Introduction
More than 1.5 billion people worldwide are affected
by hypertension, yet its etiology and pathogenic mechanisms remain
unclear (1,2). Among these patients, those with
essential hypertension (EH) account for 90–95% of the total
hypertensive patient population (3). EH is a polygenic genetic disease
mediated by interactions between genetic and environmental factors,
and is a risk factor for the development of various cardiovascular
and cerebrovascular diseases (such as myocardial infarction,
atherosclerosis and cerebral apoplexy) (4). The key component of EH morbidity is
the blood vessels (5), and the
direct etiological factor comprises an increase in peripheral
vascular resistance, which leads to endothelial dysfunction in the
heart, brain, kidney, and surrounding blood vessels (5).
It has been reported that low levels of dietary
Ca2+ represent a significant risk factor for
hypertension, whereas the intake of appropriate amounts of
Ca2+ effectively lowers blood pressure (BP), as well as
the incidence and mortality of cardiovascular disease (6,7).
This has been confirmed by animal studies (8). Calcium sensing receptor (CaSR) is the
receptor for extracellular Ca2+, and a member of the C
subfamily of the G protein coupled receptor (GPCR) family. In 1993,
Brown et al (9) first
cloned CaSR from the bovine parathyroid gland. CaSR is mainly
distributed in the parathyroid gland, bone, kidney, intestines and
other tissue cells (such as hepatocytes, vascular endothelial cells
and vascular smooth muscle cells (VSMCs) related to calcium
transfer and calcium homeostasis regulation, and it plays a key
role in maintaining calcium homeostasis (10,11).
When the extracellular calcium concentration
([Ca2+]o) rises, CaSR is activated. With Gαq
as a mediator, CaSR increases the levels of inositol
1,4,5-triphosphate (IP3) and diphosphoglycerate through
phospholipase C (PLC), inducing an increase in the intercellular
calcium concentration ([Ca2+]i) and
inhibiting parathyrin (9,12). Subsequently,
[Ca2+]o is restored to normal levels
(13).
In research on the pathogenesis of hypertension, the
renin-angiotensin system (RAS) is important, and it is the most
important regulatory factor in vivo. Renin is the first
rate-limiting enzyme of the RAS, and cyclic adenosine monophosphate
(cAMP) functions as a key effector in this system (14). Existing research shows that
[Ca2+]o and [Ca2+]i are
inversely proportional to renin secretion by the juxtaglomerular
apparatus (15). Furthermore, the
mechanism associated with this is related to the increase in
[Ca2+]o through increasing
[Ca2+]i and/or the activity of calmodulin
(CaM), and inhibiting adenylate cyclase-V (AC-V) so that cAMP
production is decreased; it is also related to the inhibition of
renin release (16,17). In freshly isolated and primary
cultured granulocytes of the glomerulus, CaSR is activated, and the
ryanodine receptor is also activated via the G
protein-PLC-IP3 pathway, which causes an increase in
[Ca2+]i, the suppression of adenylyl cyclase
V (AC-V) activity, reductions in cAMP, and the inhibition of renin
release (12). Our previous
research shows that CaSR participates in the occurrence and
development of EH. The increase in BP was found to be related to
the low expression of CaSR (18).
In addition, BP-mediated regulation of CaSR was identified via
activation of the cAMP-RAS pathway (18).
In recent years, with extensive research on CaSR,
its prominent effect on the cardiovascular system has caused this
protein to receive extensive attention. Of note, CaSR is also
expressed in the vasculature. Smajilovic and Tfelt-Hansen (19) reported a rise in
[Ca2+]o through the activation of CaSR. The
mechanism of BP reduction was also found to be linked to the
inhibition of RAS activation and consequent vasodilatation. Thus,
the changes in [Ca2+]o concentration and
activity of CaSR have a direct effect on angiotasis and thus
influences the BP directly (19).
Cow (20) revealed that increasing
[Ca2+]o and other CaSR calcimimetics (such as
neomycin, Mg2+) can mediate the vasodilatation or
exhibit two-way regulatory effect on the mesenteric artery (MA),
coronary artery, renal artery and cerebral artery; the mechanism
underlying this phenomenon does not rely on the vascular
endothelium (21), the production
of nitric oxide (NO) via CaSR activation and/or the effect of
endothelium-derived hyperpolarizing factor, and the peripheral and
central nervous systems (22–25).
The regulation of angiotasis and BP by CaSR are consequences of the
involvement of polymolecular and multiple mechanisms (19–25);
however, the specific regulatory mechanism is yet to be determined.
In addition, whether RAS participates in the regulation of CaSR
during angiotasis requires further investigation.
Based on the results of previous experiments and the
relationship between Ca2+, RAS, and CaSR, we proposed
the following research hypotheses: In spontaneously hypertensive
rats (SHRs), CaSR expression is reduced or functional defects lead
to a decrease in [Ca2+]i mediated by the
PLC-IP3 pathway, the amelioration of the inhibitory
effect on AC-V, increases in cAMP, the promotion of renin release,
and vasoconstriction mediated via the RAS pathway, which are
involved in the occurrence and development of hypertension. Thus,
the present study aimed to examine the effects of CaSR on the
vascular reactivity in hypertensive rats. This mechanism was
illustrated with respect to the PLC-IP3/AC-V/cAMP/RAS
pathway, and may provide further insight into the prevention of
hypertension and the effects of novel therapeutic agents.
Materials and methods
Animals
In total, 216 male Wistar-Kyoto rats (WKY) and SHRs
(age, 12 weeks; weight, 200–300 g) were obtained from Vital River
Laboratory Animal Science and Technology Co, Ltd. in Beijing
(license no. SCXK2012-0001), and housed in a controlled environment
with 48±2% humidity at 22±2°C under a 12 h light/dark cycle. The
SHR and WKY were divided into different groups (n=6/group), and
provided ad libitum access to food and water. The animal
study was approved by the Institutional Animal Research Committee
of Shihezi Medical University (license no. 2015-041-01), and all
animals received humane care in compliance with the Guide for the
Care and Use of Laboratory Animals published by the National
Institutes of Health (26).
Chemicals and reagents
Phenylephrine (PE; Sigma-Aldrich; Merck KGaA),
sodium nitroprusside (SNP; Sigma-Aldrich; Merck KGaA), indomethacin
(Cyclooxygenase 2 inhibitor; Sigma-Aldrich; Merck KGaA), EDTA
(Sigma-Aldrich; Merck KGaA), dimethyl sulfoxide (DMSO;
Sigma-Aldrich; Merck KGaA), acetylcholine (Ach; Apexbio
Corporation), MDL12330A (AC-V inhibitor, Apexbio Corporation).
NPS2143 (CaSR inhibitor, R&D Systems, Inc.), NPSR568 (CaSR
agonist, R&D Systems, Inc.). NG-nitro-L-arginine
methyl ester (L-NAME, NOS inhibitor; Selleck Chemicals), U73122
(PLC inhibitor, Selleck Chemicals), 2-aminoethoxydiphenyl borate
(2-APB, IP3 inhibitor; Selleck Chemicals), Captopril
(CAP, angiotensin-converting enzyme inhibitor, Selleck Chemicals),
Losartan [LOS, angiotensin II receptor type 1 (AT1R) inhibitor,
Selleck Chemicals]. All of the other chemicals were of reagent
grade. Indomethacin, L-NAME, NPS2143, NPSR568, U73122, 2-APB,
MDL12330A, CAP and LOS were dissolved in DMSO to prepare stock
solutions (10−1 mol/l), with the use of double distilled
water prepared into the corresponding working fluid. The other
agents were prepared in double distilled water. Studies have shown
no notable effects on the tension development of isolated arteries
with concentrations of DMSO<0.2% (27–29).
Blood pressure measurement
Before the experiment, the BP was measured via the
tail cuff method, which included warming the whole animal body in
the absence of anaesthesia (BP-96A-L, Softron) (30). Measurements were taken every day
for a week, at the same time of day and at a controlled temperature
of 30°C. After the rats had acclimated to the environment, the
systolic blood pressure (SBP), diastolic blood pressure (DBP) and
mean arterial pressure (MAP) were measured [MAP = (SBP+2 × DBP)/3].
The measurements were repeated three times, and the average of the
three measurements was calculated.
Western blot analysis
The rats were sacrificed via decapitation under
anesthesia with 3% sodium pentobarbital (50 mg/kg, intraperitoneal
injection). The MA was harvested and then lysed. It was then
homogenized in RIPA lysis buffer (Beijing Solarbio Science &
Technology Co., Ltd.) supplemented with PMSF (100:1) and
centrifuged at 14,000 × g for 15 min at 4°C. The supernatants were
collected and the protein concentration was determined using the
bicinchoninic acid method. Electrophoresis was performed using 15
µg of samples and 10% SDS-PAGE gels, followed by transfer of the
proteins to 0.45 mm Sequi Blot polyvinylidene fluoride membranes.
After they were incubated with 5% non-fat milk, the membranes were
then incubated overnight at 4°C with primary antibodies against
CaSR (1:1,000; Abcam; cat. no. WH0000846M1) and β-actin (1:1,000;
Wuhan Boster Biological Technology, Ltd; cat. no. A0760-41M). After
washing, the membranes were incubated with fluorescence-labelled
goat anti-mouse or anti-rabbit IgGs (1:20,000; Wuhan Boster
Biological Technology, Ltd; cat. no. ab2891) for ~2 h at room
temperature. Finally, the protein was visualized using an enhanced
chemiluminescence system (Pierce; Thermo Fisher Scientific, Inc).
The intensities of the protein bands were quantified using Bio-Rad
Quantity One 4.6.2 software (Bio-Rad Laboratories, Inc), and the
levels of the protein detected were normalized to that of
β-actin.
Preparation of isolated artery rings
(31,32)
Following sacrifice, the gastrointestinal tract with
the mesenteric arcade attached was excised rapidly from rats and
steeped in a physiological saline solution (PSS; 119 mmol/l NaCl,
4.69 mmol/l KCl, 1.17 mmol/l MgSO4·7H2O, 1.18
mmol/l KH2PO4, 2.5 mmol/l CaCl2,
25 mmol/l NaHCO3, 0.026 mmol/l EDTA and 5.5 mmol/l
glucose, which was bubbled with 95% O2 and 5%
CO2, resulting in a pH of 7.4 at 4°C) (33). Second-order small mesenteric
arteries (<400 µm internal diameter) were dissected and cleaned
of adhering fat and connective tissues (34). They were then cut into 2–3 mm blood
vessels; tissues were fixed within 45 min of sacrifice.
Pressure myograph techniques for
detecting blood vessel diameter
The blood vessel segment was placed in a perfusion
chamber filled with PSS, connected to a glass microelectrode
(diameter: ~50–100 µm), and fixed to prevent air leakage. The
perfusion chamber was moved to the microscope stage, with the
magnification set as 10 (objective) ×10 (eyepiece). During the
incubation of the blood vessels, the water bath was continuously
permeated with mixed gas (95% O2 and 5% CO2)
and the temperature was maintained at 37°C. The pressure was
increased, and the initial parameter settings were: P1 (20 mmHg),
P2 (5 mmHg), and a duration of 3 min. After that, the P2 pressure
was increased to 20 mmHg, and the duration was increased to 5 min.
Then, the pressure was increased by 10 mmHg for 5 min each time
until it stabilized to 60 mmHg. The vascular segments were
incubated for 1 h in a PSS solution maintained at 60 mmHg, 37°C,
and pH 7.4, and the PSS fluid was changed every 20 min during the
incubation period. The vascular segment was normalized after the
equilibration (35), and the
potassium physiological saline solution (KPSS, a depolarizing
stimulus) fluid was used to detect the activity of the blood
vessels. The KPSS contained: 123.70 mmol/l KCl, 1.17 mmol/l
MgSO4·7H2O, 1.18 mmol/l
KH2PO4, 2.5 mmol/l CaCl2,
NaHCO2 25 mmol/l NaHCO2, 0.026 mmol/l EDTA,
and 5.5 mmol/l glucose. A vascular contraction amplitude greater
than the diameter of 1/3 was observed, and contraction after
reaching the maximum or smooth platform with Ach (10−5
mol/l) diastolic blood vessels was achieved. The relaxation rate
was >60–80% with the endothelium-intact, and the vascular
activity was good, which could be used in the experiment or
discarded.
For denuded endothelium, the vascular tissue was
rotated once around the tip of the microelectrode of the glass, or
the gas was injected into the vessel segment slowly (36). Indomethacin (10−5 mol/l)
and L-NAME (10−4 mol/l) were pre-incubated for 20 min at
37°C (37), and persisted
throughout the experiment. Providing that Ach induced the
relaxation of blood vessels by <5% (38,39),
it was considered that the endothelium was denuded. The experiment
commenced after the specimens were rinsed three times with PSS. The
PSS solution in the water bath was controlled at 5 ml and using a
micropipette, 10−7, 10−6, 10−5,
10−4, 10−3 or 10−2 mol/l PE
(GPCR-mediated), Ach (endothelium-dependent vasodilator) and SNP
(endothelium-independent vasodilator), were respectively added.
After the vascular response reached a maximum or stable state, a
higher concentration was added. The final concentration was
10−9−10−4 mol/l, and the effect of the drug
on the blood vessel diameter was observed. The effects of
inhibitors (NPS2143, 10−5 mol/l; U73122, 10−5
mol/l; 2-APB, 10−4 mol/l; MDL12330A, 10−5
mol/l; CAP, 10−4 mol/l; LOS, 10−4 mol/l) on
the blood vessel diameter were observed. The inhibitors were
pre-incubated for 20 min at 37°C, and the inhibitors persisted
throughout the experiment (CaSR agonist and an inhibitor group
given calcium-free PSS and KPSS). Then, different concentrations of
PE, Ach, and SNP (10−9−10−4 mol/l) were
cumulatively added to observe the effects of the drug on the blood
vessel diameter. Myoview software (110P; Danish Myo Technology A/S)
was used to control the intravascular pressure and to record the
experimental data.
The vascular diameter change (D) was calculated by
the following formula: D (µm)=Dp-Dx, where Dp is the diameter of
the vascular segment of the PSS fluid when it is stable, Dx is the
diameter after vasoconstriction following the administration of
different concentrations of the drug, Dp' is the diameter of
vasodilation after the administration of different concentrations
of drugs, and Dx' is the blood vessel diameter after
pre-contraction. Vasoconstriction rate (%)=(Dp-Dx)/Dx ×100%.
Vasodilation rate (%)=(Dp'-Dx')/Dx' ×100%.
Statistical analysis
The experimental data are expressed as the mean ±
standard deviation. For comparisons between the two groups, a
Student's t-test or the rank sum test was used. Between multiple
sample means, one-way analysis of variance followed Dunnett's
post-hoc test or variance for non-parametric tests was used. The
analysis was performed with SPSS 19.0 software, in which a
bilateral P<0.05 was considered to indicate a statistically
significant difference. GraphPad Prism 5.0 was used calculate the
inhibitory concentration to exhibit 50% of the maximum contraction
and diastolic effect (IC50) and the maximum contraction
or diastolic amplitude (Emax) values as non-linear
regression curve fits.
Results
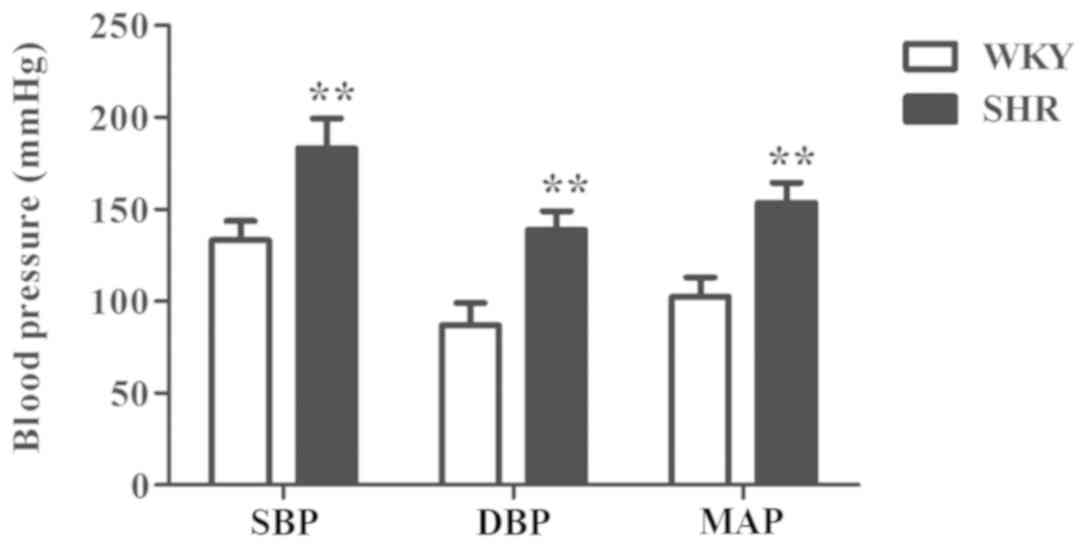
BP of SHRs is significantly higher
than that of the WKY group
The arterial BP of the rats was measured by the tail
cuff method. The results showed that the BP values (SBP, DBP and
MAP) of the SHR group were significantly higher than those of WKY
group of the same age (P<0.01; Fig.
1).
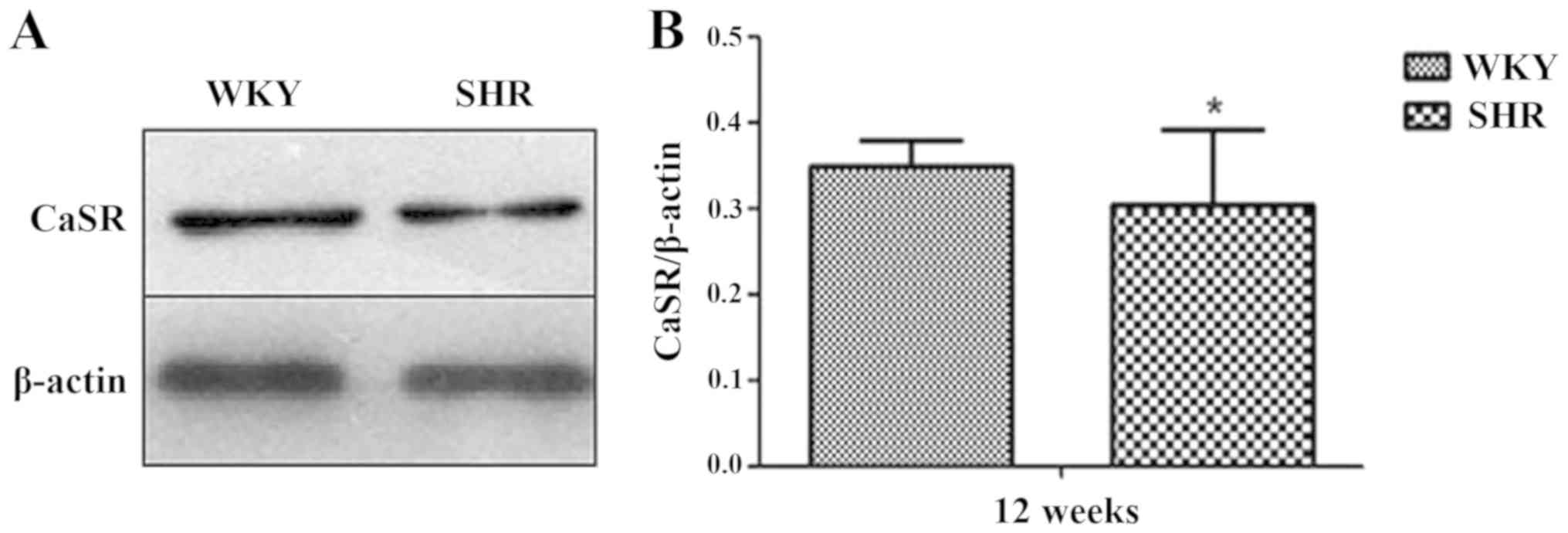
CaSR protein expression in the MA of
the WKY and SHR
Western blotting was used to analyze the expression
of the CaSR protein in the MA of the rats. The results showed that
the CaSR protein was expressed in the MA of the WKY and SHR.
Compared with that of the WKY group, CaSR total protein expression
in the SHR group was significantly reduced (P<0.05; Fig. 2).
Effects of various drugs on vascular
tension of the MA of isolated rats
Vasoconstriction of the SHR group was
higher, while vasodilation was lower than that of the WKY group,
which were endothelium-independent effects
PE-induced contraction and Ach/SNP-induced
vasodilation in the MA of the rats were measured by the pressure
myograph technique. The results showed that: PE induced
vasoconstriction of the rat MA in a dose-dependent manner (Table I; Fig.
3A), while Ach induced vasodilation (Table I; Fig.
3E). Compared with that of the WKY group, the maximal
contraction response of the PE-induced SHR was significantly
increased (Emax values: 34.78±0.90 vs. 45.32±1.41%,
respectively; P<0.05; n=6). The IC50 values were
0.44±0.14 and 0.33±0.14, respectively. The maximal relaxation
response of the Ach-induced SHR group was significantly reduced
(Emax values: 65.2±4.03 vs. 53.41±11.91%, respectively;
P<0.01; n=6). The IC50 values were −1.32±0.20 and
0.03±0.88, respectively.
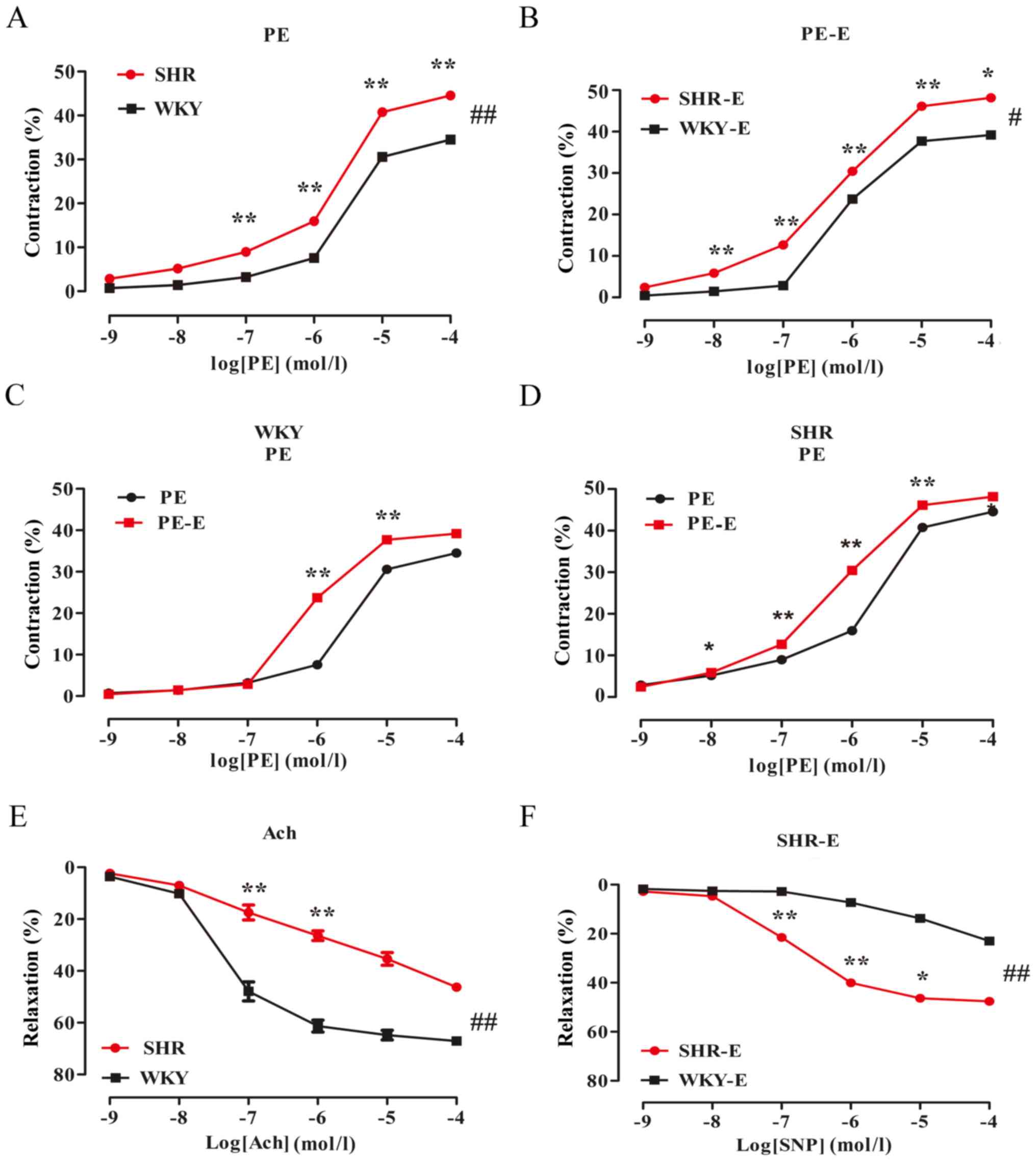 | Figure 3.Comparison of the contractions and
diastolic amplitudes of the mesenteric artery in the WKY and SHR
groups. The data are expressed as the mean ± standard deviation
(n=6). (A) SHR PE vs. WKY PE groups, ##P<0.01; (B)
SHR PE-E groups vs. WKY PE-E groups, #P<0.05. (C) WKY
PE groups vs. WKY PE-E groups, P>0.05. (D) SHR PE groups vs. SHR
PE-E groups, P>0.05. (E) SHR Ach groups vs. WKY Ach groups,
##P<0.01; (F) WKY SNP vs. SHR SNP groups,
##P<0.01. *P<0.05, **P<0.01, vs. the adjacent
concentrations in each group. -E, endothelial removal; WKY, Wistar
Kyoto rats; SHR, spontaneously hypertensive rats; PE,
phenylephrine; Ach, acetylcholine; SNP, sodium nitroprusside. |
 | Table I.Effect of pretreatment with the
calcium sensing receptor inhibitor NPS2143 on vasoconstriction and
relaxation reactivity. |
Table I.
Effect of pretreatment with the
calcium sensing receptor inhibitor NPS2143 on vasoconstriction and
relaxation reactivity.
| Rat | Group | Emax (%) | IC50
(µM) |
|---|
| WKY | PE | 34.78±0.90 | 0.44±0.14 |
|
| PE-E | 38.98±1.12 | −0.13±0.05 |
|
| PE + NPS2143 | 11.67±0.75 | 0.02±0.14 |
|
| PE + NPS2143-E | 41.53±1.34 | 0.60±0.06 |
|
| Ach | 65.2±4.03 | −1.32±0.20 |
|
| SNP | 40.24±11.26 | 1.68±0.73 |
|
| Ach + NPS2143 | 25.74±3.81 | −1.37±0.14 |
|
| SNP + NPS2143 | 41.16±1.19 | −1.46±0.11 |
| SHR | PE |
45.32±1.41a | 0.33±0.14 |
|
| PE-E |
49.81±1.33b | −0.23±0.04 |
|
| PE + NPS2143 |
35.25±1.28c | 0.46±0.05 |
|
| PE + NPS2143-E |
50.64±0.76d | −0.03±0.05 |
|
| Ach |
53.41±11.91e | 0.03±0.88 |
|
| SNP |
47.50±0.47f | −0.85±0.05 |
|
| Ach + NPS2143 |
48.98±5.96g | −1.27±0.18 |
|
| SNP + NPS2143 |
53.66±1.10h | −1.51±0.11 |
In the endothelium-denuded group, PE induced
vasoconstriction (Table I;
Fig. 3B-D), while SNP-induced
vasodilation of the rat MA in a dose-dependent manner (Table I; Fig.
3F). The PE-induced WKY and SHR groups exhibited significantly
increased vasoconstriction amplitudes; compared with the
corresponding WKY group, PE-induced endothelium-denuded SHRs had a
maximal contractile response that was significantly increased
(Emax values: 38.98±1.12 vs. 49.81±1.33%, respectively;
P<0.05; n=6). The IC50 values were −0.13±0.05 and
−0.23±0.04, respectively. The SNP-induced maximum relaxation
response in the SHR group was greater than that in the WKY group
(Emax values: 40.24±11.26 vs. 47.50±0.47%, respectively;
P<0.01; n=6). The IC50 values were 1.68±0.73 and
−0.85±0.05, respectively.
NPS2143 and NPSR568 reduce
vasoconstriction, and promote vasodilation in MA, which is
partially endothelium-dependent
The effects of the NPS2143 and NPSR568 on the
vascular tone in rats were investigated. The results showed that:
NPS2143 (10−8−10−5 mol/l) caused contraction
of the rat MA (Fig. 4A). When the
concentration of NPS2143 was 10−5 mol/l, a significant
contraction amplitude of the MA in rats was observed (P<0.01);
the contraction amplitude of the WKY group was markedly greater
than that of the SHR group (P>0.05). NPSR568
(10−8−10−5 mol/l) induced vasodilation of the
MA in the SHR group in a dose-dependent manner (Fig. 4B), and the diastolic amplitude of
the endothelium-intact group was significantly greater than that of
the endothelium-denuded group (P<0.01). Different concentrations
of NPSR568 were pre-incubated for 20 min with MA samples to study
the effect of PE, Ach, and SNP on vascular tone (Fig. 4C-F). For the endothelium-intact
group, when the concentration of NPSR568 was 10−5 mol/l,
PE-induced contractions were significantly inhibited (P<0.01),
and the vasodilatory amplitude was the most significantly increased
(P<0.01; Fig. 4B). After the
endothelium was denuded, a similar vascular reactivity was
exhibited.
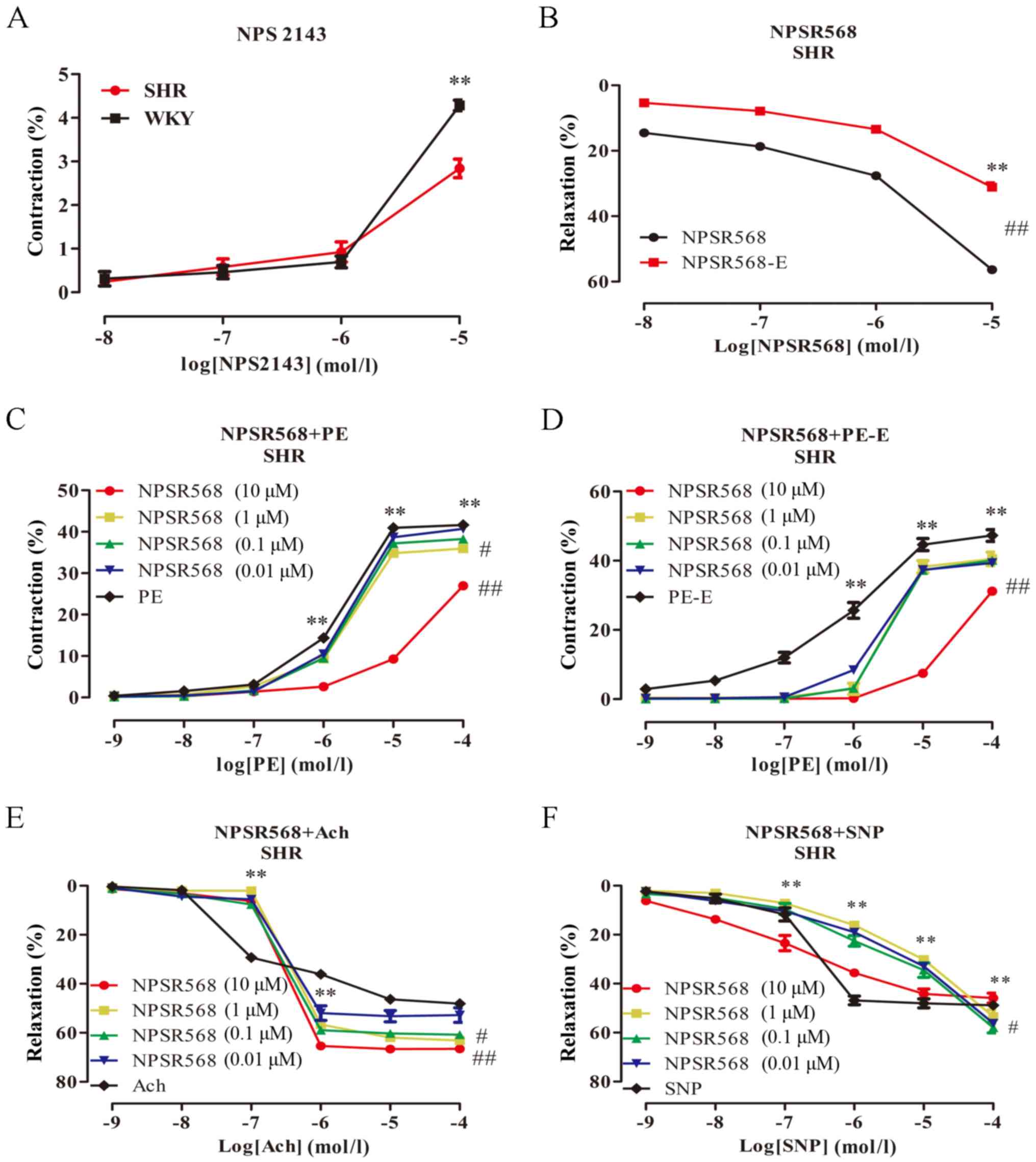 | Figure 4.Effect of NPSR568 on vascular
reactivity. The data are expressed as the mean ± standard deviation
(n=6). (A) WKY NPS2143 groups vs. SHR NPS2143 groups, P>0.05;
(B) WKY NPSR568 groups vs. SHR NPSR568 groups,
##P<0.01; (C) SHR PE groups vs. SHR PE + NPSR568 (1
µM) groups, #P<0.05; ##P<0.01, vs. SHR
PE + NPSR568 (10 µM) group. (D) SHR PE + NPSR568-E (10 µM) groups
vs. SHR PE-E groups, ##P<0.01; (E) SHR Ach group vs.
SHR Ach + NPSR568 (1 µM) group, #P<0.05;
##P<0.01, vs. SHR Ach + NPSR568 (10 µM) groups. (F)
SHR SNP group vs. SHR SNP + NPSR568 (10 µM) group,
#P<0.05. *P<0.05, **P<0.01, vs. the adjacent
concentrations in each group. -E, endothelial removal; WKY, Wistar
Kyoto rats; SHR, spontaneously hypertensive rats; PE,
phenylephrine; Ach, acetylcholine; SNP, sodium nitroprusside;
NPSR568, CaSR agonists. |
NPS2143 reduces vasoconstriction and
enhances vasodilation in MA, which is partly
endothelium-dependent
The effects of NPS2143 on the contraction and
vasodilation of the MA in rats was investigated. The results showed
that after pre-incubation of MA with NPS2143 (10−5
mol/l) for 20 min, PE-induced vasoconstriction of the rat MA
occurred in a dose-dependent manner (Table I; Fig.
5A-C). Ach also induced a relaxatory response of the rat MA in
a dose-dependent manner (Table I;
Fig. 5G-I). The inhibitory effect
of NPS2143 on the PE-WKY group was greater than that of the PE-SHR
group (Emax values: 11.67±0.75 vs. 35.25±1.28%,
respectively; P<0.01; n=6). The IC50 values were
0.02±0.14 and 0.46±0.05, respectively, while the maximum diastolic
amplitude of Ach + NPS2143-induced WKY was lower than that of the
SHR (Emax values: 25.74±3.81 vs. 48.98±5.96%,
respectively; P<0.05; n=6). The IC50 values were
−1.37±0.14 and −1.27±0.18, respectively. Compared with PE alone,
the maximal contraction response with PE + NPS2143 treatment of the
WKY and SHR were significantly reduced (Emax values:
34.78±0.90 vs. 11.67±0.75%; 45.32±1.41 vs. 35.25±1.28 respectively;
P<0.01; n=6). After NPS2143 treatment, compared with Ach group,
the diastolic amplitude of the WKY decreased (Emax
values: 65.2±4.03 vs. 25.74±3.81%, respectively; P<0.01; n=6).
The IC50 values were −1.32±0.20 and −1.37±0.14,
respectively. Compared with Ach group, the diastolic amplitude of
the Ach + NPS2143 SHR increased (Emax values:
53.41±11.91 vs. 48.98±5.96%, respectively; P>0.05; n=6). The
IC50 values were 0.03±0.88 and −1.27±0.18,
respectively.
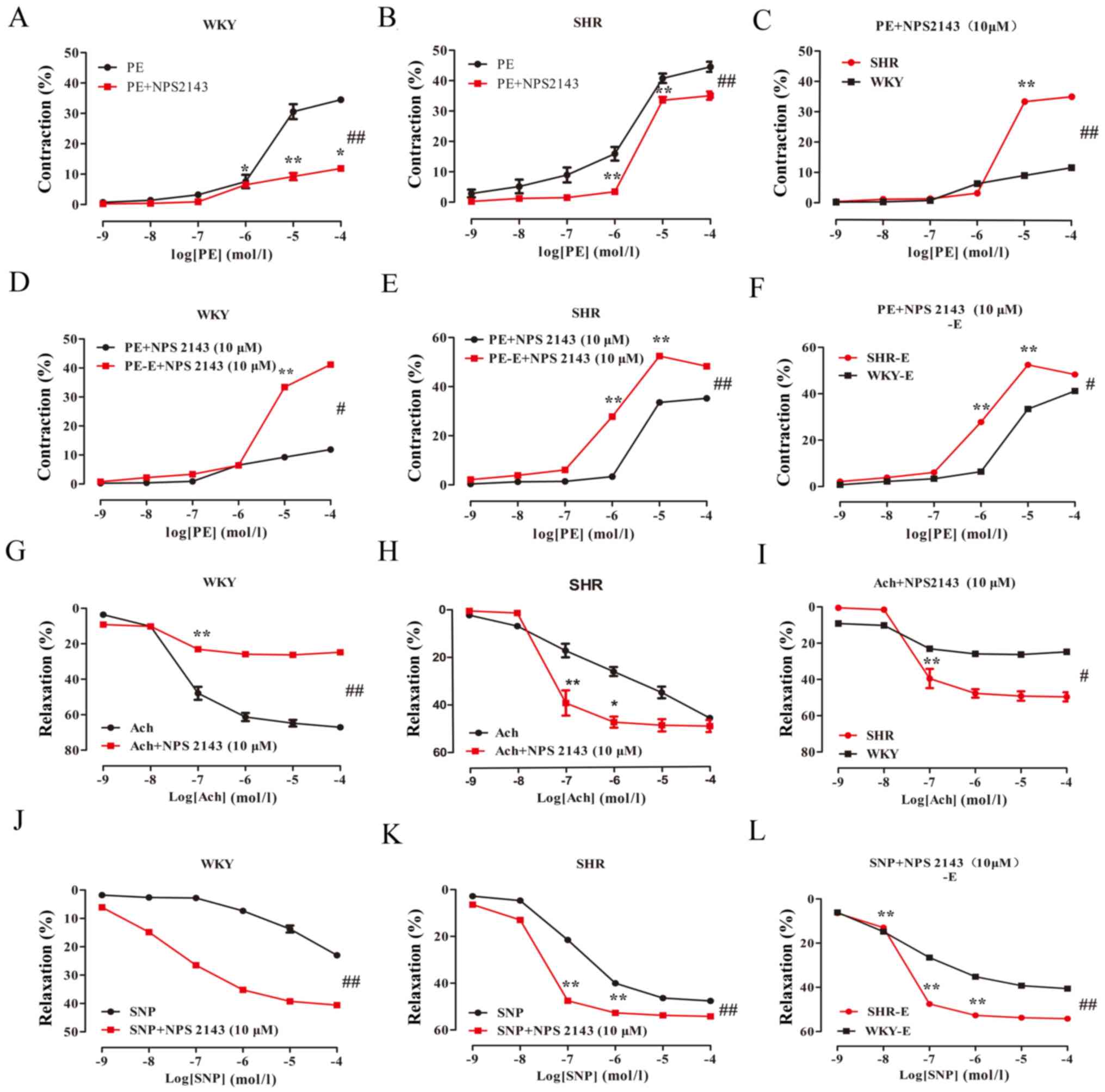 | Figure 5.Effect of the CaSR inhibitor NPS2143
on vascular reactivity of the mesenteric artery in the WKY and SHR.
The data are expressed as the mean ± standard deviation (n=6). (A)
WKY PE + NPS2143 groups vs. WKY PE groups, ##P<0.01;
(B) SHR PE groups vs. SHR PE + NPS2143 groups,
##P<0.01; (C) WKY PE + NPS2143 groups vs. SHR PE +
NPS2143 groups, ##P<0.01; (D) WKY PE + NPS2143 groups
vs. WKY PE + NPS2143-E groups, #P<0.05; (E) SHR PE +
NPS2143 groups vs. SHR PE + NPS2143-E groups,
##P<0.01; (F) WKY PE + NPS2143-E groups vs. SHR PE +
NPS2143-E groups, #P<0.05; (G) WKY Ach groups vs. WKY
Ach + NPS2143 groups, ##P<0.01; (H) SHR Ach groups
vs. SHR Ach + NPS2143 groups, ##P<0.01; (I) WKY Ach +
NPS2143 groups vs. SHR Ach + NPS2143 groups, #
P<0.05; (J) WKY SNP groups vs. WKY SNP + NPS2143 groups,
##P<0.01; (K) SHR SNP groups vs. SHR SNP+NPS2143
groups, ##P<0.01; (L) WKY SNP + NPS2143 groups vs.
SHR SNP + NPS2143 groups, ##P<0.01. *P<0.05,
**P<0.01, vs. the adjacent concentrations in each group. -E,
endothelial removal; WKY, Wistar Kyoto rats; SHR, spontaneously
hypertensive rats; PE, phenylephrine; Ach, acetylcholine; SNP,
sodium nitroprusside; NPS2143, CaSR inhibitor. |
In the endothelium-denuded group, PE induced
vasoconstriction in the rat MA in a dose-dependent manner (Table I; Fig.
5D-F). SNP induced vasodilation in the rat MA in a
dose-dependent manner (Table I;
Fig. 5J-L). The inhibitory effect
of NPS2143 on the PE-induced WKY was greater than that of the SHR
(Emax values: 41.53±1.34 vs. 50.64±0.76%, respectively;
P<0.05; n=6). The IC50 values were 0.60±0.06 and
−0.03±0.05, respectively. The maximal contraction response of PE +
NPS2143 denuded WKY was greater than that of the endothelium-intact
group (Emax values: 41.53±1.34 vs. 11.67±0.75%,
respectively; P<0.05; n=6). The IC50 values were
0.60±0.06 and 0.02±0.14, respectively. The maximum contraction
response of PE + NPS2143 denuded SHR was significantly greater than
that of the endothelial-intact group (Emax values:
50.64±0.76 vs. 35.25±1.28%, respectively; P<0.01; n=6). The
IC50 values were −0.03±0.05 and 0.46±0.05, respectively.
After the NPS2143 treatment, the SNP-induced increase in the
vasodilation in the SHR was greater than that in the WKY
(Emax values: 41.16±1.19 vs. 53.66±1.10%; P<0.01;
n=6). The IC50 values were −1.46±0.11 and −1.51±0.11,
respectively. Compared with the SNP group, SNP + NPS2143 induced a
maximal relaxation response in the WKY and SHR (Emax
values: 40.24±11.26 vs. 41.16±1.19%; 47.50±0.47 vs. 53.66±1.10%,
respectively; P<0.01; n=6).
PLC-IP3/AC-V/cAMP/RAS
pathway inhibitors reduce vasoconstriction and increase
vasodilation in the MA, which is partially
endothelium-dependent
The effect of inhibitors on the vasoconstriction and
vasodilation of the MA in rats was investigated in which
PLC-IP3/AC-V/cAMP/RAS pathway inhibitors were separately
administered. After the MA was pre-incubated for 20 min, PE induced
vasoconstriction in the rat MA in a dose-dependent manner (Table II; Fig. 6A and B). Ach
concentration-dependently induced vasodilation in the rat MA
(Table II; Fig. 6E and F). Compared with the PE
group, the vasoconstriction amplitude in response to the inhibitors
showed an overall weakening trend; the contraction amplitude with
10−5 mol/l PE was notably weakened. Therefore, a
subsequent vasodilation function test was performed in which PE
(10−3 mol/l) was selected to pre-constrict blood
vessels. U73122, 2-APB, MDL12330A, CAP and LOS acted on the blood
vessels. Compared with the PE group, the maximal contractile
responses of the PE + inhibitors WKY and SHR were significantly
lower (Table I; WKY
Emax values: 34.78±0.90 vs. 30.86±1.70, 24.05±2.25,
20.66±2.95, 30.08±3.46, 30.49±1.44%; SHR Emax values:
45.32±1.41 vs. 36.65±0.51, 12.18± 2.92, 11.89±1.44, 31.36±2.42,
31.01±1.99%, respectively; P<0.01; n=6). Compared with the Ach
group, the maximal vasodilatory responses of the Ach + inhibitors
WKY and SHR were significantly increased (WKY Emax
values: 65.2±4.03 vs. 61.12±1.64, 63.69±1.04, 73.14±4.41,
67.68±6.15, 58.22±0.90%; SHR Emax values: 53.41±11.91
vs. 67.48±3.43, 66.12±8.76, 57.97±5.82, 50.70±3.67, 52.70±1.78%,
respectively; P<0.01; n=6). However, in the SHR, U73122 and
2-APB appeared to elicit contractile reactivity when the Ach
concentration was 10−6 and 10−5 mol/l,
respectively.
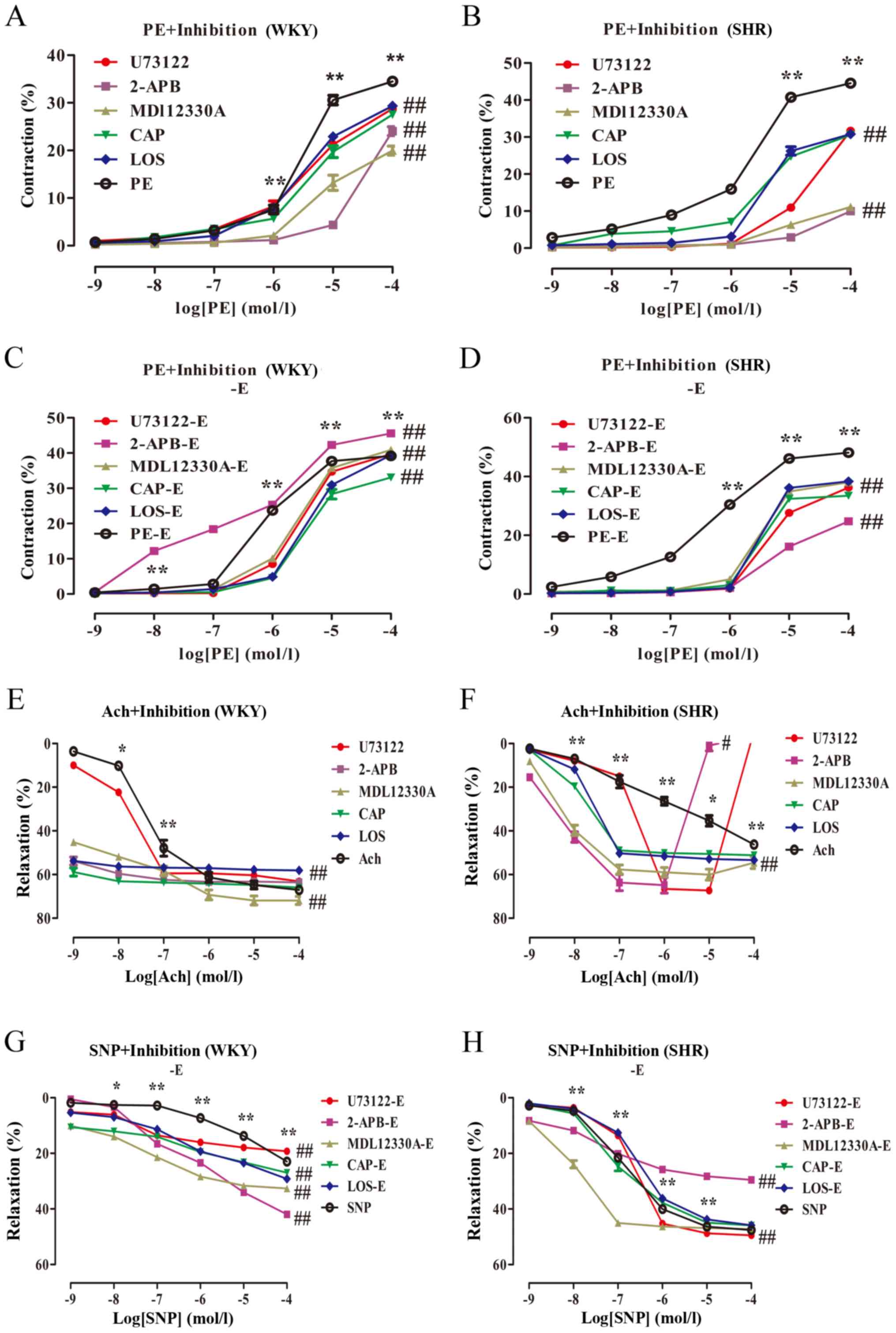 | Figure 6.Effect of
PLC-IP3/AC-V/cAMP/RAS pathway inhibitors on vascular
reactivity of the mesenteric artery in the WKY and SHR. The data
are expressed as the mean ± standard deviation (n=6). (A) WKY PE
groups vs. each signalling pathway inhibitor group,
##P<0.01; (B) SHR PE groups vs. each signalling
pathway inhibitor group, ##P<0.01; (C) WKY PE-E
groups vs. each signalling pathway inhibitor group,
##P<0.01; (D) SHR PE-E groups vs. each signalling
pathway inhibitor group, ##P<0.01; (E) WKY Ach groups
vs. each signalling pathway inhibitor group,
##P<0.01; (F) SHR Ach group vs. U73122 groups,
#P<0.05; SHR Ach group vs. MDL12330A, CAP, LOS
groups, ##P<0.01; (G) WKY SNP groups vs. each
signalling pathway inhibitor group, ##P<0.01; (H) SHR
SNP groups vs. each signalling pathway inhibitor group,
##P<0.01. *P<0.05, **P<0.01, vs. the adjacent
concentrations in each group. -E, endothelial removal; WKY, Wistar
Kyoto rats; SHR, spontaneously hypertensive rats; PE,
phenylephrine; Ach, acetylcholine; SNP, sodium nitroprusside; PLC,
phospholipase C; U73122, PLC inhibitor; 2-APB,
2-aminoethoxydiphenyl borate, IP3 inhibitor; MDL12330A, adenylyl
cyclase V inhibitor; CAP, captopril, ACE inhibitor; LOS, losartan,
AT1R inhibitor. |
| Table II.Effect of pretreatment with
PLC-IP3/AC-V/cAMP/RAS pathway inhibitors on vascular
reactivity. |
Table II.
Effect of pretreatment with
PLC-IP3/AC-V/cAMP/RAS pathway inhibitors on vascular
reactivity.
| Rat | Group | Emax (%) | IC50
(µM) |
|---|
| WKY | PE + U73122 |
30.86±1.70a | 0.61±0.15 |
|
| PE + 2APB |
24.05±2.25a | 2.86±0.33 |
|
| PE + MDL12330A |
20.66±2.95a | 0.78±0.33 |
|
| PE + CAP |
30.08±3.46a | 0.77±0.29 |
|
| PE + LOS |
30.49±1.44a | 0.51±0.07 |
|
| Ach + U73122 |
61.12±1.64b | −1.78±0.12 |
|
| Ach + 2APB |
63.69±1.04b | −2.72±0.74 |
|
| Ach +
MDL12330A |
73.14±4.41b | −1.15±0.19 |
|
| Ach + CAP |
67.68±6.15b | −2.74±4.78 |
|
| Ach + LOS |
58.22±0.90b | −4.78±3.52 |
| SHR | PE + U73122 |
36.65±0.51c | 1.32±0.03 |
|
| PE + 2APB |
12.18±2.92c | 2.13±1.16 |
|
| PE + MDL12330A |
11.89±1.44c | 0.98±0.24 |
|
| PE + CAP |
31.36±2.42c | 0.58±0.09 |
|
| PE + LOS |
31.01±1.99c | 0.61±0.14 |
|
| Ach + U73122 | 67.48±3.43 | −0.72±0.04 |
|
| Ach + 2APB |
66.12±8.76d | −2.08±0.30 |
|
| Ach +
MDL12330A |
57.97±5.82d | −2.13±0.14 |
|
| Ach + CAP |
50.70±3.67d | −1.86±0.02 |
|
| Ach + LOS |
52.70±1.78d | −1.67±0.08 |
| WKY | PE + U73122 -E |
39.98±0.92e | 0.41±0.07 |
|
| PE + 2APB -E |
53.29±1.68e | −0.19±0.08 |
|
| PE + MDL12330A
-E |
41.26±1.20e | 0.39±0.04 |
|
| PE + CAP -E |
33.25±2.18e | 0.53±0.10 |
|
| PE + LOS -E |
39.85±2.29e | 0.64±0.07 |
|
| SNP + U73122 |
18.57±18.57f | −1.16±0.21 |
|
| SNP + 2APB |
45.85±4.13f | −0.15±0.34 |
|
| SNP +
MDL12330A |
33.17±0.97f | −1.14±0.25 |
|
| SNP + CAP |
35.90±16.53f | 0.7±1.45 |
|
| SNP + LOS |
34.62±6.52f | 0.13±0.49 |
| SHR | PE + U73122 -E |
36.41±3.02g | 0.74±0.07 |
|
| PE + 2APB -E |
25.48±1.80g | 0.83±0.06 |
|
| PE +
MDL12330A-E |
38.12±0.97g | 0.46±0.07 |
|
| PE + CAP -E |
33.46±2.12g | 0.4±0.12 |
|
| PE + LOS -E |
38.35±0.73g | 0.52±0.07 |
|
| SNP + U73122 |
49.27±0.76h | −0.67±0.07 |
|
| SNP +2APB |
30.67±3.92h | −1.14±0.44 |
|
| SNP +MDL12330A | 46.91±1.59 | −1.91±0.06 |
|
| SNP +CAP | 45.45±1.89 | −1.00±0.19 |
|
| SNP +LOS | 45.68±1.21 | −0.53±0.13 |
In the endothelium-denuded group, PE induced
vasoconstriction of the rat MA in a dose-dependent manner (Table II; Fig. 6C and D); SNP induced vasodilation
in the rat MA in a similar manner (Table II; Fig. 6G and H). After the signalling
pathway inhibitors acted on the endothelium-denuded blood vessels,
the maximal vasoconstriction responses of the PE-induced WKY and
SHR were significantly reduced (Table
I; WKY Emax values: 38.98±1.12 vs. 39.98±0.92,
53.29±1.68, 41.26±1.20, 33.25±2.18, 39.85±2.29%; SHR
Emax values: 49.81±1.33 vs. 36.41±3.02, 25.48±1.80,
38.12±0.97, 33.46±2.12, 38.35±0.73%, respectively; P<0.01; n=6).
Compared with the endothelial-denuded PE group, and the attenuation
was smaller than that of the endothelium-intact group (Table II). After the signalling pathway
inhibitors acted on the blood vessels, maximal vasodilator
responses of the SNP-induced WKY were significantly increased
(Emax values: 40.24±11.26 vs. 18.57±18.57, 45.85±4.13,
33.17±0.97, 35.90±16.53, 34.62±6.52%, respectively; P<0.01;
n=6). The vasodilation of the SHR following U73122 and 2-APB
treatment was significantly increased compared with SNP alone
(Emax values: 47.50±0.47 vs. 49.27±0.76, 30.67±3.92%,
respectively; P<0.05; n=6), and the diastolic response of the
other inhibitor groups markedly changed (P>0.05). The diastolic
amplitude induced by SNP in the endothelium-denuded WKY and SHR was
lower than that in the endothelium-intact group (Table II).
CaSR inhibitors combined with
PLC-IP3/AC-V/cAMP/RAS pathway inhibitors significantly
decrease vasoconstriction and enhance vasodilation in the MA of the
rats
The effects of CaSR inhibitors and various
signalling pathways inhibitors on the vasoconstriction and
vasodilation of MA in rats were investigated. CaSR inhibitors were
pre-incubated with PLC-IP3/AC-V/cAMP/RAS pathway
inhibitors for 20 min. The results revealed that PE and Ach
concentration-dependently induced vasoconstriction and vasodilation
in the rat MA in the presence of NPS2143, respectively (Table III; Fig. 7A-D). Compared with the PE + NPS2143
group, PE induced a significant reduction in the maximal
contractile responses in the WKY and SHR (Tables I and III; WKY Emax values:
11.67±0.75 vs. 4.01±0.86, 6.14±4.59, 4.06±1.27, 3.19±0.64,
3.33±0.54%; SHR Emax values: 35.25±1.28 vs. 24.42±2.21,
5.53±2.76, 4.22±3.12, 5.06±2.36, 7.90±5.79%, respectively;
P<0.01; n=6), and some blood vessels no longer exhibited
contractile responses. Compared with that of the Ach + NPS2143
group, the maximal vasodilator response of the WKY and SHR induced
by Ach was significantly increased (WKY Emax values:
25.74±3.81 vs. 25.59±2.86, 28.02±3.02, 27.07±2.80, 21.63±1.83,
25.04±0.62%; SHR Emax values: 48.98±5.96 vs. 59.25±8.86,
67.29±7.48, 60.76±8.35, 51.17±0.65, 54.57±3.22%, respectively;
P<0.01; n=6), and a complete vasodilatory response was induced
by Ach at low concentrations (Table
III and Fig. 7C-D).
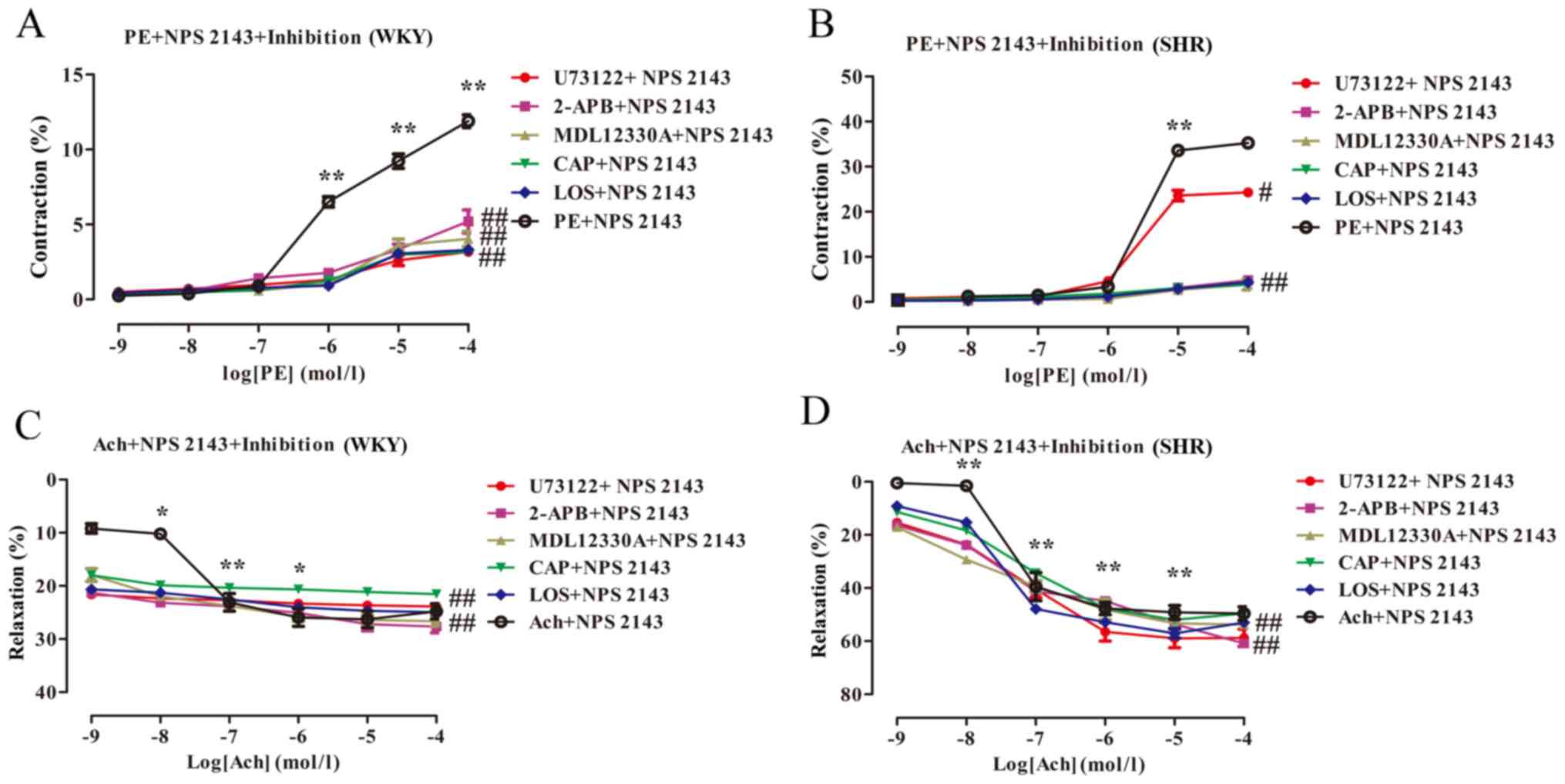 | Figure 7.Effect of CaSR inhibitors combined
with PLC-IP3/AC-V/cAMP/RAS pathway inhibitor on vascular
reactivity of the MA in the WKY and SHR. The data are expressed as
the mean ± standard deviation (n=6). (A) WKY PE + NPS2143 groups
vs. each signalling pathway blocker group, ##P<0.01;
(B) SHR PE + NPS2143 groups vs. each signalling pathway blocker
group, ##P<0.01; (C) In WKY Ach + NPS214 groups vs.
each signalling pathway blocker group, ##P<0.01; (D)
SHR Ach + NPS2143 groups vs. each signalling pathway blocker group,
##P<0.01. *P<0.05, **P<0.01, vs. the adjacent
concentrations in each group. NPS2143, CaSR inhibitor; PLC,
phospholipase C; U73122, PLC inhibitor; 2-APB,
2-aminoethoxydiphenyl borate, IP3 inhibitor; MDL12330A, adenylyl
cyclase V inhibitor; CAP, captopril, ACE inhibitor; LOS, losartan,
AT1R inhibitor. |
| Table III.Effects of CaSR inhibitors combined
with PLC-IP3/AC-V/cAMP/RAS pathway inhibitors on MA
reactivity in rats. |
Table III.
Effects of CaSR inhibitors combined
with PLC-IP3/AC-V/cAMP/RAS pathway inhibitors on MA
reactivity in rats.
| Rat | Groups | Emax (%) | IC50
(µM) |
|---|
| WKY | PE + U73122 +
NPS2143 |
4.01±0.86a | 0.56±0.70 |
|
| PE + 2APB +
NPS2143 |
6.14±4.59a | 1.14±1.24 |
|
| PE + MDL12330A +
NPS2143 |
4.06±1.27a | 0.45±0.09 |
|
| PE + CAP +
NPS2143 |
3.19±0.64a | 0.21±0.18 |
|
| PE + LOS +
NPS2143 |
3.33±0.54a | 0.44±0.18 |
|
| Ach +
U73122+NPS2143 |
25.59±2.86b | −4.37±4.42 |
|
| Ach + 2APB +
NPS2143 |
28.02±3.02b | −0.87±1.48 |
|
| Ach + MDL12330A +
NPS2143 |
27.07±2.80b | −3.74±1.13 |
|
| Ach + CAP +
NPS2143 |
21.63±1.83b | −4.51±2.59 |
|
| Ach + LOS +
NPS2143 |
25.04±0.62b | −0.84±0.46 |
| SHR | PE + U73122 +
NPS2143 |
24.42±2.21c | 0.33±0.15 |
|
| PE + 2APB +
NPS2143 |
5.53±2.76c | 0.66±0.64 |
|
| PE + MDL12330A +
NPS2143 |
4.22±3.12c | 0.58±0.50 |
|
| PE + CAP +
NPS2143 |
5.06±2.36c | 0.82±0.80 |
|
| PE + LOS +
NPS2143 |
7.90±5.79c | 1.27±1.26 |
|
| Ach +
U73122+NPS2143 |
59.25±8.86d | −1.21±0.42 |
|
| Ach + 2APB +
NPS2143 |
67.29±7.48d | −1.24±0.45 |
|
| Ach + MDL12330A +
NPS2143 |
60.76±8.35d | −1.32±0.93 |
|
| Ach + CAP+
NPS2143 |
51.17±0.65d | −1.24±0.24 |
|
| Ach + LOS +
NPS2143 |
54.57±3.22d | −1.5±0.05 |
Discussion
Hypertension has long been considered a leading
cause of cardiovascular morbidity and mortality (40). Despite the fact that there are
effective antihypertensive pharmaceutical medications available,
their side effects and negative consequences cannot be ignored.
Therefore, studying the etiology and pathogenesis of hypertension
to develop a novel method for the treatment of these patients
remains an urgent need. In this study, the role and mechanism of
CaSR in the regulation of the vascular tone in SHRs were
preliminary analyzed with respect to the
PLC-IP3/AC-V/cAMP/RAS pathway. We demonstrated the
following: i) CaSR is functionally expressed in the MA, and CaSR
expression is decreased in SHRs; ii) vasoconstriction is enhanced,
and vasodilatation is attenuated in SHRs, which is
endothelium-independent; and iii) CaSR is involved in the
regulation of BP and vascular tension in SHR and WKYs, which may be
associated with differences in the underlying mechanisms, and this
effect is partially endothelium-dependent. Furthermore, the effect
was proposed to be mediated by the PLC-IP3/AC-V/cAMP/RAS
pathway. These data may provide novel insight into the regulatory
mechanisms of CaSR in angiotasis in SHRs via the
PLC-IP3/AC-V/cAMP/RAS pathway. To the best of our
knowledge, the present study is the first to determine the
mechanism underlying the role of CaSR in angiotasis in SHRs via the
PLC-IP3/AC-V/cAMP/RAS pathway.
It has been reported that a rise in vascular tension
is caused by an increase in peripheral vascular resistance, which
is the key factor associated with the onset of hypertension
(5). The small artery determines
peripheral vascular resistance, and the microvascular segment is
the key position for forming blood circulation resistance (5). The decrease in BP reaches ≤90%, and
myogenic tension changes between the capillary and small artery are
the key factors of changes in peripheral vascular resistance and
the formation of BP (41). In the
current study, 2–3 sizes of the MA were selected (diameter: 300–500
µm), which is a resistance vessel in the circulatory system and was
in accordance with experimental requirements.
During the state of hypertension, the reactivity of
vascular smooth muscle to vasoactive substances is enhanced
(5), but this mechanism is not yet
fully understood. In the present study, an internationally
recognized animal model of hypertensive disease was used SHRs. It
has been reported that SHRs comprise a hypertensive disease model
with an enhanced vascular tone (42). Similar to human hypertension, SHRs
present with endothelial dysfunction and reduced NO bioavailability
(43). To evaluate basic
differences in angiotasis between SHR and WKY, and the effect of
the vascular endothelium, the pressure myograph technique was used
to assess changes in angiotasis in the MA of the rats. The present
study determined that, whether the endothelium was intact or
denuded, the contraction range in SHRs was higher than that in WKY.
However, the vasoconstriction of the endothelium-denuded group was
higher than that in the endothelium-intact group for the WKY. It is
worth mentioning that vasoconstriction in the endothelium-denuded
group compared with that in the endothelium-intact group markedly
differed for WKYs and SHRs. This partially suggests that
contractile enhancement in SHRs is endothelium-independent. For the
endothelium-intact rats, the vasodilatory amplitude in SHRs was
lower than that in WKYs. For the endothelium-denuded rats, the
vasodilation amplitude in SHRs was greater than that in WKYs. We
also demonstrated that the reduced vasodilation in SHRs was
endothelium-independent. In conclusion, the increased
vasoconstriction and reduced vasodilation in SHRs are
endothelial-independent. These results are similar to other recent
research reports and are in accordance with the pathophysiological
pathogenesis of hypertension (5,44).
The initiation of research on CaSR in the field of
angiogenesis occurred relatively recently. In 2003, Wang et
al (21) was the first to
confirm that CaSR is expressed in the rat myocardium. With
subsequent research, it was found that CaSR is functionally
expressed in the outer membrane of the blood vessel wall,
fibroblast, VSMCs, and endothelial cells, among others (10,17,45).
It was also found to serve a key role in the regulation of BP and
angiotasis, but the mechanism was not clear. The present study
suggests that hormones (renin, parathyroid hormone),
[Ca2+]o, CaSR, vascular calcification, the
nervous system, and certain agents are involved in the regulation
of BP and vascular tone. In 1911, Cow revealed that the relaxation
of isolated blood vessels is mediated by an increase of
[Ca2+]o. Furthermore, Wang and Bukoski
(46) reported that
[Ca2+]o can induce the relaxation of the MA,
coronary artery, renal artery and cerebral artery in rats. In
addition, other CaSR calcimimetics (like Mg2+ and
neomycin) have similar vasodilatory effects (47). In contrast, Ohanian et al
(48) found that
[Ca2+]o can induce two-way effects in
subcutaneous arterioles in rats, which suggests that low
concentrations of [Ca2+]o (0.5–2 mmol/l) can
induce the contraction of small arteries and that high
concentrations of [Ca2+]o (3–10 mmol/l) can
induce the relaxation of small arteries. It was shown that
pre-incubation with the CaSR inhibitor NPS2143 attenuates KCl and
PE-induced vasoconstriction in oxygen-poor/oxygen-rich MAs;
however, NPS2143 did not improve the endothelium-dependent
relaxation induced by Ach (49).
To evaluate the angiotasis-mediated regulation of CaSR and its
effect on the vascular endothelium, the pressure myograph technique
was used to observe changes in angiotasis with CaSR calcimimetics
and inhibitors in the MA of rats. Our results showed that in
endothelium-intact vessels, pre-incubation with NPS2143 induced
vasoconstriction in the MA and that the contraction degree of
vasoconstriction in the WKY group was higher than that in SHRs.
Pre-incubation with NPSR568 also induced vasodilation in the MA,
and the degree of relaxation in the endothelium-intact group was
higher than that in the endothelium-denuded group. In the MA of
rats, with different concentrations of NPSR568, whether the
endothelium was intact or denuded, vasoconstriction could be
inhibited and vasodilation was enhanced. The effect of NPSR568 (10
µM) was also the most obvious; however, NPS2143 could also inhibit
vasoconstriction and enhance vasodilation. Regardless of whether
the endothelium was intact or denuded, the degree of contraction in
SHRs was higher than that in WKYs. Furthermore, the enhancement of
NPS2143-induced vasodilation was significantly different between
the WKY and SHR groups. When the endothelium was intact, NPS2143
enhanced vasodilation in WKYs to a higher degree than that in SHRs.
When the endothelium was denuded, vasodilation in WKYs was lower
than that in SHRs. This suggested that NPSR568 and NPS2143 both
alleviated vasoconstriction of the MA in SHRs. Among them, for
NPS2143, the alleviating effect in SHRs was decreased compared with
that in WKYs, and this was partly endothelium-dependent. For
NPSR568, the vasodilatory inducing effect in SHRs was partly
endothelium-dependent; however, the inhibitory effect of NPS2143 on
vasodilation in WKYs was endothelium-independent. Our experimental
results were similar to the aforementioned reports (50,51).
For NPSR568, the previously mentioned two-way effects were not
observed with concentrations of 0.01–10 µM, which may be related to
the experimental animals, experimental conditions, detection
methods, types of calcimimetics (the calcimimetic used to delineate
the previously mentioned two-way effects was Ca2+), and
the concentrations of drugs. Of note, with a concentration of
10−5 mol/l, NPSR568 and NPS2143 had similar effects,
specifically the inhibition of vasoconstriction in SHRs. This
indicated that this vessel regulatory effect was not completely
induced by CaSR and that it may be due to the specific effects of
the agent itself. Our research supported this concept. When the
concentration of the NPS2143 was 10−5 mol/l, the drug
itself caused maximum vasoconstriction in SHRs and WKYs.
Conversely, for NPSR568 at a concentration of 10−5
mol/l, the drug itself caused maximum vasodilation in SHRs.
Denuding the endothelium abolished vasodilation, which indicated
that this partly relied on the NO produced by the endothelium.
Similar to our research results, other reports have proposed that,
except for changes in angiotasis induced by CaSR, the CaSR
calcimimetic can also inhibit vascular reactivity via a
CaSR-dependent mechanism by directly blocking the voltage-dependent
L-type calcium channels (VGCCs). Importantly, both the
CaSR-dependent and CaSR-independent effects are likely to occur at
similar concentrations (49).
Nakagawa et al (50) found
that the CaSR calcimimetics R-568 and S-568 have similar
BP-reducing effects in rats; however, S-568 markedly unaffects
CaSR. Thus, it is considered that the BP-reducing effect of R-568
is not induced by CaSR. Taken together, these results indicate that
the NPSR568 and NPS2143 can modulate the vascular tone in SHRs and
WKYs via different regulatory mechanisms, which is partially
endothelium-dependent and partly mediated by CaSR. The reason for
this effect may be associated with the similarities in the
molecular structure of the allosteric modulators of CaSR. The
negative allosteric modulator NPS2143 is known to be structurally
related to phenylalkylamines and has a positively charged amino
group (52,53). The positive allosteric modulator
NPSR568 is a phenylalkylamine derivative, which is a potent VGCC
blocker that exhibits weak allosteric potentiation of the CaSR
(50,53,54).
This structural similarity is highlighted by the fact that NPS2143
and NPSR568 both target a common allosteric site within the seventh
transmembrane domain of CaSR (49).
Increased [Ca2+]o induces
binding to the CaSR and activates the G-protein-PLC- IP3
pathway, promoting the influx of extracellular Ca2+; it
also participates in the regulation of myocardial
excitation-contraction coupling, which is the mechanism of
Ca2+ overload in cells. Ca2+ overload in
VSMCs can cause vasospasm, vascular wall thickening, vascular
inflammation and other diseases. Additionally, it is related to
hypertension, atherosclerosis and other diseases (9). The CaSR calcimimetic
[Ca2+]o, Gd3+ and spermine can
cause Ca2+ to be released from the myocardial
cytoplasmic reticulum of rats through the G
protein-PLC-IP3 pathway (21). In conclusion, the G
protein-PLC-IP3 pathway is involved in the regulation of
a multi-level organization system. Based on the backgrounds of
these studies, our research group proposed that CaSR might regulate
angiotasis and BP in SHRs via the G protein-PLC-IP3
pathway, in which we investigated in the present study. Based on
the alleviating effects of CaSR on vasoconstriction in SHRs and
WKYs induced by PE, U73122 and 2-APB were administered to modulate
the G protein-PLC-IP3 pathway; with this, constriction
in the two groups of rats was reduced. However, upon co-incubating
the inhibitors NPS2143 and U73122, as well as 2-APB separately,
constriction of the MA was reduced further. This result suggests
that blocking the G protein-PLC-IP3 pathway can
strengthen the aforementioned vasoconstrictive effects of NPS2143.
In other words, this pathway is the most important signalling
pathway that relieves vasoconstriction in SHRs and WKYs.
Furthermore, its mechanism is related to vasoconstriction caused by
CaSR excitation, which is inhibited by NPS2143, and the release of
reducing [Ca2+]i due to the G
protein-PLC-IP3 pathway being blocked by PLC and
IP3 inhibitors. The results of this study with respect
to the vasodilation response are as described previously:
Specifically, the NPS2143 enhanced vasodilatation in SHRs, but the
inhibition of vasodilatation in WKYs was endothelium-independent.
In the endothelium-denuded group, with the collaborative use of
L-NAME and indomethacin, NPS2143 incubated vessels, which enhanced
vasodilatation in the SHR and WKY groups. An exogenous NO donor,
SNP, induced the same vascular reaction in SHRs and reversed its
inhibitory effects on vasodilatation in WKYs, which indicated that
the exogenous NO pathway is involved in the
vasodilatation-enhancing effects of CaSR. Based on this, to further
assess the regulatory mechanism of CaSR on vasodilatation, NPS2143
was incubated with U73122 and 2-APB separately. The results showed
that the enhancing effects on vasodilatation of NPS2143 were
further promoted, and this also reversed the
vasodilatation-inhibitory effects in WKYs. It is noteworthy that in
the endothelium-denuded group, the vasodilatory growth rate in WKYs
induced by 2-APB was significantly higher than that in the other
inhibitor groups. However, when the concentration of U73122 was
10−7 mol/l, the vasodilatory growth rate in the SHRs was
the highest, after which, the vasodilatory growth rate reduced, but
remained higher than the other inhibitor groups. This result
implied that the G protein-PLC-IP3 pathway is one
endothelium-independent mechanism through which CaSR enhances
vasodilatation in SHRs and WKYs. However, it remains unknown if
other mechanisms are involved.
RAS, which is well-known, serves an important role
in the regulation of human BP and the pathogenesis of hypertension.
Renin is the first rate-limiting enzyme of the RAS system, and cAMP
functions as a key effector in this system (14). In this system, another key factor
ACE, catalyzes angiotensin I (AngI) conversion into angiotensin II
(AngII), which exerts its biological effects by binding to AT1R.
In vitro analysis suggested that AngII can increase the
catecholamine level via AT1R and can serve a key role in the
vasoconstriction induced by PE (55). Angiotensin-converting enzyme
inhibitors (ACEI) and captopril can lower the mortality rate
associated with hypertension and the complications from myocardial
infarction, diabetes, cardiac failure and other conditions
(56). Maeso et al
(57) found that losartan can
inhibit the contraction of the aorta from PE to rats and relies on
NO from the vascular endothelium. This endothelium-denuded
inhibitory effect disappeared; however, the side effects of
classical ACE inhibitors, an AngII receptor blocker, and other
antihypertensive agents, such as electrolyte imbalance, angioedema,
cough and hypotension, might limit the use of these drugs (41). It is thus particularly important to
develop a better treatment method.
The analysis of cultured MA VSMCs led to the
identification of different components of the RAS (58). Research has shown that
[Ca2+]o and [Ca2+]i are
inversely proportional to renin (15). The associated mechanism is that
CaSR causes a rise in [Ca2+]i by activating
the G protein-PLC-IP3 pathway, and inhibits AC-V and
reduces cAMP at the same time, which is related to the inhibition
of renin release (12). At first,
our research group found an inverse relationship between CaSR and
cAMP with RAS in the thoracic aorta of SHRs (18). To further assess the regulatory
mechanism of CaSR in SHR angiotasis, CaSR was separately combined
with MDL12330A, CAP and LOS, and administered to the vessels.
Similar results were reported in the blocking effects of U73122 and
2-APB on the PLC-IP3 pathway. The vasoconstriction in
SHRs and WKYs was notably diminished with mild contraction, and
even no contraction in parts of the vessels; however,
vasodilatation was enhanced significantly. Furthermore, a low
concentration of Ach was found to mediate complete vasodilatation.
Among them, a low concentration of CAP (10−9 mol/l)
induced vasodilation in WKYs, but the growth rate of vasodilation
was significantly lower than in the other inhibitor groups.
Collectively, the observations described herein support the
hypothesis that CaSR participates in angiotasis regulation in SHRs
and WKYs via the PLC-IP3/AC-V/cAMP/RAS pathway (Fig. 8). At present, the limitations of
this study were mainly that direct in vitro experiments were
conducted based on vascular administration with no in vivo
analyses. Given the differences between the in vivo and
in vitro experimental studies, our results have certain
shortcomings. Despite these limitations, our research also partly
explained the regulatory mechanism through which CaSR mediates
angiotasis during hypertension via the
PLC-IP3/AC-V/cAMP/RAS pathway.
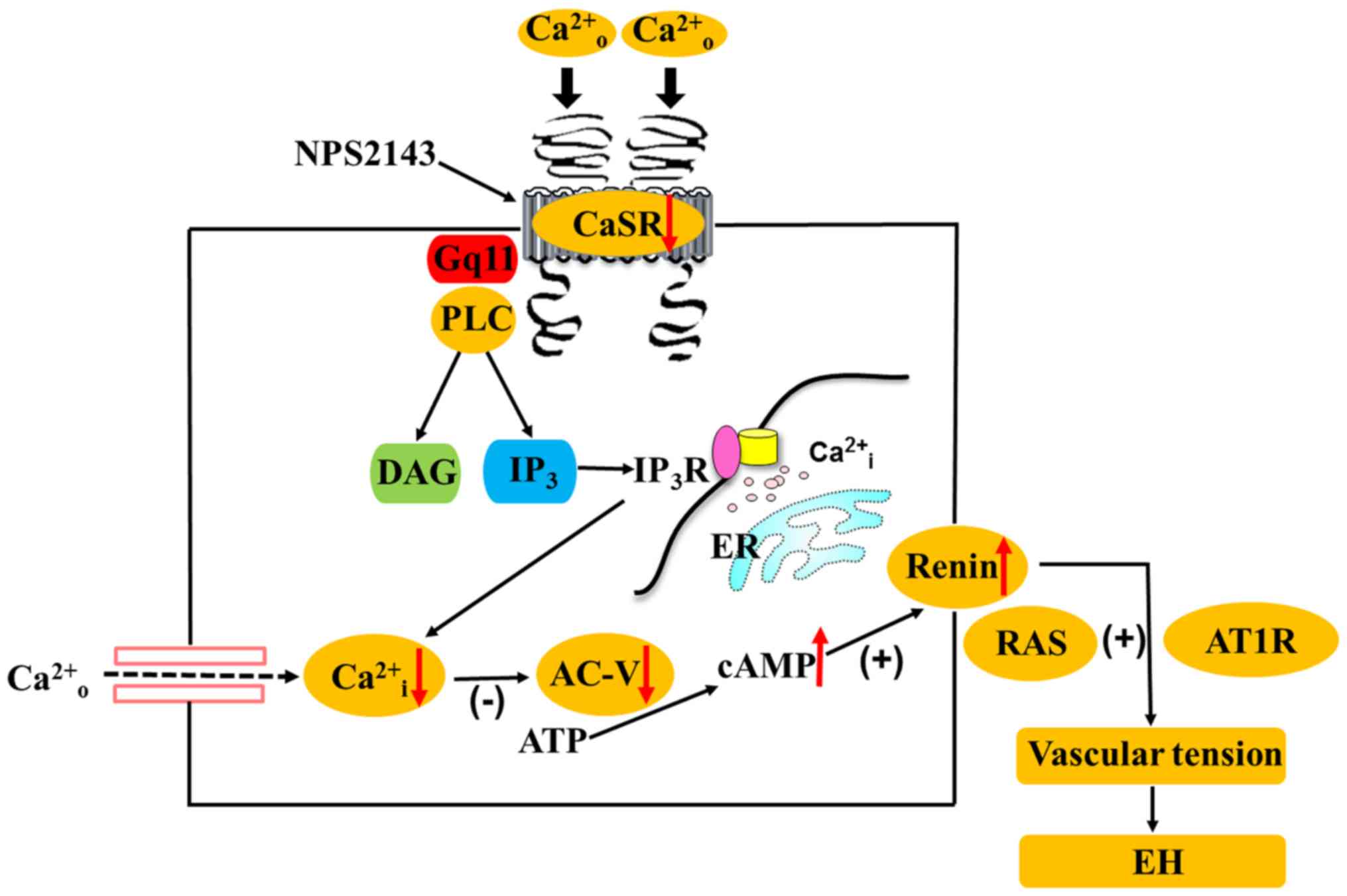 | Figure 8.Hypothetical mechanism is the calcium
sensing receptor participates in the regulation of vascular tension
in the mesentery of hypertensive rats via the PLC-
IP3/AC-V/cAMP/RAS pathway. CaSR, calcium sensing
receptor; PLC, phospholipase C; IP3, inositol
1,4,5-triphosphate; AC-V, adenylate cyclase-V; cAMP, cyclic
adenosine monophosphate; RAS, renin-angiotensin system; AT1R,
angiotensin II receptor type 1; [Ca2+]o,
increased extracellular Ca2+;
[Ca2+]i, intracellular Ca2+;
NPS2143, calcium sensing receptor inhibitor; DAG,
diphosphoglycerate; ER, endoplasmic reticulum; IP3R,
inositol 1,4,5-triphosphate receptor; ATP, adenosine triphosphate;
Gq11, G protein-coupled receptor. |
In summary, CaSR is closely related to BP and
angiotasis, and is involved in the regulation of these processes
based on multi-molecular mechanisms and different pathways. Our
research results show that CaSR is involved in the regulation of BP
and vascular tension in SHRs and WKYs, which are associated with
mechanistic differences and that this effect is partially
endothelium-dependent. Specifically, the effect is mediated by the
PLC-IP3/AC-V/cAMP/RAS pathway. Our findings provide a
novel theoretical foundation for the mechanism of action underlying
the relationship between CaSR, BP and angiotasis, and suggests drug
targets for the prevention of hypertension and vessel-related
diseases.
Acknowledgements
The authors would like to thank Dr Fang He (China
Shihezi University, Shihezi, China) for technical support and
critical review of the manuscript.
Funding
The project was supported by grants from the
National Natural Science Foundation of China (grant no.
31560287).
Availability of data and materials
The datasets used and/or analyzed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
WZ and RS contributed equally in reviewing the
publications and writing this manuscript. WZ and FH contributed to
the conception and design of the study. WZ and RS performed the
experiments. WZ and HZ and wrote the paper. YZ and TZ performed and
analyzed western blotting. NT, HZ and YL measured and analyzed
blood pressure and vascular tone values. FH revised the manuscript
and have given final approval of the version to be published.
Ethics approval and consent to
participate
All animal experiments have been approved and
supervised by Xinjiang Policy Experimental Animal Health and Use
Committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lim S, Vos T and Bruce N: ‘The burden of
disease and injury attributable to 67 risk factors and risk factor
clusters in 21 regions 1990–2010: A systematic analysis.’. Lancet.
380:2224–2260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Akpunonu BE, Mulrow PJ and Hoffman EA:
Secondary hypertension: Evaluation and treatment. Dis Mon.
42:609–722. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
WRITING GROUP MEMBERS, ; Lloyd-Jones D,
Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB,
Ford E, Furie K, et al: Heart disease and stroke statistics-2008
Update: A report from the american heart association statistics
committee and stroke statistics subcommittee. Circulation.
121:e46–e215. 2009.PubMed/NCBI
|
|
4
|
Sharp SI, Aarsland D, Day S Nnesyn H;
Alzheimer's Society Vascular Dementia Systematic Review Group, ;
Ballard C: Hypertension is a potential risk factor for vascular
dementia: Systematic review. Int J Geriatr Psychiatry. 26:661–669.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Figueroa XF, Isakson BE and Duling BR:
Vascular gap junctions in hypertension. Hypertension. 48:804–811.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Allender PS, Cutler JA, Follmann D,
Cappuccio FP, Pryer J and Elliott P: Dietary calcium and blood
pressure: A meta-analysis of randomized clinical trials. Ann Intern
Med. 124:825–831. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He K, Liu K, Daviglus ML, Morris SJ, Loria
CM, Van Horn L, Jacobs DR Jr and Savage PJ: Magnesium intake and
incidence of metabolic syndrome among young adults. Circulation.
113:1675–1682. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ayachi S: Increased dietary calcium lowers
blood pressure in the spontaneously hypertensive rat. Metabolism.
28:1234–1238. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brown EM, Gamba G, Riccardi D, Lombardi M,
Butters R, Kifor O, Sun A, Hediger MA, Lytton J and Hebert SC:
Cloning and characterization of an extracellular Ca (2+)-sensing
receptor from bovine parathyroid. Nature. 366:575–580. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Weston AH, Geraghty A, Egner I and Edwards
G: The vascular extracellular calcium-sensing receptor: An update.
Acta Physiol (Oxf). 203:127–137. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weston AH, Absi M, Ward DT, Ohanian J,
Dodd RH, Dauban P, Petrel C, Ruat M and Edwards G: Evidence in
favor of a calcium-sensing receptor in arterial endothelial cells.
Circ Res. 97:391–398. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ortiz-Capisano MC, Reddy M, Mendez M,
Garvin JL and Beierwaltes WH: Juxtaglomerular cell CaSR stimulation
decreases renin release via activation of the PLC/IP3
pathway and the ryanodine receptor. Am J Physiol Renal Physiol.
304:F248–F256. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brown EM and Macleod RJ: Extracellular
calcium sensing and extracellular calcium signaling. Physiol Rev.
81:239–297. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Churchill PC: Second messengers in renin
secretion. Am J Physiol. 249:175–184. 1985.
|
|
15
|
Atchison DK, Ortiz-Capisano MC and
Beierwaltes WH: Acute activation of the calcium-sensing receptor
inhibits plasma renin activity in vivo. Am J Physiol Regul Integr
Comp Physiol. 299:R1020–R1026. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park CS, Honeyman TW, Chung ES, Lee JS,
Sigmon DH and Fray JC: Involvement of calmodulin in mediating
inhibitory action of intracellular Ca2+ on renin
secretion. Am J Physiol. 251:F1055–F1062. 1986.PubMed/NCBI
|
|
17
|
Smajilovic S, Yano S, Jabbari R and
Tfelthansen J: The calcium-sensing receptor and calcimimetics in
blood pressure modulation. Br J Pharmacol. 164:884–893. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qu Y, Jing H, Wang LM, Tang N, Zhong H,
Liu YM, Li Z, Feng Q and He F: Reduced expression of the
extracellular calcium-sensing receptor (CaSR) is associated with
activation of the renin-angiotensin system (RAS) to promote
vascular remodeling in the pathogenesis of essential hypertension.
PLoS One. 11:e01574562016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Smajilovic S and Tfelt-Hansen J: Novel
role of the calcium-sensing receptor in blood pressure modulation.
Hypertension. 52:994–1000. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cow D: Some reactions of surviving
arteries. J Physiol. 42:125–143. 1911. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang R, Xu C, Zhao W, Zhang J, Cao K, Yang
B and Wu L: Calcium and polyamine regulated calcium-sensing
receptors in cardiac tissues. Eur J Biochem. 270:2680–2688. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Loot AE, Pierson I, Syzonenko T,
Elgheznawy A, Randriamboavonjy V, Zivković A, Stark H and Fleming
I: Ca2+-sensing receptor cleavage by calpain partially
accounts for altered vascular reactivity in mice fed a high-fat
diet. J Cardiovasc Pharmacol. 61:528–535. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ziegelstein RC, Xiong Y, He C and Hu Q:
Expression of a functional extracellular calcium-sensing receptor
in human aortic endothelial cells. Biochem Biophys Res Commun.
342:153–163. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Weston AH, Absi M, Ward DT, Ohanian J,
Dodd RH, Dauban P, Petrel C, Ruat M and Edwards G: Evidence in
favor of a calcium-sensing receptor in arterial endothelial cells:
Studies with calindol and Calhex 231. Circ Res. 97:391–398. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vizard TN, O'Keeffe GW, Gutierrez H, Kos
CH, Riccardi D and Davies AM: Regulation of axonal and dendritic
growth by the extracellular calcium-sensing receptor. Nat Neurosci.
11:285–291. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Finn R: New animal care guide leaves
details to scientists' discretion-the scientist-magazine of the
life sciences. Scientist. 10:1996.
|
|
27
|
Chen YC, Yuan TY, Zhang HF, Wang DS, Niu
ZR, Li L, Fang LH and Du GH: Fasudil evokes vasodilatation of rat
mesenteric vascular bed via Ca(2+) channels and Rho/ROCK pathway.
Eur J Pharmacol. 788:226–233. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Choi S, Ryu KH, Park SH, Jun JY, Shin BC,
Chung JH and Yeum CH: Direct vascular actions of quercetin in aorta
from renal hypertensive rats. Kidney Res Clin Pract. 35:15–21.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu XM, Fang LH, Li YJ and Du GH:
Endothelium-dependent and -independent relaxation induced by
pinocembrin in rat aortic rings. Vascul Pharmacol. 46:160–165.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saito M, Tsounapi P, Oikawa R, Shimizu S,
Honda M, Sejima T, Kinoshita Y and Tomita S: Prostatic ischemia
induces ventral prostatic hyperplasia in the SHR; possible
mechanism of development of BPH. Sci Rep. 4:38222014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wonneberger K, Scofield MA and Wangemann
P: Evidence for a calcium-sensing receptor in the vascular smooth
muscle cells of the spiral modiolar artery. J Membr Biol.
175:203–212. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chan YC, Leung FP, Wong WT, Tian XY, Yung
LM, Lau CW, Tsang SY, Yao X, Chen ZY and Huang Y: Therapeutically
relevant concentrations of raloxifene dilate pressurized rat
resistance arteries via calcium-dependent endothelial nitric oxide
synthase activation. Arterioscler Thromb Vasc Biol. 30:992–999.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Caniffi C, Cerniello FM, Gobetto MN,
Sueiro ML, Costa MA and Arranz C: Vascular tone regulation induced
by C-type natriuretic peptide: Differences in endothelium-dependent
and -independent mechanisms involved in normotensive and
spontaneously hypertensive rats. PLoS One. 11:e01678172016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sudhahar V, Shaw S and Imig JD: Mechanisms
involved in oleamide-induced vasorelaxation in rat mesenteric
resistance arteries. European J Pharmacol. 607:1432009. View Article : Google Scholar
|
|
35
|
Musha Y, Itoh S, Hanson MA and Kinoshita
K: Does estrogen affect the development of abnormal vascular
function in offspring of rats fed a low-protein diet in pregnancy?
Pediatr Res. 59:784–789. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu X, El-mahdy MA, Boslett J, Varadharaj
S, Hemann C, Abdelghany TM, Ismail RS, Little SC, Zhou D, Thuy LT,
et al: Cytoglobin regulates blood pressure and vascular tone
through nitric oxide metabolism in the vascular wall. Nat Communi.
8:148072017. View Article : Google Scholar
|
|
37
|
Schepelmann M, Yarova PL, Lopez-Fernandez
I, Davies TS, Brennan SC, Edwards PJ, Aggarwal A, Graça J, Rietdorf
K, Matchkov V, et al: The vascular Ca2+-sensing receptor
regulates blood vessel tone and blood pressure. Am J Physiol Cell
Physiol. 310:C193–C204. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Domenicali M, Ros J, Fernandez-Varo G,
Cejudo-Martín P, Crespo M, Morales-Ruiz M, Briones AM, Campistol
JM, Arroyo V, Vila E, et al: Increased anandamide induced
relaxation in mesenteric arteries of cirrhotic rats: Role of
cannabinoid and vanilloid receptors. Gut. 54:522–527. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lubomirov L, Gagov H, Petkova-Kirova P,
Duridanova D, Kalentchuk VU and Schubert R: Urocortin relaxes rat
tail arteries by a PKA-mediated reduction of the sensitivity of the
contractile apparatus for calcium. Br J Pharmacol. 134:1564–1570.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Foëx P and Sear J: Hypertension:
Pathophysiology and treatment. Continuing Edu Anaesthesia Critical
Care Pain. 4:71–75. 2004. View Article : Google Scholar
|
|
41
|
Segal SS: Regulation of blood flow in the
microcirculation. Microcirculation. 12:332005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rapp JP: Genetic analysis of inherited
hypertension in the rat. Physiol Rev. 80:135–172. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Piech A, Dessy C, Havaux X, Feron O and
Balligand JL: Differential regulation of nitric oxide synthases and
their allosteric regulators in heart and vessels of hypertensive
rats. Cardiovasc Res. 57:456–467. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sonoyama K, Greenstein A, Price A,
Khavandi K and Heagerty T: Vascular remodeling: implications for
small artery function and target organ damage. Ther Adv Cardiovasc
Dis. 1:129–137. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Molostvov G, James S, Fletcher S, Bennett
J, Lehnert H, Bland R and Zehnder D: Extracellular calcium-sensing
receptor is functionally expressed in human artery. Am J Physiol
Renal Physiol. 293:F946–F955. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang Y and Bukoski RD: Distribution of the
perivascular nerve Ca2+ receptor in rat arteries. Br J
Pharmacol. 125:1397–1404. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Northcott CA and Watts SW: Low
[Mg2+]e enhances arterial spontaneous tone via
phosphatidylinositol 3-kinase in DOCA-salt hypertension.
Hypertension. 43:125–129. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ohanian J, Gatfield KM, Ward DT and
Ohanian V: Evidence for a functional calcium-sensing receptor that
modulates myogenic tone in rat subcutaneous small arteries. Am J
Physiol Heart Circ Physiol. 288:H1756–H1762. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Greenberg HZE, Jahan KS, Jian S, Ho WSV
and Albert AP: The calcilytics Calhex-231 and NPS 2143 and the
calcimimetic Calindol reduce vascular reactivity via inhibition of
voltage-gated Ca2+ channels. Eur J Pharmacol.
791:659–668. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nakagawa K, Parekh N, Koleganova N, Ritz
E, Schaefer F and Schmitt CP: Acute cardiovascular effects of the
calcimimetic R-568 and its enantiomer S-568 in rats. Pediatr
Nephrol. 24:1385–1389. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sutherland SK and Benishin CG: Regulation
of parathyroid hypertensive factor secretion by Ca2+ in
spontaneously hypertensive rat parathyroid cells. Am J Hypertens.
17:266–272. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jensen AA and Bräuner-Osborne H:
Allosteric modulation of the calcium-sensing receptor. Curr
Neuropharmacol. 5:180–186. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Saidak Z, Brazier M, Kamel S and
Mentaverri R: Agonists and allosteric modulators of the
calcium-sensing receptor and their therapeutic applications. Mol
Pharmacol. 76:1131–1144. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Thakore P and Ho WS: Vascular actions of
calcimimetics: role of Ca2+-sensing receptors versus
Ca2+ influx through L-type Ca2+ channels. Br
J Pharmacol. 162:749–762. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Qiu HY, Henrion D and Levy BI: Endogenous
angiotensin II enhances phenylephrine-induced tone in hypertensive
rats. Hypertension. 24:317–321. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Effect of angiotensin-converting enzyme
inhibition compared with conventional therapy on cardiovascular
morbidity and mortality in hypertension, . the Captopril Prevention
Project (CAPP) randomized trial. The Captopril Prevention Project
(CAPP) Study Group. Current Hyperten Rep. 1:466–467. 1999.
|
|
57
|
Maeso R, Navarro-Cid J, Muñoz-García R,
Rodrigo E, Ruilope LM, Lahera V and Cachofeiro V: Losartan reduces
phenylephrine constrictor response in aortic rings from
spontaneously hypertensive rats role of nitric oxide and
angiotensin II type 2 receptors. Hypertension. 28:967–972. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Houillier P: Calcium-sensing in the
kidney. Curr Opin Nephrol Hypertens. 22:566–571. 2013.PubMed/NCBI
|