Introduction
Mutations of isocitrate dehydrogenase (IDH) 1 and 2
have been reported to occur in most low-grade gliomas, acute
myeloid leukemias and other solid tumors (1–2).
IDH1 and 2 mutations disrupt the normal catalytic activity of the
protein and they acquire a novel function, allowing them to reduce
α-ketoglutarate (α-KG) into R-2-hydroxyglutarate (R-2HG) (1,3–6).
Because R-2HG and α-KG are structurally similar, 2-HG acts as a
competitive inhibitor of the α-KG-dependent dioxygenases involved
in epigenetic modifications, resulting in stem cell expansion and a
cell differentiation block (4,6–9).
Therefore, 2-HG functions as an oncometabolite to mediate
tumorigenesis in cancer with mutant IDH1 and 2. Moreover, IDH1 and
2 mutations have been acknowledged as good biomarkers and potential
drug targets (6–7).
There are three different isoforms of IDH; IDH1, 2
and 3. IDH1 is predominantly localized in the cytoplasm, while IDH2
and 3 are localized exclusively in the mitochondrial matrix.
Although they catalyze a similar reaction, the oxidative
decarboxylation of isocitrate into α-KG, they play different roles
in cellular metabolism. IDH3, as a rate-limiting enzyme of the
tricarboxylic acid (TCA) cycle, irreversibly catalyzes the
NAD-dependent decarboxylation of isocitrate to generate α-KG. IDH1
and 2 are homodimeric enzymes that reversibly catalyze the
oxidative decarboxylation of isocitrate to α-KG, as well as the
reductive carboxylation of α-KG to isocitrate. Therefore, IDH 1 and
2 play important roles in the regulation of the cellular redox
status, glutamine metabolism and lipogenesis (10–13).
Lipogenesis is an important metabolic process that
provides a cellular energy source and structural components. Under
normoxia, the precursor of fatty acid synthesis, acetyl coenzyme A
(AcCoA), is predominantly generated from glucose-derived pyruvate
via the glycolysis pathway. In mitochondria, the pyruvate
dehydrogenase (PDH) complex mediates the conversion of pyruvate
into AcCoA, which is then transferred into the cytosol and
participates in lipid synthesis (14). However, under hypoxic conditions,
the PDH complex is inhibited by hypoxia-inducible factor-1α
(HIF-1α), resulting in the impaired generation of AcCoA from
glucose (15). To support rapid
proliferation, cells have to alter their metabolic pathways to
accommodate the need for precursor AcCoA for fatty acid synthesis.
Like glucose, glutamine has been recognized as another important
source of carbon and nitrogen for biosynthetic reactions in
mammalian cells. Glutamine can supply carbon to AcCoA through two
major pathways. One pathway is described as glutaminolysis, wherein
glutamine enters the TCA cycle as α-KG and traverses in the forward
direction to maintain oxidative phosphorylation (13,16).
The other pathway is called reductive carboxylation, in which
glutamine enters the TCA cycle as α-KG and is converted to
isocitrate and citrate by reverse flux through the action of IDH1
and 2 (10–11,13).
In an hypoxic microenvironment, tumor cells actively reprogram
metabolism, and IDH1 and 2 play important roles in this process by
mediating the reductive carboxylation of α-KG into isocitrate,
producing AcCoA to maintain lipid synthesis and cell survival
(10,12–13).
IDH1 mutations, the most frequent of which is the
arginine 132 to histidine mutation (R132H), inhibit not only the
oxidative decarboxylation reaction but also the reductive
carboxylation reaction (3,5). Since tumor cells need to synthesize a
large amount of lipids to support constant proliferation, IDH1
R132H was hypothesized to impair reductive carboxylation and lipid
synthesis under hypoxic conditions, leading to reduced
proliferation (5,17). However, given that both IDH1 and 2
interchangeably support reductive carboxylation and lipid synthesis
under hypoxic conditions (5,18),
the hypothesis that IDH2 might compensate for defective lipid
synthesis and compromised cell proliferation in IDH1 R132H-mutant
tumor cells requires investigation. To test this hypothesis, the
present study determined the levels of IDH2 in IDH1 R132H-mutant
patient-derived primary glioma cells and HCT116 cells, both of
which harbor monoallelic IDH1 R132H, under normoxia and hypoxia.
Furthermore, cell proliferation and reductive glutamine metabolism
was examined in these cells with or without IDH2 knockdown. The
results of the present study demonstrated that IDH2 compensates for
IDH1 mutations to maintain reductive metabolism and the survival of
IDH1-mutant cancer cells under hypoxic conditions.
Materials and methods
Primary glioma cells, HCT116 cells and
glioma cell line U251 cell culture
Primary glioma cells were derived from fresh glioma
tissue collected during brain tumor surgery from patients with
grade-II astrocytomas; informed consent was obtained from the
patients. Fresh tumor tissue was mechanically dissociated into 1
mm3 pieces in fresh DMEM (Gibco; Thermo Fisher
Scientific, Inc.). The tumor fragments were placed at the bottom of
culture flasks containing 10 ml DMEM supplemented with penicillin,
streptomycin and 10% FBS (Gibco; Thermo Fisher Scientific, Inc.),
and incubated at 37°C and 5% CO2. The medium was
replaced 3–5 days later to remove floating cells and tissues. The
two individual patient-derived primary glioma cells, which were
classified as grade-II astrocytomas, were confirmed to harbor
either wild type (WT) IDH1 or monoallelic IDH1 R132H using genomic
DNA sequencing (Sangon Biotech, Co., Ltd.) using the following
primers: 5′-gcgtcaaatgtgccactatc-3′ and 5′-cctttagctaaatgtgtgta-3′.
Experiments on these cells were performed within five passages.
Human colon cancer HCT116IDH1WT and
HCT116IDH1R132H/+ cells, which harbor WT IDH1 and one
allele of IDH1 R132H, respectively, were provided by Dr. Jing Ye
(Department of Pathology, the Fourth Military Medical University)
and cultured in McCoy's 5A modified medium (Gibco; Thermo Fisher
Scientific, Inc.) with 10% FBS. Human glioma cell lines U251 cells
(Chinese Academy of Sciences Cell Bank), were cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS.
For experiments under normoxia, cells were maintained in 21%
O2, 74% N2 and 5% CO2. For
experiments under hypoxic conditions, cells were cultured under
normoxia (21% O2 at 37°C) for 6 h to allow cells to
adhere to the flask and were then moved to a hypoxic environment
(3% O2, 92% N2 and 5% CO2 at
37°C).
Lentivirus packaging, cell infection
and selection
The pLenti6, pLenti6-IDH1 and pLenti6-IDH1R132H
vectors were generously gifted by Dr Jing Ye (Department of
Pathology, The Fourth Military Medical University, Xi'an, China).
For lentiviral packaging, 293T cells (Chinese Academy of Sciences
Cell Bank) were seeded in 10 cm cell culture dishes and on reaching
~60% confluence, the cells were transfected with the packaging
system (7.5 µg psPAX2, 3.5 µg pVSVG, 10 µg pLenti plasmid) using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol.
Forty-eight hours later, the supernatant medium containing the
virus was collected. The virus solution and fresh culture medium
(DMEM supplemented with 10% FBS) were added to U251 cells in a 1:1
mixture when the cells reached 50–70% confluence. Twenty-four hours
later, cells were selected with puromycin (0.5 µg/ml;
Sigma-Aldrich; Merck KGaA) for two weeks to generate stable U251
cell lines expressing wild-type IDH1 or IDH1R132H.
Small interfering (si) RNAs and
transfection
siRNAs that target two individual sites of the human
IDH2 coding sequence were obtained from Shanghai GenePharma Co.,
Ltd. siIDH2-1, 5′-GCAAGAACUAUGACGGAGA-3′; and siIDH2-2,
5′-CCCGUGUGGAAGAGUUCAATT-3′. Nontargeting siRNA
(5′-UUCUCCGAACGUGUCACGUTT-3′; Shanghai GenePharma Co., Ltd.) was
used as a negative control. Cells were plated at a density of
1×105 into 6-well plates in DMEM without antibiotics.
The cells were transfected 24 h after plating with 50 nM siRNA
targeting human IDH2 or nontargeting siRNA (scramble) as a negative
control. Transfections were performed with
Lipofectamine® 2000, according to the manufacturer's
protocol. Forty-eight hours following transfection, the knockdown
efficiency was determined by western blot analysis.
Proliferation assay
Cells were seeded in sextuplicate at a density of
2,000 cells/well in 96-well plates, and the medium was changed
every 3 days. The number of viable cells/well at each time point
was determined using a Cell Counting Kit-8 assay (CCK-8; Dojindo
Molecular Technologies, Inc.), according to the manufacturer's
protocol. Absorbance at 450 nm was measured 1 h after the addition
of the CCK-8 solution.
Western blot analysis
Proteins were extracted using protein lysis buffer
(Beyotime Institute of Biotechnology) in the presence of a protease
inhibitor cocktail (Roche Diagnostics). The protein concentration
was determined using the bicinchroninic acid Protein Assay kit
(Thermo Fisher Scientific, Inc.). Equal amounts of protein samples,
20 µg/lane, were separated by electrophoresis on 10% SDS-PAGE gels
and were electrotransferred to nitrocellulose membranes. The
membranes were blocked with 5% milk for 1.5 h at room temperature
and then incubated with primary antibodies against IDH2 (1:500;
cat. no. ab55271; Abcam), GAPDH (1:2,000; cat. no. 10494-1-AP;
Wuhan Sanying Biotechnology) and HIF-1α (1:1,000; cat. no. 14179;
Cell Signaling Technology, Inc.) at 4°C overnight, followed by
incubation with horseradish peroxidase-conjugated secondary
antibodies (1:5,000; cat. nos. CW0102 and CW0103; Wuhan Sanying
Biotechnology) for 1 h at room temperature. The signals were
detected with an ECL substrate kit (Thermo Fisher Scientific,
Inc.), and the immunoreactive bands were visualized using a
gel-imaging analysis system (Tanon Science and Technology Co.,
Ltd.). The band intensity was quantified using ImageJ software
(version 1.48; National Institutes of Health), wherein the relative
values, using GAPDH as an internal control, of the first bands were
designated as 1.
Metabolic flux analysis by gas
chromatography mass spectrometry (GC-MS)
HCT116IDH1R132H/+ cells, at a density of
1×106, were cultured in serum-reduced DMEM and
transfected with 50 nM IDH2-siRNA-1 or scramble using
Lipofectamine® 2000. After 48 h, the cells were cultured
in medium containing 1 mM uniformly 13C-labeled [U-13C]
glutamine under hypoxic conditions for 24 h, after which cells were
collected for metabolic extraction. GC-MS analysis of metabolic
flux was performed by Shanghai Biotech Co., Ltd., and the detailed
procedure is described in the supplementary materials and
methods.
Statistical analysis
Statistical analysis was performed using SPSS
software (version 17; IBM Corp.). Each experiment was repeated
independently a minimum of three times. Data are presented as the
mean ± SEM for multiple independent experiments or the mean ± SD
for technical replicates. Independent Student's t-test or one-way
ANOVA followed by Fisher's least significant difference test were
used to compare the continuous variables between two groups or
among more than two groups, respectively. P<0.05 was considered
to indicate a statistically significant difference.
Results
Mutant IDH1 cells grow more slowly
than WT cells under hypoxic conditions
As IDH1 mutations reduce the ability of the enzyme
to catalyze the bidirectional conversion between isocitrate and
α-KG (5), it was speculated that
an IDH1 mutation would lead to compromised reductive metabolism and
proliferation under hypoxic conditions. To test this hypothesis,
the proliferation of IDH1 R132H-harboring patient-derived primary
glioma cells was examined, which was previously established and
confirmed using DNA sequencing (Fig.
S1), H&E staining and immunohistochemical staining
(19). HCT116 cells that harbor
one allele of IDH1 R132H (Fig.
S2) were also used to test this hypothesis. Cells were cultured
under normoxia or hypoxia for 3–5 days and subjected to a CCK-8
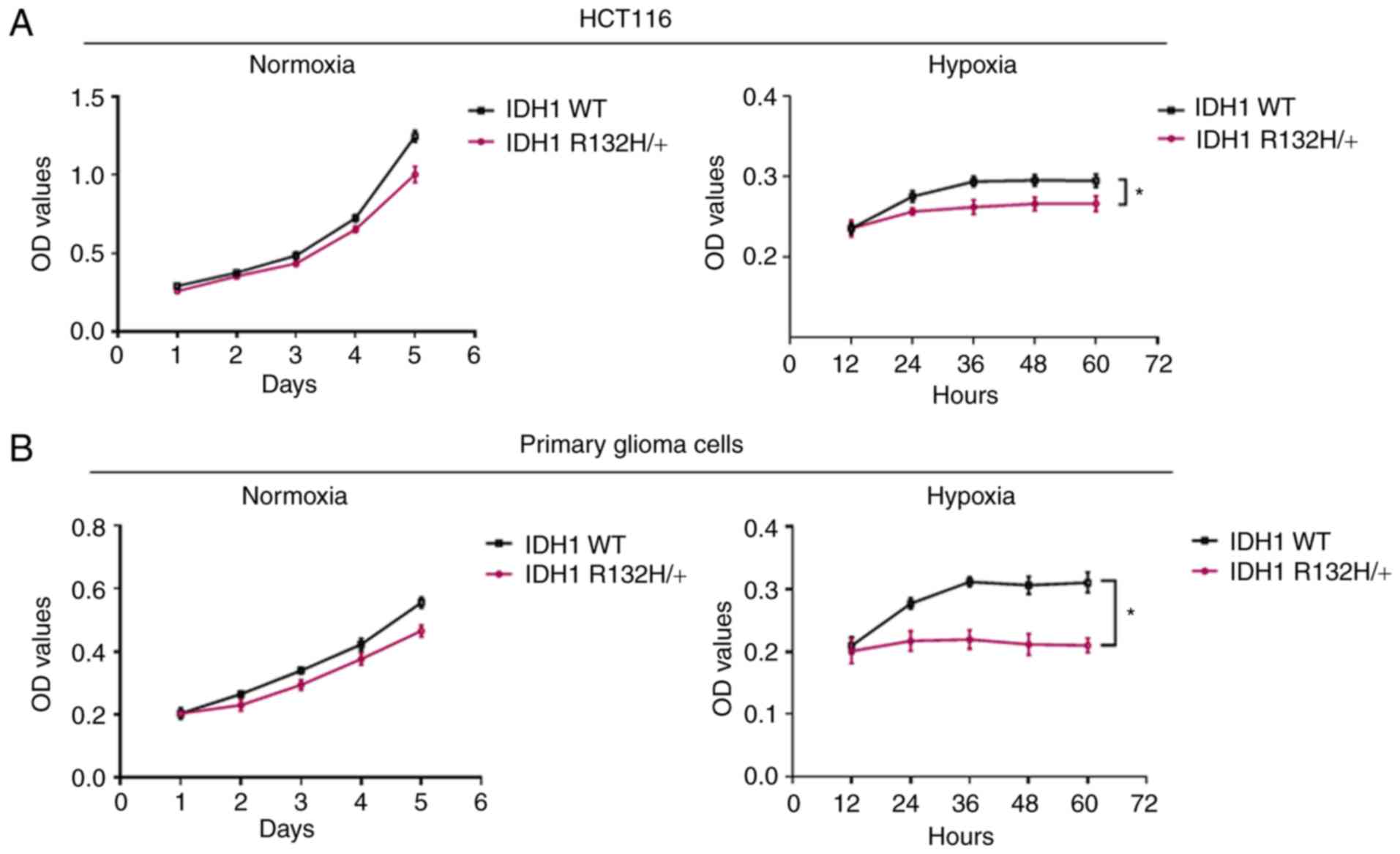
assay. Hypoxic conditions led to elevated HIF-1α levels (Fig. S3). As shown in Fig. 1, under normoxia, IDH1-mutant
primary glioma cells and HCT116R132H/+ cells grew more
slowly than WT IDH1 primary glioma cells and HCT116WT
cells, respectively; however, these IDH1-mutant cells exhibited
significantly repressed proliferation under hypoxic conditions
compared with the proliferation of the WT counterpart. These
results showed that IDH1 R132H cells are defective and exhibit
reduced growth under hypoxic conditions.
R132H-mutant cancer cells upregulate
IDH2 protein expression under hypoxic conditions
Both IDH1 and 2 are required to maintain the reverse
TCA cycle for lipogenesis under hypoxic conditions (10,13,18).
As the growth of IDH1 R132H-mutant tumor cells was markedly
inhibited under hypoxic conditions, it was hypothesized that these
cells may compensate for the impaired lipid synthesis by
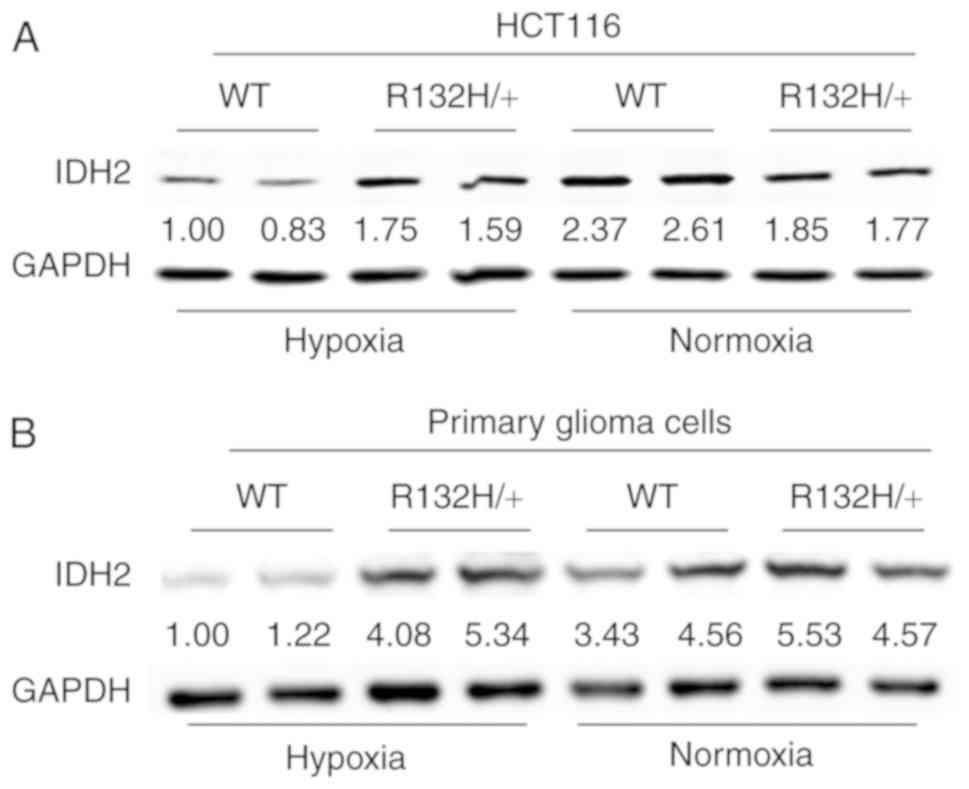
overexpressing IDH2. To test this hypothesis, the expression of
IDH2 in WT and IDH1R132H/+ HCT116 cells and primary
glioma cells was examined by western blot analysis. As shown in
Fig. 2A and B, IDH2 was
upregulated in both HCT116R132H/+ and IDH1-mutant
primary glioma cells relative to the expression in their WT
counterparts under hypoxic conditions. Stable WT IDH1- and IDH1
R132H- overexpressing U251 cell lines, widely used as a
gliobalstoma cell model (20),
were established via lentivirus infection. Ectopic expression of
IDH1 R132H consistently led to IDH2 upregulation under hypoxia
compared with WT IDH1 (Fig. S4).
These results suggested that IDH1-mutant cells may compensate for
the adverse effects of an IDH1 mutation by upregulating IDH2 under
hypoxic conditions.
Knockdown of IDH2 inhibits the growth
of IDH1-mutant cells under hypoxic conditions
Although the IDH1-mutant cells were defective and
grew more slowly under hypoxic conditions, these cells remained
viable as shown in Fig. 1
(17). In the present study, it
was also demonstrated that IDH1-mutant cells overexpressed IDH2 in
hypoxic conditions (Fig. 2),
suggesting that overexpression of IDH2 could compensate for the
adverse effects caused by mutation of IDH1. Therefore, whether the
knockdown of IDH2 would affect cell growth in these cells under
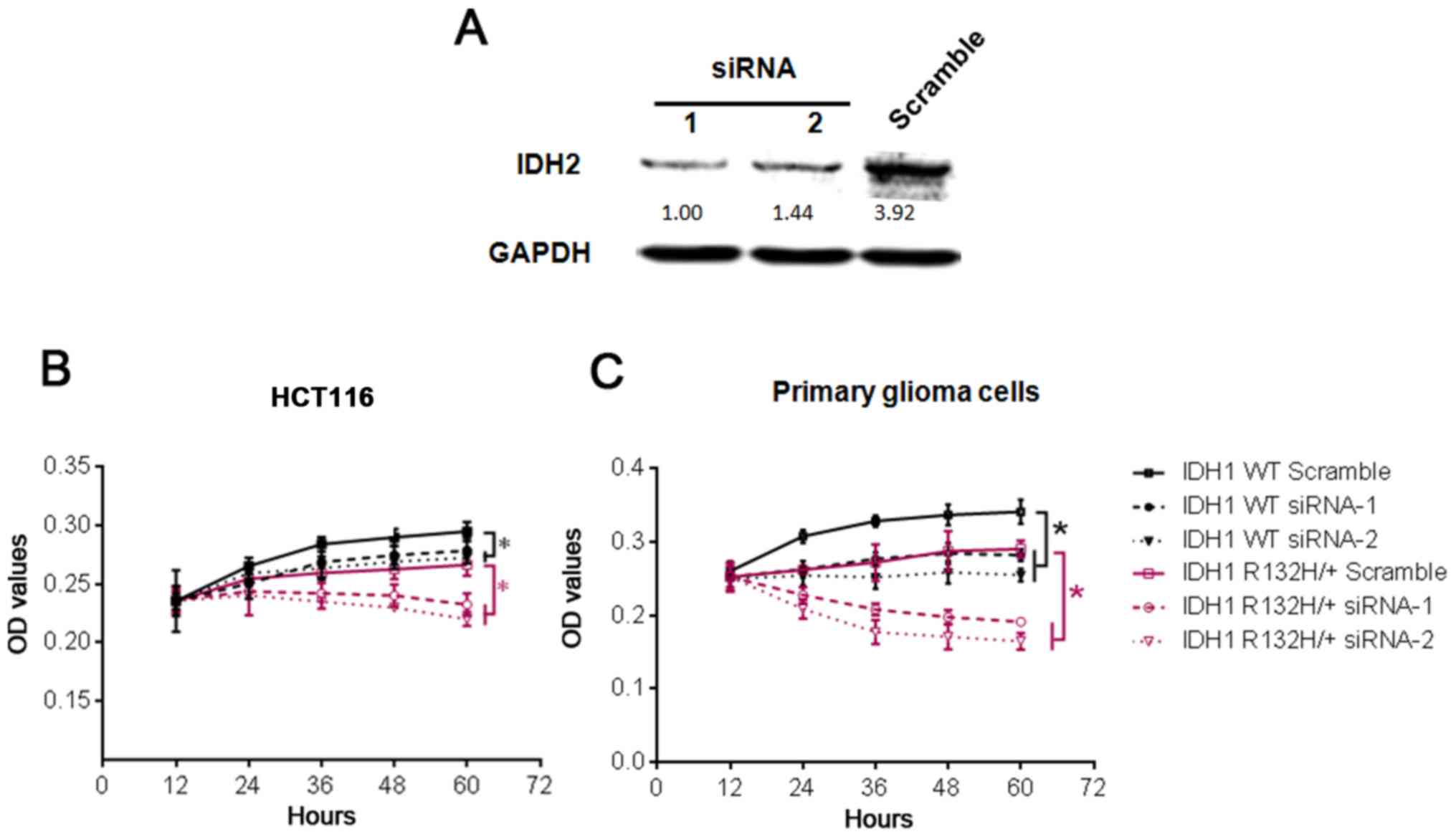
hypoxic conditions was examined. For this purpose, two individual
IDH2-siRNAs were transfected into WT HCT116 cells to determine
their efficiency; the results showed that both siRNAs reduced IDH2
protein levels by ~50% (Fig. 3A).
WT and IDH1132H/+ HCT116 cells and primary glioma cells
were transfected with IDH2-siRNA or scramble siRNA, and then
cultured under hypoxic conditions to examine proliferation. As
shown in Fig. 3B and C, the
inhibitory effect of IDH2 siRNAs on cell growth was comparable to
that of the IDH1 R132H mutation in both the HCT116 and primary
glioma cells. Moreover, IDH2 knockdown further slowed the growth of
IDH1 R132H-mutant cells under hypoxic conditions. There data
indicated that IDH2 knockdown exacerbated the growth defect in IDH1
R132H-mutant cells under hypoxic conditions.
Knockdown of IDH2 reduces the
reductive carboxylation of IDH1-mutant cells in hypoxic
conditions
As IDH1 and 2 are both involved in reductive
carboxylation and lipogenesis under hypoxic conditions, it was
reasoned that IDH2 knockdown may exacerbate the growth defect in
hypoxic IDH1-mutant cells as a result of compromised reductive
carboxylation. To detect the effect of IDH2 knockdown on reductive
carboxylation, IDH1R132H/+ HCT116 cells were transfected
with IDH2 siRNA1 or scramble siRNA and cultured in the presence of
[U-13C5]glutamine under hypoxic conditions,
and then collected for analysis of metabolic flux by GC-MS. The
labeling metabolites derived from
[U-13C5]glutamine were different between
oxidative and reductive metabolism; among the labeling metabolites,
M5 citrate (Cit), M3 aspartate (Asp), M3 malate (Mal) and M3
fumarate (Fum) represented reductive metabolism, whereas M4 Asp, M4
Mal and M4 Fum represented oxidative metabolism (Fig. 4A). As shown in Fig. 4B, knockdown of IDH2 in
IDH1R132H/+ HCT116 cells led to a significant decrease
in the percentage of Cit (M5), Asp (M3), Mal (M3) and Fum (M3).
These results suggested that IDH2 knockdown reduces reductive
carboxylation under hypoxic conditions, thus impairing lipogenesis
and cell growth in hypoxic IDH1-mutant cells.
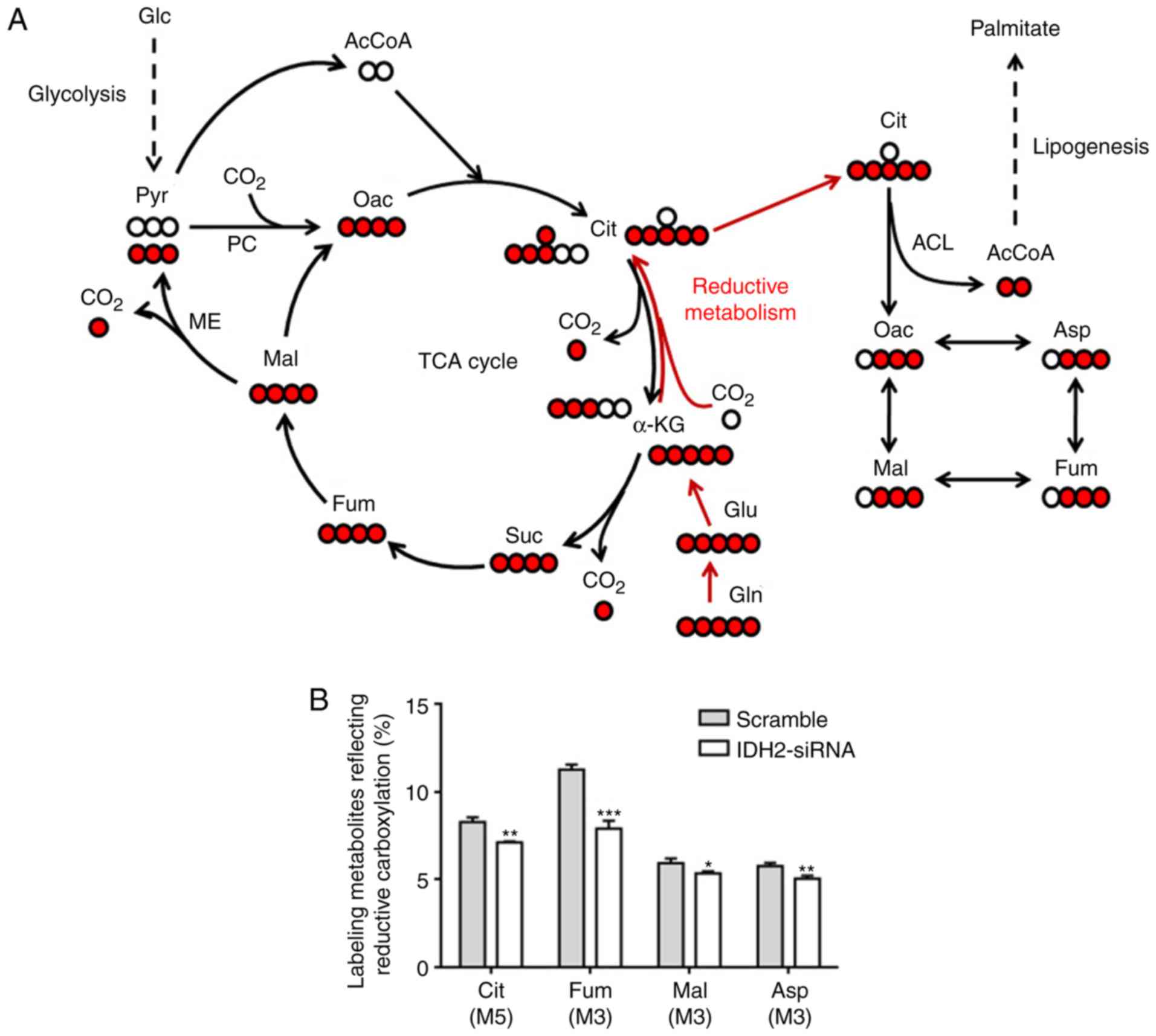 | Figure 4.Knockdown of IDH2 reduces the
reductive carboxylation of IDH1-mutant cells in hypoxic conditions.
(A) A schematic diagram of carbon atom (circles) transitions,
showing labeling metabolites derived from
[U-13C5]glutamine through oxidative (black
arrows) and reductive (red arrows) pathways. Isotopic-labeled
metabolites generated through the reductive carboxylation pathway
include M5 Cit, M3 Asp, M3 Mal and M3 Fum. Red circles represent
13C carbon atoms, white circles represent
non-13C marked carbon atoms. (B) HCT116
IDH1R132H/+ cells were transfected with IDH2 siRNA1 or
scramble siRNA and cultured in the presence of
[U-13C5] glutamine for 24 h under hypoxic
conditions. The cells were then collected for gas
chromatography-mass spectrometry analysis of the metabolites. The
relative abundance of reductive carboxylation-specific mass
isotopomers are shown as the mean ± SD (n=3). *P<0.05,
**P<0.01, ***P<0.005. IDH2, isocitrate dehydrogenase 2;
siRNA, small interfering RNA; TCA, tricarboxylic acid; Glc,
glucose; Pyr, pyruvate; AcCoA, acetyl co-enzyme A; Oac,
oxaloacetate; Cit, citrate; Mal, malate; Asp, aspartate; Fum,
fumarate; α-KG, α-ketoglutarate; Suc, succinate; Gln, glutamine;
Glu, glutamate; PC, pyruvate carboxylase; ME, malic enzyme; ACL,
ATP citrate lyase. |
Discussion
The discovery of IDH1 mutations has been an
important breakthrough in the field, with the mechanism of IDH1
mutations and targeted therapy becoming popular research topics
(2,7). Previous studies have established that
2-HG is produced by mutant-IDH1 and causes profound epigenetic
modifications and expansion of stem cells, and blocks
differentiation (1,3–4,8–9,21).
Small molecule mutant-IDH1 inhibitors have been developed and are
at different stages of clinical trials; however, the currently
available preclinical results have not been as promising as
expected (2). It has been reported
that targeting mutant IDH1 does not alter intracranial glioma
growth in murine models (22).
Although the blood-brain barrier or the irreversibility of the IDH
mutation-induced epigenetic changes may account for the limited
efficacy of IDH1 inhibitors (2),
other alterations in cellular processes, including metabolism and
signaling pathways, caused by mutant-IDH1, also play important
roles in tumor progression. Therefore, it is important to
understand the mechanism of mutant-IDH1 and to develop other
therapeutic strategies. For the first time, to the best of our
knowledge, the present study demonstrated that IDH1 R132H-mutant
cells upregulate IDH2 as a compensatory mechanism to maintain
reductive glutamine metabolism-dependent lipogenesis and cell
proliferation under hypoxic conditions. As a result, the knockdown
of IDH2 significantly inhibits the proliferation of IDH1
R132H-mutant cells under hypoxic conditions. Thus, IDH2 inhibition
may serve as a new approach for the treatment of
mutant-IDH1-harboring tumors.
Although mutations in IDH1 have been hypothesized to
cause tumorigenesis, IDH1 is also a positive prognostic marker for
patients with glioma (23). The
finding that IDH1 R132H inhibited the growth of glioma cell lines
(24) and murine glioma progenitor
cells (25) was proposed to result
in a positive effect on patient survival (24). However, a previous study postulated
that the survival benefit gained by IDH1 mutation is a result of
sensitivity to temozolomide, rather than biological behavior
(26). By contrast, Koivunen et
al (27) reported that R-2HG
produced by mutant-IDH1 enhanced the proliferation and soft agar
growth of human astrocytes. A recent study showed that IDH1
R132H-knock-in in the mouse subventricular zone promoted the
self-renewal and proliferation of neural stem cells (8). As such, the current opinions about
mutant-IDH1 are contradictory, which may be due to the different
cell types and contexts examined in previous studies.
In the present study, two IDH1 R132H-mutant cell
models were used as models, including primary glioma cells and
HCT116R132H/+ cells, both of which harbor a single
allelic IDH1 R132H mutation and consistently reflect the genetic
IDH1 mutation status in patients. In the present study, IDH1-mutant
cells were found to grow more slowly than their WT counterparts,
particularly under hypoxic conditions. As mutations in IDH1
mitigate the oxidative and reductive activities of the enzyme
(3,5), it was concluded that the defective
reductive glutamine metabolism caused by IDH1 mutation leads to
reduced cell proliferation. This view is consistent with that of
Grassian et al (17), who
showed that cancer cells with a mutant IDH1 allele are unable to
induce reductive glutamine metabolism under hypoxic conditions,
thus compromising AcCoA and lipid production, leading to decreased
cell growth. Therefore, it is proposed that the defective cell
growth may account for the better prognosis of patients with IDH1
R132H mutation compared with those with WT-IDH1. Large-scale
studies have established that IDH mutations together with other
molecules are important markers for glioma classification and
prognosis (6). A recent study,
using genome-wide gene expression profiling, established a six-gene
signature as an independent prognostic factor for IDH1-mutant
glioma, and showed that the effect of IDH1 mutation is conserved
across histological classifications; by combining histology grade,
IDH1 status and the six-gene signature in all grades of glioma,
patients could be stratified into six subgroups with distinct
prognoses (28). Therefore, it
would be interesting to observe the growth attributes of primary
glioma cells derived from these different groups.
In the present study, data presented suggest that
IDH2 was upregulated in both HCT116 IDH1R132H/+ cells
and IDH1-mutant primary glioma cells under hypoxic conditions. This
finding suggests that cancer cells can compensate for the adverse
effects caused by mutation of IDH1 by upregulating IDH2 under
hypoxic conditions. This allows processes, including reductive
carboxylation and lipogenesis, that are required for cell
proliferation, to continue. This is consistent with the findings of
Mustafa et al (29), who
examined the genes participating in the TCA cycle and anaerobic
glycolysis in 33 IDH1 mutated and 39 IDH1 WT glioma samples and
found that the expression levels of several genes were different,
among which IDH2 was reported to be upregulated. However, Chen
et al (25) reported no
change in IDH2 expression in the expression profiling data of 46
IDH1 R132H high-grade gliomas compared with 163 WT-IDH1 high-grade
gliomas. Such a discrepancy may result from the different datasets
analyzed or the different oxygen concentrations the glioma samples
were exposed to. The findings of the present study showed that IDH2
was upregulated in IDH1-mutant cells under hypoxic conditions;
therefore, IDH2 may serve as a therapeutic target for the treatment
of IDH1-mutant cancer. Consistent with this proposed treatment
strategy, the present study found that targeting IDH2 with siRNAs
significantly decreased reductive carboxylation and inhibited the
proliferation of cancer cells with a monoallelic IDH1 mutation.
A limitation of the present study is the use of a
limited sample number and tumor grading, a WT and mutant grade-II
astrocytoma. However, similar results were obtained with WT and
IDH1 R132H/+ HCT116 cells. IDH2 upregulation was also observed in
the stable U251 glioblastoma cell line, which overexpresses IDH1
R132H, but not WT IDH1, under hypoxic conditions. Furthermore, it
was proposed in a genome-wide gene expression profiling study that
the effect of mutations in IDH1 is conserved across histological
classifications (28). Therefore,
the compensatory role played by IDH2 could apply to a wider range
of cancer cells containing mutations in IDH1. Previous studies have
shown that mutation of IDH1 disrupts the reductive activity used to
generate citrate for lipid synthesis (5), and also increases glutaminolysis to
form lipids (6,25,30),
indicating the presence and importance of compensatory mechanisms.
Glioma cells with mutant IDH1 were found to be sensitive to the
inhibition of glutaminase or glutamate dehydrogenase (25,31).
Therefore, the results of the present study together with data
presented in the literature provide potential and promising
therapeutic strategies that exploit the compensatory mechanisms
caused by the mutation of IDH1. Further systematic studies,
including in vivo examination of the effects of siRNAs
targeting the compensatory molecules, are required in the future.
In addition, future studies should investigate whether IDH1 can
compensate for mutations of IDH2 in IDH2 mutant malignancies.
In conclusion, the present study reports, for the
first time, to the best of our knowledge, that IDH2 compensates for
the IDH1 R132H mutation to maintain cell survival under hypoxic
conditions in IDH1-mutant tumor cells. Thus, IDH2 may serve as a
potential antitumor target for IDH1 mutant tumors.
Supplementary Material
Supporting Data
Acknowledgements
The authors thank Dr Jing Ye and Mr. Chao Wang (The
Fourth Military Medical University) for their kind assistance with
the IDH1R132H/+ HCT116 cells and other experimental
materials used in this present study.
Funding
This study was supported by grants from the National
Natural Science Foundation of China (grant nos. 81572504 and
81572469), the Natural Science Foundation of Shaanxi Province
(grant no. 2016JZ028), and the Science and Technology Reseach and
Development Program of Shaanxi Province (grant no. 2016SF-094).
Availability of data and materials
The materials used in the current study are
available from the corresponding author on reasonable request.
Authors' contributions
YZ, WLv and QL performed the primary glioma cell
culture and cellular experiments. QW and YR conducted the
statistical analysis. FY, TP and XX performed routine cell culture
transfections. WLi and XL designed the study and wrote the
manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Xijing Hospital, the Fourth Military Medical
University (approval no. 20180122). All human subjects recruited
for this study provided written informed consent prior to
participation.
Patient consent for publication
All human subjects whose samples were used for this
study provided written informed consent for publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yan H, Parsons DW, Jin G, McLendon R,
Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ,
et al: IDH1 and IDH2 mutations in gliomas. N Engl J Med.
360:765–773. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Waitkus MS, Diplas BH and Yan H:
Biological role and therapeutic potential of IDH mutations in
cancer. Cancer Cell. 34:186–195. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dang L, White DW, Gross S, Bennett BD,
Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et
al: Cancer-associated IDH1 mutations produce 2-hydroxyglutarate.
Nature. 462:739–744. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang
P, Yu W, Li Z, Gong L, Peng Y, et al: Glioma-derived mutations in
IDH1 dominantly inhibit IDH1 catalytic activity and induce
HIF-1alpha. Science. 324:261–265. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Leonardi R, Subramanian C, Jackowski S and
Rock CO: Cancer-associated isocitrate dehydrogenase mutations
inactivate NADPH-dependent reductive carboxylation. J Biol Chem.
287:14615–14620. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Waitkus MS, Diplas BH and Yan H:
Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol.
18:16–26. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang H, Ye D, Guan KL and Xiong Y: IDH1
and IDH2 mutations in tumorigenesis: Mechanistic insights and
clinical perspectives. Clin Cancer Res. 18:5562–5571. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bardella C, Al-Dalahmah O, Krell D,
Brazauskas P, Al-Qahtani K, Tomkova M, Adam J, Serres S, Lockstone
H, Freeman-Mills L, et al: Expression of Idh1R132H in
the murine subventricular zone stem cell niche recapitulates
features of early gliomagenesis. Cancer Cell. 30:578–594. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan
S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et
al: IDH mutation impairs histone demethylation and results in a
block to cell differentiation. Nature. 483:474–478. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Filipp FV, Scott DA, Ronai ZA, Osterman AL
and Smith JW: Reverse TCA cycle flux through isocitrate
dehydrogenases 1 and 2 is required for lipogenesis in hypoxic
melanoma cells. Pigment Cell Melanoma Res. 25:375–383. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Koh HJ, Lee SM, Son BG, Lee SH, Ryoo ZY,
Chang KT, Park JW, Park DC, Song BJ, Veech RL, et al: Cytosolic
NADP+-dependent isocitrate dehydrogenase plays a key role in lipid
metabolism. J Biol Chem. 279:39968–39974. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wise DR, Ward PS, Shay JE, Cross JR,
Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC and
Thompson CB: Hypoxia promotes isocitrate dehydrogenase-dependent
carboxylation of α-ketoglutarate to citrate to support cell growth
and viability. Proc Natl Acad Sci USA. 108:19611–19616. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Metallo CM, Gameiro PA, Bell EL, Mattaini
KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L,
et al: Reductive glutamine metabolism by IDH1 mediates lipogenesis
under hypoxia. Nature. 481:380–384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hatzivassiliou G, Zhao F, Bauer DE,
Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA and
Thompson CB: ATP citrate lyase inhibition can suppress tumor cell
growth. Cancer Cell. 8:311–321. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim JW, Tchernyshyov I, Semenza GL and
Dang CV: HIF-1-mediated expression of pyruvate dehydrogenase
kinase: A metabolic switch required for cellular adaptation to
hypoxia. Cell Metab. 3:177–185. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wise DR, DeBerardinis RJ, Mancuso A, Sayed
N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon
SB and Thompson CB: Myc regulates a transcriptional program that
stimulates mitochondrial glutaminolysis and leads to glutamine
addiction. Proc Natl Acad Sci USA. 105:18782–18787. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Grassian AR, Parker SJ, Davidson SM,
Divakaruni AS, Green CR, Zhang X, Slocum KL, Pu M, Lin F, Vickers
C, et al: IDH1 mutations alter citric acid cycle metabolism and
increase dependence on oxidative mitochondrial metabolism. Cancer
Res. 74:3317–3331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mullen AR, Wheaton WW, Jin ES, Chen PH,
Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS and
DeBerardinis RJ: Reductive carboxylation supports growth in tumour
cells with defective mitochondria. Nature. 481:385–388. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Y, Wei X, Wang Q, Zhang M, L X and
Lin W: Culture of primary human astrocytoma cell with isocitrate
dehydrogenase1 (IDH1) R132H mutation. J Modern Oncol. 25:843–847.
2017.(In Chinese).
|
|
20
|
Fujikawa A, Sugawara H, Tanaka T,
Matsumoto M, Kuboyama K, Suzuki R, Tanga N, Ogata A, Masumura M and
Noda M: Targeting PTPRZ inhibits stem cell-like properties and
tumorigenicity in glioblastoma cells. Sci Rep. 7:56092017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Turcan S, Rohle D, Goenka A, Walsh LA,
Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, et al: IDH1
mutation is sufficient to establish the glioma hypermethylator
phenotype. Nature. 483:479–483. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pusch S, Krausert S, Fischer V, Balss J,
Ott M, Schrimpf D, Capper D, Sahm F, Eisel J, Beck AC, et al:
Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of
IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 133:629–644.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Leu S, von Felten S, Frank S, Vassella E,
Vajtai I, Taylor E, Schulz M, Hutter G, Hench J, Schucht P, et al:
IDH/MGMT-driven molecular classification of low-grade glioma is a
strong predictor for long-term survival. Neuro Oncol. 15:469–479.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bralten LB, Kloosterhof NK, Balvers R,
Sacchetti A, Lapre L, Lamfers M, Leenstra S, de Jonge H, Kros JM,
Jansen EE, et al: IDH1 R132H decreases proliferation of glioma cell
lines in vitro and in vivo. Ann Neurol. 69:455–463. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen R, Nishimura MC, Kharbanda S, Peale
F, Deng Y, Daemen A, Forrest WF, Kwong M, Hedehus M, Hatzivassiliou
G, et al: Hominoid-specific enzyme GLUD2 promotes growth of
IDH1R132H glioma. Proc Natl Acad Sci USA. 111:14217–14222. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Houillier C, Wang X, Kaloshi G, Mokhtari
K, Guillevin R, Laffaire J, Paris S, Boisselier B, Idbaih A,
Laigle-Donadey F, et al: IDH1 or IDH2 mutations predict longer
survival and response to temozolomide in low-grade gliomas.
Neurology. 75:1560–1566. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Koivunen P, Lee S, Duncan CG, Lopez G, Lu
G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, et al:
Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked
to EGLN activation. Nature. 483:484–488. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cheng W, Ren X, Zhang C, Cai J, Han S and
Wu A: Gene expression profiling stratifies IDH1-mutant glioma with
distinct prognoses. Mol Neurobiol. 54:5996–6005. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mustafa DA, Swagemakers SM, Buise L, van
der Spek PJ and Kros JM: Metabolic alterations due to IDH1 mutation
in glioma: Opening for therapeutic opportunities? Acta Neuropathol
Commun. 2:62014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reitman ZJ, Duncan CG, Poteet E, Winters
A, Yan LJ, Gooden DM, Spasojevic I, Boros LG, Yang SH and Yan H:
Cancer-associated isocitrate dehydrogenase 1 (IDH1) R132H mutation
and d-2-hydroxyglutarate stimulate glutamine metabolism under
hypoxia. J Biol Chem. 289:23318–23328. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Seltzer MJ, Bennett BD, Joshi AD, Gao P,
Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS,
Rabinowitz JD, et al: Inhibition of glutaminase preferentially
slows growth of glioma cells with mutant IDH1. Cancer Res.
70:8981–8987. 2010. View Article : Google Scholar : PubMed/NCBI
|