Introduction
Chronic myeloid leukemia (CML) is a
myeloproliferative disorder characterized, in the vast majority of
cases, by the presence of Philadelphia chromosome (Ph) formed by
translocation of sections between chromosomes 9 and 22. Abnormally
short chromosome 22 encodes the chimeric p210 BCR-ABL tyrosine
kinase protein, a product of the oncogene BCR-ABL,
constitutively active enzyme that drives uncontrolled cellular
growth and differentiation of CML cells. The Ph chromosome with the
BCR-ABL fusion gene is also present in 25–50% of adult
patients with acute lymphoblastic leukemia (ALL) and rare cases of
acute myeloid leukemia (AML) (1,2).
BCR-ABL is the target of tyrosine kinase inhibitors
(TKIs) introduced, with great success, for the treatment of CML
patients at the end of the last century. Despite the high
therapeutic efficacy of TKIs, around 25% of CML patients develop
resistance to 1st (Imatinib) and 2nd (Desatinib, Nilotinib) line of
TKIs. This resistance may result from mutations within the kinase
domain of BCL-ABL, although other mechanisms of primary or acquired
resistance to TKIs have been investigated as well (2–5).
Apart from these genetic abnormalities also epigenetic alterations
may contribute to CML pathogenesis and drug resistance (6,7).
TKIs effectively inhibit BCR-ABL kinase, although CML stem cell
survival has been observed (5).
Thus, it is reasonable to seek a novel epigenetic approach to
improve CML treatment.
Epigenetic alterations regulate gene expression
via DNA methylation, histone modifications and activity of
non-coding RNAs (8,9). Interference between these epigenetic
processes affects chromatin accessibility for transcription
(8). Although, it is still DNA
methylation that is the most stable epigenetic reaction modulating
gene expression. It consists of the attachment of methyl group to
cytosine mainly in CpG islands within gene promoters. Dysregulated
epigenetic code, including aberrant methylation patterns, is often
observed and considered to be one of the causes, in addition to
genetic changes, of the development and progression of neoplastic
diseases (10,11). In cancer cells, a certain pool of
genes (mainly tumor suppressor genes) is silenced by methylation of
their promoter regions while other genes are activated (oncogenes
and prometastatic genes) through the hypomethylation of their
regulatory regions. Methylation patterns of DNA are controlled by
enzymes named DNA methyltransferases (DNMTs). DNMTs family include
methyltransferases DNMT3a and DNMT3b responsible for the de novo
methylation and the major DNMT1 which maintains and ensures the
fidelity of replication of inherited epigenetic marks and shows a
preference for hemi-methylated DNA (12).
As we have shown in our previous studies
deoxyadenosine analog-clofarabine
(2-chloro-2′-fluoro-2′-deoxyarabinosyladenine, CIF), apart from its
anticancer activity resulting from inhibition of ribonucleotide
reductase and DNA polymerases, and apoptosis induction by altering
mitochondrial activity, can also modulate gene expression
via redesigning DNA methylation patterns within gene
regulatory regions in cancer cells (13,14).
All these molecular mechanisms of CIF anticancer action contributed
to the FDA-approved therapeutic usage of this drug in ALL and some
AML cases (15,16).
Natural phytochemicals have raised considerable
interest not only as chemopreventive agents but also as
chemotherapeutic adjuvants because of their anticancer properties
demonstrated in a large number of studies (17). Resveratrol
(3,4′,5-trihydroxystilbene, RSV), the polyphenol from red grapes
and peanuts, has been shown to modulate cell cycle, survival and
apoptosis also through altering gene methylation patterns (18–22).
Other possible molecular targets of RSV are AMPK and SIRT1, mTOR,
NF-kB, PI3K/AKT, MAPK signaling pathways (23).
ATRA (all-trans retinoic acid) is a natural,
physiologically active, predominant metabolite of vitamin A. ATRA
acts as a hormone and impacts many physiological processes. ATRA
through its binding to specific nuclear retinoic acid receptors
RARs (RARA, RARB and RARG) that form heterodimers with retinoid X
receptors RXRs can regulate transcription of some genes (24). Within promoters of these genes, the
retinoic acid response elements (RAREs) have been found. According
to present knowledge, the transcriptional activity of RAR/RXR
complex results from the incorporation of ATRA to RAR receptors.
This model of interaction is known as a classical or genomic
pathway that regulates cell differentiation, cell cycle, and
apoptosis (25). RARs and RXRs are
able to create heterodimers with other receptors, such as vitamin D
receptor (VDR), steroid receptors or peroxisome
proliferator-activated receptor (PPAR). There is evidence that ATRA
can also regulate the gene expression independently of the presence
of RAREs. Furthermore, ATRA and its receptors may affect other
critical signaling pathways, including NF-κB, IFN-G, TGFB, VEGF,
and MAPK pathways, as well as cause chromatin remodeling (24,26).
Because of ATRA importance in cell physiology, the antitumor
activity of retinoids has been broadly studied. Consequently, ATRA
heretofore has gained FDA approval for treatment of APL (acute
promyelocytic leukemia) and cutaneous T-cell lymphoma. There are
some suggestions that the cause of the lack of ATRA anticancer
activity in other types of leukemia and solid tumors might be
associated with aberrant epigenetic marks, for example, frequent
DNA methylation-mediated silencing of retinoic acid receptor beta
(RARB) (26,27).
Interestingly, the growing body of literature
demonstrates that some natural bioactive compounds, including ATRA
and RSV, might be indirectly involved in the regulation of
DNMT1 expression and/or DNMT1 activity. DNMT1 has been shown
to be overexpressed in many types of cancer (28). The following mechanisms responsible
for ATRA or RSV-mediated DNMT1 downregulation in cancer
cells have been detected, i.e., cyclin-dependent kinase inhibitor
1A (CDKN1A) transcriptional reactivation (18,29)
followed by decreased activity of E2F (elongation factor 2)
transcription factor, as well as re-expression of DNA
methylation-silenced tumor suppressor genes, phosphatase and tensin
homologue (PTEN) and RARB, encoding proteins that may
inhibit activity of AP-1 (activator protein-1) transcriptional
complex (29,30). E2F and AP-1 transcription factors
activate DNMT1 expression due to the presence of binding
sites in DNMT1 regulatory region (31,32).
Moreover, CDKN1A (p21) belongs to
tumor suppressor genes and encodes a protein that competes with
DNMT1 for the same binding site on proliferating cell nuclear
antigen (PCNA, the homotrimeric ring surrounding DNA) during DNA
replication. It disrupts the forming of DNMT1/PCNA complex and
subsequently may lead to inhibition of DNA methylation reaction
(33,34). PTEN was shown to be mutated
or DNA methylation-silenced in a large number of malignancies. PTEN
protein as a phosphatase negatively regulates intracellular levels
of phosphatidylinositol-3,4,5-trisphosphate in cells which is
crucial for its tumor suppressive activity. The dephosphorylated
phosphoinositide through negative regulation of PI3K/AKT and
MAPK/AP-1 signaling pathways modulates cell cycle progression and
cell survival (35).
The promising results of combining nucleoside
analogues, such as cladribine and fludarabine (CIF precursors),
with ATRA or RSV in breast cancer cells, including
methylation-mediated PTEN and RARB transcriptional
reactivation (18,30), indicate that the combination of CIF
with these phytochemicals (ATRA or RSV) may exhibit a new effective
approach in anticancer epigenetic therapy.
As mentioned above, alterations in DNA methylation
marks are common in cancer cells, including different leukemia
cells. Thus, the present study aimed to evaluate anticancer
potential of CIF combined with natural bioactive compounds, RSV or
ATRA, in K562 cells representing an experimental in vitro
model of CML cells. This is the first study to investigate the
influence of CIF-phytochemical combination exposures on the
regulation of DNA methylation machinery in CML cells. We focused on
determining any changes in DNMT1 and CDKN1A
expression, as well as in promoter methylation and expression of
tumor suppressor genes PTEN and RARB.
Materials and methods
Compounds and chemicals
All tested compounds CIF, ATRA, and RSV were
purchased from Sigma-Aldrich. CIF was dissolved in sterile water (1
mM) and stored in −20°C. Solutions of ATRA (10 mM) and RSV (5 mM)
were prepared in 96% ethanol and stored in the dark in −20°C.
Subsequent dilutions were made in growth fresh medium with a final
ethanol concentration of 0.1% (v/v), and this ethanol concentration
was used as vehicle control in all experiments.
Cell culture, growth and viability
assay
Human erythroleukemic cell line K562 (American Type
Culture Collection, ATCC) was cultured in RPMI 1640 medium with
HEPES (Lonza) supplemented with 2 mM L-glutamine, 10% foetal bovine
serum (FBS), 1 U/ml penicillin and 1 µg/ml streptomycin
(Sigma-Aldrich), at 37°C and a humidified atmosphere of 5%
CO2. K562 cell line was routinely verified by
morphology, invasion and growth rate. The tested cell line was
authenticated by DNA profiling using the short tandem repeat
(ATCC), in 2018. In all experiments the cells were seeded at the
amount of 40×103 cells per ml, and were cultured for 72
h with three different compounds, CIF, ATRA and RSV, used
separately, at concentrations equal to GI50
concentrations (i.e., doses leading to 50% inhibition of cell
growth), respectively: 8 nM (CIF), 30 µM (ATRA) and 11.5 µM (RSV).
Additionally, the cells were treated for 72 h with the compounds
administered in two combinations: CIF + ATRA (both at
GI50 concentrations, i.e., 8 nM for CIF and 30 µM for
ATRA) and CIF + RSV (both at GI50 concentrations, i.e.,
8 nM for CIF and 11.5 µM for RSV).
Cell growth and viability were determined using the
trypan blue (Sigma-Aldrich) exclusion test, to estimate
GI50 values. The number of viable cells in culture
treated with the tested compounds was expressed as a percentage of
viable cells in control untreated culture (without the compounds,
vehicle control). The following calculation has been used: (viable
exposed/viable vehicle control)*100%. The number of dead cells that
took up trypan blue was specified as the percentage of the total
cell number.
The number of viable, necrotic, early and late
apoptotic cells were determined after 72 h compound exposure by
flow cytometry analysis using annexin V/propidium iodide (PI) (FITC
Annexin V Apoptosis Detection Kit II, BD Pharmingen) staining,
according to the manufacturer's protocol (13). The following excitation/emission
wavelengths have been used: FITC 488/519 nm and PI 488/617.
Caspase-3 assay (PE Active Caspase-3 Apoptosis Kit, BD Pharmingen)
was performed to estimate its activity as a marker of the early
stage of the caspase-dependent apoptotic pathway. The
excitation/emission wavelengths of 488/578 nm have been applied.
The flow cytometry analysis was carried out using BD FACSuite™
version 1.2.1 software.
Methylation-sensitive restriction
analysis (MSRA)
The methylation level of the proximal promoter of
PTEN and RARB in K562 cells was estimated using
methylation-sensitive restriction analysis according to the method
of Iwase et al (36). The
MSRA included four steps: i) digestion of cellular DNA with
endonuclease that recognizes only non-methylated sequence, ii) PCR
amplification of digested DNA with PCR primers shown in Table I, iii) electrophoretic analysis of
amplified promoter fragments, and iv) densitometric quantitative
analysis of the band intensity. The analysis was performed as
described previously (13).
 | Table I.PCR primer sequences. |
Table I.
PCR primer sequences.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Product (bp) |
|---|
| PTEN |
gcggaagcagccgttcggag |
gtcatgtctgggagcctgtg | 286 |
| RARB |
ctcgctgcctgcctctctgg |
gcgttctcggcatcccagtc | 295 |
Reverse transcription quantitative
(RT-q) PCR
Total RNA was isolated using TRIZOL®
(Invitrogen, USA). cDNA was synthesized using 2 µg of total RNA, 6
µl of random hexamers, 5 µl of oligo(dT)15, and
ImProm-II reverse transcriptase (Promega, USA). All RT-qPCR
reactions were carried out in a Rotor-Gene TG-3000 machine (Corbett
Research, Australia) as we previously described (13,14).
RPS17 (40S ribosomal protein S17), RPLP0 (60S acidic
ribosomal protein P0), H3F3A (H3 histone family 3A), and
BMG (β2-microglobulin) were used as housekeeping
control genes. The relative expression of each tested gene
(DNMT1, CDKN1A, PTEN, and RARB) was normalized to the
geometric mean of these four housekeeping genes, according to the
method of Pfaffl et al (37). Primers sequences for RT-qPCR are
shown in Table II.
| Table II.SYBR-Green-based reverse
transcription-quantitative PCR primer sequences. |
Table II.
SYBR-Green-based reverse
transcription-quantitative PCR primer sequences.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Product (bp) |
|---|
| DNMT1 |
accgcccctggccaaagccattg |
agcagcttcctcctcctttattttagctgag | 100 |
| CDKN1A |
gctcaggggagcaggctgaag |
cggcgtttggagtggtagaaatctgt | 103 |
| PTEN |
cgaactggtgtaatgatatgt |
catgaacttgtcttcccgt | 330 |
| RARB |
ttcaagcaagcctcacatgtttcca |
aggtaattacacgctctgcacctttag | 292 |
Measuring the amount of DNMT1
protein
Protein nuclear extracts were isolated using the
EpiQuik Nuclear Extraction Kit (Epigentek), according to
manufacturer's protocol. The ELISA-like EpiQuik DNMT1 Assay Kit
(Epigentek) was used for quantification of DNMT1 (DNA
methyltransferase) in 10 µg of the total protein content. Each
measurement was performed in triplicates according to the
instructions in the manual. The absorbance at 450 nm was measured
on a microplate reader (GloMax-Multi+ Microplate Multimode Reader,
Promega) within 2–10 min.
Statistical analysis
Results from three independent experiments are
presented as the mean ± standard deviation (SD). Statistical
analysis of cell viability, apoptosis, MSRA, qPCR and ELISA-like
EpiQuik DNMT1 assays was performed using two-way analysis of
variance (ANOVA) followed by Bonferroni post hoc test. The results
were considered statistically significant when P<0.05.
Results and Discussion
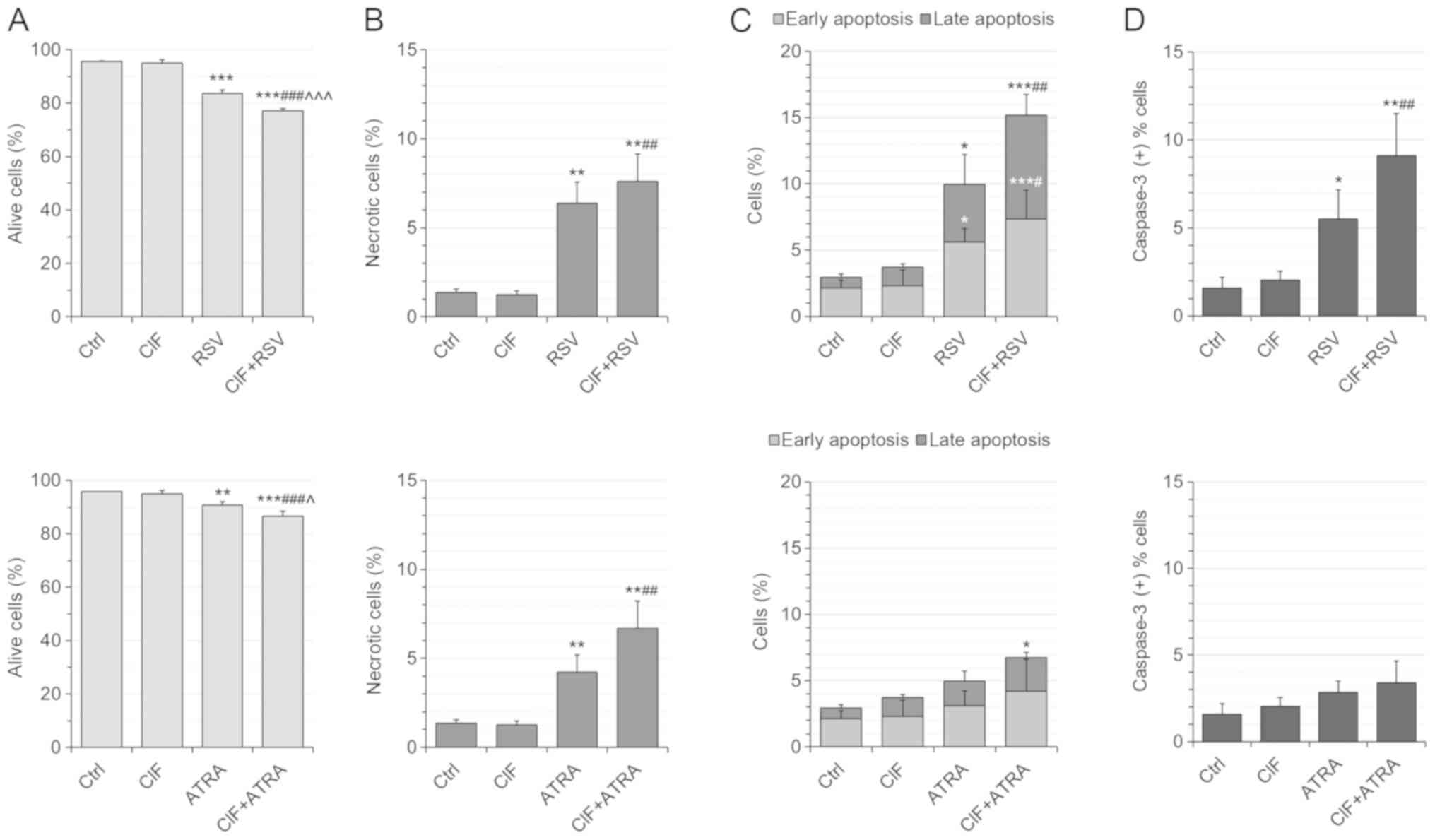
Effects of RSV and ATRA combined with
CIF on inhibition of CML cell growth and apoptosis induction
Following 72 h-exposure, all the tested compounds
used alone, CIF, RSV, and ATRA, inhibited K562 cell growth in a
dose-dependent manner with low cytotoxicity (Fig. 1A-C and F). The trypan blue
exclusion test was carried out to determine concentrations leading
to 50% inhibition of cell growth (GI50) (Fig. 1A-C). The GI50
concentration for CIF was determined as equal to 8 nM in K562 cells
(Fig. 1A), as we showed previously
(13). GI50 values for
RSV and ATRA were determined as equal to 11.5 and 30 µM,
respectively (Fig. 1B and C). The
number of dead cells upon exposure to the tested compounds at
GI50 concentrations did not exceed 10% (Fig. 1A-C), which support the use of all
the compounds at GI50 concentrations in the
combinatorial administrations, CIF and RSV, or CIF and ATRA
(Fig. 1D-F).
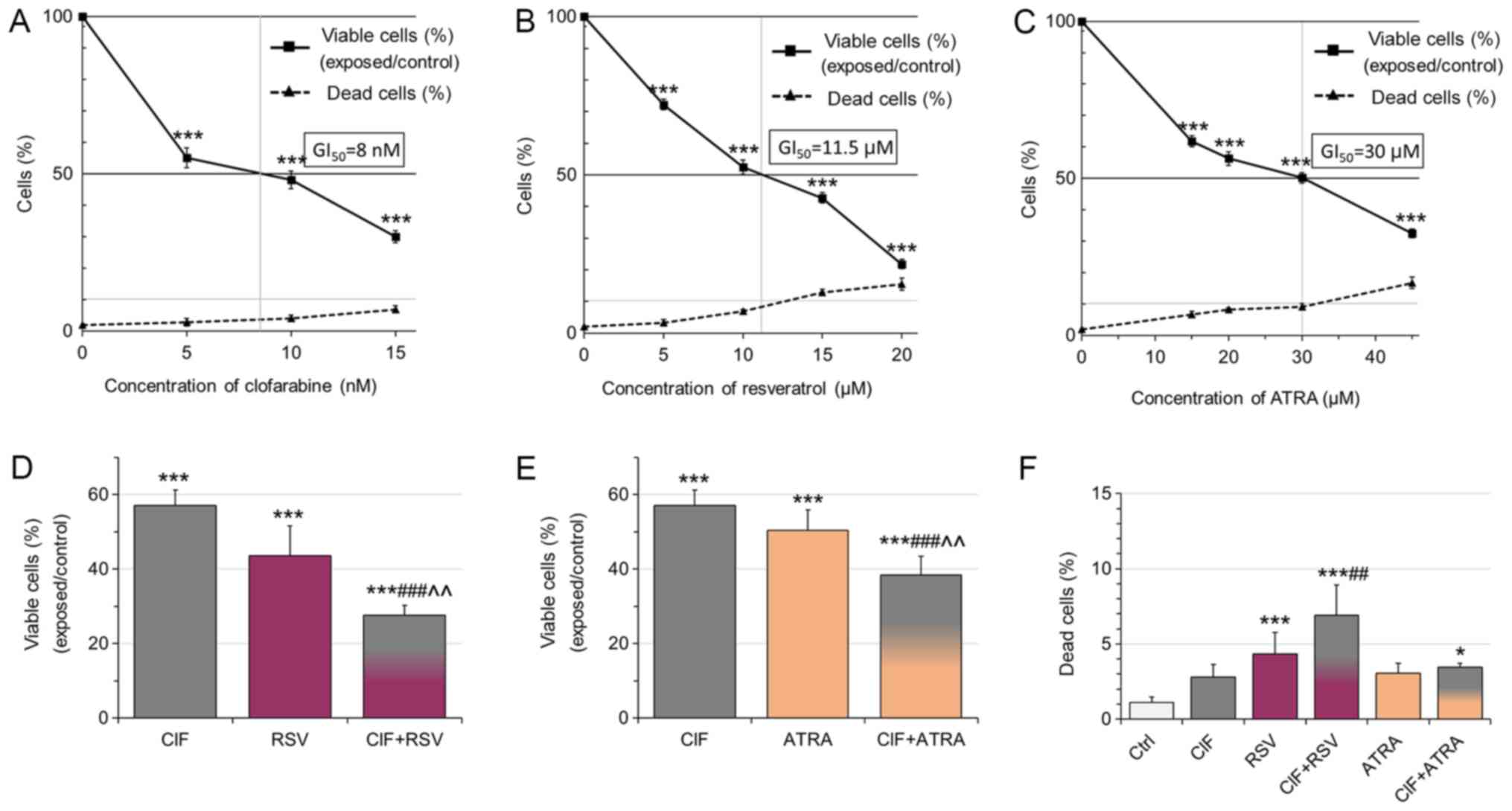 | Figure 1.Effects of (A) CIF, (B) RSV and (C)
ATRA on K562 cell growth and viability. Data represent the mean ±
standard deviation of three independent experiments. The number of
viable cells after 3 days exposure to CIF, RSV and ATRA at
GI50 concentrations was expressed as a percentage of
viable cells in the vehicle control [(viable exposed/viable vehicle
control)*100%]. GI50 values were determined as equal to:
8 nM for CIF, 11.5 µM for RSV, and 30 µM for ATRA. The number of
dead cells in either vehicle control or exposed group was
calculated as a percentage of the total cell number [(dead
cells/all cells)*100%]. Effects of (D) CIF, RSV and CIF+RSV, as
well as (E) CIF, ATRA and CIF+ATRA at GI50
concentrations on K-562 cell viability [(viable exposed/viable
vehicle control) × 100%]. (F) The number of dead cells in either
vehicle control or exposed groups was calculated as a percentage of
the total cell number. Exposure (CIF alone, RSV alone, ATRA alone,
CIF+RSV or CIF+ATRA) versus vehicle control, *P<0.05 and
***P<0.001 vs. vehicle control. ##P<0.01 and
###P<0.001 vs. CIF alone. ^^P<0.01 vs.
RSV or ATRA alone. CIF, clofarabine; RSV, resveratrol; ATRA,
all-trans retinoic acid. |
Next, the cytotoxicity of all the compounds
administered individually and in combinations was determined by
employing flow cytometric assay (Fig.
2). The number of necrotic (Ann-/PI+) cells did not exceed 10%
of all the cells upon any of the exposures, supporting low
cytotoxicity of the tested concentrations (Fig. 2B, top and bottom panels). The use
of CIF+RSV combination resulted in the most severe induction of
apoptosis in K562 cells (Fig. 2C,
upper panel). The number of apoptotic cells increases from nearly
4% after CIF alone and 10% after RSV alone to 15% after combined
administration CIF+RSV (Fig. 2C,
upper panel). This enhanced pro-apoptotic effect of combinatorial
CIF and RSV was associated with caspase-3 activation (Fig. 2D, upper panel). Upon 72
h-incubation with this combination over 9% of all K562 cells showed
active caspase 3, whereas after CIF or RSV alone approximately 2%
or 5.5% of all K562 bound antibodies against caspase 3,
respectively (Fig. 2D, upper
panel). The extent of the effects of ATRA alone and CIF+ATRA on
cell viability and caspase-dependent apoptosis was not as robust as
for RSV used alone or in combination with CIF (Fig. 2C, bottom panel). The number of
apoptotic cells increases from 4–5% after CIF or ATRA used alone to
slightly more than 6% after combined administration, CIF+ATRA
(Fig. 2C, bottom panel). The
percentage of K562 cells with active caspase-3 was similar after
the individual (2–3%, CIF or ATRA) and combinatorial (3.5%,
CIF+ATRA) exposures (Fig. 2D,
bottom panel).
Hitherto, only Lee and colleagues demonstrated that
RSV in combination with CIF induces relevant anti-proliferative
effects in malignant mesothelioma MSTO-211H and H-2452 cells. This
observation was linked to multi-targeted anticancer effects,
including inhibition of AKT activity (20,21).
Sui et al (38) showed that RSV indicates significant
cytotoxic effect and induces apoptosis in K562 cells in a dose and
time-dependent manner. The authors suggested that downregulation of
the PI3K/AKT/mTOR signaling cascades (through the attenuated
phosphorylation) may be a crucial mediator in the inhibition of
proliferation and induction of apoptosis by resveratrol in K562
cells.
Results of Wang et al (39) also indicated that resveratrol
significantly decreases cell viability and triggers cell apoptosis
in K562 cells. They observed up-regulation of Bax/Bcl-2 ratio, the
activation of caspase-3 and increased PARP cleavage in K562 cells
treated with resveratrol (39).
Interdependence between DNMT1 and
CDKN1A expression upon combinatorial exposures in K562 cells
Aberrant methylation pattern is a common feature of
cancer cells. The purpose of the study was to investigate the
interdependence between DNA methylation and expression of selected
tumor suppressor genes and the expression of the main DNA
methyltransferase, DNMT1, after treatment of model CML cells with a
chemotherapeutic agent, CIF, combined with natural bioactive
compounds, RSV and ATRA.
First of all, we analyzed the publicly available
data from Oncomine for DNMT1 expression in different types
of leukemia, as DNMT1 overexpression has been observed in
many types of cancer (28). As
depicted in Fig. 3A, in almost all
types of leukemia DNMT1 expression is significantly higher
compared to healthy individuals. Only in CML, the level of
DNMT1 expression is lower than in normal blood cells,
although the only available microarray data of CML, presented in
Fig. 3A, are not statistically
significant, so it is difficult to draw clear conclusions about the
level of DNMT1 in CML cells. However, Mizuno et al
(40) reported relevant
DNMT1 up-regulation in AML and CML cells as compared to
normal blood cells.
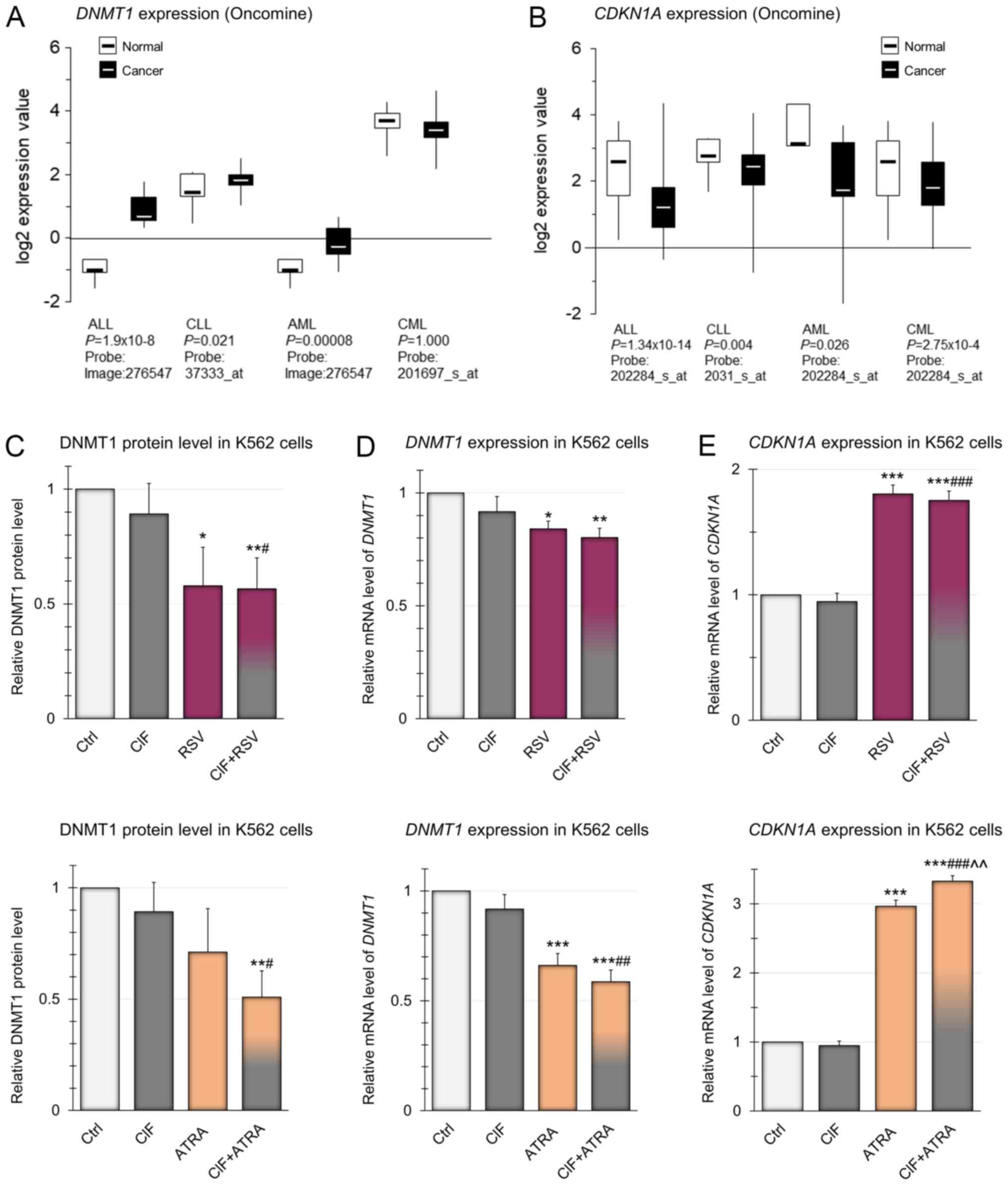 | Figure 3.Expression of DNMT1 and
CDKN1A genes in different types of leukemia and in K562
cells. (A) Gene expression microarray data for DNMT1 in
different types of leukemia. The normal vs. cancer gene expression
data were obtained from Oncomine and are presented as
log2-transformed median centered per array, and SD-normalized to 1
per array. The presented changes are statistically significant
(P<0.05) apart from the one for CML leukemia (lack of
statistically significant data). (B) Gene expression microarray
data for CDKN1A in different types of leukemia. The normal
vs. cancer gene expression data were obtained from Oncomine and are
presented as log2-transformed median centered per array, and
SD-normalized to 1 per array. The presented changes are
statistically significant (P<0.05). Effects of CIF, RSV and
ATRA, as well as CIF in combination with RSV (upper panels) or ATRA
(bottom panels) on: (C) DNMT1 protein level, (D) mRNA level of
DNMT1 and (E) mRNA level of CDKN1A in K562 cells. All
compounds used at GI50 concentrations in all
experiments. Data represent the mean ± SD of three independent
experiments. *P<0.05, **P<0.01 and ***P<0.001 vs. vehicle
control; #P<0.05, ##P<0.01 and
###P<0.001 vs. CIF alone; ^^P<0.01 vs.
RSV or ATRA alone. DNMT1, DNA methyltransferase 1; CDKN1A, Cyclin
dependent kinase inhibitor 1A; CIF, clofarabine; RSV, resveratrol;
ATRA, all-trans retinoic acid; SD, standard deviation. |
In our study, in K562 cells treated with CIF at
GI50 concentration (8 nM) for 72 h, slight almost 10%
reduction in DNMT1 gene expression, in comparison to control
unexposed cells, was estimated using RT-qPCR and Pfaffl's method
(37) (Fig. 3D). The effects of exposure to RSV
or ATRA administered alone, also at GI50 concentrations,
caused an even greater diminution in DNMT1 mRNA levels by 15
and 35%, respectively. However, the most robust, over 40% decrease
in DNMT1 expression was noticed as the effect of combined
exposure to CIF and ATRA (Fig. 3D,
bottom panel). These changes in the expression of DNMT1 at
the mRNA level correspond to changes in gene expression at the
protein level, determined using ELISA-like commercial immunoassays
(Fig. 3C). The combination
CIF+ATRA caused almost 50% reduction in DNMT1 protein level as
compared to control K562 cells (Fig.
3C, bottom panel). CIF and ATRA used alone led only to 11 and
29% decrease in DNMT1 protein levels, respectively (Fig. 3C). It has been shown that
manifestation of the catalytic function of DNMT1 enzyme requires
its binding to PCNA during DNA replication (33,34).
Moreover, CDKN1A, as an antagonist of DNMT1, binds to the same
domain of PCNA. Thus, CDKN1A polypeptide may disturb the formation
of PCNA-DNMT1 complex, and then leads to repression of DNA
methylation processes (41). The
estimation of CDKN1A expression on mRNA level (in connection
and comparison with DNMT1 expression) allows defining the
potential interrelations between DNA methylation processes and
expression of DNMT1 and CDKN1A genes in cells exposed
to natural bioactive compounds and CIF, also in combined therapy.
So we found that changes in DNMT1 expression are associated
with concomitant changes in CDKN1A mRNA level (Fig. 3D and E). Upon 72 h-incubation of
K562 cells with ATRA at GI50 concentration,
CDKN1A transcript level increased almost three times, and
almost two times in cells exposed to GI50 concentration
of RSV (Fig. 3E), in comparison to
control unexposed cells. Since ATRA binds to nuclear RARs that
heterodimerize with RXRs, it may further modulate transcription
through cognate response elements in the promoters of the target
genes including CDKN1A (42,43).
Due to structural similarity of RSV to estradiol and its binding to
estrogen receptors (ERs) it may elicit similar responses as upon
endogenous estrogens and modulate the expression of
estrogen-responsive genes, such as CDKN1A (44).
Combination of ATRA and CIF resulted in a 3.3-fold
increase in CDKN1A mRNA level. CIF used alone did not
influence the CDKN1A expression, and the combination of
CIF+RSV did not increase the level of CDKN1A above that
achieved with RSV used alone (Fig.
3E, upper panel). Our findings suggest that especially
CIF+ATRA-mediated concomitant CDKN1A induction and DNMT1
downregulation in K562 cells may decrease DNA methylation
efficiency of TSGs.
According to Oncomine publicly available data in all
types of leukemia, CDKN1A expression is significantly
decreased as compared to normal blood cells (Fig. 3B). Thus, reactivation of
CDKN1A gene encoding protein capable of cell cycle arrest is
one of the goals of anti-leukemic therapy (45).
DNA methylation-mediated PTEN
reactivation in K562 cells exposed to CIF combined with RSV or
ATRA
PTEN is a multifunctional tumor suppressor
gene, encoding a phosphatase with dual specificity for lipid and
protein substrates, has been shown to be silenced in multiple
cancers, including different types of leukemia (Fig. 4A). The PTEN downregulation
in cancer cells may be related to genetic changes, but also it may
result from hypermethylation of its promoter region, which partly
implies epigenetic regulation of PTEN transcription
(46–48). DNA methylation-mediated regulation
of PTEN expression was observed for example in ALL (46), breast cancer (47) and colorectal cancer (48).
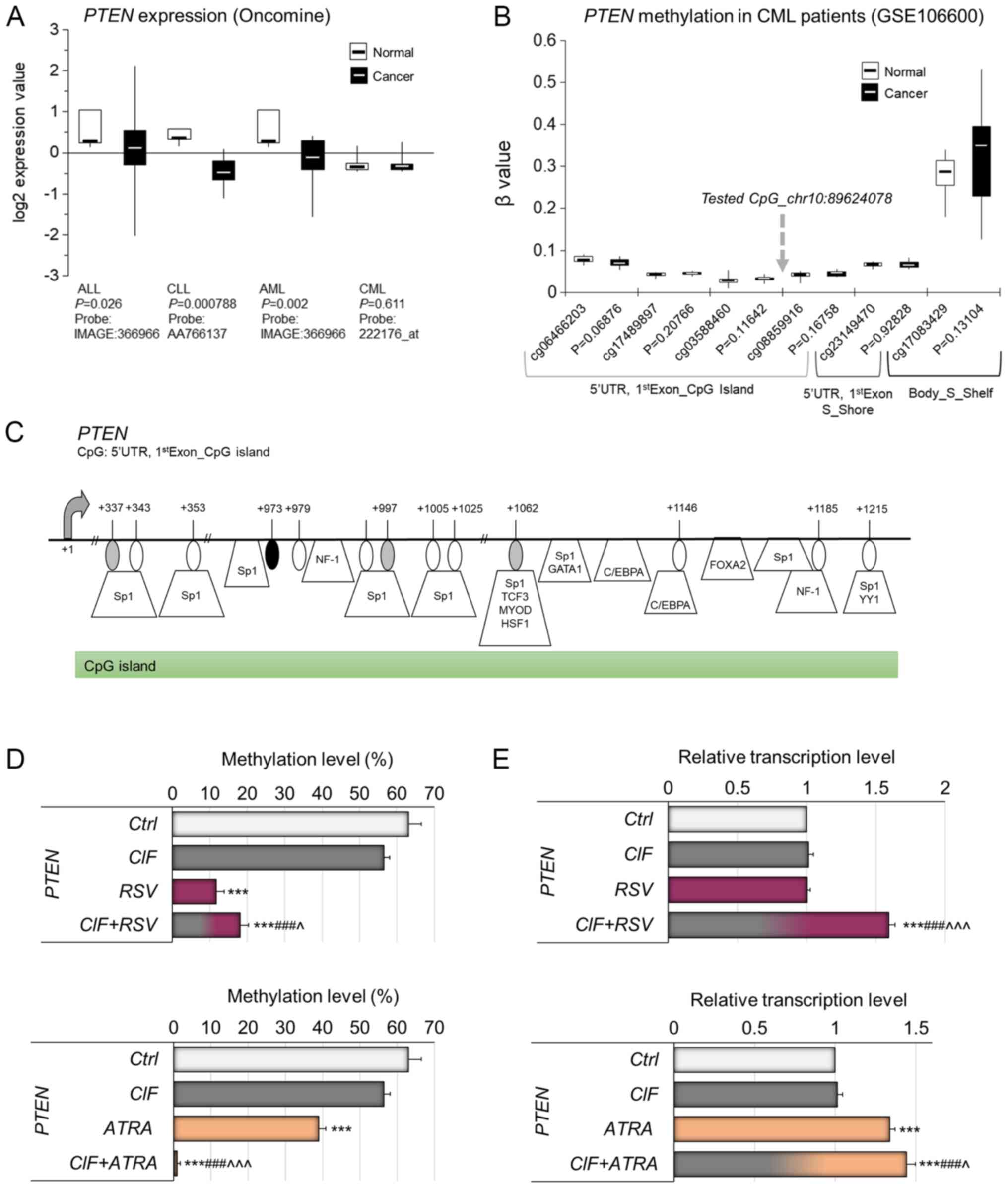 | Figure 4.Relevance of DNA methylation-mediated
silencing of PTEN in human leukemia in vivo. (A) Gene
expression microarray data for PTEN in different types of
leukemia. The normal vs. cancer gene expression data were obtained
from Oncomine and are presented as log2-transformed median centered
per array, and SD-normalized to 1 per array. The presented changes
are statistically significant (P<0.05) apart from the one
for CML leukemia (lack of statistically significant data). (B)
Methylation status of the CpG sites located in the neighborhood of
CpG site tested by MSRA (marked with gray arrow) within PTEN
CpG island, covered on Illumina 450K array and expressed as beta
value in CML and normal blood cells, based on NCBI's Gene
Expression Omnibus GEO (publicly available datasets, no.
GSE106600). Beta value, the methylation score for a specific CpG
site according to the fluorescent intensity ratio with any values
between 0 (unmethylated) and 1 (completely methylated). (C) A map
of the PTEN CpG island within gene first exon (Human
GRCh37/hg19 Assembly). The CpG site [+973 bp from transcription
start site (TSS)], which methylation state was tested by MSRA, is
indicated by a black oval shape. The CpG sites located nearby,
covered on Illumina 450K microarray platform, are depicted by gray
ovals. Putative transcription factor binding sites are marked as
predicted using TransFac. Effects of CIF, RSV and ATRA, as well as
CIF in combination with RSV (upper panels) or ATRA (bottom panels)
on (D) methylation of PTEN proximal promoter, and (E)
expression on mRNA level of PTEN gene in K562 cells (72 h
exposure). All compounds used at GI50 concentrations in
all experiments. Data represent the mean ± SD of three independent
experiments. ***P<0.001 vs. vehicle control;
###P<0.001 vs. CIF alone; ^P<0.05 and
^^^P<0.001 vs. RSV or ATRA alone. PTEN, phosphatase
and tensin homologue; SD, standard deviation. |
Oncomine data indicate that PTEN is
transcriptionally silenced in three types of leukemia, including
ALL, CLL and AML (Fig. 4A).
According to the results of one available study with CML patients,
no significant difference in PTEN expression has been
noticed between cancer and normal blood cells (Fig. 4A). The proximal promoter region
including CpG island of PTEN has been depicted in Fig. 4C. According to publicly available
Illumina 450K data (GSE106600), currently the only available study
for CML patients, any significant changes have not been observed in
PTEN promoter methylation within CpGs covered on Illumina
450K microarray in CML cells as compared to normal blood cells
(Fig. 4B). The detailed map in
Fig. 4C shows the exact position
of the tested CpG site, that is the CpG site within PTEN
proximal promoter CpG island, i.e., 5′UTR and/or first exon (+973
bp from transcription start site, TSS), not covered on Illumina
450K array (marked in black), located between two CpGs from this
microarray platform, i.e., cg03588460 (+337 bp from TSS, marked in
gray) and cg08859916 (+997 bp from TSS, marked in gray) (Fig. 4C). In our previous studies, this
CpG (chr10: 89624078, according to Human GRCh37/hg19 Assembly) has
been shown to be differentially methylated between breast cancer
cell lines with different level of invasiveness, suggesting its
regulatory role in PTEN transcription (14,18,30).
Putative transcription factor binding sites are demonstrated on the
PTEN gene map (Fig. 4C), as
predicted using TransFac. The multiple binding sites for DNA
methylation-sensitive transcription factors within the tested
PTEN promoter fragment support its potential regulatory role
in PTEN transcription (Fig.
4C) (18,47,48).
Previously, we identified the role of CIF in the
regulation of promoter methylation and expression of PTEN in
K562 (CML) cells (13). In the
present study, we checked if RSV and ATRA used alone can also
affect the transcriptional activity of these genes through the
remodeling of their promoter methylation.
72-hour exposure of K562 cells to RSV used alone at
GI50=11.5 µM, and ATRA used alone at GI50=30
µM concentration led to significant decreases in PTEN
promoter methylation by 51 and 24%, respectively (Fig. 4D), comparing to control unexposed
cells (63%). CIF administrated alone mediated 7% diminution in
PTEN promoter methylation level, although no significant
changes in DNMT1 expression have been observed. Our initial
unpublished studies in K562 cells indicate that CIF exposure leads
to inhibition of the activity of two enzymes important for
2′-deoxyadenosine metabolism, deoxyadenosine deaminase (ADA) and
S-adenosyl-L-homocysteine (SAH) hydrolase. CIF used at 5 nM
concentration caused decreases in ADA and SAH-hydrolase activities
by 30 and 15%, respectively. The CIF-mediated repression of ADA
activity may lead to 2′-deoxyadenosine accumulation up to the level
of toxic concentration in exposed cells. The raised levels of
2′-deoxyadenosine in cells can indirectly disrupt DNA methylation
reaction via SAH-hydrolase inhibition leading to SAM pool
depletion. A similar effect was shown by Wyczechowska and
Fabianowska-Majewska (49) in
K562 cells exposed to cladribine (49).
Upon exposure of K562 cells to CIF combined with
ATRA, we observed almost complete demethylation of PTEN
promoter compared to control K562 cells (Fig. 4D, bottom panel), whereas the extent
of PTEN hypomethylation followed by CIF+RSV administration
was similar to that caused by RSV alone (by approximately 50%)
(Fig. 4D, upper panel). These
alterations in the PTEN methylation pattern in K562 cells
were accompanied by enforced expression of this gene (Fig. 4E). The robust PTEN
upregulation was detected after both combinatorial administrations,
CIF+RSV or CIF+ATRA, that caused increases in PTEN
transcript level by 59 and 44%, when compared to control K562
cells, respectively (Fig. 4E).
Surprisingly, although CIF and RSV used alone did not lead to any
significant changes in PTEN expression in K562 cells upon 72
h of exposure, those compounds together exerted significant 59%
PTEN upregulation (Fig. 4E,
bottom panel).
Possibly, different concentration of CIF and RSV
used alone as well as other exposure time could benefit in stronger
PTEN re-expression (13).
It may also suggest that these compounds may cooperate in other
unknown mechanisms driving changes in PTEN expression.
Our findings suggest partial involvement of DNA
methylation in the regulation of PTEN transcriptional
activity, although other mechanisms can play an additional role as
well (46–48).
RARB transcriptional reactivation
followed by combinatorial exposures in K562 cells partly related to
its promoter hypomethylation
Expression of some tumor suppressor genes might be
indirectly regulated by PTEN, one of them is RARB. Lefebvre
et al (50) reported that
PTEN via negative regulation of PI3K/AKT signaling pathway
could affect RARB expression by blocking of SMRT
co-repressor recruitment to RARB promoter region, which
enhances histone acetylation and promotes RARB transcription
(50). Moreover, according to
publicly available data (Oncomine), tumor suppressor gene
RARB is downregulated in all types of leukemia (Fig. 5A). In Fig. 5B, the methylation status of CpG
sites at TSS200 promoter region of RARB enhancer in CML and
healthy individuals has been depicted (analyzed by Illumina 450K
Human Methylation Array, publicly available datasets from NCBI's
Gene Expression Omnibus GEO no. GSE106600). Among the 5 CpG sites
within the demonstrated fragment of the RARB promoter, the
CpG site located −139 bp from TSS, cg06720425 (chr3:25469694, Human
GRCh37/hg19 Assembly) was examined by MSRA (Fig. 5C). Similarly to PTEN, the
methylation state of the tested RARB CpG site has been shown
to distinguish between non-invasive and highly invasive breast
cancer cell lines, implying its potential regulatory role in
RARB transcription (14).
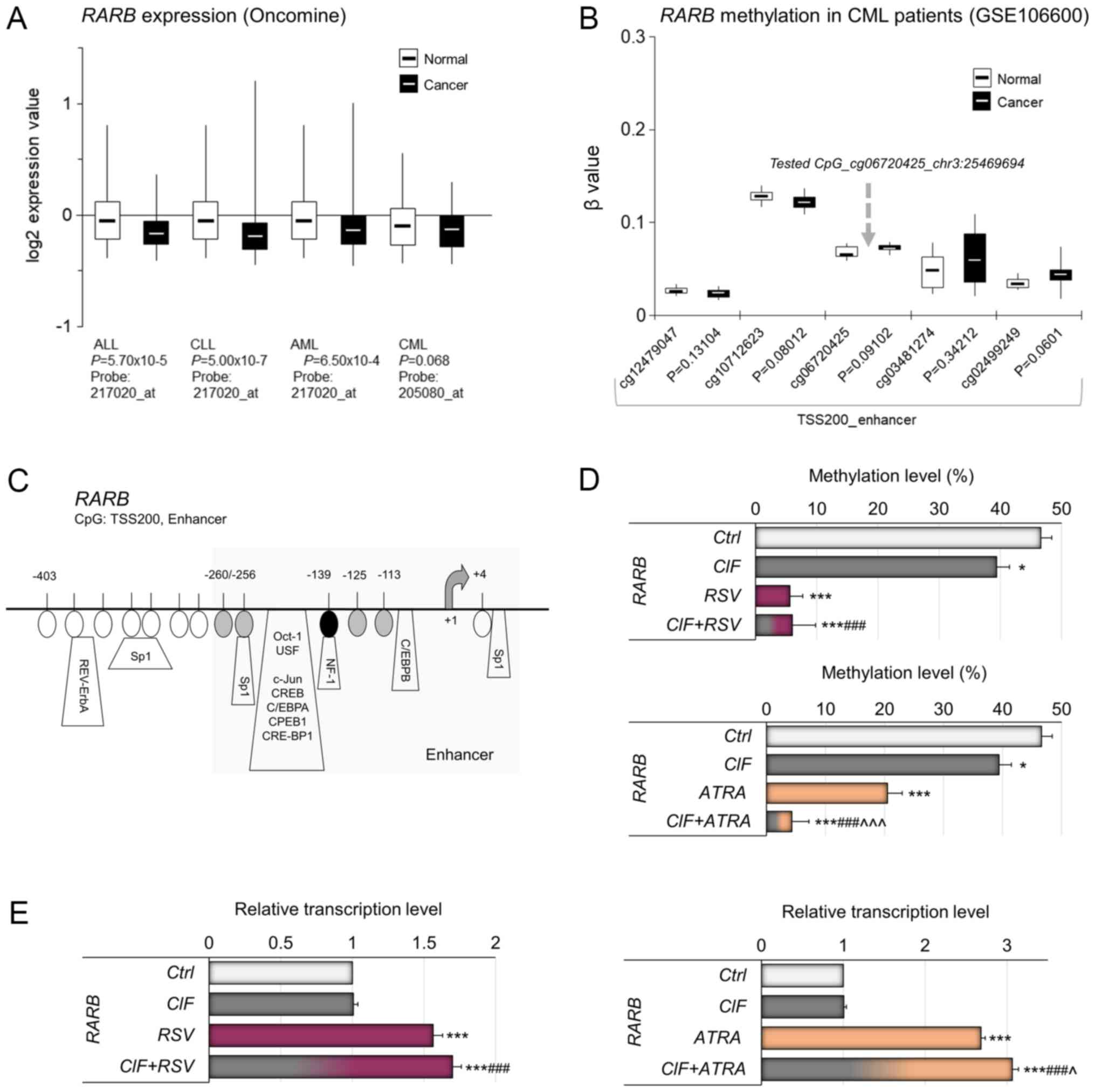 | Figure 5.Relevance of DNA methylation-mediated
silencing of RARB in human leukemia in vivo. (A) Gene
expression microarray data for RARB in different types of
leukemia. The normal vs. cancer gene expression data were obtained
from Oncomine and are presented as log2-transformed median centered
per array, and SD-normalized to 1 per array. The presented changes
are statistically significant (P<0.05) apart from the one
for CML leukemia (P=0.068). (B) Methylation status of the
CpG sites located in the neighborhood of CpG site tested by MSRA
(marked with gray arrow) covered on Illumina 450K array and
expressed as beta value in CML and normal blood cells, based on
NCBI's Gene Expression Omnibus GEO (publicly available datasets,
no. GSE106600). Beta value, the methylation score for a specific
CpG site according to the fluorescent intensity ratio with any
values between 0 (unmethylated) and 1 (completely methylated). (C)
A map of the RARB enhancer within TSS200 promoter region
(Human GRCh37/hg19 Assembly). The CpG site [-139 bp from
transcription start site (TSS)], which methylation state was tested
by MSRA, is indicated by a black oval shape. The CpG sites located
nearby, covered on Illumina 450K microarray platform, are depicted
by gray ovals. Putative transcription factor binding sites are
marked as predicted using TransFac. The effects of CIF, RSV and
ATRA used alone, as well as CIF in combination with RSV or ATRA on
(D) methylation of RARB promoter, and (E) expression on mRNA
level of RARB gene in K562 cells. All compounds used at
GI50 concentrations in all experiments. Data represent
the mean ± SD of three independent experiments. *P<0.05 and
***P<0.001 vs. vehicle control; ###P<0.001 vs. CIF
alone; ^P<0.05 and ^^^P<0.001 vs. RSV
or ATRA alone. RARB, retinoic acid receptor beta; SD, standard
deviation; CIF, clofarabine; RSV, resveratrol; ATRA, all-trans
retinoic acid. |
In K562 cells exposed to CIF, RSV or ATRA, used
alone, as well as to CIF+RSV and CIF+ATRA, statistically
significant demethylation of RARB gene promoter was
observed. CIF reduced the RARB promoter methylation level by
approximately 10% in comparison to control cells, RSV by 41% and
ATRA by 26% (Fig. 5D).
Combinational treatment with CIF+ATRA caused almost total
demethylation of RARB promoter with concomitant over 3-fold
increase in gene expression (Fig. 5D
and E, bottom panels). Interestingly, the exposure to RSV and
CIF+RSV, despite robust alteration in promoter methylation led to
less pronounced RARB up-regulation, by 60–70% (Fig 5D and E, upper panels).
It is worth pointing out, that the higher extent of
changes mediated by CIF+ATRA combination in exposed K562 cells,
i.e., CDKN1A transcriptional reactivation that may result in
decreased E2F activity, as well as re-expression of PTEN and
RARB encoding proteins that inhibit AP-1 activity, strongly
support enhanced DNMT1 downregulation (Fig. 6) observed upon this combined
exposure as compared to the compounds used alone and CIF+RSV
combination (Figs. 3, 4 and 5).
Mechanisms underlying the observed effects are interesting and
remain to be elucidated in future experiments.
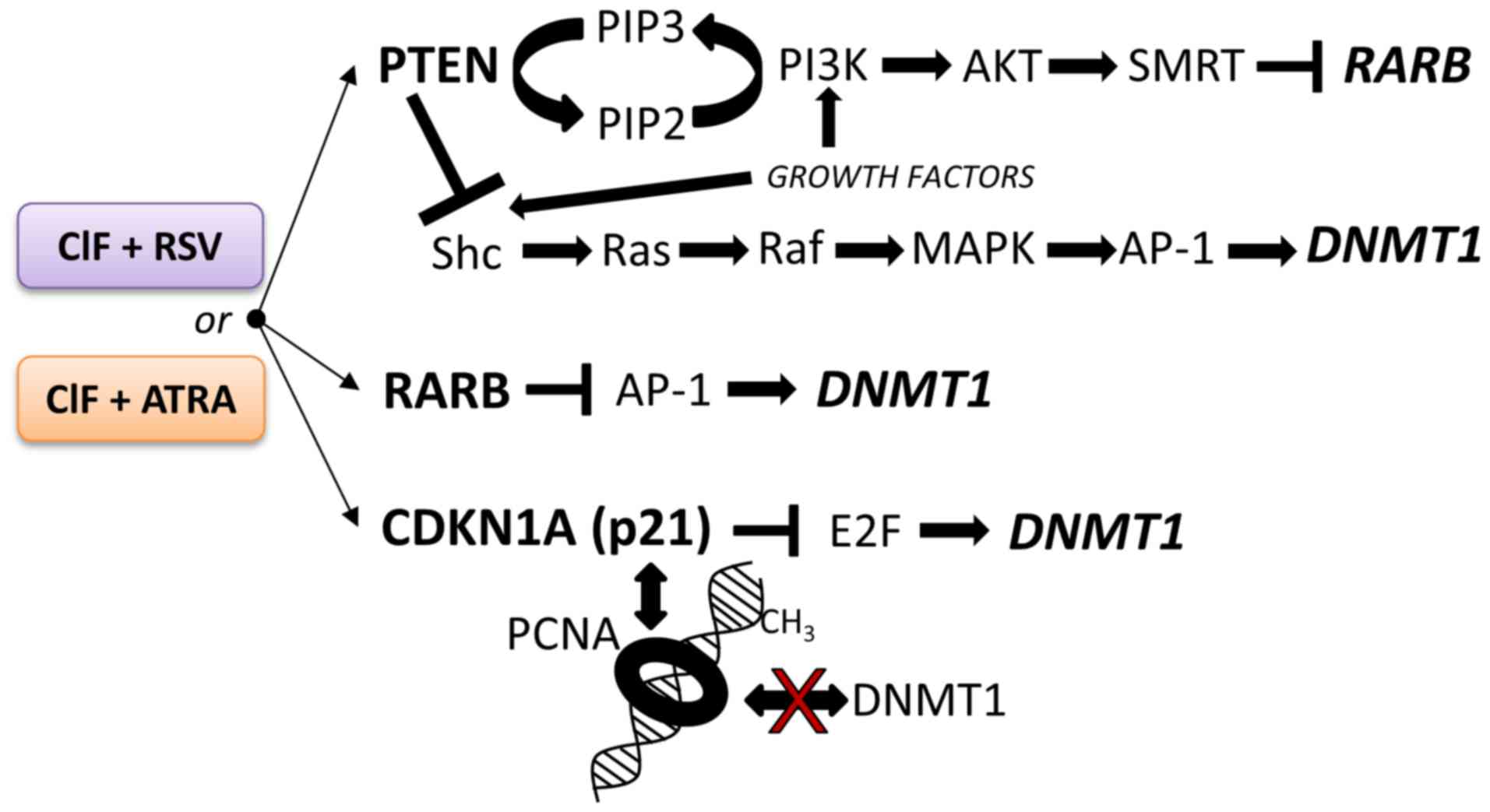 | Figure 6.The potential repressive effects of
the tested combinatorial exposures of CIF with RSV or ATRA on
modulation of DNMT1 transcription and/or DNMT1 activity in
K562 leukemia cells. Implications of PTEN-mediated negative
regulation of intracellular oncogenic signaling pathways, including
PI3K/AKT and MAPK/AP-1. RARB and p21 (CDKN1A) proteins are negative
regulators of AP-1 and E2F. These transcription factors (AP-1 and
E2F) activate DNMT1 expression due to the presence of
binding sites in DNMT1 regulatory region. A competition of
CDKN1A (p21) with DNMT1 for the same binding site on proliferating
cell nuclear antigen. Shc, SH2-containing collagen-related
proteins; PIP2, phosphatidylinositol (4,5)-bisphosphate; PIP3,
phosphatidylinositol (3,4,5)-trisphosphate; SMRT, thyroid-,
retinoic-acid-receptor-associated corepressor. CIF, clofarabine;
DNMT1, DNA methyltransferase 1; RSV, resveratrol; ATRA, all-trans
retinoic acid; PTEN, phosphatase and tensin homologue; CDKN1A,
Cyclin dependent kinase inhibitor 1A. |
As some authors suggest, CML is the ‘poster child’
for targeted cancer therapy. The identified target, a product of
abnormal gene BCR-ABL became the aim of drug development and as
mentioned above the TKIs inhibitors were introduced to CML therapy.
Nowadays the first- or second-generation TKIs are the first-line
treatment of patients with newly diagnosed CML. Although initial
responses are high, in more than 25% of patients the therapy fails
and/or they develop resistance to the treatment. For several years,
intensive work has been underway to explain treatment failure and
to identify different mechanisms of the drug resistance. The
resistance to TKIs based on clinical outcomes can be explained by
genomic mechanisms (mutations in the BCR-ABL domain), but also by
BCR-ABL-independent mechanisms (poor compliance, drug influx and
efflux, activation of alternative signaling pathways, plasma TKI
concentration, insensitivity of quiescent stem cells) (51). However, epigenetic dysregulation of
the expression of the CML-associated genes has been reported as
well (7). Thus, in order to
improve the effectiveness of CML therapies, there is a strong need
to develop new treatment strategies.
Nishioka et al reported that
hypermethylation of PTEN promoter is associated with this
gene downregulation and activation of pro-survival signaling
mediated by AKT in leukemia cells. According to the other authors'
findings, the PTEN silencing induced by DNA methylation
requires EZH2 and DNA methylation enzymes. Moreover, the authors
claim that the epigenetic silencing of PTEN is one of the
mechanisms that cause drug resistance in individuals with leukemia
after exposure to Imatinib (52,53).
In this context demethylation and re-expression of PTEN seem
to be a promising way to achieve long term therapeutic response.
Our initial results show that natural bioactive compounds, mainly
ATRA but also RSV, especially in combination with CIF, might
positively modulate PTEN expression. Additionally, a
combination of RSV with CIF indicates high pro-apoptotic activity
in K562 CML cells. In work of Can et al (54) RSV (used alone in high dose) has
also effectively induced apoptosis of K562/IMA-3 cells (resistant
CML cells). These results may suggest the potential use of ATRA
and/or RSV in CML therapy not only in patients with primary but
also with acquired resistance to TKIs.
In summary, our study is the first to demonstrate
the epigenetic anticancer capacity of the combinatorial exposures
of CIF and ATRA or RSV in CML cells. Upon 72 h-treatment, the
tested combinations led to significant cell growth inhibition and
greater induction of caspase-3-dependent apoptosis. These
observations may be related to accompanied relevant DNMT1
downregulation and robust CDKN1A upregulation, with a
concomitant, enhanced decrease in DNMT1 protein level, especially
after CIF with ATRA. Concurrent methylation-mediated RARB
and PTEN reactivation have been detected. The proteins
encoded by these genes are crucial for the regulation of important
intracellular oncogenic signaling pathways, including PI3K/AKT and
MAPK/AP-1 pathways. Taken together, our results reveal that CIF
used in combination with the tested phytochemicals, RSV or ATRA,
has the higher ability to remodel DNA methylation marks and promote
cell death in CML cells.
Future studies will focus on assessing the efficacy
of clofarabine-phytochemical combination exposures in other CML
in vitro and in vivo models. We believe that further
extensive studies of this new combinatorial strategy, the CIF
combinations with ATRA or RSV, may support its translational
application as a therapeutic epigenetic approach against CML.
Acknowledgements
The authors would like to thank Dr Justyna
Jakubowska from The Department of Pediatrics, Oncology and
Hematology, as well as Professor Piotr Smolewski and Dr Barbara
Cebula-Obrzut from Department of Experimental Hematology, Medical
University of Lodz, Poland, for their support in performing and
analyzing flow cytometry experiments.
Funding
This study was supported by Medical University of
Lodz in Poland (grant nos. 503/6-099-01/503-61-001,
502-03/6-099-01/502-64-007, 502-03/6-099-01/502-64-089 and
502-03/6-099-01/502-64-133) granted to The Department of Biomedical
Chemistry, Faculty of Health Sciences at the Medical University of
Lodz (Lodz, Poland).
Availability of data and materials
All data generated or analyzed during this study
are included in this published article.
Authors' contributions
AKS and KM conducted the experiments. AKS, KM, ASM,
KFM and KL performed the analysis and contributed to writing and
editing the manuscript. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen Y, Peng C, Li D and Li S: Molecular
and cellular bases of chronic myeloid leukemia. Protein Cell.
1:124–132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Soverini S, de Benedittis C, Mancini M and
Martinelli G: Mutations in the BCR-ABL1 kinase domain and elsewhere
in chronic myeloid leukemia. Clin Lymphoma Myeloma Leuk. 15
(Suppl):S120–S128. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Druker BJ, Talpaz M, Resta DJ, Peng B,
Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R,
Ohno-Jones S and Sawyers CL: Efficacy and safety of a specific
inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid
leukemia. N Engl J Med. 344:1031–1037. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bhatia R, Holtz M, Niu N, Gray R, Snyder
DS, Sawyers CL, Arber DA, Slovak ML and Forman SJ: Persistence of
malignant hematopoietic progenitors in chronic myelogenous leukemia
patients in complete cytogenetic remission following imatinib
mesylate treatment. Blood. 101:4701–4707. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chomel JC, Bonnet ML, Sorel N, Bertrand A,
Meunier MC, Fichelson S, Melkus M, Bennaceur-Griscelli A, Guilhot F
and Turhan AG: Leukemic stem cell persistence in chronic myeloid
leukemia patients with sustained undetectable molecular residual
disease. Blood. 118:3657–3660. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Leo E and Martinelli G: DNA methylation in
chronic myeloid leukemia. J Mol Genet Med. 8:1182014.
|
|
7
|
Koschmieder S and Vetrie D: Epigenetic
dysregulation in chronic myeloid leukaemia: A myriad of mechanisms
and therapeutic options. Semin Cancer Biol. 51:180–197. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Robertson KD: Epigenetic mechanisms of
gene regulationDNA Methylation and Cancer Therapy. Medical
Intelligence Unit. Springer; Boston, MA: pp. 13–30. 2005,
View Article : Google Scholar
|
|
9
|
Jaenisch R and Bird A: Epigenetic
regulation of gene expression: How the genome integrates intrinsic
and environmental signals. Nat Genet. 33 (Suppl):S245–S254. 2003.
View Article : Google Scholar
|
|
10
|
Jones PA and Baylin SB: The epigenomics of
cancer. Cell. 128:683–692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chik F, Szyf M and Rabbani SA: Role of
epigenetics in cancer initiation and progression. Adv Exp Med Biol.
720:91–104. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen T and Li E: Structure and function of
eukaryotic DNA methyltransferases. Curr Top Dev Biol. 60:55–89.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Majda K, Kaufman-Szymczyk A,
Lubecka-Pietruszewska K, Bednarek A and Fabianowska-Majewska K:
Influence of clofarabine on transcriptional activity of PTEN, APC,
RARB2, ZAP70 genes in K562 cells. Anticancer Res. 30:4601–4606.
2010.PubMed/NCBI
|
|
14
|
Lubecka-Pietruszewska K, Kaufman-Szymczyk
A, Stefanska B, Cebula-Obrzut B, Smolewski P and
Fabianowska-Majewska K: Clofarabine, a novel adenosine analogue,
reactivates DNA methylation-silenced tumour suppressor genes and
inhibits cell growth in breast cancer cells. Eur J Pharmacol.
723:276–287. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ghanem H Jabbour E, Faderl S, Ghandhi V,
Plunkett W and Kantarjian H: Clofarabine in leukemia. Expert Rev
Hematol. 3:15–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ghanem H, Kantarjian H, Ohanian M and
Jabbour E: The role of clofarabine in acute myeloid leukemia. Leuk
Lymphoma. 54:688–698. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stefanska B, Karlic H, Varga F,
Fabianowska-Majewska K and Haslberger A: Epigenetic mechanisms in
anti-cancer actions of bioactive food components-the implications
in cancer prevention. Br J Pharmacol. 167:279–297. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stefanska B, Salamé P, Bednarek A and
Fabianowska-Majewska K: Comparative effects of retinoic acid,
vitamin D and resveratrol alone and in combination with adenosine
analogues on methylation and expression of phosphatase and tensin
homologue tumour suppressor gene in breast cancer cells. Br J Nutr.
107:781–790. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lubecka K, Kurzava L, Flower K, Buvala H,
Zhang H, Teegarden D, Camarillo I, Suderman M, Kuang S, Andrisani
O, et al: Stilbenoids remodel the DNA methylation patterns in
breast cancer cells and inhibit oncogenic NOTCH signaling through
epigenetic regulation of MAML2 transcriptional activity.
Carcinogenesis. 37:656–668. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee YJ, Lee YJ, Im JH, Won SY, Kim YB, Cho
MK, Nam HS, Choi YJ and Lee SH: Synergistic anti-cancer effects of
resveratrol and chemotherapeutic agent clofarabine against human
malignant mesothelioma MSTO-211H cells. Food Chem Toxicol.
52:61–68. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee YJ, Hwang IS, Lee YJ, Lee CH, Kim SH,
Nam HS, Choi YJ and Lee SH: Knockdown of Bcl-xL enhances
growth-inhibiting and apoptosis-inducing effects of resveratrol and
clofarabine in malignant mesothelioma H-2452 cells. J Korean Med
Sci. 29:1464–1472. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee YJ, Lee YJ and Lee SH: Resveratrol and
clofarabine induces a preferential apoptosis-activating effect on
malignant mesothelioma cells by Mcl-1 down-regulation and caspase-3
activation. BMB Rep. 48:166–171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kulkarni SS and Cantó C: The molecular
targets of resveratrol. Biochim Biophys Acta. 1852:1114–1123. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Theodosiou M, Laudet V and Schubert M:
From carrot to clinic: An overview of the retinoic acid signaling
pathway. Cell Mol Life Sci. 67:1423–1445. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tang XH and Gudas LJ: Retinoids, retinoic
acid receptors, and cancer. Annu Rev Pathol. 6:345–364. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Connolly R, Nguyen NK and Sukumar S:
Molecular pathways: Current role and future directions of the
retinoic acid pathway in cancer prevention and treatment. Clin
Cancer Res. 19:1651–1659. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schenk T, Stengel S and Zelent A:
Unlocking the potential of retinoic acid in anticancer therapy. Br
J Cancer. 111:2039–2045. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang W and Xu J: DNA methyltransferases
and their roles in tumorigenesis. Biomark Res. 5:12017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu Q, Chen ZM and Su WJ: Anticancer effect
of retinoic acid via AP-1 activity repression is mediated by
retinoic acid receptor alpha and beta in gastric cancer cells. Int
J Biochem Cell Biol. 34:1102–1114. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stefanska B, Rudnicka K, Bednarek A and
Fabianowska-Majewska K: Hypomethylation and induction of retinoic
acid receptor beta 2 by concurrent action of adenosine analogues
and natural compounds in breast cancer cells. Eur J Pharmacol.
638:47–53. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
McCabe MT, Davis JN and Day ML: Regulation
of DNA methyltransferase 1 by the pRb/E2F1 pathway. Cancer Res.
65:3624–3632. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Delavaine L and La Thangue NB: Control of
E2F activity by p21Waf1/Cip1. Oncogene. 18:5381–5392. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chuang LS, Ian HI, Koh TW, Ng HH, Xu G and
Li BF: Human DNA-(cytosine-5) methyltransferase-PCNA complex as a
target for p21WAF1. Science. 277:1996–2000. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Iida T, Suetake I, Tajima S, Morioka H,
Ohta S, Obuse C and Tsurimoto T: PCNA clamp facilitates action of
DNA cytosine methyltransferase 1 on hemimethylated DNA. Genes
Cells. 7:997–1007. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yamada KM and Araki M: Tumor suppressor
PTEN: Modulator of cell signaling, growth, migration and apoptosis.
J Cell Sci. 114:2375–2382. 2001.PubMed/NCBI
|
|
36
|
Iwase H, Omoto Y, Iwata H, Toyama T, Hara
Y, Ando Y, Ito Y, Fujii Y and Kobayashi S: DNA methylation analysis
at distal and proximal promoter regions of the oestrogen receptor
gene in breast cancers. Br J Cancer. 80:1982–1986. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pfaffl MW, Horgan GW and Dempfle L:
Relative expression software tool (REST) for group-wise comparison
and statistical analysis of relative expression results in
real-time PCR. Nucleic Acids Res. 30:e362002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sui T, Ma L, Bai X, Li Q and Xu X:
Resveratrol inhibits the phosphatidylinositide 3-kinase/protein
kinase B/mammalian target of rapamycin signaling pathway in the
human chronic myeloid leukemia K562 cell line. Oncol Lett.
7:2093–2098. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang B, Liu J and Gong Z: Resveratrol
induces apoptosis in K562 cells via the regulation of mitochondrial
signaling pathways. Int J Clin Exp Med. 8:16926–16933.
2015.PubMed/NCBI
|
|
40
|
Mizuno S, Chijiwa T, Okamura T, Akashi K,
Fukumaki Y, Niho Y and Sasaki H: Expression of DNA
methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in
acute and chronic myelogenous leukemia. Blood. 97:1172–1179. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tan HH and Porter AG: p21(WAF1) negatively
regulates DNMT1 expression in mammalian cells. Biochem Biophys Res
Commun. 382:171–176. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu M, Iavarone A and Freedman LP:
Transcriptional activation of the human p21(WAF1/CIP1) gene by
retinoic acid receptor. Correlation with retinoid induction of U937
cell differentiation. J Biol Chem. 271:31723–31728. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yu Z, Li W, Lu Q, Wang L, Zhang X, Han P,
Chen P and Pei Y: p21 is required for atRA-mediated growth
inhibition of MEPM cells, which involves RAR. J Cell Biochem.
104:2185–2192. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bowers JL, Tyulmenkov VV, Jernigan SC and
Klinge CM: Resveratrol acts as a mixed agonist/antagonist for
estrogen receptors alpha and beta. Endocrinology. 141:3657–3667.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Parveen A, Akash MS, Rehman K and Kyunn
WW: Dual role of p21 in the progression of cancer and its
treatment. Crit Rev Eukaryot Gene Expr. 26:49–62. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Montiel-Duarte C, Cordeu L, Agirre X,
Román-Gómez J, Jiménez-Velasco A, José-Eneriz ES, Gárate L, Andreu
EJ, Calasanz MJ, Heiniger A, et al: Resistance to Imatinib
mesylate-induced apoptosis in acute lymphoblastic leukemia is
associated with PTEN down-regulation due to promoter
hypermethylation. Leuk Res. 32:709–716. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
García JM, Silva J, Peña C, Garcia V,
Rodríguez R, Cruz MA, Cantos B, Provencio M, España P and Bonilla
F: Promoter methylation of the PTEN gene is a common molecular
change in breast cancer. Genes Chromosomes Cancer. 41:117–124.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Goel A, Arnold CN, Niedzwiecki D,
Carethers JM, Dowell JM, Wasserman L, Compton C, Mayer RJ,
Bertagnolli MM and Boland CR: Frequent inactivation of PTEN by
promoter hypermethylation in microsatellite instability-high
sporadic colorectal cancers. Cancer Res. 64:3014–3021. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wyczechowska D and Fabianowska-Majewska K:
The effects of cladribine and fludarabine on DNA methylation in
K562 cells. Biochem. Pharmacol. 65:219–225. 2003.
|
|
50
|
Lefebvre B, Brand C, Flajollet S and
Lefebvre P: Down-regulation of the tumour suppressor gene retinoic
acid receptor beta2 through the phosphoinositide 3-knase/Akt
signaling pathway. Mol Endocrinol. 20:2109–2121. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lussana F, Intermesoli T, Stefanoni P and
Rambaldi A: Mechanisms of resistance to targeted therapies in
chronic myeloid leukemia. Handb Exp Pharmacol. 249:231–250. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nishioka C, Ikezoe T, Yang J and Yokoyama
A: Long-term exposure of leukemia cells to multi-targeted tyrosine
kinase inhibitor induces activations of AKT, ERK and STAT5
signaling via epigenetic silencing of the PTEN gene. Leukemia.
24:1631–1640. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Nishioka C, Ikezoe T, Yang J, Udaka K and
Yokoyama A: Imatinib causes epigenetic alterations of PTEN gene via
upregulation of DNA methyltransferases and polycomb group proteins.
Blood Cancer J. 1:e482011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Can G, Cakir Z, Kartal M, Gunduz U and
Baran Y: Apoptotic effects of resveratrol, a grape polyphenol, on
imatinib-sensitive and resistant K562 chronic myeloid leukemia
cells. Anticancer Res. 32:2673–2678. 2012.PubMed/NCBI
|