Introduction
Triple negative breast cancer (TNBC) is one of the
most aggressive subtypes of breast cancer, which is characterized
by the lack of estrogen receptor, progesterone receptor and human
epidermal growth factor receptor 2 (1–4). Due
to these clinical features, the response of patients with TNBC to
conventional therapies remains unsatisfactory. Therefore,
identifying novel drugs that may be applied in the treatment of
TNBC and furthering our understanding of the underlying molecular
mechanisms are urgently required.
Aberrant gene expression contributes substantially
to the initiation and development of cancer in humans. It is well
documented that epigenetic alternations regulate gene expression
(5). In particular, the
acetylation of histone proteins, which is catalyzed by histone
acetyltransferases and histone deacetylases (HDACs), is the most
extensively investigated epigenetic modification (6,7).
HDACs have been revealed to be upregulated in a variety of human
cancer types, promoting the progression of cancer (6–12).
An increased expression of HDACs has been correlated with a poorer
prognosis in patients with cancer. Previous studies have also
demonstrated that disrupting the expression of HDACs with an HDAC
inhibitor (HDACi) markedly suppressed the progression of cancer
(13–16). These HDACis included quisinostat,
entinostat and chidamide (17–22).
It has been reported that chidamide is an inhibitor of class I
HDACs, with specificity in targeting HDAC 1, 2, 3 and 10. The
anticancer effects of chidamide have been reported in pancreatic
cancer, non-small cell lung cancer, colon cancer and NK/T lymphoma
cells (19–23). However, the function of chidamide
and the underlying molecular mechanisms by which chidamide
regulates the proliferation of TNBC cells remain to be fully
elucidated.
MicroRNAs (miRNAs/miRs) are a class of small,
non-coding RNAs with a length of 18–24 nucleotides, which modulate
gene expression by binding to the 3′-untranslated region (3′-UTR)
of downstream targets (24–26).
The interactions between miRNAs and the 3′-UTR of target genes
result in the degradation or translational inhibition of mRNAs
(25). The aberrant expression of
miRNAs is involved in the progression of human diseases. A recent
study showed that renal miR-214-3p serves a functional role in the
development of hypertension (27).
miR-192-5p in the kidney protects against the development of
hypertension (28). An increasing
body of evidence demonstrates that miRNAs serve important roles in
regulating the development of cancer by acting as tumor suppressors
or oncogenes (29,30). The aberrant expression of miRNAs
confers resistance to drug treatment, therefore, modulating the
expression of miRNAs has become a critical target of drugs in order
to suppress the growth of cancer cells. As a specific
characteristic of cancer, cancer cells metabolize glucose via
aerobic glycolysis rather than mitochondrial aerobic respiration,
even in conditions with sufficient oxygen (31,32).
The process of glycolysis is sequentially catalyzed by several
enzymes, including glucose transporters, glucose-6-phosphate
dehydrogenase and lactate dehydrogenase A (LDHA) (33). To regulate the growth of cancer
cells, miRNAs have been found to modulate the expression of enzymes
associated with glycolysis, which in turn regulates the metabolism
of cancer cells. For example, miR-142-3p was shown to inhibit the
aerobic glycolysis of hepatocellular carcinoma by targeting LDHA
(34). Additionally, miR-30a-5p
was reported to suppress the LDHA-mediated glucose metabolism of
breast cancer cells (35). These
studies indicate that miRNAs target LDHA and modulate the
progression of cancer.
miR-33a-5p has been reported to be downregulated in
melanoma, osteosarcoma and hepatocellular carcinoma, acting as a
tumor suppressor in regulating the growth of cancer cells (36,37).
miR-33a-5p has been shown to increase the radiosensitivity of
melanoma cells by inhibiting glycolysis (37). However, the expression and function
of miR-33a-5p in TNBC remain unknown. In the present study, the
results revealed that chidamide treatment suppressed the
proliferation of TNBC cells. Further molecular investigations
revealed that chidamide upregulated the expression of miR-33a-5p,
which targeted LDHA and suppressed the glucose metabolism of TNBC
cells. The results also demonstrated the possible functional
mechanism of chidamide in inhibiting the growth of TNBC, which may
be considered as a promising drug in the treatment of patients with
TNBC.
Materials and methods
Clinical cancer tissues and cell
lines
A total of 20 paired TNBC tissues and adjacent
normal tissues were collected from patients with TNBC who had
undergone surgical resection at Beijing Chaoyang Hospital between
April 2013 and August 2014 (age: 29–66 years old; all patients were
female and diagnosed with TNBC). The tissue samples were confirmed
by three pathologists independently. None of these patients had
received treatment prior to surgery. Written informed consent was
obtained from all of participants prior to tissue collection. The
samples were maintained at −80°C until subsequent use. The Ethics
Committee of The Affiliated Hospital of Capital Medical University
(Beijing, China) approved the present study.
The MDA-MB-231 and BT-20 TNBC cell lines were
purchased from American Type Culture Collection. The cells were
cultured in RPMI-1640 medium containing 10% fetal bovine serum
(FBS; Thermo Fisher Scientific, Inc.) and were maintained at 37°C
in a humidified atmosphere with 5% CO2. For
transfection, 20 µM miRNA was transfected into the TNBC cells using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific,
Inc.).
Cell proliferation assay
The MDA-MB-231 and BT-20 cells were seeded into
96-well plates with 2,000 cells per well in 100 µl of medium. After
24 h, the cells were treated with increasing concentrations (0, 5,
20, 40, 80, 160 and 320 nM) of chidamide for 24, 48 and 72 h at
37°C, respectively. To measure cell proliferation under different
concentrations of chidamide, 20 µl of Cell Counting Kit (CCK)-8
reagent (Beyotime Institute of Biotechnology) was added to the
cells and incubated at 37°C for 3 h. The absorbance of each well
was measured at 450 nm with a microplate reader.
In vitro colony formation
A total of 3,000 cells/well were cultured in a
6-well plate with RPMI-1640 medium and treated with or without 80
nM chidamide at the indicated concentration. Following incubation
for 10 days at 37°C, the medium was discarded and the cells were
washed twice with PBS. The colonies were first fixed with 4% of
paraformaldehyde at room temperature for 15 min and then stained
with 1% crystal violet for 10 min at room temperature. The colonies
were washed with PBS and counted via light microscopy.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
RNA extraction from the tissues or cells was
performed using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). The concentration of RNA was evaluated using the
NanoDrop 2000 instrument (Thermo Fisher Scientific, Inc.). The RNA
was first reverse transcribed into cDNA using the first-strand cDNA
synthesis kit (Takara Biotechnology Co., Ltd.) with following the
conditions: 25°C for 5 min; 42°C for 20 min and 95°C for 1 min.
qPCR of the miR-33a-5p was conducted with SYBR Super mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.) on the 7900 Fast
Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The expression of GAPDH was detected as the endogenous
control. The thermocycling conditions were as follows: 95°C for 10
min, followed by 40 cycles at 95°C for 15 sec and 60°C for 1
min.
Targets prediction
The targets of miR-33a-5p were predicted using the
TargetScan database (http://www.targetscan.org/vert_72/). Input
‘miR-33a-5p’ in the ‘Enter a microRNA name’ box and the targets
were displayed following submission.
Luciferase reporter assay
The TNBC cells (10,000 per well) were cultured in
24-well plates and co-transfected with 20 nM miR-33a-5p mimics
(5′-GUGCAUUGUAGUUGCAUUGCA) or mimic control miRNA
(5′-UUUGUACUACACAAAAGUACUG) (Guangzhou RiboBio Co., Ltd.) and 250
ng luciferase reporter vector (Promega Corporation) containing the
wild-type or mutant 3′-UTR of LDHA. Transfection was performed with
Lipofectamine 2000™ (Thermo Fisher Scientific, Inc.) according to
the manufacturer's instructions. Following transfection for 48 h,
the cells were harvested and the luciferase reporter activity was
measured using a Dual-Luciferase Assay kit (Promega Corporation)
and normalized to Renilla luciferase activity. The
experiment was performed in triplicate.
Western blot analysis
The MDA-MB-231 and BT-20 cells were collected and
lysed with RIPA lysis buffer containing the protease inhibitor PMSF
(Beyotime Institute of Biotechnology). The protein concentration
was determined using a BCA assay (Beyotime Institute of
Biotechnology). A total of 20 µg of protein was loaded into 15%
SDS-PAGE and then transferred onto polyvinylidene fluoride
membranes (EMD Millipore). The membrane was first blocked with 5%
non-fat milk at room temperature (RT) for 1 h and then incubated
with the primary antibody overnight at 4°C. Subsequently, the
membrane was incubated with a secondary antibody conjugated to
horseradish peroxidase (HRP) at RT for 1 h and the protein bands
were visualized with an ECL detection kit (Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. The
antibodies used in the present study included anti-LDHA (1:2,000
dilution; cat. no. 2012; Cell Signaling Technology, Inc.),
anti-GAPDH (1:2,000 dilution; cat. no. 5174; Cell Signaling
Technology, Inc.) and HRP-conjugated secondary antibody (1:10,000
dilution; cat. no. ZDR 5306; OriGene Technologies, Inc.).
Statistical analysis
The data are expressed as the mean ± standard
deviation. Statistical significance was analyzed using Student's
t-test or one-way analysis of variance followed by Tukey's post hoc
test. Statistical analyses were performed using SPSS 13.0 software
(SPSS, Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
Chidamide treatment suppresses the
growth of TNBC cells
To evaluate the effect of chidamide on the growth of
TNBC cells, the MDA-MB-231 and BT-20 cells were treated with an
increasing concentrations of chidamide for 24, 48 and 72 h,
respectively. Cell proliferation was measured using a CCK-8 assay.
As shown in Fig. 1A and B,
chidamide significantly decreased the proliferation of MDA-MB-231
and BT-20 cells in a dose- and time-dependent manner. To further
confirm these results, the influence of chidamide on the growth of
TNBC cells was evaluated via an in vitro colony formation
assay. The results indicated that chidamide treatment suppressed
colony formation of the MDA-MB-231 and BT-20 cells (Fig. 1C). Additionally, the inhibitory
effect of chidamide on the progression of TNBC cells was
investigated using a cell invasion assay, which revealed that
exposure to chidamide significantly attenuated the invasion of
MDA-MB-231 and BT-20 cells (Fig.
1D). Collectively, these results demonstrated that chidamide
treatment negatively regulated the growth of TNBC cells.
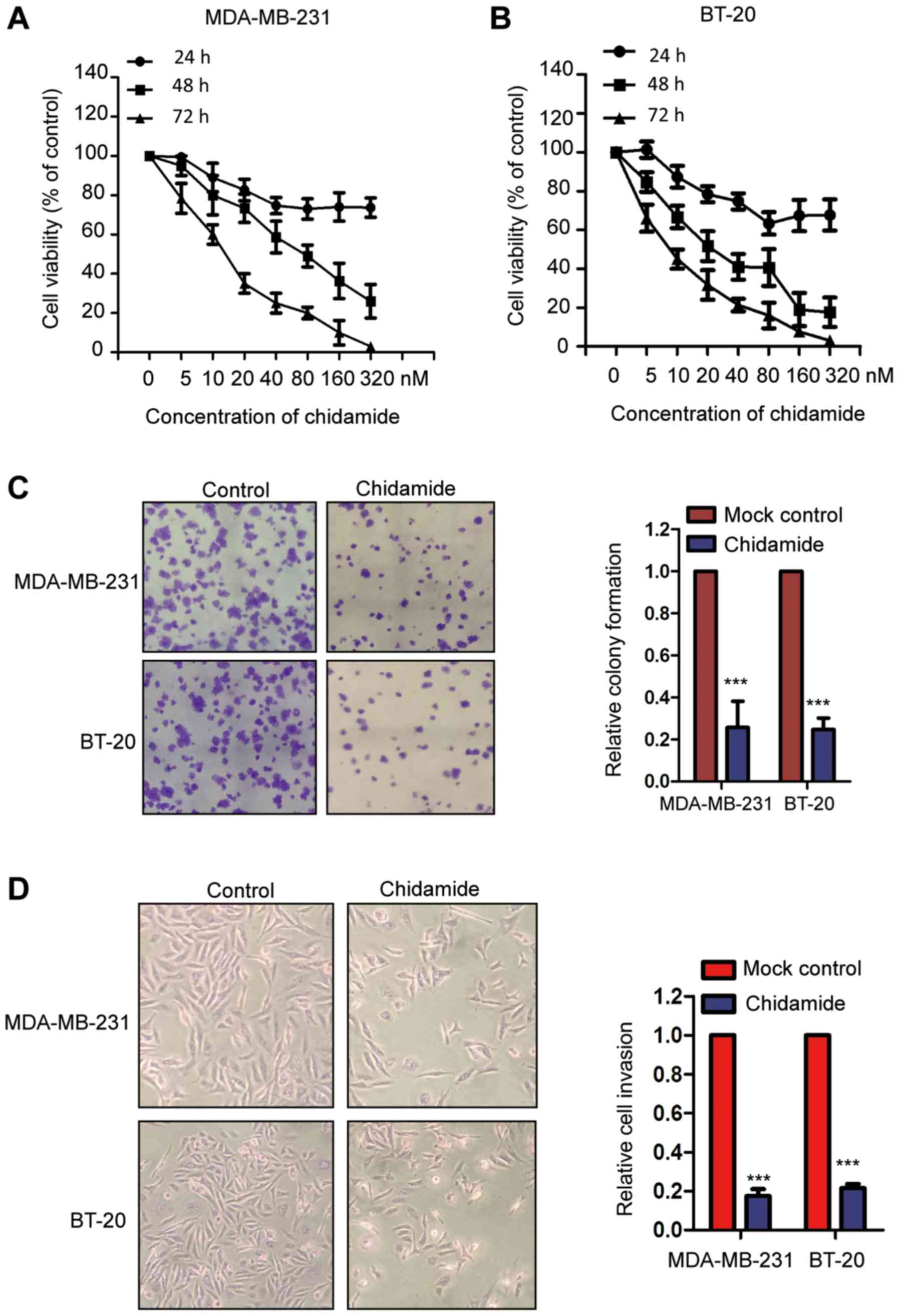 | Figure 1.Chidamide suppresses the growth of
TNBC cells. (A) MDA-MB-231 and (B) BT-20 cells were treated with
increasing doses of chidamide for the 24, 48 and 72 h. Data were
obtained from three independent experiments (n=3). (C) Colony
formation of TNBC cells with or without chidamide exposure (n=3).
Left-hand panel, representative image of the colonies
(magnification, ×40); right-hand panel, quantitative data of the
colony numbers in each group. ***P<0.001, chidamide vs. Mock
control. (D) Invasion of TNBC cells following treatment with
chidamide was evaluated using a Transwell assay (n=3). Left-hand
panel, representative image of the invasion of TNBC cells
(magnification, ×40); right-hand panel, quantitative data of the
invasion numbers in each group. ***P<0.001, Chidamide vs. Mock
control. TNBC, triple-negative breast cancer. |
Chidamide upregulates the expression
of miR-33a-5p in TNBC cells
Considering the significant suppressive effect of
chidamide on the growth of TNBC cells, the possible underlying
molecular mechanisms mediating the inhibitory function of chidamide
were further investigated. An increasing body of evidence has
suggested that miRNAs serve important roles in modulating drug
sensitivity and cancer cell growth. To evaluate whether miRNAs are
involved in chidamide-mediated cell growth inhibition in TNBC
cells, our previous study screened the expression of miRNAs with
MDA-MB-231 cells that were treated with chidamide. The data
indicated that the expression level of miR-33a-5p was significantly
increased the most with exposure to chidamide (Table SI). The present study confirmed
this observation via the incubation of MDA-MB-231 and BT-20 cells
with an increasing dose of chidamide, and the levels of miR-33a-5p
were detected by RT-qPCR analysis. As presented in Fig. 2A, chidamide treatment significantly
upregulated the expression of miR-33a-5p in a dose-dependent
manner. To support this result, the expression of miR-33a-5p in
TNBC cell lines and tissues was detected. The results revealed that
the level of miR-33a-5p was decreased in the TNBC cell lines
compared with that in the normal MCF-10A cells (Fig. 2B). Consistent with this, the
expression of miR-33a-5p in TNBC tissues was significantly reduced
compared with that in the paired adjacent normal tissues (Fig. 2C). The decreased expression of
miR-33a-5p suggested the potential involvement of miR-33a-5p in the
development of TNBC. To obtain further evidence, the MDA-MB-231 and
BT-20 cells were transfected with miR-33a-5p mimics or control
miRNA. The ectopic expression of miR-33a-5p was confirmed using an
RT-qPCR assay (Fig. 2D). The
proliferation of MDA-MB-231 and BT-20 cells was then detected using
a CCK-8 assay. The results indicated that the overexpression of
miR-33a-5p significantly decreased the proliferation rate of the
TNBC cells (Fig. 2E and F).
Consistently, transfection of the MDA-MB-231 and BT-20 cells with
miR-33a-5p significantly inhibited colony formation and cell
invasion (Fig. 2G and H). To
further confirm the regulation of miR-33a-5p on the growth of TNBC
cells, the MDA-MB-231 and BT-20 cells were transfected with
miR-33a-5p inhibitor to deplete the expression of miR-33a-5p
(Fig. S1A). The CCK-8 assay
revealed that the knockdown of miR-33a-5p significantly promoted
the proliferation of TNBC cells (Fig.
S1B and C). Consistently, the downregulation of miR-33a-5p
enhanced the colony formation and invasion of the MDA-MB-231 and
BT-20 cells (Fig. S1D and E).
These results were suggestive of the tumor suppressive function of
miR-33a-5p in the progression of TNBC.
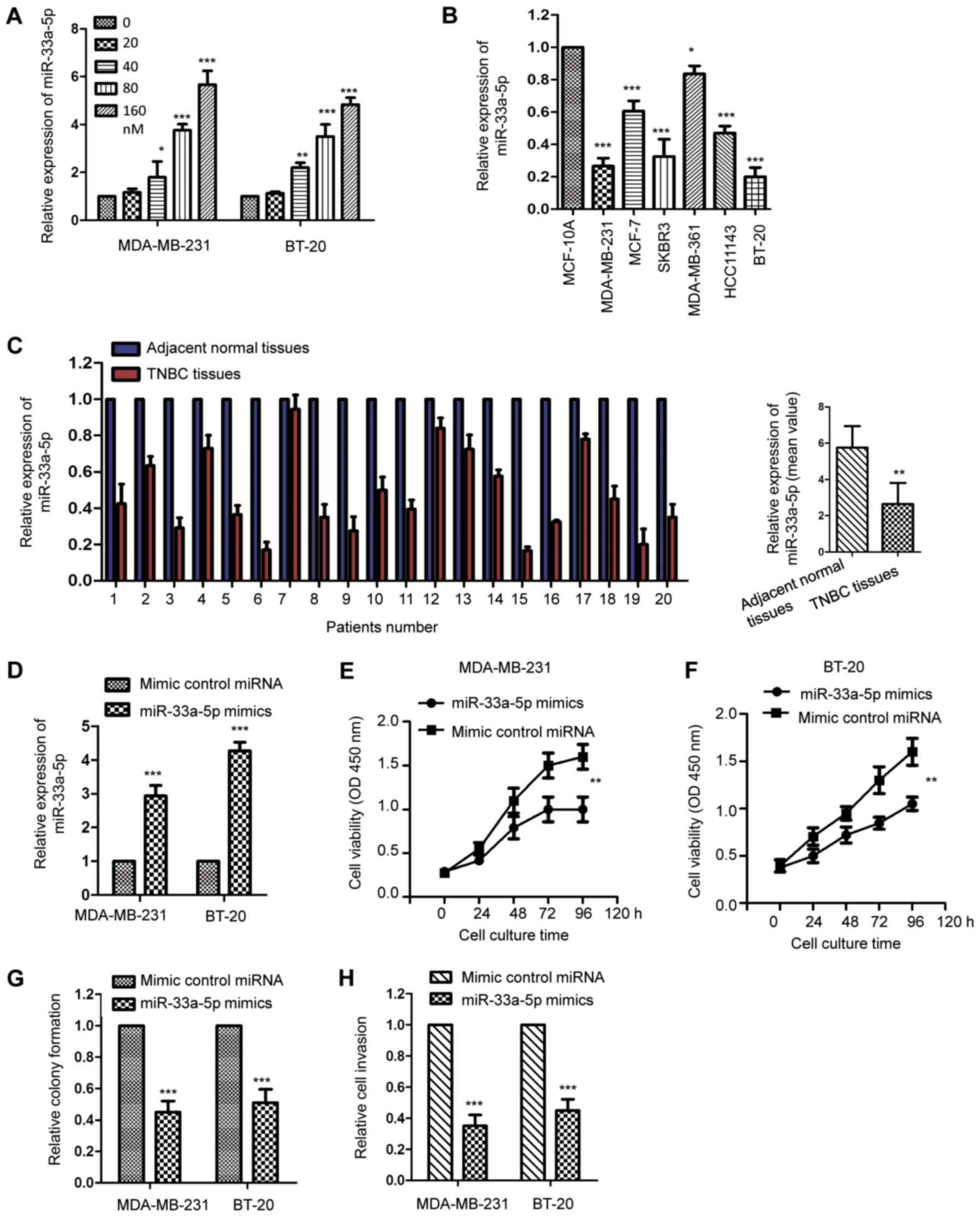 | Figure 2.Chidamide upregulates the expression
of miR-33a-5p in TNBC cells. (A) MDA-MB-231 and BT-20 cells were
treated with increasing concentrations of chidamide for 48 h and
the expression of miR-33a-5p was determined by RT-qPCR analysis
(n=3). *P<0.05, **P<0.01, ***P<0.001, chidamide (20, 40,
80 and 160 nM) vs. Mock control (0 nM). (B) Expression levels of
miR-33a-5p in normal breast cells (MCF-10A) and different breast
cancer cell lines were compared (n=3). *P<0.05; ***P<0.001,
compared with the expression of miR-33a-5p in MCF-10A. (C) Level of
miR-33a-5p in 20 paired TNBC tissues and adjacent normal tissues
were detected by RT-qPCR analysis. The mean values for the
expression of miR-33a-5p in TNBC tissue and normal tissues are
shown in the right-hand panel. **P<0.01 TNBC tissues vs.
Adjacent normal tissues. (D) MDA-MB-231 and BT-20 cells were
transfected with miR-33a-5p mimics or control miRNA and the ectopic
expression of miR-33a-5p was determined via RT-qPCR analysis (n=3).
***P<0.001, miR-33a-5p mimics vs. control miRNA group.
Proliferation of (E) MDA-MB-231 and (F) BT-20 cells transfected
with miR-33a-5p or control miRNA was detected using a Cell Counting
Kit-8 assay (n=3). **P<0.01 miR-33a-5p mimics vs. control miRNA
group. Overexpression of miR-33a-5p inhibited (G) colony formation
and (H) cell invasion (n=3). ***P<0.001 miR-33a-5p mimics vs.
control miRNA group. TNBC, triple-negative breast cancer; RT-qPCR,
reverse transcription-quantitative polymerase chain reaction; miR,
microRNA. |
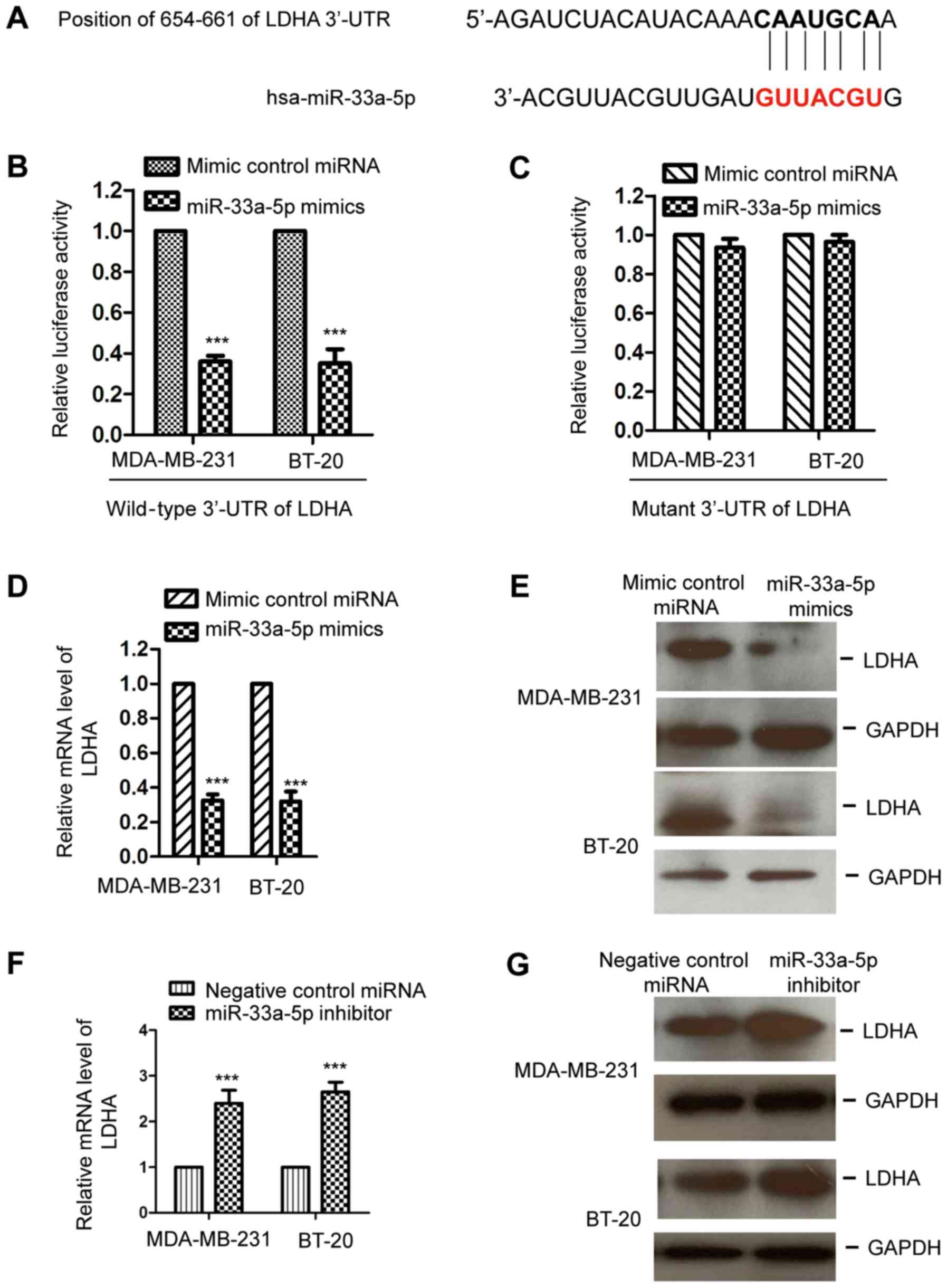
miR-33a-5p targets LDHA in TNBC
cells
To further understand the functional mechanisms by
which miR-33a-5p modulated the growth of TNBC cells, the downstream
targets of miR-33a-5p were predicted using the TargetScan database.
Notably, LDHA was predicted as the putative downstream target of
miR-33a-5p. The binding sites of miR-33a-5p at the 3′-UTR of LDHA
are presented in Fig. 3A. To
confirm the prediction, the MDA-MB-231 and BT-20 cells were
co-transfected with miR-33a-5p mimics or control miRNA and a
luciferase reporter vector containing the wild-type or mutant
3′-UTR of LDHA. The results revealed that transfection with
miR-33a-5p significantly decreased the luciferase activity of the
wild-type, but not the mutant, 3′-UTR of LDHA (Fig. 3B and C). To investigate whether the
binding between miR-33a-5p with the 3′-UTR of LDHA affected the
mRNA stability of LDHA, the MDA-MB-231 and BT-20 cells were
transfected with miR-33a-5p mimics or control miRNA and the mRNA
levels of LDHA were detected by RT-qPCR analysis. The results
demonstrated that the overexpression of miR-33a-5p significantly
decreased the mRNA expression of LDHA in the MDA-MB-231 and BT-20
cells (Fig. 3D). Additionally, to
further characterize the importance of the binding between
miR-33a-5p and the 3′-UTR of LDHA, nucleotides in miR-33a-5p were
mutated. The results showed that mutated miR-33a-5p lost its
ability to bind to the 3′-UTR of LDHA compared with the wild-type
miR-33a-5p (Fig. S2A).
Additionally, the expression of LDHA was unchanged by transfection
with mutated miR-33a-5p in TNBC cells (Fig. S2B). To further characterize the
negative regulation of miR-33a-5p on LDHA, the protein level of
LDHA with ectopic expression of miR-33a-5p was also examined by
western blotting. As shown in Fig.
3E, compared with the control group, a high expression of
miR-33a-5p reduced the protein level of LDHA in the MDA-MB-231 and
BT-20 cells. To further confirm the negative regulation of
miR-33a-5p on the expression of LDHA, the expression of LDHA was
detected in TNBC cells transfected with miR-33a-5p inhibitor. The
results showed that the downregulation of miR-33a-5p increased the
expression of LDHA at the mRNA and protein levels (Fig. 3F and G). These results demonstrated
that miR-33a-5p bound the 3′-UTR of LDHA and suppressed the
expression of LDHA in TNBC cells.
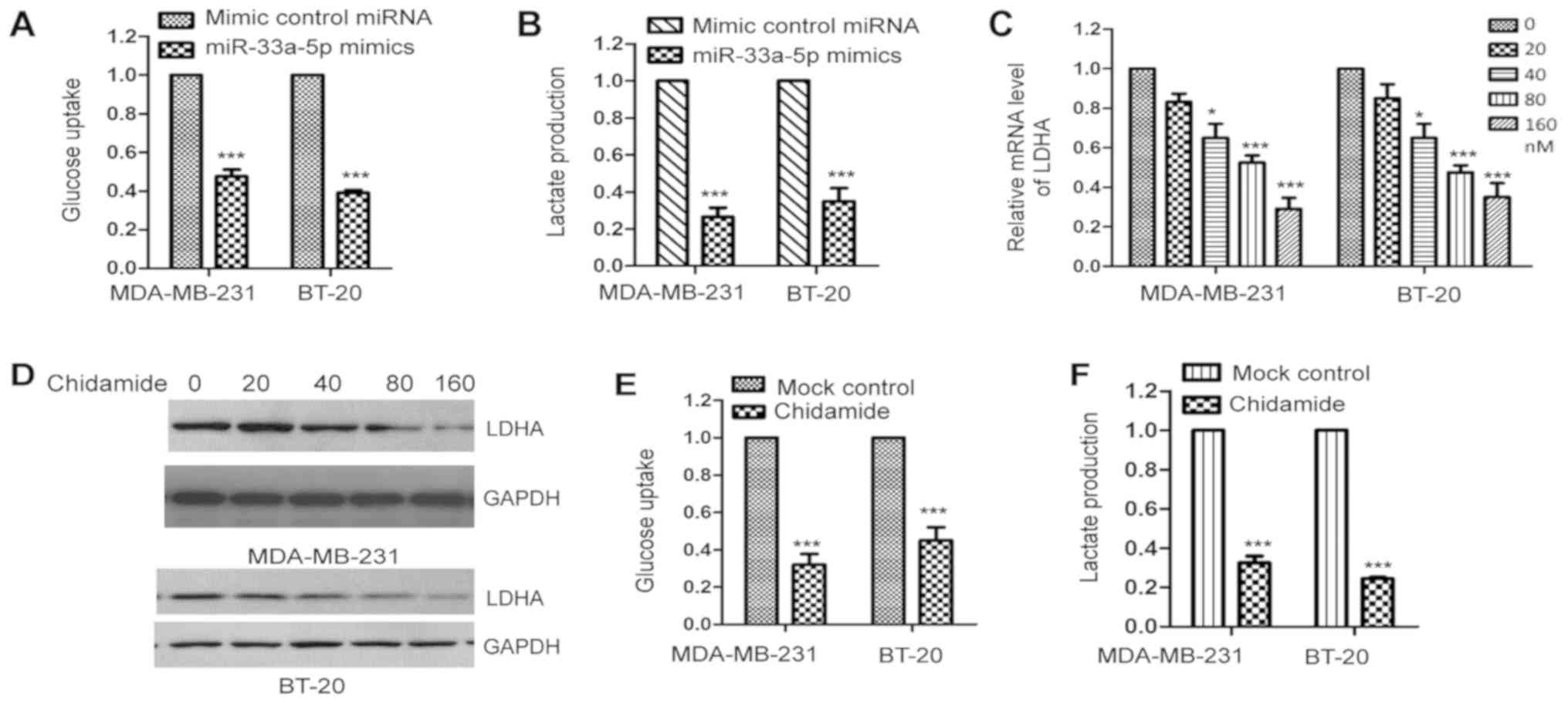
Chidamide reduces the glycolysis of
TNBC cells
LDHA is an essential enzyme in the glycolysis of
cancer cells. As overexpression suppressed the expression of LDHA
in TNBC cells, to detect whether the aberrant expression of
miR-33a-5p affected the glucose metabolism of TNBC cells, the
MDA-MB-231 and BT-20 cells were transfected with miR-33a-5p mimics
or control miRNA and glycolysis was evaluated. The results
demonstrated that the ectopic expression of miR-33a-5p
significantly decreased glucose uptake and lactate production in
the MDA-MB-231 and BT-20 cells (Fig.
4A and B), which indicated that the overexpression of
miR-33a-5p suppressed the glycolysis of TNBC cells.
As chidamide upregulated the expression of
miR-33a-5p in TNBC cells, to further analyze the effect of
chidamide on the glycolysis of TNBC cells, the MDA-MB-231 and BT-20
cells were treated with increasing concentrations of chidamide and
the mRNA and protein levels of LDHA were examined by RT-qPCR
analysis and western blotting, respectively. The results revealed
that exposure to chidamide decreased the levels of LDHA in these
two cell lines (Fig. 4C and D).
The present study also measured glucose consumption and lactate
generation by treating MDA-MB-231 and BT-20 cells with chidamide.
The results suggested that chidamide significantly reduced glucose
uptake and lactate production in TNBC cells (Fig. 4E and F). These results demonstrated
that chidamide upregulated miR-33a-5p, which consequently decreased
the expression of LDHA and resulted in defects in the glycolysis of
TNBC cells.
Discussion
Patients with TNBC respond poorly to the currently
available chemotherapeutic strategies, which indicates the need for
novel, efficient treatments to improve the outcomes for these
patients. Due to the critical involvement of epigenetic
modifications in modulating gene expression and cell growth,
targeting enzymes that mediate epigenetic changes have been
reported to suppress the progression of cancer. The promising
anticancer effect of HDAC has emerged in a recent study (38). In the present study, exposure to
chidamide significantly decreased the proliferation of TNBC cells
by modulating the glycolysis of TNBC cells, which highlights the
potential application of chidamide in the treatment of TNBC.
Chidamide has been identified as a class I HDACi,
which was generated and has been approved for use in clinical
practice in China (39,40). Previous studies revealed that
chidamide treatment led to defects in the growth of a variety of
cancer cells. For example, chidamide reduced proliferation and
induced apoptosis in NK/T lymphoma cells by regulating the
serine/threonine kinase-checkpoint kinase 2-p53-p21 signaling
pathway (23). The inhibitory
effect of chidamide was also observed in blastic plasmacytoid
dendritic cell neoplasia (41).
However, to the best of our knowledge, the function of chidamide in
breast cancer, particularly in TNBC, has not been illustrated. In
the present study, MDA-MB-231 and BT-20 cells were treated with
chidamide and the results revealed that chidamide suppressed the
proliferation, colony formation and migration of the TNBC cells.
Further in vivo experiments may elucidate the growth
inhibitory role of chidamide in TNBC. Additionally, it would be of
interest to examine whether chidamide also reduces the progression
of non-TNBC breast cancer cells.
An increasing body of evidence has suggested the
critical involvement of miRNAs in the development of human cancer
(42,43). In the present study, the results
demonstrated that miR-33a-5p was upregulated following chidamide
treatment in TNBC cells. Previous studies demonstrated that the
expression of miR-33a-5p was decreased, and inhibited the
proliferation of lung adenocarcinoma cells and osteosarcoma cells
(36,44). In the present study, the
overexpression of miR-33a-5p inhibited the proliferation of
MDA-MB-231 and BT-20 cells. Further molecular investigations
revealed that miR-33a-5p targeted LDHA and suppressed the
expression of LHDA in TNBC cells. Consistent with the upregulation
of miR-33a-5p, exposure to chidamide significantly decreased the
levels of LDHA, which consequently suppressed the glycolysis of
TNBC cells. A previous study indicated the overexpression of LDHA
in TNBC tissues (45). It is
noteworthy that miR-33a-5p was also observed to inhibit the
glycolysis of melanoma by targeting hypoxia-inducible factor-1α,
and the defects in glycolysis caused by the overexpression of
miR-33a-5p conferred radiosensitivity to melanoma cells (37). These results demonstrate that
miR-33a-5p is a novel regulator of glucose metabolism by modulating
the expression of enzymes that are essential for the glycolysis of
cancer cells. It will be useful to examine whether the negative
regulation of miR-33a-5p and chidamide on glucose metabolism also
occurs in other types of cancer. Additionally, other miRNAs that
may be involved in the anticancer effect of chidamide warrant
further investigation. There were several limitations of the
present study that require further improvements. For example, most
of the results were obtained from in vitro experiments,
indicating the need for in vivo experiments to further
validate the effects of chidamide on the growth of TNBC cells. It
may also be necessary to investigate the involvement of miRNAs
other than miR-33a-5p in the suppressive function of chidamide in
the progression of TNBC. The complete molecular mechanism by which
chidamide regulates the expression of miR-33a-5p in TNBC cells
remains to be fully elucidated.
In conclusion, the results of the present study
revealed that chidamide inhibited the proliferation of TNBC cells,
likely by upregulating the expression of miR-33a-5p. A high
expression of miR-33a-5p decreased the expression of LDHA and
suppressed the glycolysis of TNBC cells. These data indicate a
possible mechanism by which chidamide inhibited the growth of TNBS
cells, which suggests the potential application of chidamide in the
treatment of patients with TNBC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by a research grant from
China's National Natural Science Foundation (grant no. 81471590)
and by Chipscreen Biosciences Co., Ltd.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XB and QH designed the study. XB performed the
experiments. HJ collected the tissues and performed the RT-qPCR. GH
helped with the data analysis. XB and QH wrote the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all of
participants prior to tissue collection. The Ethics Committee of
The Affiliated Hospital of Capital Medical University (Beijing,
China) approved the present study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kandula M, Ch KK and Ys AR: Molecular
mechanism and targeted therapy options of triple-negative (ER, PgR,
HER-2/neu) breast cancer: Review. World J Oncol. 4:137–141.
2013.PubMed/NCBI
|
|
2
|
Jamdade VS, Sethi N, Mundhe NA, Kumar P,
Lahkar M and Sinha N: Therapeutic targets of triple-negative breast
cancer: A review. Br J Pharmacol. 172:4228–4237. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zeichner SB, Terawaki H and Gogineni K: A
Review of systemic treatment in metastatic triple-negative breast
cancer. Breast Cancer (Auckl). 10:25–36. 2016.PubMed/NCBI
|
|
4
|
Grubb W, Young R, Efird J, Jindal C and
Biswas T: Local therapy for triple-negative breast cancer: A
comprehensive review. Future Oncol. 13:1721–1730. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu Z, Gao Y and Li X: Cancer epigenetics
and the potential of epigenetic drugs for treating solid tumors.
Exp Rev Anticancer Ther. 2018.(Epud ahead of print).
|
|
6
|
Perri F, Longo F, Giuliano M, Sabbatino F,
Favia G, Ionna F, Addeo R, Della Vittoria Scarpati G, Di Lorenzo G
and Pisconti S: Epigenetic control of gene expression: Potential
implications for cancer treatment. Crit Rev Oncol Hematol.
111:166–172. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Haberland M, Montgomery RL and Olson EN:
The many roles of histone deacetylases in development and
physiology: Implications for disease and therapy. Nat Rev Genet.
10:32–42. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mei S, Ho AD and Mahlknecht U: Role of
histone deacetylase inhibitors in the treatment of cancer (Review).
Int J Oncol. 25:1509–1519. 2004.PubMed/NCBI
|
|
9
|
Bruserud O, Stapnes C, Ersvaer E, Gjertsen
BT and Ryningen A: Histone deacetylase inhibitors in cancer
treatment: A review of the clinical toxicity and the modulation of
gene expression in cancer cell. Curr Pharm Biotechnol. 8:388–400.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hrabeta J, Stiborova M, Adam V, Kizek R
and Eckschlager T: Histone deacetylase inhibitors in cancer
therapy. A review. Biomed Pap Med Fac Univ Palacky Olomouc Czech
Repub. 158:161–169. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Valente S and Mai A: Small-molecule
inhibitors of histone deacetylase for the treatment of cancer and
non-cancer diseases: A patent review (2011–2013). Expert Opin Ther
Pat. 24:401–415. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ali SR, Humphreys KJ, McKinnon RA and
Michael MZ: Impact of histone deacetylase inhibitors on microRNA
expression and cancer therapy: A review. Drug Dev Res. 76:296–317.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hiriyan J, Shivarudraiah P, Gavara G,
Annamalai P, Natesan S, Sambasivam G and Sukumaran SK: Discovery of
PAT-1102, a novel, potent and orally active histone deacetylase
inhibitor with antitumor activity in cancer mouse models.
Anticancer Res. 35:229–237. 2015.PubMed/NCBI
|
|
14
|
Min A, Im SA, Kim DK, Song SH, Kim HJ, Lee
KH, Kim TY, Han SW, Oh DY, Kim TY, et al: Histone deacetylase
inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances
anti-tumor effects of the poly (ADP-ribose) polymerase (PARP)
inhibitor olaparib in triple-negative breast cancer cells. Breast
Cancer Res. 17:332015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schech A, Kazi A, Yu S, Shah P and Sabnis
G: Histone deacetylase inhibitor entinostat inhibits
tumor-initiating cells in Triple-negative breast cancer Cells. Mol
Cancer Ther. 14:1848–1857. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang L, Liang Q, Shen K, Ma L, An N, Deng
W, Fei Z and Liu J: A novel class I histone deacetylase inhibitor,
I-7ab, induces apoptosis and arrests cell cycle progression in
human colorectal cancer cells. Biomed Pharmacother. 71:70–78. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhong L, Zhou S, Tong R, Shi J, Bai L, Zhu
Y, Duan X, Liu W, Bao J, Su L and Peng Q: Preclinical assessment of
histone deacetylase inhibitor quisinostat as a therapeutic agent
against esophageal squamous cell carcinoma. Invest New Drugs. Aug
31–2018.(Epub ahead of print). doi: 10.1007/s10637-018-0651-4.
View Article : Google Scholar
|
|
18
|
Connolly RM, Rudek MA and Piekarz R:
Entinostat: A promising treatment option for patients with advanced
breast cancer. Future Oncol. 13:1137–1148. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu L, Chen B, Qin S, Li S, He X, Qiu S,
Zhao W and Zhao H: A novel histone deacetylase inhibitor Chidamide
induces apoptosis of human colon cancer cells. Biochem Biophys Res
Commun. 392:190–195. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qiao Z, Ren S, Li W, Wang X, He M, Guo Y,
Sun L, He Y, Ge Y and Yu Q: Chidamide, a novel histone deacetylase
inhibitor, synergistically enhances gemcitabine cytotoxicity in
pancreatic cancer cells. Biochem Biophys Res Commun. 434:95–101.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou Y, Pan DS, Shan S, Zhu JZ, Zhang K,
Yue XP, Nie LP, Wan J, Lu XP, Zhang W and Ning ZQ: Non-toxic dose
chidamide synergistically enhances platinum-induced DNA damage
responses and apoptosis in non-small-cell lung cancer cells. Biomed
Pharmacother. 68:483–491. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao B and He T: Chidamide, a histone
deacetylase inhibitor, functions as a tumor inhibitor by modulating
the ratio of Bax/Bcl-2 and P21 in pancreatic cancer. Oncol Rep.
33:304–310. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou J, Zhang C, Sui X, Cao S, Tang F, Sun
S, Wang S and Chen B: Histone deacetylase inhibitor chidamide
induces growth inhibition and apoptosis in NK/T lymphoma cells
through ATM-Chk2-p53-p21 signalling pathway. Invest New Drugs.
36:571–580. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fabian MR, Sonenberg N and Filipowicz W:
Regulation of mRNA translation and stability by microRNAs. Annu Rev
Biochem. 79:351–379. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mohr AM and Mott JL: Overview of microRNA
biology. Semin Liver Dis. 35:3–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu Y, Usa K, Wang F, Liu P, Geurts AM, Li
J, Williams AM, Regner KR, Kong Y, Liu H, et al: MicroRNA-214-3p in
the kidney contributes to the development of hypertension. J Am Soc
Nephrol. 29:2518–2528. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Baker MA, Wang F, Liu Y, Kriegel AJ,
Geurts AM, Usa K, Xue H, Wang D, Kong Y and Liang M: miR-192-5p in
the kidney protects against the development of hypertension.
Hypertension. 73:399–406. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kwak PB, Iwasaki S and Tomari Y: The
microRNA pathway and cancer. Cancer Sci. 101:2309–2315. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Farazi TA, Spitzer JI, Morozov P and
Tuschl T: miRNAs in human cancer. J Pathol. 223:102–115. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zheng J: Energy metabolism of cancer:
Glycolysis versus oxidative phosphorylation (Review). Oncol Lett.
4:1151–1157. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Akram M: Mini-review on glycolysis and
cancer. J Cancer Educ. 28:454–457. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li XB, Gu JD and Zhou QH: Review of
aerobic glycolysis and its key enzymes-new targets for lung cancer
therapy. Thorac Cancer. 6:17–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hua S, Liu C, Liu L and Wu D: miR-142-3p
inhibits aerobic glycolysis and cell proliferation in
hepatocellular carcinoma via targeting LDHA. Biochem Biophys Res
Commun. 496:947–954. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li L, Kang L, Zhao W, Feng Y, Liu W, Wang
T, Mai H, Huang J, Chen S, Liang Y, et al: miR-30a-5p suppresses
breast tumor growth and metastasis through inhibition of
LDHA-mediated Warburg effect. Cancer Lett. 400:89–98. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang J, Wang D, Xiong J, Chen L and Huang
J: MicroRNA-33a-5p suppresses growth of osteosarcoma cells and is
downregulated in human osteosarcoma. Oncol Lett. 10:2135–2141.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cao K, Li J, Chen J, Qian L, Wang A, Chen
X, Xiong W, Tang J, Tang S, Chen Y, et al: microRNA-33a-5p
increases radiosensitivity by inhibiting glycolysis in melanoma.
Oncotarget. 8:83660–83672. 2017.PubMed/NCBI
|
|
38
|
Tsilimigras DI, Ntanasis-Stathopoulos I,
Moris D, Spartalis E and Pawlik TM: Histone deacetylase inhibitors
in hepatocellular carcinoma: A therapeutic perspective. Surg Oncol.
27:611–618. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lu X, Ning Z, Li Z, Cao H and Wang X:
Development of chidamide for peripheral T-cell lymphoma, the first
orphan drug approved in China. Intractable Rare Dis Res. 5:185–191.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shi Y, Jia B, Xu W, Li W, Liu T, Liu P,
Zhao W, Zhang H, Sun X, Yang H, et al: Chidamide in relapsed or
refractory peripheral T cell lymphoma: A multicenter real-world
study in China. J Hematol Oncol. 10:692017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang S, Guo W, Wan X, Teng Y, Zhou X and
Bai O: Exploring the effect of chidamide on blastic plasmacytoid
dendritic cell neoplasm: A case report and literature review. Ther
Clin Risk Manag. 14:47–51. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gentilin E, Degli Uberti E and Zatelli MC:
Strategies to use microRNAs as therapeutic targets. Best Prac Res
Clin Endocrinol Metab. 30:629–639. 2016. View Article : Google Scholar
|
|
43
|
Hosseinahli N, Aghapour M, Duijf PHG and
Baradaran B: Treating cancer with microRNA replacement therapy: A
literature review. J Cell Physiol. 233:5574–5588. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pan J, Zhou C, Zhao X, He J, Tian H, Shen
W, Han Y, Chen J, Fang S, Meng X, et al: A two-miRNA signature
(miR-33a-5p and miR-128-3p) in whole blood as potential biomarker
for early diagnosis of lung cancer. Sci Rep. 8:166992018.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Huang X, Li X and Xie X, Ye F, Chen B,
Song C, Tang H and Xie X: High expressions of LDHA and AMPK as
prognostic biomarkers for breast cancer. Breast. 30:39–46. 2016.
View Article : Google Scholar : PubMed/NCBI
|