACACCGTGAACCACAAGCA-3′, reverse 5′-GACCACAGCCAGGAAACCC-3′
(24); and NEP, forward
5′-TGAACTTTGCCCAGGTGTG-3′, and reverse 5′-GCAAAGTCCCAATGATCCTG-3′
(28).
Detection of APP cleavage
production
The expression levels of Aβ, sAPPα and sAPPβ were
detected to evaluate the effects of Navβ2-kd on APP amyloidogenic
processing. Following 5 days of culturing, medium was obtained from
the cell cultures of WT, APP/PS1, Navβ2-kd and APP/PS1/Navβ2-kd
mice. Amyloid β ELISA kits (Demeditec Diagnostics GmbH) were used
to evaluate Aβ40 (cat. no. JP27718) and Aβ42 (cat. no. JP27719)
expression levels, according to the manufacturer's protocol. The
expression levels of Aβ40/42 in cell culture media were compared
between the different genotypes.
To determine sAPPα and sAPPβ expression levels, a
sAPPα/sAPPβ multiplex electrochemiluminescence assay kit (cat. no.
K15120E; 96-well MULTI-SPOT sAPPα/sAPPβ Assay; Meso Scale
Discovery) was used. Cells were cultured in serum-free medium for
16 h at 37°C prior to harvesting. The cell medium was obtained to
quantify sAPPα and sAPPβ expression levels, according to the
manufacturer's protocol. Briefly, 150 µl/well of 3% Blocker A
solution (included in the kit) was added in the plate and incubated
at room temperature with shaking for 1 h. The plate was washed with
300 µl/well of 1X Tris buffer for three times. The 25 µl/well of
cell medium were added in the plate and incubated at room
temperature for 1 h, followed by washing for three times with 300
µl/well of 1X Tris buffer. Then, 25 µl/well of sAPPα or sAPPβ
Calibrator (included in the kit) were added and incubated for 1 h
at room temperature with shaking. The plate was washed again with
300 µl/well 1X Tris buffer for three times. In total, 150 µl/well
1X Read Buffer T (included in the kit) was then added and incubated
for 10 min at room temperature without shaking. The plate was
analyzed with a SECTOR Imager 2400 (Meso Scale Discovery).
Detection of protein expression
Samples from cultured cells were collected to
evaluate Navβ2 and NEP expression as previously described (25). Lysis buffer (10 mM Tris-HCl buffer
pH 7.4, 10 mM EDTA, 30% Triton-100, 10% SDS and 150 mM NaCl)
supplemented with protease inhibitor cocktails (Roche Diagnostics
GmbH) was used to lyse cells. A total of 50 µg protein (quantified
using the bicinchoninic acid method) was used for SDS-PAGE on 4–12%
gels, followed by transfer to nitrocellulose membranes
(Sigma-Aldrich; Merck KGaA). The membrane was subsequently
incubated with antibodies specific for Navβ2 (1:500; cat. no.
ASC-007; Alomone Labs) or NEP (1:800; cat. no. ab951; Abcam) at 4°C
for 12 h. β-actin (mouse monoclonal anti-β-actin; 1:800; cat. no.
sc-69879; Santa Cruz Biotechnology, Inc.) was used as the loading
control. The membranes were washed and incubated with the
peroxidase-conjugated anti-mouse secondary antibody (1:10,000; cat.
no. PI-2000; Vector Laboratories, Inc.) or the horseradish
peroxidase-conjugated anti-rabbit antibody (1:2,000; cat. no.
PI-1000; Vector Laboratories, Inc.) at 20–25°C for 2 h. Enhanced
chemiluminescence reagent (Pierce; Thermo Fisher Scientific, Inc.)
was used to visualize the bands. Densitometric analysis of the
target protein bands was performed using a ChemiDoc™ RS+ imaging
system and Image Lab™ software v5.2.1 (Bio-Rad, Laboratories,
Inc.).
NEP activity evaluation
According to Grimm et al (28), the activity of NEP was evaluated.
Briefly, 1×106 cells were treated with 500 µl lysis
buffer, containing 0.5% Triton X-100, 20 mM Tris (pH 7.4) and 10%
Sucrose at 4°C for 25 min. A total of 2 µl anti-NEP antibody
(1:500; cat. no. AF1182; R&D Systems, Inc.) and 5 µM
MCA-RPPGFSAFK(DNP)-OH fluorogenic peptide substrate (R&D
Systems, Inc.) were dissolved in HEPES buffer (pH 7.4) (Invitrogen;
Thermo Fisher Scientific, Inc.). Cell lysates were incubated with
anti-NEP antibody and fluorogenic substrate mixture (pH 7.4) for 30
min at room temperature. Using a iMark Fluorometer Microplate
Reader (Bio-Rad, Laboratories, Inc.), the fluorescence was
evaluated with an excitation wavelength of 320±10 nm and an
emission wavelength of 405±10 nm.
Cell viability assay
The effects of Navβ2-knockdown on the viability of
primary cultured neurons were evaluated by measuring lactate
dehydrogenase (LDH) activity using a Cytotoxicity Detection Kitplus
(cat. no. 04744926001; Roche Diagnostics) according to the
manufacturer's protocols.
Aβ degradation detection
Total Aβ degradation was determined as previously
described (28) with adjustments
to the protocol. Lysis buffer (150 mM NaCl, 50 mM Tris/HCl pH 7.4,
2 mM EDTA, 0.1% NP-40 and 0.1% Triton-X 100) was used to lyse cells
at 20–25°C for 15 min. In total, 60 µg protein from each sample was
incubated with 1 µg/ml synthetic human Aβ40 peptide (cat. no.
A9810; Sigma-Aldrich; Merck KGaA) in 100 µl PBS for 1 h at 37°C.
Additionally, 50 µM thiorphan was added into the samples to
determine the specificity of NEP-mediated Aβ degradation.
Quantification of the remaining Aβ was performed using an
anti-β-amyloid 40/42 (Aβ40/42) polyclonal antibody (1:800; cat. no.
AB5076; EMD Millipore). The degrading activity in the lysates was
evaluated and presented as the reciprocal value of the remaining
quantified Aβ peptides. The Alexa Fluor 350-labeled secondary
antibody anti-rabbit IgG (1:500; cat. no. A0408; Beyotime Institute
of Biotechnology, Shanghai, China) was incubated at 37°C for 1.5 h.
The signals were analyzed using the SECTOR Imager 2400 (Meso Scale
Discovery) (28).
ChIP analysis
ChIP was performed as previously described to
determine the AICD enrichment of the promotor of the NEP gene
(31–33). Briefly, Cells (20×106)
were harvested with trypsin treatment and washed three times with
PBS and then treated with 1% formaldehyde (Sigma-Aldrich) in PBS
for 8–10 min at 4°C. Fixation was stopped by addition of glycine to
a final concentration of 125 mM. Extracts were sonicated at 20 kHz
for 2 min at room temperature repeated three times, and incubated
with protein-G-Sepharose (GE Healthcare) for 1 h with rotation at
room temperature, following treatment overnight at 4°C with
anti-AICD (Y188; 1 µg; cat. no. ab32136; Abcam) or mouse
immunoglobulin G (IgG; 1 µg; cat. no. ab18413; Abcam) primary
antibody. Subsequently de-crosslinking and DNA isolation was
performed prior to qPCR analysis. The PCR results were presented as
the fold enrichment of DNA pulled down with the anti-AICD antibody
compared with the IgG negative control.
Statistical analysis
The results are presented as the mean ± standard
deviation of the means of triplicate measurements of five
independent experiments. One-way ANOVA followed by Fisher's least
significant difference test were employed to analyze data using
SPSS 11.5 (SPSS, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Detection of Navβ2 expression in
cultured cells
Navβ2 expression in cultured cells isolated from the
different mice was evaluated by RT-qPCR and western blot analyses.
Consistent with previous findings (25), Navβ2 knockdown decreased the
expression levels of the Navβ2 gene (Fig. 1A) and protein (Fig. 1B) by ~61% compared with the WT.
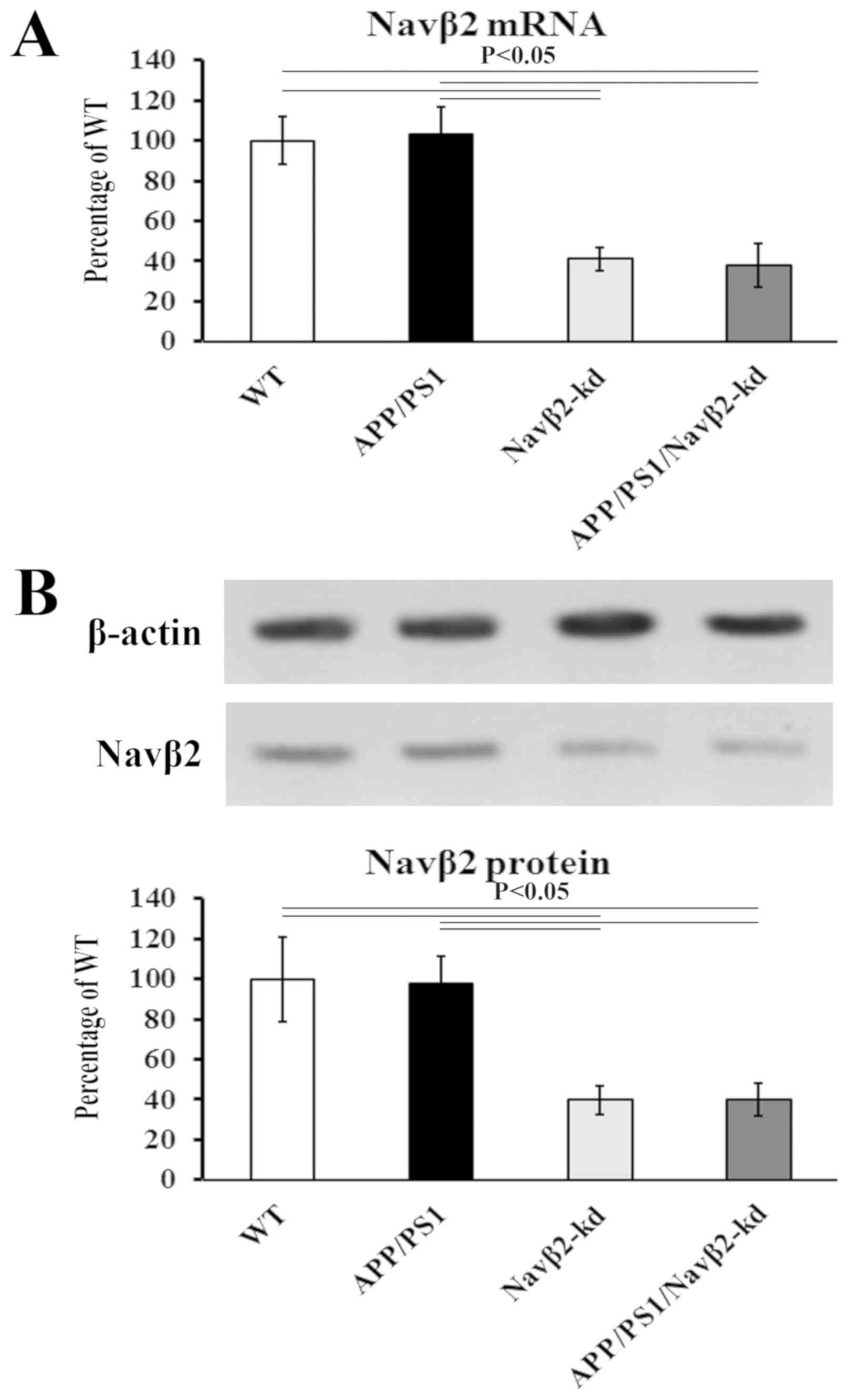 | Figure 1.Navβ2 expression in neurons. (A) mRNA
levels of Navβ2 evaluated by reverse transcription-quantitative PCR
analysis in APP/PS1/Navβ2-kd (0.25±0.08), Navβ2-kd (0.27±0.06),
APP/PS1 (0.67±0.14) and WT (0.65±0.12) mouse-derived cells. (B)
Protein levels of Navβ2 evaluated by western blotting in
APP/PS1/Navβ2-kd (0.16±0.08), Navβ2-kd (0.18±0.07), APP/PS1
(0.45±0.13) and WT (0.46±0.09) mouse-derived cells, respectively.
Navβ2, voltage-gated sodium channel β2; APP, amyloid precursor
protein; PS1, presenilin 1; kd, knockdown; WT, wild-type. |
Expression and activity of NEP in cell
culture
Compared with the WT group, NEP expression was
significantly decreased in cultured cells derived from APP/PS1
mice; NEP gene expression levels decreased by 32.7±4.3%, whereas
NEP protein expression levels decreased by 35±5.1% (Fig. 2A and C). Navβ2 knockdown induced a
partial restoration of the expression of NEP in the
APP/PS1/Navβ2-kd group. There was no significant difference between
the WT and Navβ2-kd groups (P>0.05; Fig. 2).
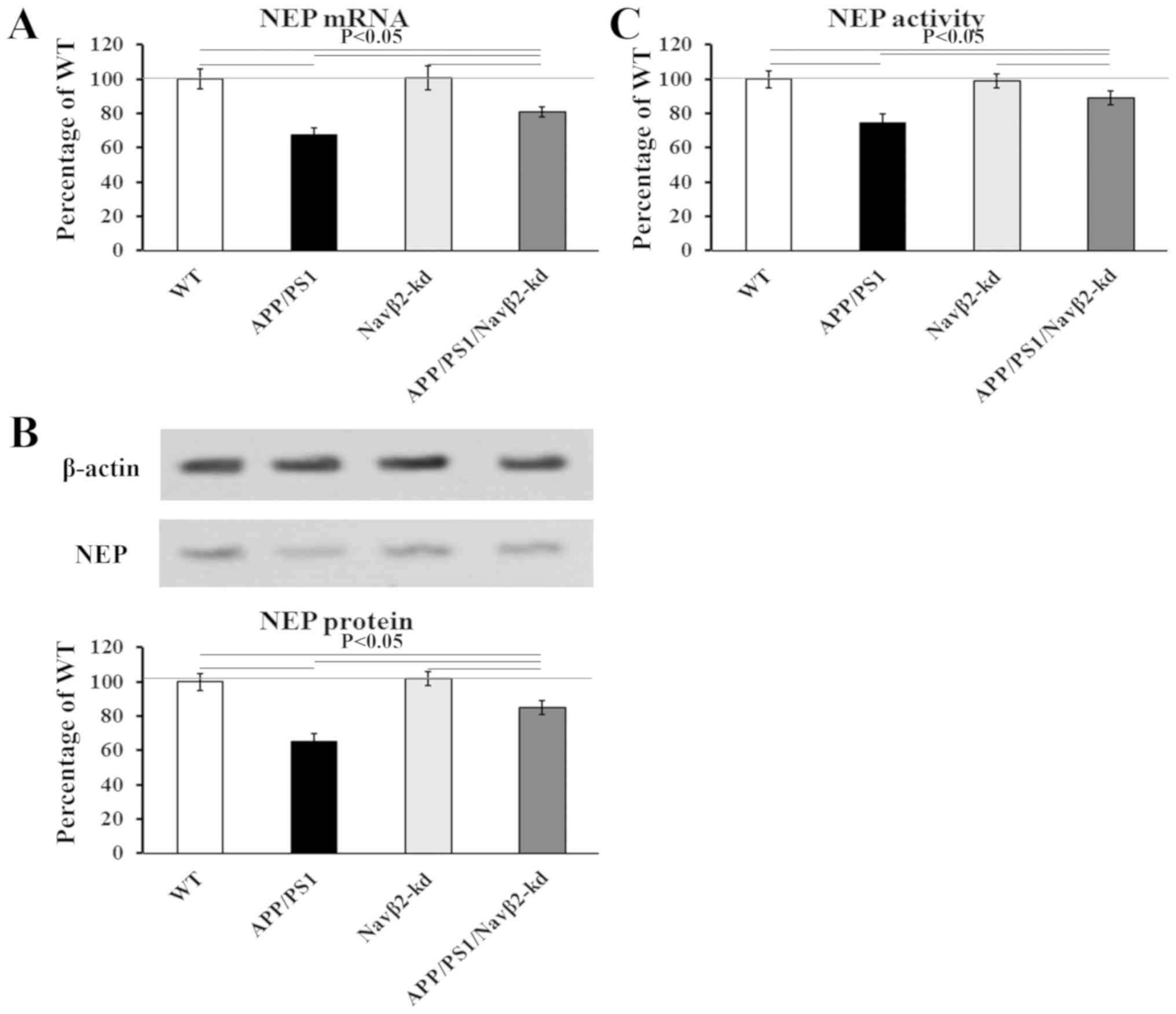 | Figure 2.Effects of Navβ2 knockdown on the
expression and activity of NEP. (A) mRNA levels of NEP as
determined via reverse transcription-quantitative PCR analysis in
APP/PS1/Navβ2-kd, Navβ2-kd, APP/PS1 and WT mouse-derived neurons.
(B) Activity of NEP in cells derived from APP/PS1/Navβ2-kd,
Navβ2-kd, APP/PS1 and WT mice. (C) Protein levels of NEP as
determined via western blotting in APP/PS1/Navβ2-kd, Navβ2-kd,
APP/PS1 and WT mouse-derived cells. Navβ2, voltage-gated sodium
channel β2; APP, amyloid precursor protein; PS1, presenilin 1; kd,
knockdown; WT, wild-type; NEP, neprilysin. |
Consistent with the NEP expression analysis, NEP
activity was reduced to 74.5±3.9% in the APP/PS1 group, and
recovered to 89±2.8% in APP/PS1/Navβ2-kd cells compared with the
WT. Navβ2 knockdown induced no effects on NEP activity compared
with WT cells (P>0.05; Fig.
2B).
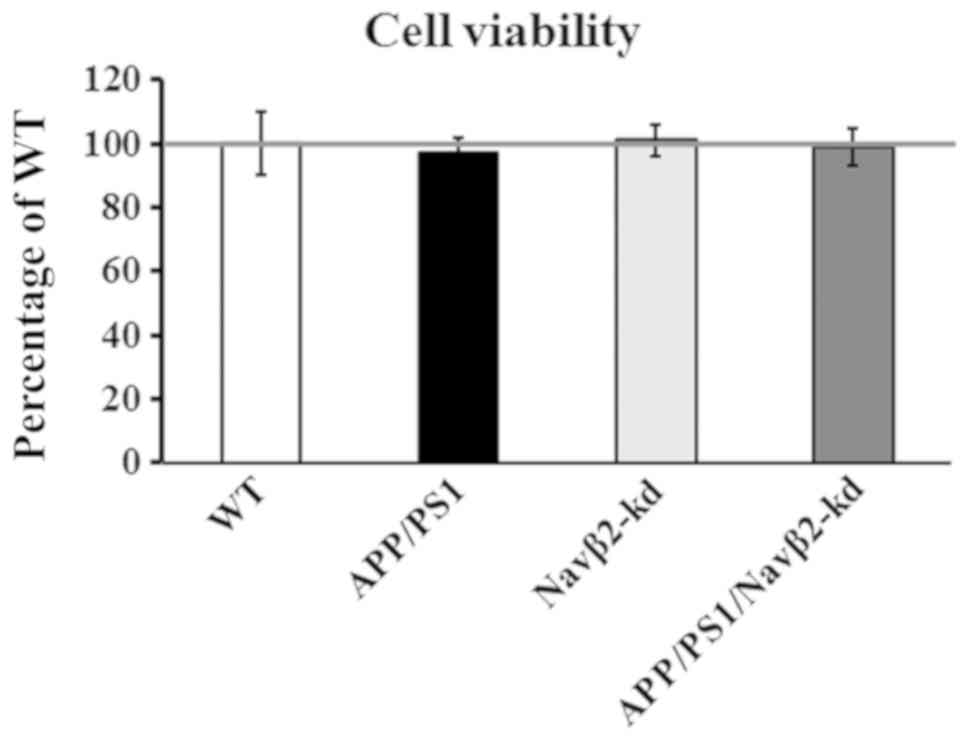
Measurement of cell viability
No difference in cell viability was detected in
cells from APP/PS1 mice compared with WT mice, as determined by an
LDH assay (P>0.05; Fig. 3).
Additionally, compared with the WT group, Navβ2-kd cells exhibited
no significant difference in viability (P>0.05). Furthermore,
there was no significant difference in the viability of cells
obtained from the APP/PS1/Navβ2-kd mice, and those from APP/PS1 or
Navβ2-kd mice.
Evaluation of APP processing and Aβ
degradation
To evaluate the role of Navβ2 expressional knockdown
in the amyloidogenic processing of APP, the levels of Aβ, sAPPα and
sAPPβ, and total Aβ degradation were measured. Significant
increases in Aβ40 and Aβ42 levels, and the ratio of Aβ42/Aβ40 were
observed in cultured cells derived from APP/PS1 mice compared with
the WT group (P<0.05; Fig. 4A and
B). In the APP/PS1/Navβ2-kd group, the expression levels of
Aβ40 and Aβ42, and the ratio of Aβ42/Aβ40 were partially decreased
compared with the APP/PS1 group. Additionally, reduced sAPPα and
increased sAPPβ expression levels as determined via ECLIA were
detected in cells derived from APP/PS1 mice, compared with the WT
or Navβ2-kd groups (P<0.05; Fig. 4C
and D). The APP/PS1/Navβ2-kd transgene induced significant
sAPPα upregulation, sAPPβ downregulation and an increased
sAPPα/sAPPβ ratio in mouse cells compared with the APP/PS1 group
(P<0.05; Fig. 4C and D).
Conversely, Navβ2 knockdown alone induced no significant effects on
Aβ40/42, sAPPα or sAPPβ expression levels compared with the WT
group (P<0.05; Fig. 4).
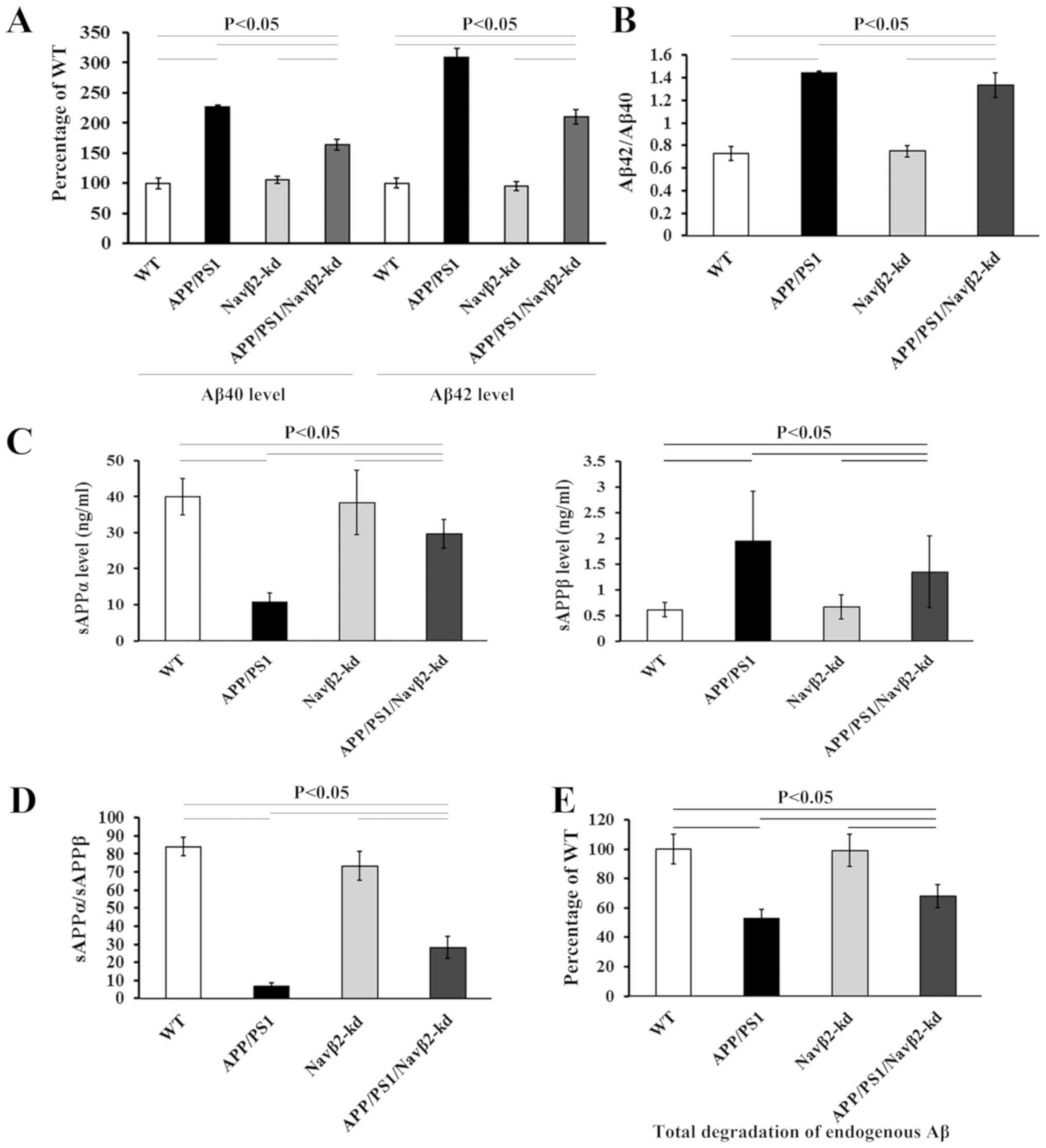 | Figure 4.Navβ2 knockdown affects APP
processing and Aβ degradation. (A) Aβ40/42 levels, (B) Aβ42/Aβ40
ratio, (C) sAPPα and sAPPβ levels, (D) sAPPα/sAPPβ ratio and (E)
total Aβ degradation in lysates of APP/PS1/Navβ2-kd, Navβ2-kd,
APP/PS1 and WT mouse-derived neurons. Navβ2, voltage-gated sodium
channel β2; APP, amyloid precursor protein; PS1, presenilin 1; kd,
knockdown; WT, wild-type; s, soluble. |
Total degradation of endogenous Aβ was measured in
the cell lysates of the different groups. Total degradation of Aβ
40/42 in the lysates of APP/PS1 cells significantly decreased to
53.1±6.4% compared with the WT (Fig.
4E). Navβ2 knockdown induced a partial recovery of Aβ
degradation in the APP/PS1/Navβ2-kd group compared with the APP/PS1
group, without reaching the levels of the WT group. There was no
significant difference between WT and Navβ2-kd cells.
Navβ2 knockdown enhances the
enrichment of AICD in the NEP promoter in cells with APP/PS1
ChIP analysis demonstrated that the APP/PS1
transgene significantly decreased the enrichment of AICD in the NEP
promoter compared with WT or Navβ2-kd cells, (P<0.05; Fig. 5). Navβ2 knockdown significantly
increased AICD binding to the promoter of the NEP gene in
APP/PS1/Navβ2-kd cells compared with the APP/PS1 group (P<0.05;
Fig. 5); however, it did not
restore AICD enrichment to WT levels (P<0.05 vs. WT; Fig. 5). No statistical difference was
detected between WT- and Navβ2-kd-derived cells (P>0.05;
Fig. 5).
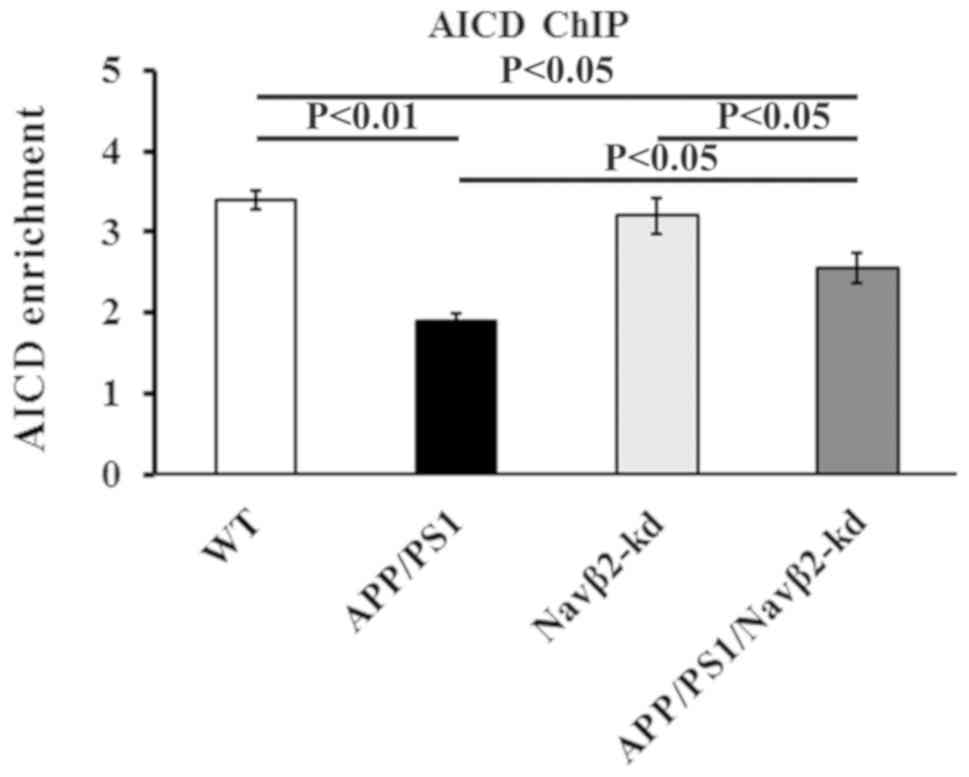 | Figure 5.Navβ2 knockdown increases the levels
of AICD binding to the NEP promotor. Effects of Navβ2 knockdown on
AICD enrichment of the NEP gene promoter in APP/PS1/Navβ2-kd,
Navβ2-kd, APP/PS1 and WT mouse-derived neurons, as determined by a
ChIP assay. AICD enrichment is presented as the fold enrichment
compared with that obtained with non-specific immunoglobulin G.
Navβ2, voltage-gated sodium channel β2; APP, amyloid precursor
protein; PS1, presenilin 1; kd, knockdown; WT, wild-type; NEP,
neprilysin; ChIP, chromatin immunoprecipitation; AICD, APP
intracellular domain. |
Navβ2-kd promotes neurite outgrowth in
APP/PS1 cells
The APP/PS1 transgene induced a significant decrease
in the neurite extension (P<0.05) and areas (P<0.05) of
cultured cells compared with the WT group (Fig. 6). Navβ2-knockdown rescued the
reduced neurite outgrowth in APP/PS1/Navβ2-kd cells (P<0.05;
Fig. 6); however, Navβ2 knockdown
alone induced no effects on the extension or the area of cells
compared with the WT (P>0.05).
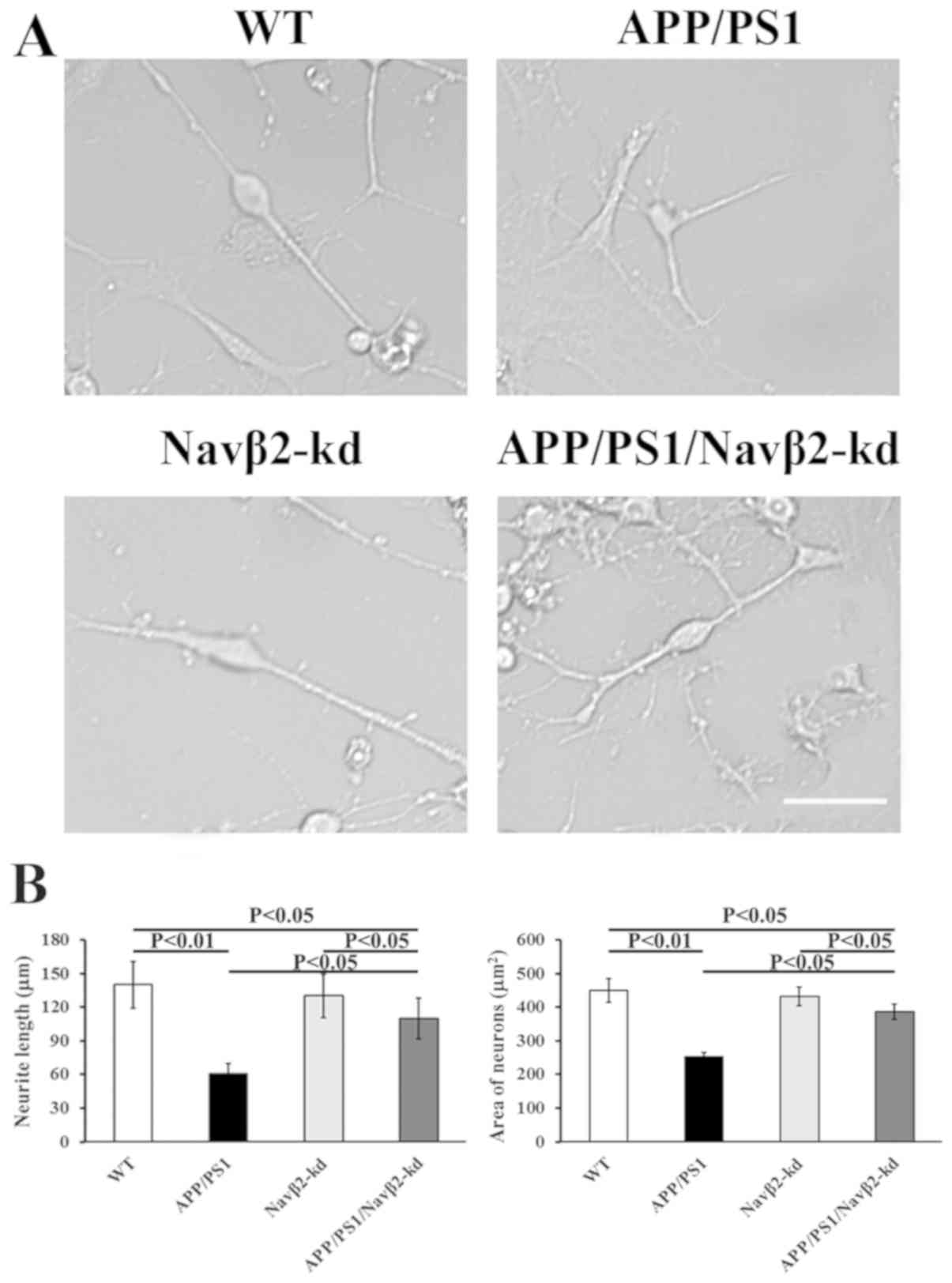 | Figure 6.Effects of Navβ2 knockdown on
morphological changes in APP/PS1 mutant neurons. (A) Micrograph of
cultured cells derived from APP/PS1/Navβ2-kd, Navβ2-kd, APP/PS1 and
WT mice. Magnification, ×200. Scale bar, 100 µm. (B) Quantitation
of the neurite length and area of APP/PS1/Navβ2-kd, Navβ2-kd,
APP/PS1 and WT mouse-derived neurons. Navβ2, voltage-gated sodium
channel β2; APP, amyloid precursor protein; PS1, presenilin 1; kd,
knockdown; WT, wild-type. |
Discussion
Knockdown of Navβ2 expression in APP/PS1 mice has
been reported to convert the amyloidogenic processing of APP
induced by APP/PS1 mutation toward non-amyloidogenic metabolism
(25); however, the underlying
mechanisms remain unclear. The present study investigated whether
Navβ2 knockdown altered APP amyloidogenic metabolism via the
regulation of NEP. The data demonstrated that Navβ2 knockdown
induced a partial recovery of NEP expression and activity, an
increased level of AICD binding to the promoter of NEP gene, and
decreased Aβ generation in APP/PS1 mutant mice. Additionally, Navβ2
knockdown promoted neurite extension and increased the neuronal
area in cultured cells derived from APP/PS1-mutated mice. These
data suggested that expressional knockdown of Navβ2 partially
rectified the APP/PS1 mutation induced-amyloid clearance deficit,
and attenuated the effects on neurites and neuron area induced by
APP/PS1 mutation via regulation of the expression and activity of
NEP.
Primary hippocampal neurons derived from APP/PS1 Tg
mouse were used in the present study; these cells are frequently
used to investigate the pathological conditions and mechanisms of
AD (34). It was suggested that
the Swedish mutation of APP (APPswe) may be processed by the
β-secretase-mediated amyloidogenic pathway instead of
α-secretase-dependent non-amyloidogenic processing; the double
mutation of APPswe at the crucial cleavage site targeted by
β-secretase is proposed to result in an increase in Aβ production
(35,36).
The present findings revealed that increased Aβ40,
−42, and sAPPβ expression levels, and decreased sAPPα levels were
detected in APP/PS1 mutant cells. Additionally, the ratio of
sAPPα/sAPPβ decreased, and Aβ degradation was reduced. In cells
derived from APP/PS1/Navβ2-kd mice, Navβ2 knockdown induced
enhanced Aβ degradation, decreased Aβ40/42 and sAPPβ production,
and increased the expression levels of sAPPα and the ratio of
sAPPα/sAPPβ. During α-secretase processing, APP is cleaved and
releases sAPPα into the extracellular space; the remaining 83 amino
acids in the membrane are subsequently processed by γ-secretase to
generate an Aβ truncated fragment p3 (37,38).
As indicated by ectodomain shedding, sAPPα production is the
initial procedure in non-amyloidogenic processing, whereas sAPPβ
generation is the initial step in amyloidogenic processing
(39). Increased cleavage of APP
by α-secretase resulted in decreased Aβ production (40). Consistent with a previous in
vivo study, the present findings indicated that expressional
knockdown of Navβ2 attenuated amyloidogenic metabolism of APP
induced by APP/PS1 mutation in favor of non-amyloidogenic
processing in vitro (25).
It was previously demonstrated that brain Aβ
expression levels represent a dynamic balance between APP-derived
Aβ production and elimination by a series of amyloid clearance
proteins (6–8). At present, the group of known enzymes
capable of Aβ degradation comprises >20 members (41,42),
including the NEP family (NEP and NEP2), endothelin-converting
enzyme-1 and −2, and insulin-degrading enzyme (41–43).
Aβ may be metabolized by these enzymes, producing metabolites with
reduced neurotoxicity (44). NEP,
a member of the neutral zinc metalloendopeptidase family, is a
plasma membrane glycoprotein of 90–110 kDa (45,46).
The decline in expression levels and activity of NEP leads to a
clearance deficit of amyloid, which is considered as a principal
pathogenic factor in sporadic AD (47,48).
Therefore, recovery of NEP expression and activity may be a
promising therapeutic approach for AD.
A constitutive regulatory processing (49) and an epigenetically-regulated
component (32,50) are responsible for the complexity of
NEP expressional regulation. Among them, the
epigenetically-regulated component involves an unstable
transcription factor, AICD, which is derived from β-secretase
amyloidogenic processing of APP. AICD is a competitive binding
component to the NEP gene promoter (51). By binding to the promoter of NEP,
AICD induces activation of NEP transcription, leading to increased
NEP activity and Aβ clearance; histone deacetylases inhibit this
process (52,53).
In the present study, it was observed that
APP/PS1/Navβ2-kd neurons exhibited reduced Aβ40/42 generation and
increased total Aβ degradation compared with the APP/PS1 mutation
group. To determine whether Navβ2 knockdown enhanced Aβ degradation
via NEP regulation, the levels and activity of NEP in APP/PS1
mutant neurons with or without Navβ2 knockdown were evaluated. AICD
enrichment in the promoter region of NEP was detected by a ChIP
assay. As hypothesized, the data demonstrated that Navβ2 knockdown
induced increased expression and activity levels of NEP, and
increased AICD binding to the NEP gene, which suggested that Navβ2
knockdown rectified the APP/PS1 mutation induced-amyloid clearance
deficit in a NEP-dependent manner. Considering that Navβ2 is a
substrate of BACE1, which is a key APP-cleaving enzyme for APP
hydrolyzation via the amyloidogenic pathway, it is hypothesized
that Navβ2 knockdown may alter the activity of BACE1 and thus
interfere with the amyloidogenic pathway of APP, leading to reduced
Aβ production. Li et al (54) identified that APP modulated Nav1.6
sodium channels via a Go-coupled JNK-mediated pathway,
which is dependent on phosphorylation of APP at Thr668. Navβ2
causes activation and inactivation of Nav1.1 and Nav1.6 in a
voltage-dependent manner. O'Malley et al (16) demonstrated that Navβ2 knockout
attenuated the upregulation of Nav1.6 protein expression in the
brain of an animal model of EAE (16). Therefore, it is additionally
proposed that Navβ2 knockdown may interact with APP associated with
Nav1.6 and influence APP processing; however, further studies are
required to verify these hypotheses.
Additionally, Navβ2 knockdown induced no effects on
cellular LDH activity; however, it prolonged neurite extension in
APP/PS1/Navβ2-kd Tg neurons. It is hypothesized that Navβ2, as an
associated auxiliary subunits of VGSCs, which modulate channel
activity, may serve roles in regulating neuronal activity via
interactions with Nav1.1/1.6, stabilizing the sodium current
density and maintaining channel expression on the cell surface of
neurons as opposed to altering the cell viability (12,13,17).
A previous in vivo study reported that
knockdown of Navβ2 in APP/PS1 mice induced partial restoration of
neuronal activity of hippocampal neurons and improvement of spatial
cognitive function (25). The
present in vitro data demonstrated that Navβ2 knockdown
induced neurite extension and restoration of the neuronal area,
which may be associated with the recovery of neuroplasticity and
contribute to cognitive improvement in APP/PS1 mice.
Additionally, this previous study reported decreased
levels of Navβ2 CTF and a reversal of abnormal Navβ2 cleavage by
BACE1 and γ-secretase following Navβ2 knockdown in a APP/PS1
transgene model of AD (25).
Upregulation of the Navβ2 gene in the brains of aged SAMP8 mice was
previously observed (24).
MicroRNAs (miRNAs/miRs) are small noncoding RNAs that bind via base
pair interactions with the 3′-untranslated region of target mRNAs
to either degrade the mRNA or repress its translation (55). Aberrant miRNA expression may
contribute to the progression of neurodegenerative diseases
(56,57). A previous study additionally
identified different expressional profiles of miRNAs between the
brains of SAMP8 mice and SAMR1 control mice. It was demonstrated
that miR-9 and miR-9* serve important roles during aging via
interactions with target genes in SAMP8 mice; one of the target
genes of miR-9 is SCN2B, a Navβ2-coded gene (58). Therefore, it is hypothesized that
hydrolysis status and/or miR levels may make important
contributions to the regulation of the Navβ2 gene; however, further
investigation is required to verify this hypothesis.
In conclusion, the present findings demonstrated
that Navβ2 knockdown in cultured APP/PS1-derived neurons induced a
partial recovery of NEP expression and activity, an increase in
AICD enrichment of the NEP promoter, a decrease in Aβ generation,
and restoration of neurite length and area. These findings
suggested that Navβ2 knockdown partially recovered the deficiency
of Aβ cleavage and retarded neurite outgrowth induced by the
APP/PS1 mutation, by protecting the expression and activity of NEP,
and enhancing the levels of ACID binding to the NEP gene.
Acknowledgements
Not applicable.
Funding
This research was supported by the National Natural
Science Foundation of China (grant nos. 81560238, 81502377 and
81701212), Yunnan Applied Basic Research Foundation of Yunnan
Province in China (grant nos. 2016FB139 and 2016FB123), Special
Fund of the Applied Basic Research Programs of Yunnan Province
associated with Kunming Medical University in China (grant no.
2015FB001), Foundation of Science and Technology Innovative Team
Building of Kunming Medical University (grant no. CXTD201807), and
the Medical Reserve Talents Cultivation Project of the Health and
Family Planning Commission of Yunnan Province (grant no.
H-2017026).
Availability of data and materials
The datasets used and analyzed in the present study
are available from the corresponding author on reasonable
request.
Authors' contributions
TH and YBX made substantial contributions towards
the design and conception of the present study. YBX and SSL drafted
and critically revised the manuscript. LZ, SSL, MNL, RMa, SL and BC
performed cell and animal model preparation. LZ, MNL, RMa, YXT,
SSL, RMe, SL and TH performed RT-qPCR, APP cleavage detection and
western blot analyses. LZ, MNL, YXT, SSL and YBX performed the NEP
activity evaluation. TH, SSL, MNL, BC, RMa and YBX performed NEP
activity evaluation and the cell viability assay. LZ and YBX
conducted the ChIP analysis. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
All experiments and care of animal were in
compliance with the Guide for the Care and Use of Laboratory
Animals published by the US National Institutes of Health. This
study was conducted in accordance with the Care and Use Guidelines
of Experimental Animals established by the Research Ethics
Committee of Kunming University of China, who approved the present
study (permit no. kmu-eac-2017008). The purchase, storage and use
of the diethyl ether in the present study were approved by the
associated departments and Kunming Medical University (license no.
kmykdx-D-A00258).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jia J, Wei C, Chen S, Li F, Tang Y, Qin W,
Zhao L, Jin H, Xu H, Wang F, et al: The cost of Alzheimer's disease
in China and re-estimation of costs worldwide. Alzheimers Dement.
14:483–491. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yiannopoulou KG and Papageorgiou SG:
Current and future treatments for Alzheimer's disease. Ther Adv
Neurol Disord. 6:19–33. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chu J, Li JG, Hoffman NE, Madesh M and
Praticò D: Degradation of gamma secretase activating protein by the
ubiquitin-proteasome pathway. J Neurochem. 133:432–439. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Esler WP and Wolfe MS: A portrait of
Alzheimer secretases-new features and familiar faces. Science.
293:1449–1454. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Glenner GG and Wong CW: Alzheimer's
disease: Initial report of the purification and characterization of
a novel cerebrovascular amyloid protein. Biochem Biophys Res
Commun. 120:885–890. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Agostinho P, Pliássova A, Oliveira CR and
Cunha RA: Localization and trafficking of amyloid-β protein
precursor and secretases: Impact on Alzheimer's disease. J
Alzheimers Dis. 45:329–347. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ashe KH and Zahs KR: Probing the biology
of Alzheimer's disease in mice. Neuron. 66:631–645. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sakono M and Zako T: Amyloid oligomers:
Formation and toxicity of Abeta oligomers. FEBS J. 277:1348–1358.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu LJ, Leung KH, Chan DS, Wang YT, Ma DL
and Leung CH: Identification of a natural product-like STAT3
dimerization inhibitor by structure-based virtual screening. Cell
Death Dis. 5:e12932014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ito M, Tanaka T, Toita A, Uchiyama N,
Kokubo H, Morishita N, Klein MG, Zou H, Murakami M, Kondo M, et al:
Discovery of
3-Benzyl-1-(trans-4-((5-cyanopyridin-2-yl)amino)cyclohexyl)-1-arylurea
derivatives as novel and selective cyclin-dependent kinase 12
(CDK12) inhibitors. J Med Chem. 61:7710–7728. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma DL, Lin S, Wang W, Yang C and Leung CH:
Luminescent chemosensors by using cyclometalated iridium(iii)
complexes and their applications. Chem Sci. 8:878–889. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dhar Malhotra J, Chen C, Rivolta I, Abriel
H, Malhotra R, Mattei LN, Brosius FC, Kass RS and Isom LL:
Characterization of sodium channel alpha- and beta-subunits in rat
and mouse cardiac myocytes. Circulation. 103:1303–1310. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vijayaragavan K, Powell AJ, Kinghorn IJ
and Chahine M: Role of auxiliary beta1-, beta2-, and beta3-subunits
and their interaction with Na (v) 1.8 voltage-gated sodium channel.
Biochem Biophys Res Commun. 319:531–540. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bechtold DA and Smith KJ: Sodium-mediated
axonal degeneration in inflammatory demyelinating disease. J Neurol
Sci. 233:27–35. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Waxman SG: Axonal conduction and injury in
multiple sclerosis: The role of sodium channels. Nat Rev Neurosci.
7:932–941. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
O'Malley HA, Shreiner AB, Chen GH,
Huffnagle GB and Isom LL: Loss of Na+ channel beta2
subunits is neuroprotective in amouse model of multiple sclerosis.
Mol Cell Neurosci. 40:143–155. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen C, Bharucha V, Chen Y, Westenbroek
RE, Brown A, Malhotra JD, Jones D, Avery C, Gillespie PJ III,
Kazen-Gillespie KA, et al: Reduced sodium channel density, altered
voltage dependence of inactivation, and increased susceptibility to
seizures in mice lacking sodium channel beta 2-subunits. Proc Natl
Acad Sci USA. 99:17072–17077. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lopez-Santiago LF, Pertin M, Morisod X,
Chen C, Hong S, Wiley J, Decosterd I and Isom LL: Sodium channel
beta2 subunits regulate tetrodotoxin-sensitive sodium channels in
small dorsal root ganglion neurons and modulate the response to
pain. J Neurosci. 26:7984–7994. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim DY, Carey BW, Wang H, Ingano LA,
Binshtok AM, Wertz MH, Pettingell WH, He P, Lee VM, Woolf CJ and
Kovacs DM: BACE1 regulates voltage-gated sodium channels and
neuronal activity. Nat Cell Biol. 9:755–764. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wong HK, Sakurai T, Oyama F, Kaneko K,
Wada K, Miyazaki H, Kurosawa M, De Strooper B, Saftig P and Nukina
N: beta subunits of voltage-gated sodium channels are novel
substrates of beta-site amyloid precursor protein-cleaving enzyme
(BACE1) and gamma-secretase. Biol Chem. 280:23009–23017. 2005.
View Article : Google Scholar
|
|
21
|
Kim DY, Ingano LA, Carey BW, Pettingell WH
and Kovacs DM: Presenilin/gamma-secretase-mediated cleavage of the
voltage-gated sodium channel beta2-subunit regulates cell adhesion
and migration. J Biol Chem. 280:23251–23261. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huth T, Schmidt-Neuenfeldt K, Rittger A,
Saftig P, Reiss K and Alzheimer C: Non-proteolytic effect of
beta-site APP cleaving enzyme 1 (BACE1) on sodium channel function.
Neurobiol Dis. 33:282–289. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Corbett BF, Leiser SC, Ling HP, Nagy R,
Breysse N, Zhang X, Hazra A, Brown JT, Randall AD, Wood A, et al:
Sodium channel cleavage is associated with aberrant neuronal
activity and cognitive deficits in a mouse model of Alzheimer's
disease. J Neurosci. 33:7020–7026. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
XiYang YB, Wang YC, Zhao Y, Ru J, Lu BT,
Zhang YN, Wang NC, Hu WY, Liu J, Yang JW, et al: Sodium
channel-voltage-gated-beta 2 plays a vital role in brain aging
associated with synaptic plasticity and expression of COX5A and
FGF-2. Mol Neurobiol. 53:955–967. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hu T, Xiao Z, Mao R, Chen B, Lu MN, Tong
J, Mei R, Li SS, Xiao ZC, Zhang LF and Xiyang YB: Navβ2 knockdown
improves cognition in APP/PS1 mice by partially inhibiting seizures
and APP amyloid processing. Oncotarget. 8:99284–99295. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals, . Guide for the Care and Use of Laboratory Animals. 8th.
National Academies Press; Washington, DC: 2011
|
|
27
|
Hu T, Li YS, Chen B, Chang YF, Liu GC,
Hong Y, Chen HL and Xiyang YB: Elevated glucose-6-phosphate
dehydrogenase expression in the cervical cancer cases is associated
with the cancerigenic event of high-risk human papillomaviruses.
Exp Bio Med (Maywood). 240:1287–1297. 2015. View Article : Google Scholar
|
|
28
|
Grimm MO, Mett J, Stahlmann CP, Grösgen S,
Haupenthal VJ, Blümel T, Hundsdörfer B, Zimmer VC, Mylonas NT,
Tanila H, et al: APP intracellular domain derived from
amyloidogenic β- and γ-secretase cleavage regulates neprilysin
expression. Front Aging Neurosci. 7:772015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hu T, Chang YF, Xiao Z, Mao R, Tong J,
Chen B, Liu GC, Hong Y, Chen HL, Kong SY, et al: miR-1 inhibits
progression of high-risk papillomavirus-associated human cervical
cancer by targeting G6PD. Oncotarget. 7:86103–86116. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zuccato C, Belyaev N, Conforti P, Ooi L,
Tartari M, Papadimou E, MacDonald M, Fossale E, Zeitlin S, Buckley
N and Cattaneo E: Wide spread disruption of repressor element-1
silencing transcription factor/neuron-restrictive silencer factor
occupancy at its target genes in Huntington's disease. J Neurosci.
27:6972–6983. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Belyaev ND, Nalivaeva NN, Makova NZ and
Turner AJ: Neprilysin gene expression requires binding of the
amyloid precursor protein intracellular domain to its promoter:
Implications for Alzheimer disease. EMBO Rep. 10:94–100. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kerridge C, Kozlova DI, Nalivaeva NN and
Turner AJ: Hypoxia affects neprilysin expression through caspase
activation and an APP intracellular domain-dependent Mechanism.
Front Neurosci. 9:4262015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mullan M, Crawford F, Axelman K, Houlden
H, Lilius L, Winblad B and Lannfelt L: A pathogenic mutation for
probable Alzheimer's disease in the APP gene at the N-terminus of
beta-amyloid. Nat Genet. 1:345–347. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Citron M, Oltersdorf T, Haass C,
McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I and
Selkoe DJ: Mutation of the beta-amyloid precursor protein in
familial Alzheimer's disease increases beta-protein production.
Nature. 360:672–674. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Felsenstein KM, Hunihan LW and Roberts SB:
Altered cleavage and secretion of a recombinant beta-APP bearing
the Swedish familial Alzheimer's disease mutation. Nat Genet.
6:251–255. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Esch FS, Keim PS, Beattie EC, Blacher RW,
Culwell AR, Oltersdorf T, McClure D and Ward PJ: Cleavage of
amyloid beta peptide during constitutive processing of its
precursor. Science. 248:1122–1124. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lichtenthaler SF: α-secretase in
Alzheimer's disease: Molecular identity, regulation and therapeutic
potential. J Neurochem. 116:10–21. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hardy J and Selkoe DJ: The amyloid
hypothesis of Alzheimer's disease: Progress and problems on the
road to therapeutics. Science. 297:353–356. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xie Z and Xu Z: General anesthetics and
β-amyloid protein. Prog Neuropsychopharmacol Biol Psychiatry.
47:140–146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nalivaeva NN, Beckett C, Belyaev ND and
Turner AJ: Are amyloid-degrading enzymes viable therapeutic targets
in Alzheimer's disease? J Neurochem. 120 (Suppl 1):S167–S185. 2012.
View Article : Google Scholar
|
|
42
|
Nalivaeva NN, Belyaev ND, Kerridge C and
Turner AJ: Amyloid- clearing proteins and their epigenetic
regulation as a therapeutic target in Alzheimer's disease. Front
Aging Neurosci. 6:2352014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Marr RA and Spencer BJ: NEP-like
endopeptidases and Alzheimer's disease. Curr Alzheimer Res.
7:223–229. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pacheco-Quinto J and Eckman EA:
Endothelin-converting enzymes degrade intracellular β-amyloid
produced within the endosomal/lysosomal pathway and autophagosomes.
J Biol Chem. 288:5606–5615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Miners JS, Baig S, Palmer J, Palmer LE,
Kehoe PG and Love S: Abeta-degrading enzymes in Alzheimer's
disease. Brain Pathol. 18:240–252. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Turner AJ, Brown CD, Carson JA and Barnes
K: The neprilysin family in health and disease. Adv Exp Med Biol.
477:229–240. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Turner AJ, Isaac RE and Coates D: The
neprilysin (NEP) family of zinc metalloendopeptidases: Genomics and
function. Bioessays. 23:261–269. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Selkoe DJ: Preventing Alzheimer's disease.
Science. 337:1488–1492. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Pluta R, Jabłoński M, Ułamek-Kozioł M,
Kocki J, Brzozowska J, Januszewski S, Furmaga-Jabłońska W,
Bogucka-Kocka A, Maciejewski R and Czuczwar SJ: Sporadic
Alzheimer's disease begins as episodes of brain ischemia and
ischemically dysregulated Alzheimer's disease genes. Mol Neurobiol.
48:500–515. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li C, Booze RM and Hersh LB:
Tissue-specific expression of rat neutral endopeptidase
(neprilysin) mRNAs. J Biol Chem. 270:5723–5728. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pardossi-Piquard R, Petit A, Kawarai T,
Sunyach C, Alves da Costa C, Vincent B, Ring S, D'Adamio L, Shen J,
Müller U, et al: Presenilin-dependent transcriptional control of
the Abeta-degrading enzyme neprilysin by intracellular domains of
betaAPP and APLP. Neuron. 46:541–554. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Belyaev ND, Kellett KA, Beckett C, Makova
NZ, Revett TJ, Nalivaeva NN, Hooper NM and Turner AJ: The
transcriptionally active amyloid precursor protein (APP)
intracellular domain is preferentially produced from the 695
isoform of APP in a {beta}-secretase-dependent pathway. J Biol
Chem. 285:41443–41454. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kerridge C, Belyaev ND, Nalivaeva NN and
Turner AJ: The Aβ-clearance protein transthyretin, like neprilysin,
is epigenetically regulated by the amyloid precursor protein
intracellular domain. J Neurochem. 130:419–431. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li S, Wang X, Ma QH, Yang WL, Zhang XG,
Dawe GS and Xiao ZC: Amyloid precursor protein modulates Nav1.6
sodium channel currents through a Go-coupled JNK pathway. Sci Rep.
6:393202016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hansen TB, Jensen TI, Clausen BH, Bramsen
JB, Finsen B, Damgaard CK and Kjems J: Natural RNA circles function
as efficient microRNA sponges. Nature. 495:384–388. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hébert SS and De Strooper B: Alterations
of the microRNA network cause neurodegenerative disease. Trends
Neurosci. 32:199–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lukiw WJ: Micro-RNA speciation in fetal,
adult and Alzheimer's disease hippocampus. Neuroreport. 18:297–300.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Liu W, Liu C, Yin B and Peng XZ: Functions
of miR-9 and miR-9* during aging in SAMP8 mice and their possible
mechanisms. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 37:253–258.
2015.PubMed/NCBI
|