Introduction
Lung cancer is one of most lethal cancer types
worldwide with an estimate of 1.8 million new cases and 1.5 million
mortalities in 2012 (1). As a
major subtype of lung cancer, non-small cell lung cancer (NSCLC)
accounted for >80% of all lung cancer cases (2). Although development of concomitant
chemoradiotherapy has improved patient outcome, patients with NSCLC
can develop resistance, reducing the 5-year survival rate to ~30%
(3). Therefore, further
investigation of the molecular mechanism driving NSCLC cell
proliferation is required in order to develop new effective
therapeutic approaches to improve NSCLC patient survival.
MicroRNAs (miRNAs) are small, non-coding,
single-stranded RNA molecules that are ubiquitously expressed in
various types of cells (4).
Mechanistically, miRNAs can bind to the 3′ untranslated region
(UTR) of multiple target genes, thus inducing mRNA degradation or
translational inhibition (5). By
regulating expression of multiple genes, miRNAs are involved in
many physiological processes including cell proliferation,
differentiation, cell cycle and metastasis (6). Accumulating evidences showed that
miRNA dysregulation is responsible for cancer initiation and
progression (7). In NSCLC, using
microarray analysis, 15 differentially expressed miRNAs were
identified between normal lung and squamous cell carcinoma, one
subtype of NSCLC, and these miRNAs were proved to be good
predictors of patient survival (8). Downregulation of miR-138-5p was first
identified in human anaplastic thyroid carcinoma cells and was
found to regulate cell proliferation by targeting telomerase
reverse transcriptase (9). A
previous study showed that downregulation of miR-138-5p is involved
in the development of many cancer types (10,11).
A previous study identified that miR-138-5p is pivotal for
gefitinib sensitivity in NSCLC cells (12). However, little is known about the
role of miR-138-5p in mediating NSCLC cell proliferation and
survival.
Cyclin-dependent kinases (CDKs) form complexes with
cyclins to regulate cell cycle progression in human cells, which is
maintained by the tight control of CDK activity (13). In cancer cells, aberrant expression
of CDKs causes an accelerated cell cycle, leading to uncontrolled
cell proliferation (14). Among
the CDK family members, various studies observed that CDK8 is
upregulated in cancer cells and may represent a promising target
for cancer therapy (15).
Interestingly, the mechanism underlying CDK8 overexpression remains
unclear.
In the present study, a decrease in miR-138-5p was
observed in NSCLC tumor tissues compared with normal tissues.
Reverse transcription-quantitative PCR (RT-qPCR) and western blot
analysis suggested that CDK8 was regulated by miR-138-5p in A549
cells. Furthermore, CDK8 was identified as a target gene of
miR-138-5p. In addition, miR-138-5p overexpression inhibited A549
cell growth, leading to cell apoptosis and accumulation of cells in
G0/G1 phase. Notably, the effect of miR-138-p could be reversed by
CDK8 overexpression.
Materials and methods
Patient samples
Tumor tissues and matched normal tissues were
collected from 30 patients with NSCLC (17 males and 13 females;
age, 62. 21±5.23 years) in The Central Hospital of Linyi between
February 2015 and January 2016. Written consents were obtained from
all participants. Ethical approval was obtained for the use of
human tissues prior to the start of the study from an Ethics
Committee based at The Central Hospital of Linyi. All tissues were
subjected to RNA extraction.
Cell lines
Normal epithelial lung cell line BEAS-2B and the
NSCLC cell lines A549, H358 and H1299 were purchased from The
American Type Culture Collection and used within 6 months. All cell
lines were maintained in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS (HyClone; GE Healthcare
Life Sciences) in a humidified incubator at 37°C with 5%
CO2.
Western blot analysis
Protein lysates were prepared using RIPA lysis
buffer (Sigma-Aldrich; Merck KGaA) according to the manufacturer's
protocol. The protein concentration was determined using the method
bicinchoninic acid method. Antibodies for anti-CDK8 (cat. no 17395;
dilution, 1:1,000) and β-catenin (cat. no 8480; dilution, 1:1,000)
were purchased from Cell Signaling Technology, Inc. β-actin
antibody (cat. no SAB5500001; dilution, 1:1,000) was bought from
Sigma-Aldrich (Merck KGaA). Western blotting was performed as
follows: Protein lysates (20 µg) were loaded into each lane of a
10% SDS gel and separated by electrophoresis. The proteins were
transferred to a PVDF membrane. After that, membranes were blocked
at room temperature for 2 h using 5% non-fat milk followed by
incubation with primary antibodies overnight at 4°C. On the next
day, the membranes were washed and incubated with the appropriate
anti-rabbit horseradish peroxidase-conjugated secondary antibody
(cat. no. 7074; 1:10,000; Cell Signaling Technology, Inc.) for 1 h
at room temperature. The bands were visualized using an enhanced
chemiluminescence (ECL) western blotting kit (Pierce; Thermo Fisher
Scientific, Inc.). The quantification of bands was carried out
using ImageJ (version 1.51; National Institutes of Health).
RNA extraction and RT-qPCR
RNA was extracted from cells or tissues using TRIzol
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. The RNA concentration was measured using a
NanoDrop 2000 (Thermo Fisher Scientific, Inc.). For detection of
miRNA expression, RNA was reverse transcribed into cDNA using the
Mir-X miRNA First Strand Synthesis Kit (Takara Bio, Inc.) at 37°C
for 1 h and 85°C for 5 min. The RT-qPCR was performed using the
Mir-X miRNA RT-qPCR SYBR kit (Thermo Fisher Scientific, Inc.) using
the following thermocycling conditions: 95°C for 30 sec followed by
40 cycles of 95°C for 5 sec and 60°C for 30 sec. For mRNA
expression analysis, RNA was reverse transcribed into first strand
cDNA using PrimeScript RT Master Mix (Takara Bio, Inc.) at 37°C for
15 min followed by 85°C for 5 sec qPCR with SYBR Premix Ex Taq II
(Thermo Fisher Scientific, Inc.) was performed using the following
thermocycling conditions: 95°C for 30 sec followed by 40 cycles of
95°C for 5 sec and 60°C for 30 sec. U6 and β-actin served as
internal controls to normalize the expression of miRNAs and mRNAs,
respectively. The primers used were the following: miR-138 forward,
5′-GGTGTCGTGGAGTCGGCAA-3′ and reverse, 5′-AACTTCACAACACCAGCTTA-3′;
U6 forward, 5′-CTCGCTTCGGCAGCACA-3′ and reverse,
5′-AACGCTTCACGAATTTGCGT-3′; CDK8 forward,
5′-ACCTGTTTGAATACGAGGGCT-3′ and reverse,
5′-TGCCGACATAGAGATCCCAGT-3′; and β-actin forward,
5′-CATGTACGTTGCTATCCAGGC-3′ and reverse,
5′-CTCCTTAATGTCACGCACGAT-3′. Relative gene expression was analyzed
using the 2−ΔΔCq method (16).
CDK8 overexpression
The full length of the open reading frame of CDK8
was amplified using Taq DNA polymerase (Invitrogen; Thermo Fisher
Scientific, Inc.) from the cDNA of BEAS-2B (forward
5′-AAGCTTGGGCTCCGGCCTCAGAGGCT-3′ and reverse,
5′-CTCGAGACCACATACAAAGACAAATGCT-3′) using the following
thermocycling conditions: 94°C for 2 min followed by 35 cycles of
94°C for 2 sec, 60°C for 60 sec and 72°C for 1 min. The product was
digested with HindIII and XhoI and cloned into a
pcDNA3.1 plasmid (Invitrogen; Thermo Fisher Scientific, Inc.). For
CDK8 overexpression, pcDNA3.1-CDK8 (2 µg) was transfected into A549
cells using Lipofectamine 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. The
medium was changed after 8 h and the cells were cultured for a
further 48 h prior to subsequent experiments.
Dual luciferase reporter assay
TargetScan (release 7.1, http://www.targetscan.org/vert_71/) was used to
predict the interaction between miR-138-5p and CDK8 3′UTR. The
3′UTR of CDK8 was amplified using the Taq DNA polymerase
(Invitrogen; Thermo Fisher Scientific, Inc.) from the cDNA of
BEAS-2B (forward, 5′-GGTACCAAGGAGACAAAAAGAACC-3′ and reverse,
5′-CTCGAGGTAGTGGTAGGAGGAACAAC-3′) using the following thermocycling
conditions: 94°C for 2 min followed by 35 cycles of 94°C for 2 sec,
60°C for 60 sec and 72°C for 1 min. The product was ligated into
pGL3 plasmid (Promega Corporation) between the KpnI and
Xhol restriction enzyme sites. In total, two site mutations
were introduced into pGL3-CDK8 3′UTR-wild-type (WT) to construct
the pGL3-CDK8 3′UTR-mutant (Mut) plasmid using the Quick
Site-directed Mutagenesis kit (Agilent Technologies, Inc.) using
the following primers: Forward,
5′-GAGACAAAAAGAACGTGCAGCAGCAGCAGGG-3′ and reverse,
5′-CCCTGCTGCTGCTGCACGTTCTTTTTGTCTC-3′. The following thermocycling
conditions were used: 95°C for 30 sec followed by 35 cycles of 95°C
for 30 sec, 55°C for 60 sec and 78°C for 1 min. In A549 cells,
pGL3-CDK8 3′UTR-WT (0.4 mg) or pGL3-CDK8 3′UTR-Mut (0.4 mg) and
miR-negative control (NC; 5′-UUGUACUACACAAAAGUACUG-3′; 20 nM) mimic
or miR-138-5p mimic (5′-AGCUGGUGUUGUGAAUCAGGCCG-3′; 20 nM) were
cotransfected into A549 cells using Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.). After 24 h, the
relative luciferase activity in each well was measured using the
dual-luciferase reporter assay system (Promega Corporation).
Renilla luciferase activity was used for normalization.
Cell cycle and cell apoptosis
analysis
For cell cycle analysis, cells (1×105
cells/ml) were harvested and treated with 75% ethanol overnight at
4°C. On the next day, cells were stained with propidium iodide (PI;
Sigma-Aldrich; Merck KGaA). Cell cycle was then analyzed on a
FACSCalibur flow cytometer (BD Biosciences). Cell apoptosis was
detected using an Annexin V/PI apoptosis detection kit
(Sigma-Aldrich; Merck KGaA). Briefly, cells were washed with PBS
and resuspended in Annexin binding buffer. Subsequently, Annexin
V-FITC and PI were added and incubated for 15 min at 4°C. Cells
were then subjected to flow cytometry analysis on a FACSCalibur
flow cytometer (BD Biosciences). Cell cycle distribution and cell
apoptotic rates were analyzed with FlowJo software (version 7.2;
FlowJo LLC).
Cell proliferation assay
Cell proliferation was measured using a Cell
Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.).
Briefly, 1,000 cells were plated in each well of a 96-well plate.
After 24, 48 and 72 h, 10 µl CCK-8 solution was added into each
well and maintained for 2 h at 37°C. The medium containing CCK-8
was removed and added into another well. The absorbance at 450 nm
was detected in each well using a microplate reader (Bio-Rad
Laboratories, Inc.).
Statistical analysis
All experiments were repeated three times. All data
were analyzed using GraphPad Prism (version 7.0; GraphPad Software,
Inc.). Data are presented as the mean ± SD. Differences between two
groups were compared using a paired or unpaired Student's t-test.
Differences among three groups were calculated using one-way ANOVA
followed by Newman-Keuls test. Pearson's correlation analysis was
used to analyze the correlation between miR-138-5p and CDK8.
P<0.05 was considered to indicate a statistically significant
difference.
Results
miR-138-5p is downregulated in NSCLC
tissues and cell lines
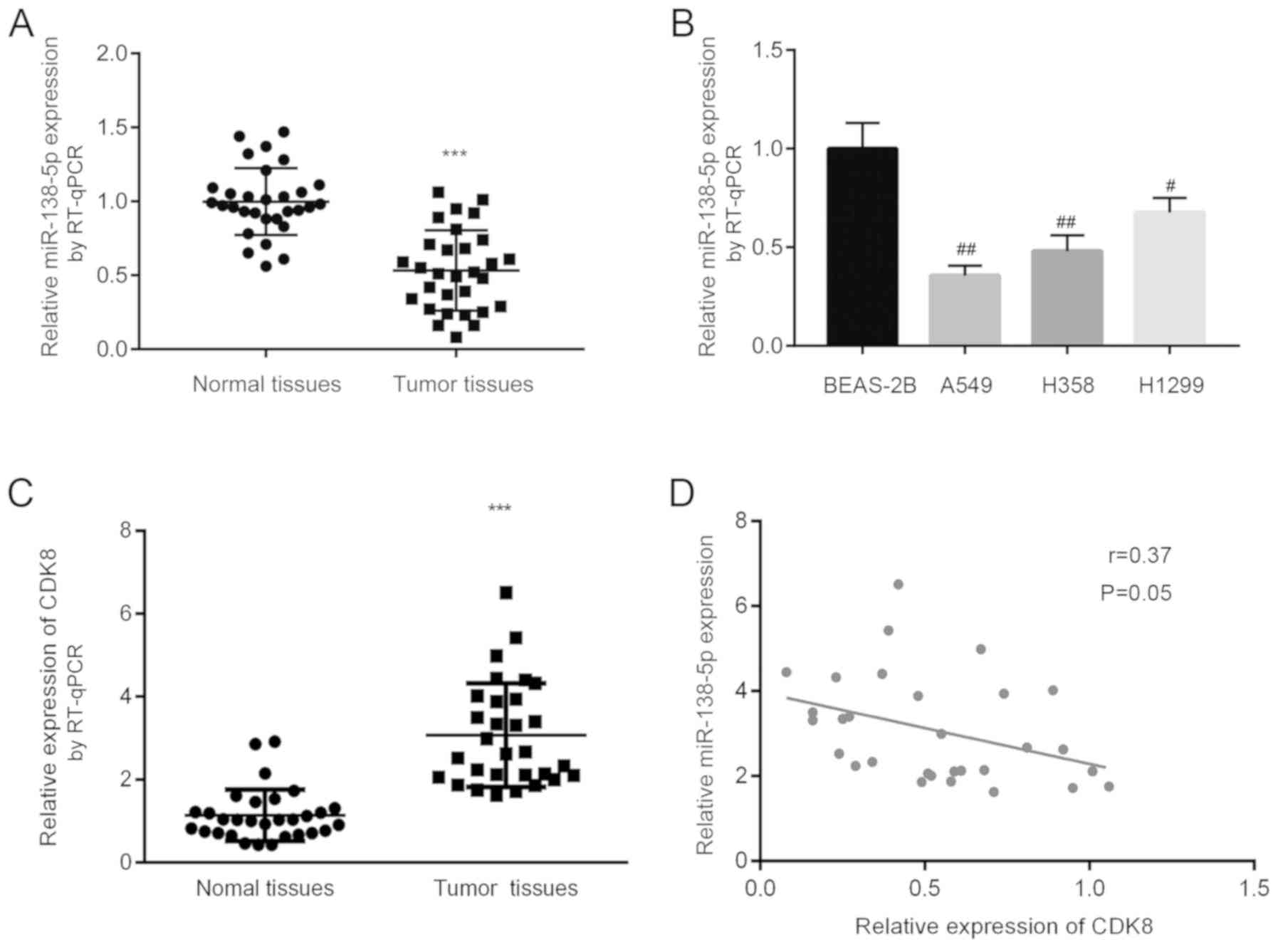
To investigate the potential biological function of
miR-138-5p in NSCLC, miR-138-5p expression was measured in 30 pairs
of tumor tissues and normal tissues from 30 patients with NSCLC.
RT-qPCR results showed that miR-138-5p levels were significantly
decreased in tumor tissues compared with normal tissues (Fig. 1A). Subsequently, miR-138-5p levels
were analyzed in normal epithelial lung cell line BEAS-2B and three
NSCLC cell lines, including A549, H358 and H1299. In comparison
with BEAS-2B, miR-138-5p expression was significantly lower in the
three NSCLC cell lines tested (Fig.
1B). CDK8 mRNA expression was increased in tumor tissues
compared with matched normal tissues (Fig. 1C). There was an inverse correlation
between miR-138-5p and CDK8 (Fig.
1D). The present findings suggested that miR-138-5p may be
associated with CDK8 and may play a role in NSCLC.
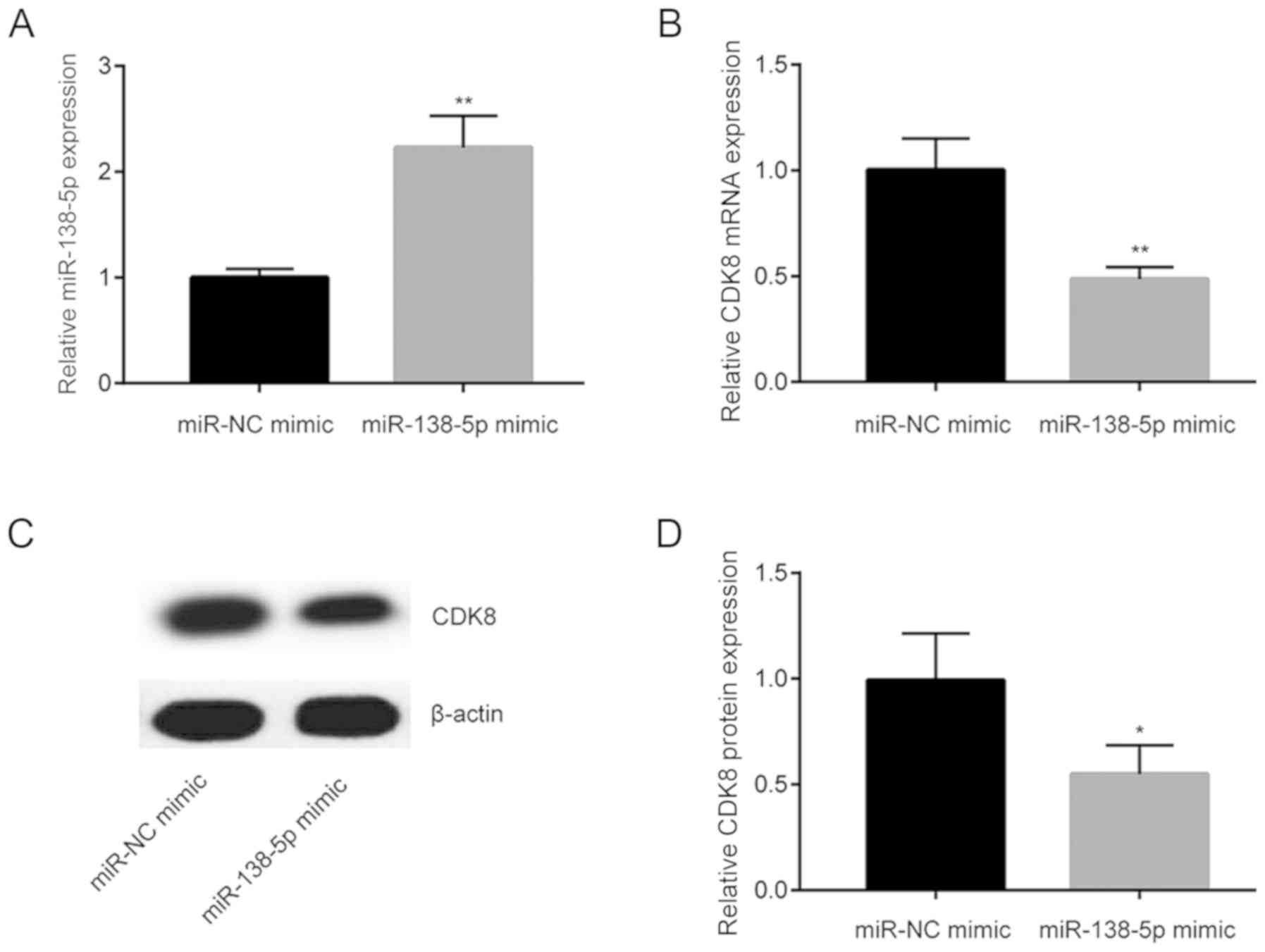
miR-138-5p negatively regulates CDK8
in NSCLC cells
CDK8 was upregulated in NSCLC tissues. To evaluate
whether miR-138-5p regulated CDK8 expression, miR-138-5p mimic was
transfected into A549 cells. Transfection of miR-138-5p mimic
increased miR-138-5p level in A549 cells (Fig. 2A). In addition, miR-138-5p mimic
significantly reduced CDK8 mRNA level (Fig. 2B). Furthermore, western blot
analysis indicated that CDK8 protein levels were also decreased
following miR-138-5p overexpression (Fig. 2C and D).
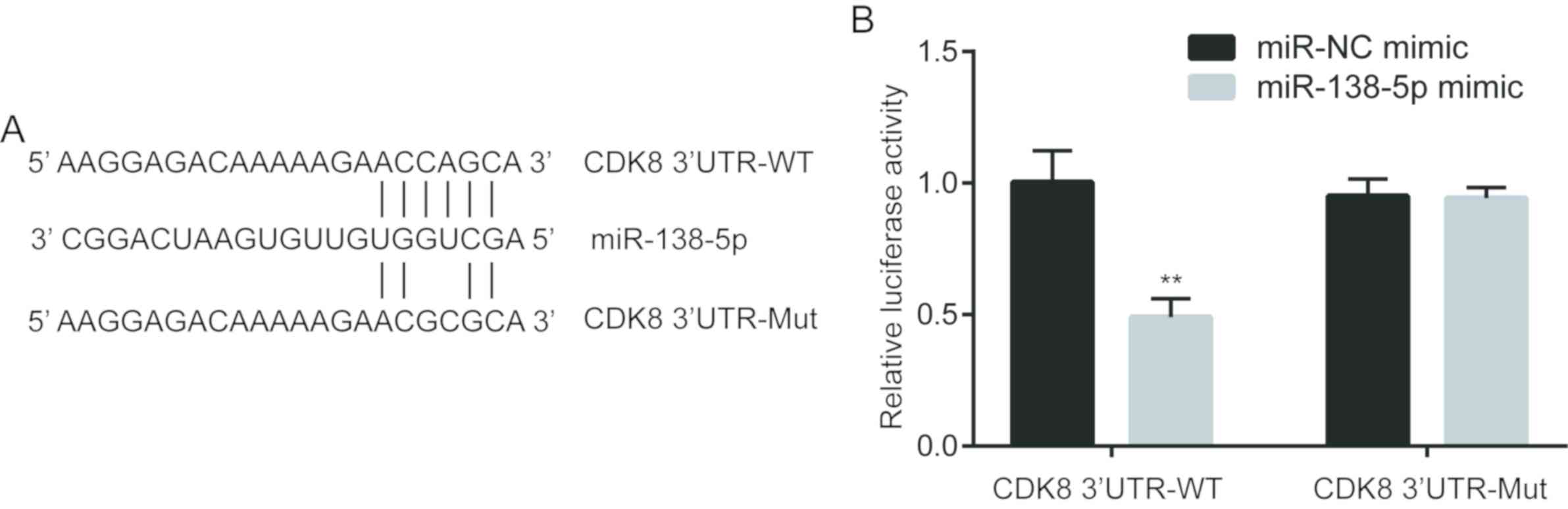
CDK8 is a target gene of
miR-138-5p
To further investigate whether CDK8 was a target
gene of miR-138-5p, TargetScan was used to predict the interaction
between miR-138-5p and CDK8 3′UTR. Bioinformatic analysis indicated
that there was a complementary binding site between miR-138-5p and
the 3′UTR of CDK8 mRNA (Fig. 3A).
To determine their direct binding, a portion of the 3′UTR of CDK8
mRNA was inserted into a firefly luciferase reporter. A dual
luciferase assay was performed, and miR-138-5p mimic decreased the
relative luciferase activity of cells transfected with CDK8
3′UTR-WT, whereas luciferase activity was not changed in cells
transfected with CDK8 3′UTR-Mut and miR-138-5p mimic compared with
cells transfected with CDK8 3′UTR-Mut and miR-NC (Fig. 3B). Collectively, the present
results indicated that CDK8 was directly regulated by miR-138-5p in
NSCLC cells.
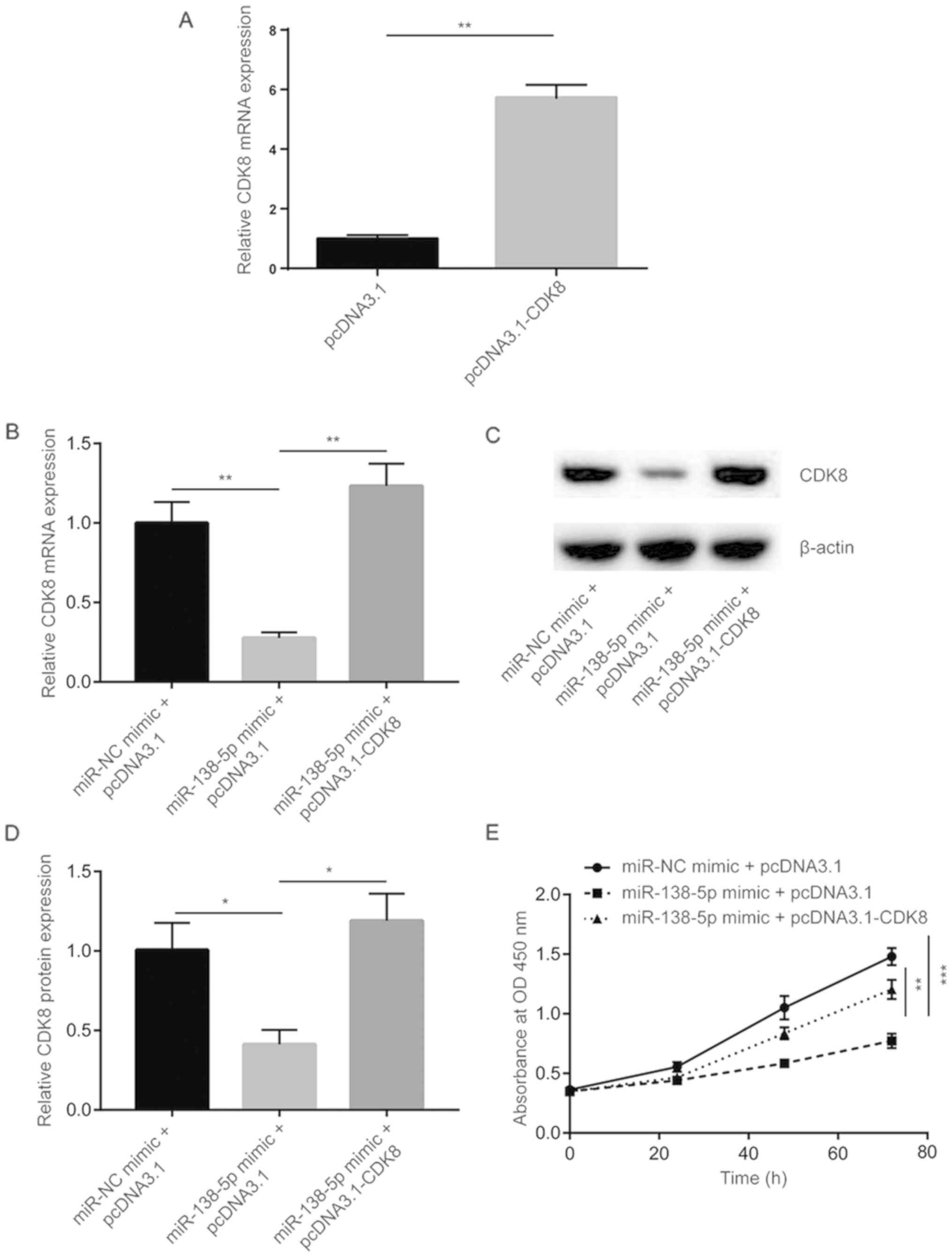
Overexpression of miR-138-5p inhibits
NSCLC cell proliferation by regulating CDK8
The function of miR-138-5p in NSCLC cells was
examined. A pcDNA3.1 plasmid expressing CDK8 was transfected into
A549 cells to increase CDK8 expression. Using RT-qPCR, CDK8 mRNA
level was identified to be increased following pcDNA3.1-CDK8
transfection (Fig. 4A). Using
RT-qPCR and western blot analysis, miR-138-5p mimic was found to
decrease CDK8 mRNA and protein level, which could be recovered
following pcDNA3.1-CDK8 transfection (Fig. 4B-D). Cell proliferation was
investigated, and overexpression of miR-138-5p inhibited cell
proliferation of A549 cells, whereas transfection of pcDNA3.1-CDK8
partially reversed the cell proliferation inhibition induced by
miR-138-5p (Fig. 4E).
Overexpression of miR-138-5p
influences cell cycle and cell apoptosis by regulating CDK8
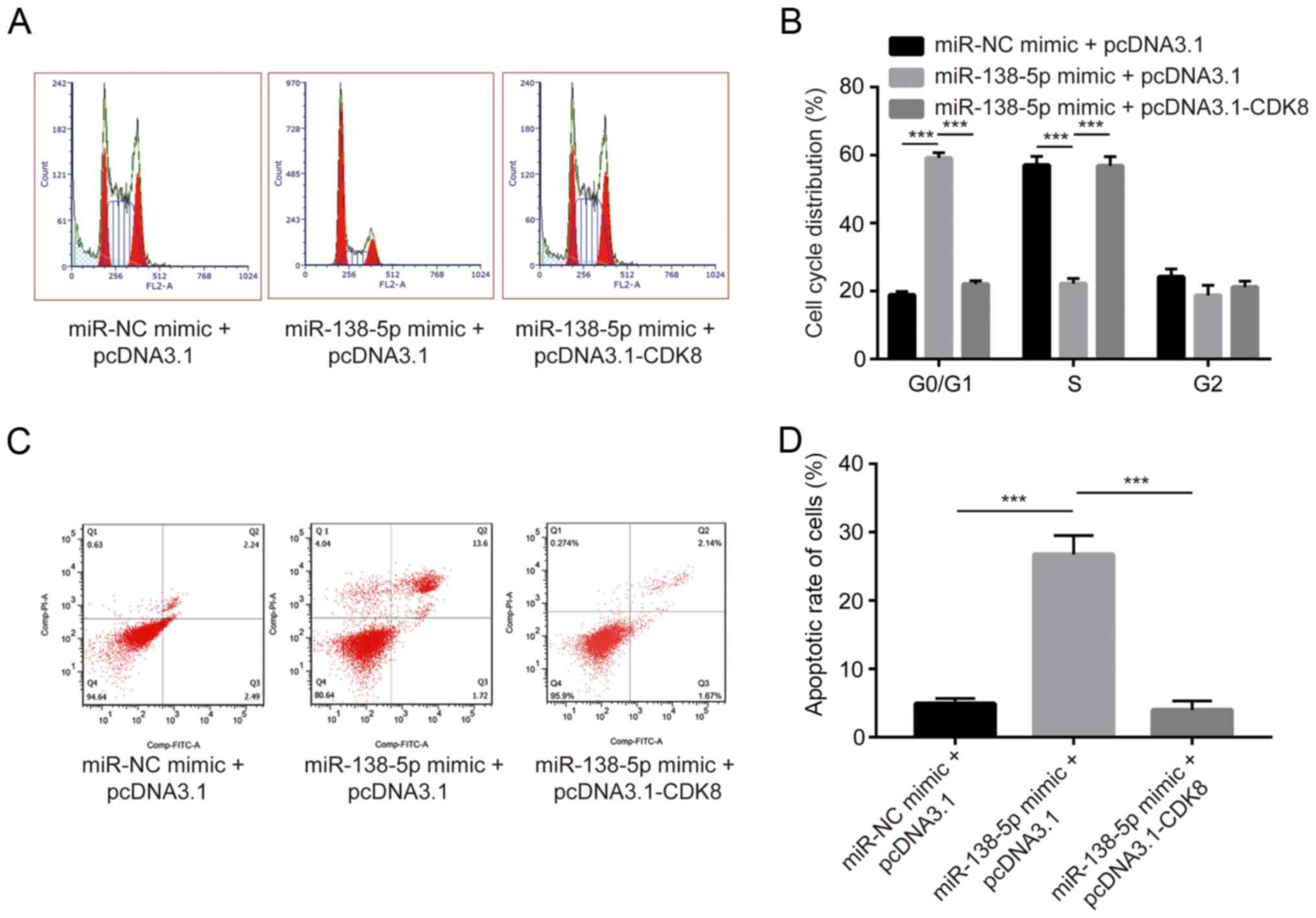
CDK8 is a major regulator of cell cycle (13). Using flow cytometry, miR-138-5p
mimic was found to induce an accumulation of cells in G0/G1 phase.
In addition, overexpression CDK8 reduced the number of cells in
G0/G1 phase (Fig. 5A and B). A
cell apoptosis assay was performed, and transfection of miR-138-5p
significantly increased cell apoptosis rate, whereas CDK8
overexpression partially reversed cell apoptosis (Fig. 5C and D). The present results
suggested that miR-138-5p promoted cell cycle arrest and cell
apoptosis by repressing CDK8 expression in NSCLC cells.
Discussion
NSCLC is a major type of lung cancer, and is one of
most lethal cancer types worldwide (17). Previous studies identified that
many miRNAs serve critical roles in cancer progression (18,19).
In cancer cells, miRNAs can act as oncogenes or tumor suppressors
(20). In addition, several miRNAs
were identified as good prognostic predictors in patients with
NSCLC (21,22).
Accumulating evidence showed that miR-138-5p is a
tumor suppressor in many cancer types, including bladder and
colorectal cancer (23,24). In NSCLC, microarray screening
identified that miR-138-5p is significantly downregulated in
gefitinib-resistant NSCLC cells compared with parental cells and
may regulate gefitinib sensitivity by regulating G protein-coupled
receptor 124 (12). However,
whether and how miR-138-5p may be involved in cell proliferation,
cell cycle and cell apoptosis remain unclear. In the present study,
RT-qPCR results suggested that miR-138-5p was decreased in NSCLC
tumor tissues compared with matched normal tissues. Additionally, a
decreased expression of miR-138-5p was observed in NSCLC cell lines
compared with normal epithelial lung cell line BEAS-2B. More
importantly, overexpression of miR-138-5p induced cell
proliferation arrest, cell apoptosis and increased the number of
cells in G0/G1 phase. The present data suggested that miR-138-5p
may serve as a tumor suppressor gene in NSCLC.
CDK8 is a well-characterized oncogene in various
cancer types (25,26). Several reports showed that the
downregulation of various miRNAs contribute to the upregulation of
CDK8 in cancer cells (27,28). In NSCLC, miR-10a targets CDK8,
regulating cisplatin chemosensitivity (29). The present bioinformatic analysis
indicated that miR-138-5p may bind to the 3′UTR of CDK8 mRNA. In
addition, overexpression of miR-138-5p decreased CDK8 expression in
A549 cells. Moreover, dual luciferase assay confirmed that CDK8 was
a target gene of miR-138-5p. CDK8 is a regulator of cell cycle, and
alteration of CDK8 activity or expression was identified to
influence cell cycle, leading to cell apoptosis and cell
proliferation arrest (30,31). In the present study, miR-138-5p
mimic was found to induce cell growth arrest, increasing the number
of cells in G0/G1 phase and increasing the cell apoptotic rate.
Moreover, transfection of CDK8 partially reversed the effects of
miR-138-5p overexpression on cell proliferation inhibition, cell
cycle redistribution and cell apoptosis increase. Therefore, the
present study suggested that the downregulation of miR-138-5p may
be responsible for the upregulation of CDK8 in NSCLC, and
miR-138-5p was found to inhibit NSCLC cell proliferation by
regulating cell cycle and cell apoptosis.
Collectively, miR-138-5p was found to be
downregulated in NSCLC tumor tissues and inhibited NSCLC cell
proliferation by directly targeting CDK8. The present results
suggested miR-138-5p may be a promising therapeutic target for
treating patients with NSCLC.
However, the present study presented certain
limitations; since miR-138-5p was downregulated in NSCLC tumor
tissues, the effects of miR-138-5p overexpression were investigated
to examine whether miR-138-5p mimics could reverse the abnormal
phenotype of NSCLC. Therefore, further studies are required to
investigate the effect of miR-138-5p inhibition. In addition, the
present study did not investigate the effects of CDK8 on the
regulation of β-catenin activity, Notch signaling and on additional
downstream effectors of CDK8. Moreover, the role of miR-138-5p on
cell migration and invasion were not examined in the present study,
and further studies are required to investigate these aspects.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
FT conceived the study, analyzed the data, and wrote
the manuscript. SX, QX, XF, and SW carried out the experiments and
analyzed the data.
Ethics approval and consent to
participate
Ethical approval was obtained for the use of human
tissues prior to the start of the study from an Ethics Committee
based at The Central Hospital of Linyi.
Patient consent for publication
Each patient provided written consent for
publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Heist RS and Engelman JA: SnapShot:
Non-small cell lung cancer. Cancer Cell. 21:e448–e442. 2012.
View Article : Google Scholar
|
|
3
|
Auperin A, Le Pechoux C, Rolland E, Curran
WJ, Furuse K, Fournel P, Belderbos J, Clamon G, Ulutin HC, Paulus
R, et al: Meta-analysis of concomitant versus sequential
radiochemotherapy in locally advanced non-small-cell lung cancer. J
Clin Oncol. 28:2181–2190. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao Y and Srivastava D: A developmental
view of microRNA function. Trends Biochem Sci. 32:189–197. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hao J, Zhao S, Zhang Y, Zhao Z, Ye R, Wen
J and Li J: Emerging role of microRNAs in cancer and cancer stem
cells. J Cell Biochem. 115:605–610. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Raponi M, Dossey L, Jatkoe T, Wu X, Chen
G, Fan H and Beer DG: MicroRNA classifiers for predicting prognosis
of squamous cell lung cancer. Cancer Res. 69:5776–5783. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mitomo S, Maesawa C, Ogasawara S, Iwaya T,
Shibazaki M, Yashima-Abo A, Kotani K, Oikawa H, Sakurai E, Izutsu
N, et al: Downregulation of miR-138 is associated with
overexpression of human telomerase reverse transcriptase protein in
human anaplastic thyroid carcinoma cell lines. Cancer Sci.
99:280–286. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ding J, Yeh CR, Sun Y, Lin C, Chou J, Ou
Z, Chang C, Qi J and Yeh S: Estrogen receptor β promotes renal cell
carcinoma progression via regulating LncRNA
HOTAIR-miR-138/200c/204/217 associated CeRNA network. Oncogene.
37:5037–5053. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peng J, Hou F, Feng J, Xu SX and Meng XY:
Long non-coding RNA BCYRN1 promotes the proliferation and
metastasis of cervical cancer via targeting microRNA-138 in
vitro and in vivo. Oncol Lett. 15:5809–5818.
2018.PubMed/NCBI
|
|
12
|
Gao Y, Fan X, Li W, Ping W, Deng Y and Fu
X: miR-138-5p reverses gefitinib resistance in non-small cell lung
cancer cells via negatively regulating G protein-coupled receptor
124. Biochem Biophys Res Commun. 446:179–186. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: A review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nakamura A, Nakata D, Kakoi Y, Kunitomo M,
Murai S, Ebara S, Hata A and Hara T: CDK8/19 inhibition induces
premature G1/S transition and ATR-dependent cell death in prostate
cancer cells. Oncotarget. 9:13474–13487. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clin Proc. 83:584–594.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu C, Zhao Y, Zhang Z, Ni Y, Li X and
Yong H: MicroRNA-33a inhibits lung cancer cell proliferation and
invasion by regulating the expression of β-catenin. Mol Med Rep.
11:3647–3651. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Z, Zhang Y, Sun XX, Ma X and Chen
ZN: microRNA-146a inhibits cancer metastasis by downregulating VEGF
through dual pathways in hepatocellular carcinoma. Mol Cancer.
14:52015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang B, Pan X, Cobb GP and Anderson TA:
microRNAs as oncogenes and tumor suppressors. Dev Biol. 302:1–12.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lu G, Fu D, Jia C, Chai L, Han Y, Liu J,
Wu T, Xie R, Chang Z, Yang H, et al: Reduced miR-105-1 levels are
associated with poor survival of patients with non-small cell lung
cancer. Oncol Lett. 14:7842–7848. 2017.PubMed/NCBI
|
|
22
|
Li Y, Zhang H, Dong Y, Fan Y, Li Y, Zhao
C, Wang C, Liu J, Li X, Dong M, et al: MiR-146b-5p functions as a
suppressor miRNA and prognosis predictor in non-small cell lung
cancer. J Cancer. 8:1704–1716. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang R, Liu M, Liang H, Guo S, Guo X, Yuan
M, Lian H, Yan X, Zhang S, Chen X, et al: miR-138-5p contributes to
cell proliferation and invasion by targeting Survivin in bladder
cancer cells. Mol Cancer. 15:822016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao L, Yu H, Yi S, Peng X, Su P, Xiao Z,
Liu R, Tang A, Li X, Liu F and Shen S: The tumor suppressor
miR-138-5p targets PD-L1 in colorectal cancer. Oncotarget.
7:45370–45384. 2016.PubMed/NCBI
|
|
25
|
Serrao A, Jenkins LM, Chumanevich AA,
Horst B, Liang J, Gatza ML, Lee NY, Roninson IB, Broude EV and
Mythreye K: Mediator kinase CDK8/CDK19 drives YAP1-dependent
BMP4-induced EMT in cancer. Oncogene. 37:4792–4808. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Philip S, Kumarasiri M, Teo T, Yu M and
Wang S: Cyclin-dependent kinase 8: A new hope in targeted cancer
therapy? J Med Chem. 61:5073–5092. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Luo Q, Zhang Z, Dai Z, Basnet S, Li S, Xu
B and Ge H: Tumor-suppressive microRNA-195-5p regulates cell growth
and inhibits cell cycle by targeting cyclin dependent kinase 8 in
colon cancer. Am J Transl Res. 8:2088–2096. 2016.PubMed/NCBI
|
|
28
|
Li M, Tian L, Ren H, Chen X, Wang Y, Ge J,
Wu S, Sun Y, Liu M and Xiao H: MicroRNA-101 is a potential
prognostic indicator of laryngeal squamous cell carcinoma and
modulates CDK8. J Transl Med. 13:2712015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Z, Zhang L, Yin ZY, Fan XL, Hu B,
Wang LQ and Zhang D: miR-107 regulates cisplatin chemosensitivity
of A549 non small cell lung cancer cell line by targeting cyclin
dependent kinase 8. Int J Clin Exp Pathol. 7:7236–7241.
2014.PubMed/NCBI
|
|
30
|
Liu ZJ, Ueda T, Miyazaki T, Tanaka N, Mine
S, Tanaka Y, Taniguchi T, Yamamura H and Minami Y: A critical role
for cyclin C in promotion of the hematopoietic cell cycle by
cooperation with c-Myc. Mol Cell Biol. 18:3445–3454. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
He SB, Yuan Y, Wang L, Yu MJ, Zhu YB and
Zhu XG: Effects of cyclin-dependent kinase 8 specific siRNA on the
proliferation and apoptosis of colon cancer cells. J Exp Clin
Cancer Res. 30:1092011. View Article : Google Scholar : PubMed/NCBI
|