Introduction
A mobile genetic element (MGE) is a fragment of
genetic material that encodes an enzyme capable of moving or
inserting it in another location in the same or another host
genome. DNA transposases and retroviral integrases are among the
most extensively studied MGEs. MGEs are present in all genomes
sequenced thus far and comprise 3% of the human genome (1). MGEs play an essential role in genome
evolution, actively driving rearrangements, insertions, deletions
and translocations, while some transposases have evolved to be
‘domesticated’ by the host and perform functional specific tasks
within the cell. As a result of selection pressure across millions
of years, some parts of MGEs have evolved as non-coding gene
modulators at the DNA or RNA level or code for proteins employed as
defensive mechanisms against genetic instabilities or with an
entirely new and sophisticated function, such as DNA repair
(2,3).
A transposase is typically a multidomain enzyme,
usually organized as a multimer in its active form. It possesses
domains capable of specific interaction with DNA sequences flanking
the mobilized fragment [terminal inverted repeats (TIRs)] and a
catalytic core domain with nuclease activity, which performs a
two-step process of DNA cleavage and transfer into the target DNA
(4).
DNA transposons were first identified by Barbara
McClintock almost 70 years ago while studying mutable loci in maize
(5). Since then a wide variety of
elements have been identified and classified. There are two major
strategies for DNA transposition: ‘Copy and paste’, in which the
transposon remains in place, but inserts a copy of itself into
another location and ‘cut and paste’, in which the DNA fragment is
excised from the original location and moves into a new one. DNA
transposons can be classified according to the folding of their
catalytic region and key mechanistic similarities: DDE(D)
transposons, HUH transposons (6),
serine transposons and tyrosine transposons (7). There are 19 superfamilies of ‘cut and
paste’ elements and 11 of these share a DDE(D) triad (Asp, Asp and
Glu) (8). DDE(D) elements include
prokaryotic transposons, such as Tn5, Tn10, bacteriophage MuA and
eukaryotic transposon families, such as hAT (Ac/Ds, Hermes),
Transib, Tc1/Mariner (Mos1, Sleeping Beauty, Himar, Hsmar1 and 2,
transposase domain of SETMAR) (7).
Several transposase systems [such as Flp/FRT (9) or piggyBac (10)] are now part of the molecular
toolkit for manipulating genes in vitro and in
vivo.
DDE(D) transposons belong to the polynucleotide
transferase superfamily, along with other evolutionarily-related
metal-ion dependent enzymes, such as retroviral INs, RuvC
resolvase, RNaseH, the Argonaut component of the RNA-induced
silencing complex, reverse transcriptase (RT) and
recombination-activating gene protein 1 (RAG1). Their reaction
mechanism is driven by three negatively charged amino acids (Asp,
Asp and Glu/Asp), which coordinate divalent metal ions and are
located within a RNase H-like fold region of the catalytic core
domain (4,11).
The precise positioning of the two metal ions in the
DDE/(D) coordination system enables the nucleophilic substitution
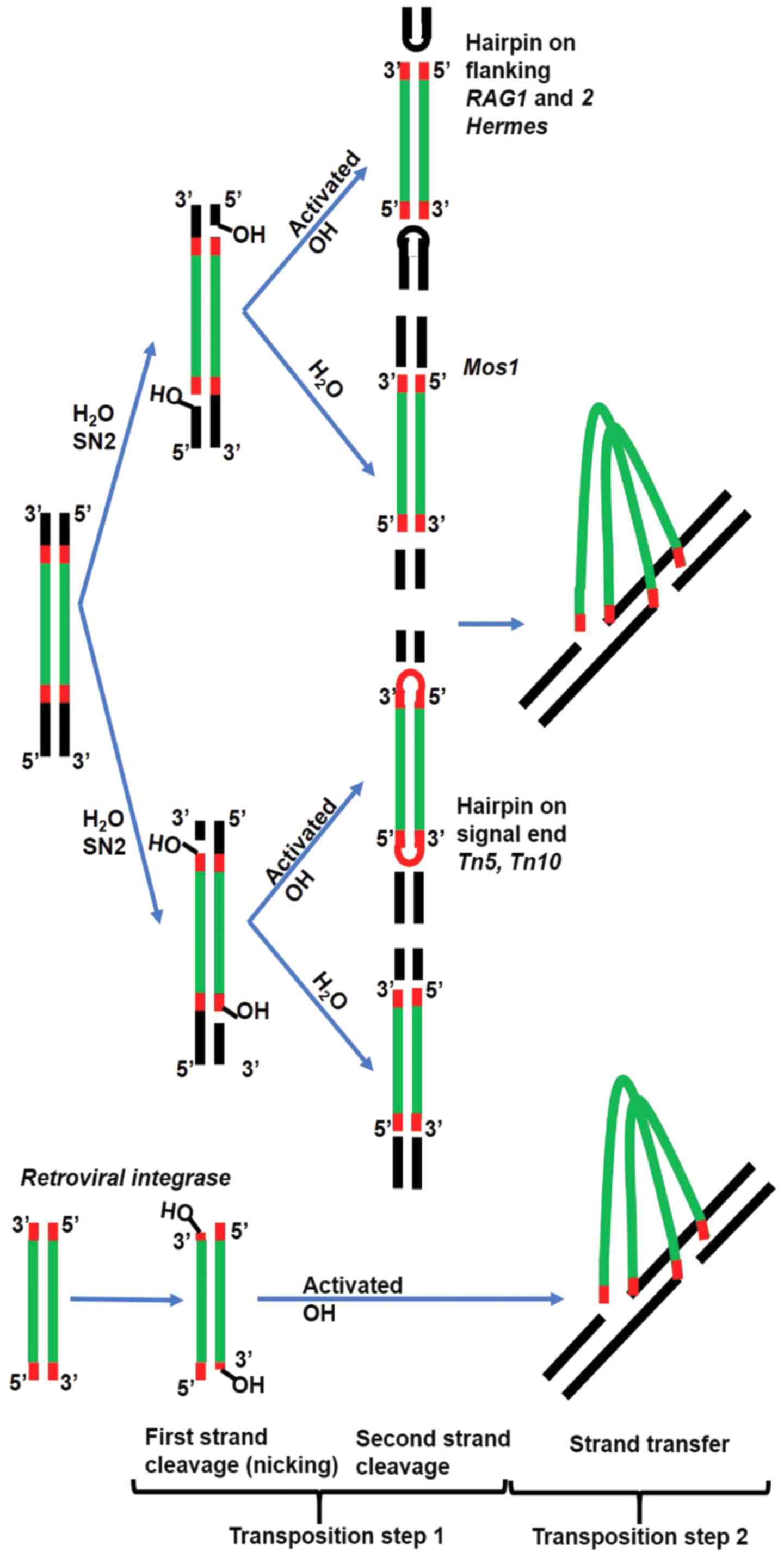
through a two-step mechanism. The first step (nicking), results in
a reactive 3′OH group at both ends of the MGE. Each active hydroxyl
will attack in the second step one strand of the target DNA [strand
transfer (ST)] (Fig. 1). Reaction
intermediates vary between DDE(D) polynucleotidyl transferases.
Some cut only one DNA strand followed by joining the 3′donor end at
the target site, while the 5′donor end remains in place, resulting
in a branched DNA product that is resolved by DNA replication
machinery (e.g., MuA and Tn3). In the case of retroviral INs, the
5′ flanking DNA is constituted only of a few bases and can be
removed by repair enzymes. Others cut both strands with a hairpin
intermediate on the excised fragment (e.g., Tn5 and Tn10) or on the
flanking DNA (RAG1 and 2, and Hermes). The hairpin is opened
through another hydrolytic reaction to free the 3′OH for ST, but in
the case of RAG1 and 2, transposition does not normally occur and
the hairpin is resolved by the non-homologous end joining (NHEJ)
system by joining the flanking ends (Fig. 1) (12).
The ST occurs in trans, only in the context
of a protein-DNA paired complex, known as a synaptic complex
(transpososome or intasome), featuring an oligomeric state of the
protein bound to both ends of the mobile element. Accessory
proteins are usually needed to stabilize the complex, and introduce
bending and conformational changes in the DNA, thus making it
accessible for the nuclease or guide the complex to the target
site, by interacting with open chromatin (13).
The similarities between HIV-1 IN and domesticated
transposases in terms of catalytic domain organization and the
mechanism of DNA cleavage function as a double-edged sword. The
non-specific inhibition of RAG1 by HIV-1 IN inhibitors can
interfere with the assembly of immune system receptors (14). On the other hand, HIV-1 IN
inhibitor interaction with the catalytic domain of metnase or other
enzymes critical for viral replication [HTLV-1 IN (15), HMCV terminase (16)] offers new alternatives for
medicinal chemistry.
This review summarizes the general common
characteristics of DDE(D) polynucleotidyl transferases with
emphasis on the particular case of RAG1 recombination and HIV-1
integration systems. In this context, we outline the off-target
effects of HIV-1 IN inhibitors either with potential deleterious
effects or on the contrary which can be exploited for the
identification and optimization of novel treatment strategies.
Moreover, we argue that computational analysis is a useful tool to
predict and analyze possible off target interactions. This is
illustrated herein by docking simulations revealing different
affinities of HIV-1 IN inhibitors towards RAG1 catalytic domain.
The calculated docking conformations explain the possible
interaction between IN inhibitors already suggested by reported
experimental data and illustrates the behavior of compounds with
different IN inhibition mechanisms on RAG1 RNaseH. Such
computational analysis is helpful for predicting the possible
extent of compound interaction, not only with RAG1, but also with
other DDE systems.
Docking simulations and conformations
Docking simulations were performed using Autodock
Vina software (17) on the RAG1
dimer structure selected from the published crystal of RAG1&2
complex (PDB accession code 4WWX) (18). The 3D structure of the ligands was
constructed with Avogadro (19).
The protein structure was maintained rigid and the ligand was
flexible having all the rotatable bonds set free. The protein and
ligand were prepared using AutoDock Tools and Gasteiger partial
charges were computed. The search space was defined by the
coordinates x=21.488, y=63.789, z=58.445 and the dimensions 100 Å
on × scale, 96 Å on y scale and 112 Å on z scale. To improve the
sampling of the energy landscape and to increase the probability of
finding deep energy minima, the exhaustiveness parameter was set to
50, given a default value of the program of only 8. Five rounds of
simulation were performed for each compound, each resulting in 20
output conformations. The conformations were sorted and clustered
based on the docking regions in the protein and their energies.
Only conformations within 5 Å of the DDE motif were selected for
analysis. The results were rendered in PyMol (20).
Recombination protein RAG: A domesticated
transposase
One illustrating example of the transposase
domestication is the emergence of RAG1 and RAG2, key enzymes for
generating the diverse repertoire of adaptive immune system
effectors. The gene encoding the variable region of immunoglobulins
and T cell receptors (TCR) is assembled in a combinatorial manner
from 3 segments, namely V (variable), D (diversity) and J
(joining), each originating from clusters or loci located at large
distances in the genome. This process is termed V(D)J recombination
and is catalyzed by RAG1 in complex with RAG2 and other accessory
proteins, which recognize and cleave at specific sequences termed
recombination signal sequence (RSS) bordering the V, D and J
segments. An RSS sequence has a 9 bp region (nonamer) rich in A/T
and a conserved 7 bp region (heptamer) separated by less conserved
12 or 23 bp (12 RSS and 23 RSS). The heptamer is followed by the
coding V, D or J segment and recombination occurs in an orderly and
tightly regulated manner, always between a segment flanked by a
12RSS and another flanked by a 23RSS (12/23 rule). The Ig heavy
chain and TCR β chain loci are the first to be recombined by
bringing together a D next to a J, followed by a V next to the
pre-assembled DJ. Subsequently, the variable region of the Ig light
chain and TCR α genes are assembled from the V and J segments. The
functional unit for concerted recombination is a heterotetramer,
containing two RAG1 subunits each in association with a RAG2. The
heterotetramer binds a pair of 12/23 RSS and forms the synaptic or
paired complex, facilitated by DNA bending performed by accessory
protein high mobility group protein B (HMGB)1 or 2.
Transesterification is performed by the RAG1 catalytic domain,
while RAG2 is involved in heptamer binding, chromatin targeting and
the stability of the reaction intermediate complexes. RAG1
introduces at both 12 and 23 RSS, a single strand cleavage between
the heptamer and the V, D or J coding sequence with the release of
a 3′OH on the coding flank and the free phosphate on the heptamer
(signal end). In the following step, the active hydroxyl becomes a
nucleophile and attacks the second strand of the coding strand,
forming a cyclic phosphate ester intermediate structure termed a
‘hairpin’. In terms of the DDE(D) transposases classic mechanism,
this is considered the ST step (Fig.
2). The RAG1 and 2 post-cleavage complex maintains the recessed
DNA ends in close proximity, while repair enzymes belonging to NHEJ
pathway open the hairpins and attach to each other the two coding
ends with the deletion or addition of nucleotides and also the two
signal ends, respectively, typically without modifications (signal
end joint) (12). In the
description below, we refer to mouse RAG residue numbers (14,15).
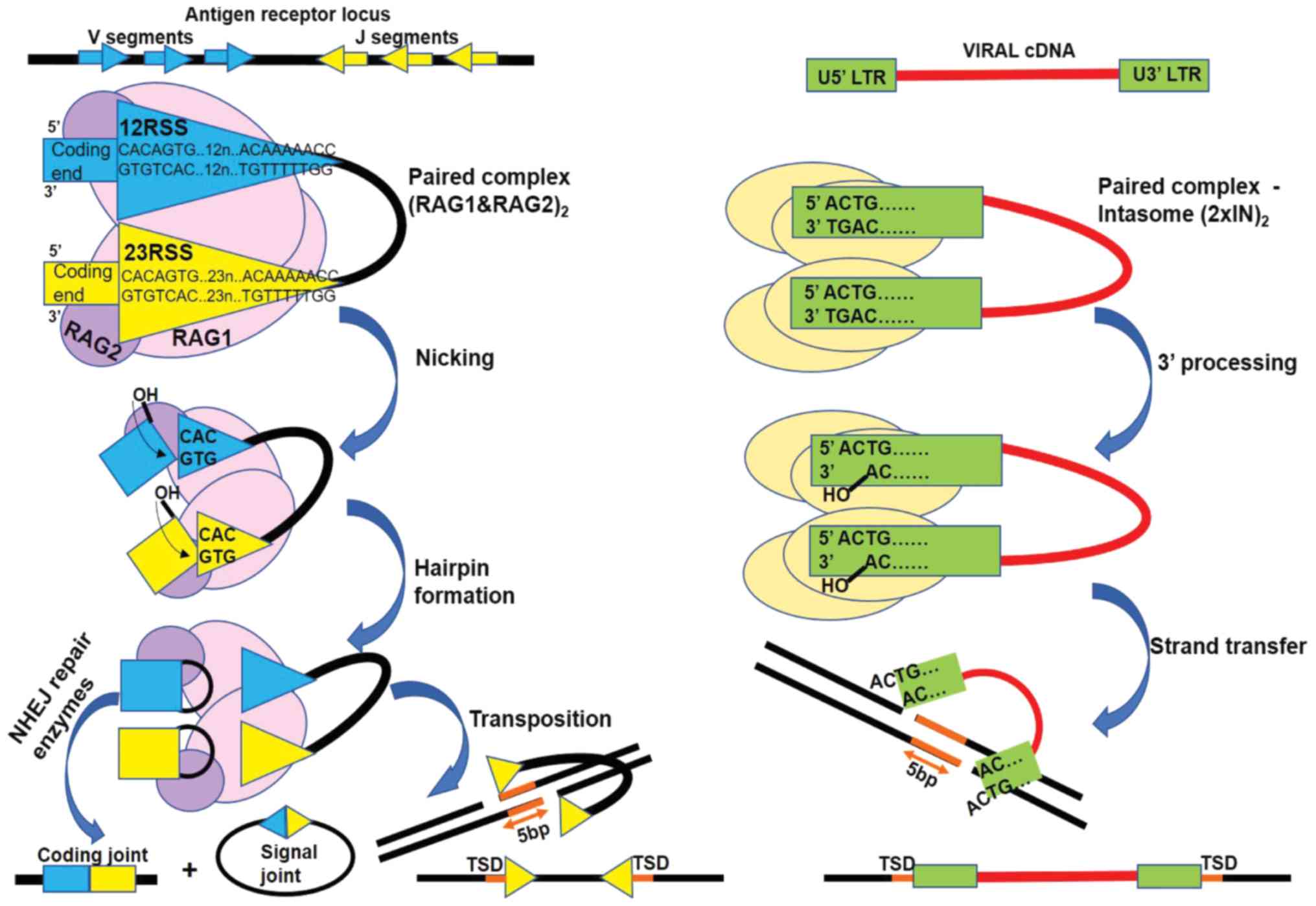 | Figure 2.RAG-mediated recombination (left) and
HIV-1 IN mediated retroviral integration (right) share a two-step
transesterification mechanism. The RAG complex recognizes specific
sequences named recognition signal sequences (RSS). RSS flank the
segments (coding segments) to be recombined in the antigen receptor
loci. IN recognizes sequences flanking the long terminal repeats of
the viral cDNA (U5′LTR and U3′LTR). Both enzymes are active in the
context of a paired complex with DNA: synaptic complex
heterotetrameric (RAG1 and 2)2 or intasome
homotetrameric (2×IN)2. In the first step RAG1 and 2
cleaves one DNA strand between the heptamers and the coding
segments ends, with the release of a reactive 3′ hydroxyl on the
coding end. Similarly, IN cleaves one DNA strand at the end of LTR,
near the conserved CA dinucleotide, with the removal of the last
two 3′ nucleotides of LTR ends and the release of a reactive 3′
hydroxyl (3′ processing). In the second step the viral cDNA
reactive ends attack the double stranded target DNA and the 3′
viral DNA ends are united with 3′ target strand. By contrast, RAG
generated reactive hydroxyl, attacks the second coding end strand
forming a structure called hairpin and detaching the RSS ends
(signal ends). Cellular repair enzymes open the hairpins and fuse
together the coding segments and in a similar fashion they repair
the gaps near the inserted viral DNA. In vivo RAG mediated
transposition events of signal ends are highly uncommon, but they
result in a 5-bp target site duplication (TSD) similarly to HIV-1
IN strand transfer product. The TSD arises from the 5 nucleotides
in the target DNA separating the insertion sites of LTRs and RSS,
respectively. NHEJ, non-homologous end joining; RAG,
recombination-activating gene protein. |
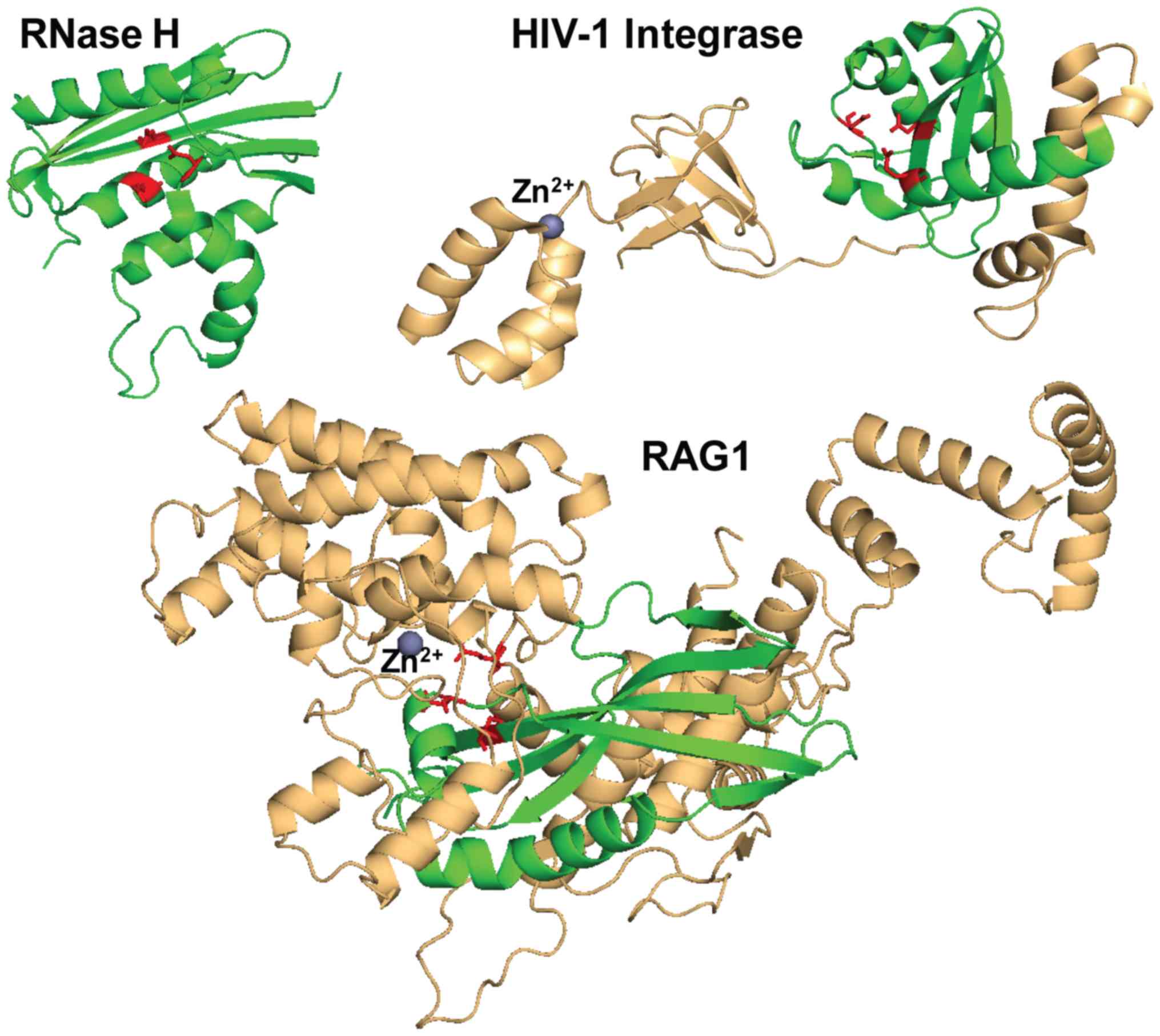
RAG1 is a 1,040 amino acid protein divided into
three main domains: The N-terminal domain (1–383), core domain (384–1008) and a short
C-terminal domain (1009–1040). RAG2 is a 527 amino acids protein,
essential for the proper function of RAG1, comprised of a core
region (1–387) and a C-terminal
domain (388–527). The most extensively studied regions of RAG
proteins are the core domains, defined as the minimum portion of
the proteins capable of performing V(D)J recombination. Their
structure and conformational changes have been recently illustrated
by X-ray and cryo-EM studies (16,17).
The N-terminal (NTD) and C-terminal (CTD) domains have regulatory
functions and stabilize the protein-DNA complex. RAG1 NTD contains
a RING finger domain (264–389), which has E3 ubiquitin-ligase
properties and ubiquitylates histone H3 (24). It also has three conserved cysteine
pairs that form a Zn2+ binding site (ZnA). RAG1
possesses a complex core region further subdivided into functional
subdomains. At the NTD, a series of three helices from each monomer
intertwine to form the nonamer binding domain, essential for
catalysis (NBD, 391–459) connected via a linker to the dimerization
and DNA binding domain (DDBD, 460–515). This is followed by pre
RNaseH (515–588) and the RNaseH domains (589–719). The highly
helical region separating the last Glu962 from the rest of the
catalytic triad contains a pair of cysteines (Cys727 and Cys730)
and a pair of histidines (His937 and His942), forming the second
Zn2+ binding site (ZnB). RAG2 folds into a 6-bladed
β-propeller structure. RAG2 establishes contacts with the RAG1
preR, RNaseH and ZnB domains, through a well conserved interface.
RAG2 CTD contains a plant homeodomain finger (PHD) thought to guide
the complex to accessible DNA areas of open chromatin by binding to
the lysine 4 of the trimethylated histone H3 (25).
RAG1 shares a number of similarities with DNA DDE(D)
transposases and retroviral INs in terms of reaction mechanism,
intermediates and functional motifs. Double strand cleavage via a
hairpin intermediate on the flanking DNA ends is also performed by
hAT transposases (Hermes). Following its recruitment, the rag1 gene
evolves under positive selection away from transposase origins,
losing the ability to perform transposition, but instead developing
as part of a strictly regulated recombination machinery which
minimizes random and deleterious cleavage within the genome. This
argument is further supported by recent research which identified
ProtoRAG in cephalochordate amphioxus, a transposon intermediate in
the evolution and molecular taming of RAG (26). During chordate development, the RAG
transposase ancestor undergoes critical changes that transform it
in jawed vertebrates into a recombinase, which favors the joining
of excised DNA rather than its insertion. It has been demonstrated
that RAG1 residues Arg848, Glu649 and RAG2 acidic hinge
(amino-acids 362–383) suppress transposition in vivo
(27).
RAG-mediated double strand breaks have been found to
be involved in generating translocations responsible for T cell and
B cell lymphomas, along with activation-induced deaminase (AID)
(28). In human T cells, deletions
can arise between two RAG-mediated DSB or translocations between
one RAG-mediated DSB and a DSB from another source. Despite its
high substrate specificity, RAG is known to also rarely bind to
RSS-mimicking sequences (cryptic RSS), all of which have in common
the first three 5′-CAC-3′ nucleotides of the heptamer. It has been
estimated that there are around 10 million such RAG cleavages in
the human genome, which may play an important role in lymphoid
tumor development (29).
Off-target cleavage at the CAC motif leads to genomic instability
(deletions, insertions and translocations) in acute lymphoblastic
leukemia pre-B cells, due to the continuous expression of RAG
(30). However, it is very
uncommon for RAG to perform transposition in vivo in normal
cells (31). RAG transposition,
identified as the reinsertion of the DNA piece flanked by signal
ends (Fig. 2), has been
demonstrated to occur in vitro. Reddy et al reported
a transposition frequency of one to every 50,000 V(D)J
recombination events, in a murine preB cell line (32).
RAG recombination versus retroviral
integration
The survival and proliferation of retroviruses
depends on the integration of their genetic material into the host
genome and the exploitation of cellular enzymatic equipment to
synthesize viral proteins. The genetic material of retroviruses is
represented by two molecules of plus sense RNA, which is released
into the cytoplasm following capsid fusion with the cellular
membrane, as part of a nucleoprotein complex along with
reverse-transcriptase and IN enzymes and other viral proteins.
Viral cDNA (vDNA) synthesis occurs through reverse transcription in
the cytoplasm, resulting in a pre-integration complex (PIC), which
will be further be transported into the nucleus for subsequent
insertion into the chromosomal DNA (33). IN is one of the main components of
PIC responsible for coordinated DNA integration, which make it an
attractive pharmacological target for antiretroviral therapy (ART).
Recent studies have demonstrated that IN also plays critical roles
in the viral life cycle, apart from integration (34,35).
It has been shown that certain mutations in the IN gene can disrupt
viral particle assembly and nuclear import. IN follows the
classical transesterification process of DDE(D) transposases with
the difference that it can only use linear DNA as a substrate
flanked by sequences called long terminal repeats (LTRs) and cannot
engage DNA already inserted in the genome. Concerted integration of
the two vDNA ends takes place in the context of a homotetrameric
complex (intasome), in which one IN dimer binds one LTR end (U5′LTR
and U3′LTR respectively). 3′Processing (3′P) occurs invariably at a
conserved 5′-CAGT-3′, using a water molecule as a nucleophile, with
the removal of GT end and the release of adenosine's reactive 3′OH.
For the ST PIC is transported to the nucleus, where 3′OH becomes
the nucleophile for the single strand hydrolysis of the target DNA
(tDNA). The two vDNA ends are inserted simultaneously in a
staggered fashion. Similar to RAG1-mediated transposition, a 5 bp
TSD emerges after the repair of the resulting branched
intermediate. Interestingly, in the case of RAG1, only the first
three nucleotides (CAC) of the heptamer sequence establish specific
protein-nucleotide interactions via hydrogen bonds. The CA- and
TG-rich sequences observed in the RSS consensus heptamer, U5′LTR
and U3′LTR promote backbone deformation, DNA unwinding and
facilitate cleavage which can explain their high degree of
conservation among polynucleotidyl transferases recognition
sequences (Fig. 2).
HIV-1 IN is a 288 amino acid protein, divided into
three main functional domains: Amino acids 1–49 (NTD), amino acids
50–212 catalytic core domain (CCD) and amino acids 213–288 (CTD).
For concerted integration, IN is organized as a dimer of dimers,
each dimer binding to one LTR end and tDNA. Only inner protomers
are involved in catalysis, while outer protomers are mostly
contacting vDNA. NTD contains three consecutive, antiparallel
α-helices (α1, α2 and α3), slightly tilted with respect to each
other, which harbor a Zn binding motif His12, His16, Cys40 and C43
at their tip. The inner protomer NTD contacts the CCD of the other
inner protomer, creating the tetramerization interface in a
configuration which wraps the two IN dimers around the DNA. CCD
adopts a SH3 like fold (Src homology 3 domain), which was initially
described in the tyrosine kinase of Rous sarcoma virus (36). This fold brings together 5
antiparallel β-sheets packed around each other in a barrel
configuration. CTD establishes contacts mainly with vDNA through
Glu246, Ala248, Lys266 and a series of arginines (Arg228, Arg231
and Arg263).
The hallmark RNase H-like fold of DDE(D)
polynucleotidyl transferases catalytic domain contains a series of
β-sheets, the first 3 antiparallel and consecutive, the others
parallel, separated and flanked by 4 α-helices positioned around
the β-sheets. This fold was first identified in 1990 by Yang et
al, in ribonuclease H from E. coli,
β1-β2-β3-α1-β4-α2-α3-β5-α4 (37).
IN closely follows this fold in both distribution and orientation
of the α and β structures (β1-β2-β3-α4-β4-α5-α6-β5-α7), while in
the case of RAG1, there are some differences: There are only two
α-helices and four initial consecutive β-sheets, 3 of which are
antiparallel, while the following two β-sheets are not separated by
α-helices and are instead parallel (β3-β4-β5-β6-α8-β7-β8-α9)
(Fig. 3). The DDE(D) motif is the
common denominator for this superfamily, imposing the catalytic
mechanism of transesterification; however, the spacing of the
acidic residues within the RNase H-like fold and overall catalytic
core varies according to the protein domain organization and
intasome functional assembly. Various intasome structures have
evolved different ways to accommodate the substrate DNA with the
same purpose of precisely aligning the scissile phosphate in the
Mg2+ coordination sphere. Thus, HIV-1 IN Asp64, Asp116
and Glu152 are spaced in a similar manner to other transposases.
However, the last catalytic residue of the triad Asp600, Asp708 and
Glu962 of mouse RAG1 is separated by a sequence of α-helices from
the main RNase H-like fold. The same particularity can be observed
in Hermes transposase, where the last Glu is separated by an
α-helical insertion domain of almost 300 amino acids from the rest
of the catalytic residues.
IN inhibitors
The search for more efficient antiretroviral drugs
is focusing lately on molecules able to prevent viral infection and
spread such as fusion inhibitors (38) and INs. IN has emerged as an
important antiretroviral therapeutic target due to its key role in
the early steps of infection, immediately after the virus enters
the cell. IN inhibitors prevent the insertion of the genetic
material into the infected cell genome and thus limit the number of
latent reservoirs and the viral spread. α,γ-diketo acid derivatives
were discovered independently by Shinoghi and Merck in 1999
(39). These compounds provide a
successful strategy for inhibition via chelation, metal-dependent
viral endonucleases, such as HIV IN, RT RNase H and hepatitis C
virus polymerase. The first selective IN ST inhibitors (INSTI)
developed were two α,γ-diketo acid derivatives, L-731,988 and
L-708,906, that proved to be active against virus spread in cell
culture assays (40). A large
series of derivatives based on the acetylpyruvic acid scaffold were
developed as less toxic, highly active IN inhibitors, and with
improved biopharmaceutical properties. These compounds are
generally known as diketo acids, even if chemically most of them
have a keto-enol structure or even lack this chemical moiety
(Fig. 4A).
The majority of INSTIs are characterized by a
chelating moiety, usually containing three coplanar O or N atoms.
They also have an aromatic hydrophobic group thought to displace
the activated adenine after 3′P, rendering it less accessible for
ST (Fig. 4A). The binding of
diketo acid INSTIs occurs more significantly in the assembled
intasome, after the 3′P step, due to their π stacking contacts with
the bases adjacent to the activated adenine. Thus, they are more
potent inhibitors of ST than of 3′P step.
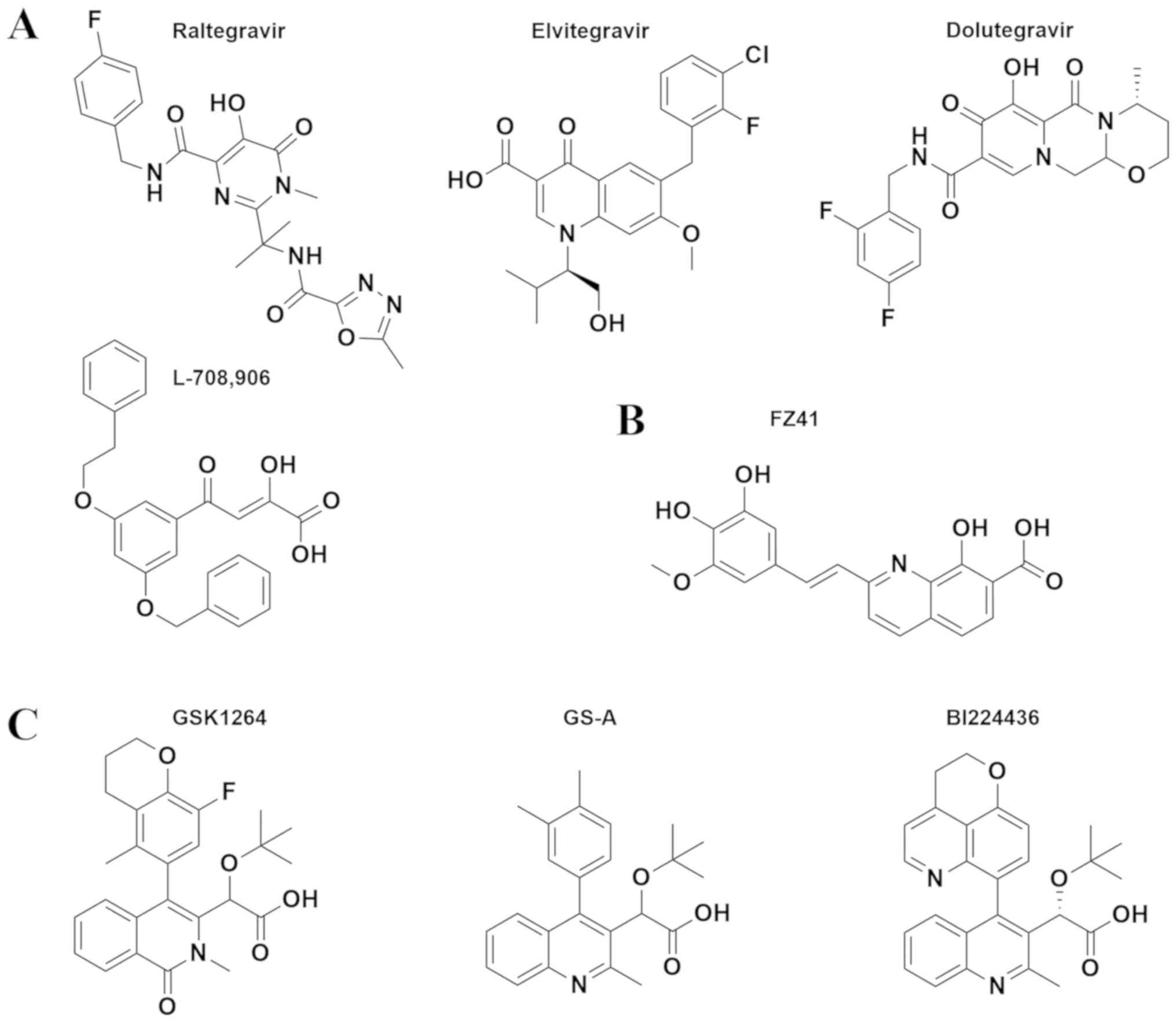
Raltegravir (RAL) (41) was developed based on the diketo
acids, although chemically it has a distinct structure being a
N-methyl-4-hydroxypyrimidinone-carboxamide derivative. It was the
first FDA-approved INSTI for treatment-experienced adult patients
and treatment-naive patients (42). Despite its effectiveness, RAL has a
low genetic barrier as a large number of resistance mutations, such
as E92QV/N155H, T97A/Y143CHR, and G140CS/Q148HKR have been
described (43). The structural
simplification of the diketo acid scaffold led to the development
of 4-quinolone-3-carboxylic acids which selected elvitegravir
(EVG), a potent, once-daily dosing INSTI (44). EVG was first approved in 2012 by
the FDI as part of a fixed dose combination and in 2014 as
independent formulation for the treatment of HIV-1 infection in
treatment-experienced adults in combination with other
antiretrovirals (42). EVG is
metabolized by CYP3A and thus it is usually associated with CYP3A
inhibitors, such as ritonavir or cobicistat (45). EVG shares a similar spectrum of
resistance mutation as RAL with some exceptions: Y143/CR,
T97A/Y143CR respond to EVG, but are resistant to RAL, while
E138K/Q148H and T66I/R263K are susceptible to RAL, but moderately
to highly resistant to EVG (43).
Second generation INSTIs were developed in order to have a high
barrier genetic resistance and reduced cross-resistance (46). The first representative of this
category was dolutegravir (DTG), approved by the FDA in 2013, in
ARV regimens of both treatment-experienced and treatment-naïve
patients (42). DTG acts in a
similar manner to first generation INSTIs via three coplanar oxygen
atoms which coordinate the active Mg2+ (47) and a difluorobenzyl group which
dislocates the 3′activated nucleotide. DTG displays higher potency
and is active on most previously reported resistance mutants due to
intimate interactions within the catalytic core binding pocket
involving a cytidine base next to the activated nucleotide and
E152, Q146 residues (40,41). This is evidenced by a 8-fold longer
dissociation rate from the IN-DNA complex compared to RAL (50). The most relevant resistance
pathways are E138 and Q148 substitutions, where E138K/Q148K in
particular is associated with low DTG susceptibility (43). Several other dolutegravir analogues
are in current development: S/GSK-1265744 (cabotegravir),
formulated as long-acting injectable drug (51) is now under clinical trials
(ClinicalTrials.gov Identifier:
NCT00920426) and GS-9883 (bictegravir) (52) with similar antiviral potency and
improved activity on INSTI-associated IN variants is undergoing
phase III clinical trials (ClinicalTrials.gov Identifier: NCT02607930).
Initially designed as polyhydroxylated metal
chelators, styrylquinoline derivatives (SQLs) were identified as
potent in vitro IN inhibitors of both 3′P and ST steps and
reduced HIV-1 replication in cell culture (53). Unlike INSTIs which have a much more
potent and specific inhibition on ST compared to 3′P and only bind
to IN in complex with vDNA, the SQL in vitro IC50 values for
3′P and ST are usually in the same range or lower for 3′P and they
are inactive if added before the assembly of IN-LTR complex
(54). SQLs, such as FZ41, KHD161
or FZ55 are competitors of the 3′P reaction and for FZ41 it has
been demonstrated that it interferes with DNA binding (Fig. 4B) (55). The SQL binding pocket has not been
confirmed by crystallographic data; however, resistance mutations
and docking studies have indicated they interact within the CCD
with residues involved in vDNA binding. V164I (CCD), V249I and
C280Y (CTD) have been reported as resistance mutations to FZ41
(56). Docking studies of KHD161
and FZ55 (56) pointed to 3
possible locations for the compounds, two of which were situated
around V165 where the compounds establish contacts with critical
residues contacting vDNA (57)
(K156, K160, S119, N120, N117) and also located at dimer interface
(K186, K188) (57). A later
docking study on FZ41 also confirmed interaction with CCD residues
situated near protein-protein interface (58).
In parallel with developing inhibitors of the
catalytic site, the dimer interface between the two CCDs has been
acknowledged as a valid target for small molecule inhibitors.
Tetra-acetylated chicoric acid (59) and 1-pyrrolidineacetamide (60) are some of the first compounds
discovered to engage residues involved in protein-protein
interaction (K103, K173 and T174) and prevent vDNA binding.
Moreover, at the IN dimer interface lays the binding site for lens
epithelium-derived growth factor (LEDGF), one of the most relevant
cellular binding partners of IN. LEDGF, also known as human
transcriptional activator p75 is a 530-amino acid nuclear protein
associated with transcriptional activation (61). Interaction with LEDGF is a specific
property of lentiviruses and it has been demonstrated that it plays
a role in the chromatin tropism of PIC (62), viral replication and protects IN
from proteasome degradation (63).
LEDGF also plays an important role in IN oligomerization. IN
binding to LEDGF before vDNA results in inactive tetramers via
allosteric alterations, which is supported by studies in which the
overexpression of LEDGF has been shown to inhibit viral infectivity
(64). LEDGF contains a small
domain of interaction with IN (amino acids 347–429), made out of a
bundle of 5 helices which contacts a pocket at the IN CCD
dimerization interface, in the vicinity of RNaseH domain,
establishing contacts with Q168, T174, R166, E170, H171 of inner
monomer and W131, W132, A128, T125, L102, Q95 of the outer monomer
(65).
This inspired researchers into finding an
alternative inhibitory mechanism to INSTIs, based on the LEDGF
binding pocket, leading to the development of allosteric IN
inhibitors (ALLINIs). Phenoxymethyl-benzoic acid derivative D77 was
among the first for which this mechanism of inhibition was
demonstrated (66). The most
successful class to date is quinoline-based acetic acid
derivatives. Structure-based virtual screening approach targeted at
LEDGF-IN binding pocket has yielded 2-(quinolin-3-yl) acetic acid
derivatives that block the IN-LEDGF interaction (LEDGINs) (67).
Tert-butoxy-(4-aryl-quinolin-3-yl)-acetic acid derivative (tBQA)
and chemically related compounds, such as BI224436 (68), GSK1264, GSK002 (34) and GS-A (69) derivatives, are among the most
successful ALLINIs (Fig. 4C).
Although thought in the beginning to act differently, LEDGINs and
tBQAs share the same complex mechanism beyond disrupting IN-LEDGF
interaction. Both inhibit the integration of competent IN-DNA
complex assembly, promote IN aggregation by the formation of high
order oligomers and at a later stage, inhibit viral particle
assembly and maturation (34).
Crystal structures of the compounds at their binding site, as well
as resistant IN mutants have revealed critical residues for their
activity, such as A128, E170, H171, W131, T174 and T125 which
facilitate stronger interactions between the two dimers and also
between dimer units, leading to IN aggregation (61,62).
Notably, ALLINI resistance mutations at residues 226, 235, 264 and
266 have highlighted the role CTD plays in protein assembly in the
absence of vDNA (34).
HIV-1 IN inhibitors potentially interfere
with RAG-mediated recombination
Before RAL was introduced as part of highly active
ART, the prospective that IN inhibitors could also interfere with
RAG activity, due to its similarities to IN, was considered. In
2002 Melek et al published an in vitro study on the
effects of 5-CITEP and one of the most active β diketo acids to
date, L-708,906 (70). 5-CITEP was
demonstrated to be a less efficient RAG inhibitor than L-708,906 by
10-fold in all assays. The former has exhibited an IC50 value of
2.1 µM in a ST assay, while the later has exhibited an IC50 value
of 0.1 µM (71) and a 2.5 µM IC50
value in a HIV-1 infectivity assay (40). L-708,906 inhibits several steps of
RAG catalysis in vitro: Nicking, subsequent hairpin
formation and the disintegration reaction, with an IC50 value of 20
µM, when the compound is incubated with IN prior to the addition of
the divalent cation. The authors concluded that L-708,906
interferes with RAG binding to its RSS substrate when added prior
to the metal ion cofactor and alters the complex if added after,
although in this case, the enzyme remains partially competent for
cleavage in the presence of Mg2+. Hairpin formation has
been found to be more sensitive to the compound presence than
nicking. However, in contrast with the IN inhibitory mechanism, the
compound does not interfere with the transposition, the equivalent
of ST step, nor with non-specific target DNA binding (70).
A later study indicated a similar inhibition pattern
on enzymatic 12RSS binding, nicking and hairpin formation for EVG,
which displayed a Kd=32.53+2.9 µM corresponding to 1:1 binding to
RAG1 central domain (amino acids 528–760) as determined by biolayer
interferometry assay. EVG induces the impairment of RAG function as
also confirmed in the context of an episomal assay on Nalm6 cell
culture (B cell precursor leukemia cell line) transfected with
plasmids bearing an artificial 12/23RSS substrate and an antibiotic
resistance reporter gene. In cells exposed to 1 µM EVG signal joint
formation was reduced by 5.9-fold and coding joint formation by
8.2-fold, respectively. Moreover, the percentage of the
CD45+CD25+ B cell population displayed a
significant decrease in Balb/c mice treated with EVG and cobicistat
(30 mg/kg body weight), but only in 11 out of 16 mice, while the
other 5 remained unaffected. This decrease could be linked to RAG
activity, although the results warrant further investigation as
other mechanisms may also be involved. The same authors reported
only limited enzymatic inhibitory activity for RAL, which was not
pursued further in cell culture and in vivo assays. However,
they reported RAL binding to IN central domain (amino acids
528–760) using the same circular dichroism assay as for EVG
(14).
The safety and efficacy of INSTIs currently
available for ART is monitored by clinical studies and case
reports. This class of antiretrovirals has been considered well
tolerated and highly effective in reducing viral loads in adults
(72). However, a study conducted
between 2006–2008 involving 78 patients following new ART revealed
an increase in the incidence of non-Hodgkin lymphoma (NHL) by
20-fold and a rapid onset of NHL following treatment initiation; he
virologic response was greater than in patients without NHL which
could facilitate abnormal proliferation pathways in lymphocytes
(73). The development of NHL
could be associated with severe immunosuppression or complications
from immune reconstitution inflammatory syndrome in
treatment-experienced patients receiving new ART. Nucleos(t)ide
reverse transcriptase inhibitors and RAL were the only common
antiretrovirals taken by all patients with NHL included in the
study. Previous studies have also indicated a high risk of
malignancy associated with RAL, NHL being the most common (74). However, NHL could also be
associated in these cases with the presence of EBV, which is
responsible for dysfunctional T cell activity, or due to other
HIV-1 induced oncogenic mechanisms (75). Huhn et al suggested that an
enhanced cytokine response and/or impaired T cell function in
conjunction with alterations in the RAG1 and 2 recombination
induced by INSTIs could account for the proliferation of abnormal
lymphocytes (73).
Possible immunological adverse reactions associated
with V(D)J recombination interference and associated with INSTI
therapy should also be investigated in the context of developing
immune response in newborns and children. IN inhibitors are
approved for children beginning from 4 weeks old (RAL) or 6 years
old (EVG/cobicistat/emtricitabine/tenofovir and DTG). Through their
mechanism of preventing viral DNA integration, INSTIs could
potentially prevent infections of newborns from mothers infected
with HIV-1. The significant benefit, safety and pharmacokinetics of
RAL in this case are being evaluated during a phase I clinical
trial initiated in 2013 and expected to be completed in 2020
(ClinicalTrials.gov Identifier:
NCT01780831). The GS-US-292-0106 ongoing study (ClinicalTrials.gov Identifier: NCT01854775) is
currently evaluating the pharmacokinetics, antiviral activity and
safety of the EVG/Cobicistat/Emtricitabine/Tenofovir alafenamide
fumarate (GENVOYA®) in pediatric patients <18 years
of age. Based on preliminary results, the FDA reported a decline in
CD4+ counts in 23 children between 6 and 12 years of
age, beginning with the second week of treatment and persisting up
to 24 weeks. After 48 weeks the CD+ decline is less than
the one observed initially. The etiology remains unclear and
further data are required for a conclusive evaluation (76).
Docking simulations of IN inhibitors on RAG1
dimer
Computational approaches are useful to estimate and
compare the affinities of different compounds towards an enzyme and
also to predict a potential binding pocket and an associated
mechanism. Docking analysis can be applied to verify the IN
inhibitor predilection for binding within RNase H-like fold
catalytic domains and associate it with potential off-target
effects observed in vivo.
To exemplify this argument, we selected four diketo
acid INSTIs (RAL, EVG, DTG and L-708,906), one styrylquinoline
(FZ41) and three allosteric inhibitors (GSK1264, GS-A and BI224436)
and performed docking simulations on RAG1 dimer (amino acids
391–1006) in apo form, from the previously reported crystal
structure of RAG1 and 2 tetrameric complex at resolution 3.2 Å (PDB
accession code 4WWX) (18). For
each compound 5 rounds of simulation were performed, corresponding
to 100 possible binding configurations. We focused on the
configurations located in RNase H-like fold, in the vicinity of the
DDE catalytic motif, within maximum 5 Å of any of the catalytic
residues and the following analysis will refer exclusively to this
binding pocket. Docking configurations within this pocket also
displayed the highest calculated affinity corresponding to the
lowest free energy (kcal/mol) of all 100 configurations, in the
case of INSTIs and FZ41, but not in the case of ALLINIs. The pocket
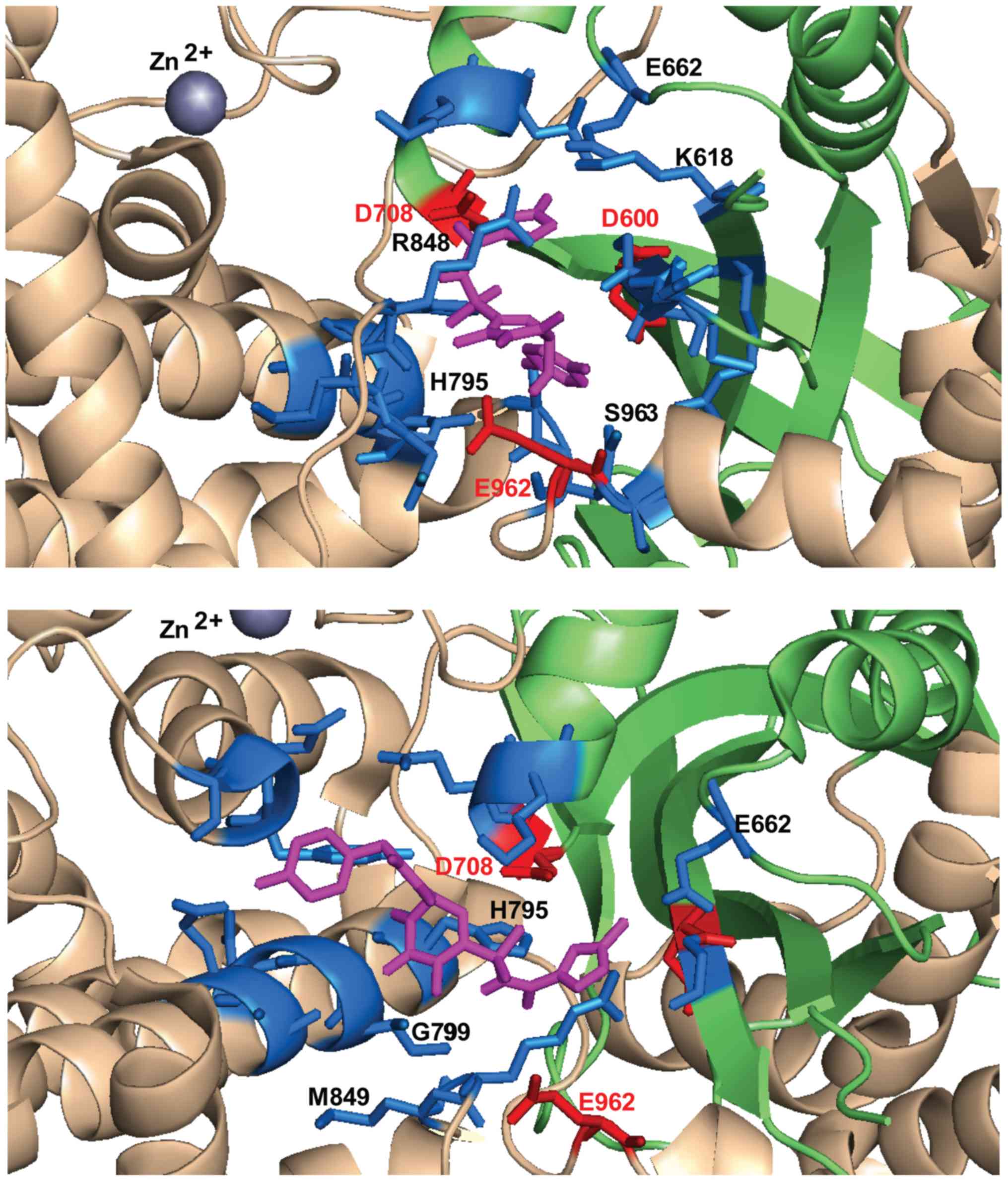
involves residues located on the first β-sheet of the RNase H fold,
near D600 (between 599 and 604), on the last helix of the the RNase
H fold, near D708 (708–711), on the helical region between RNase H
domain and the last catalytic residue E962 (795–806, 848–852,
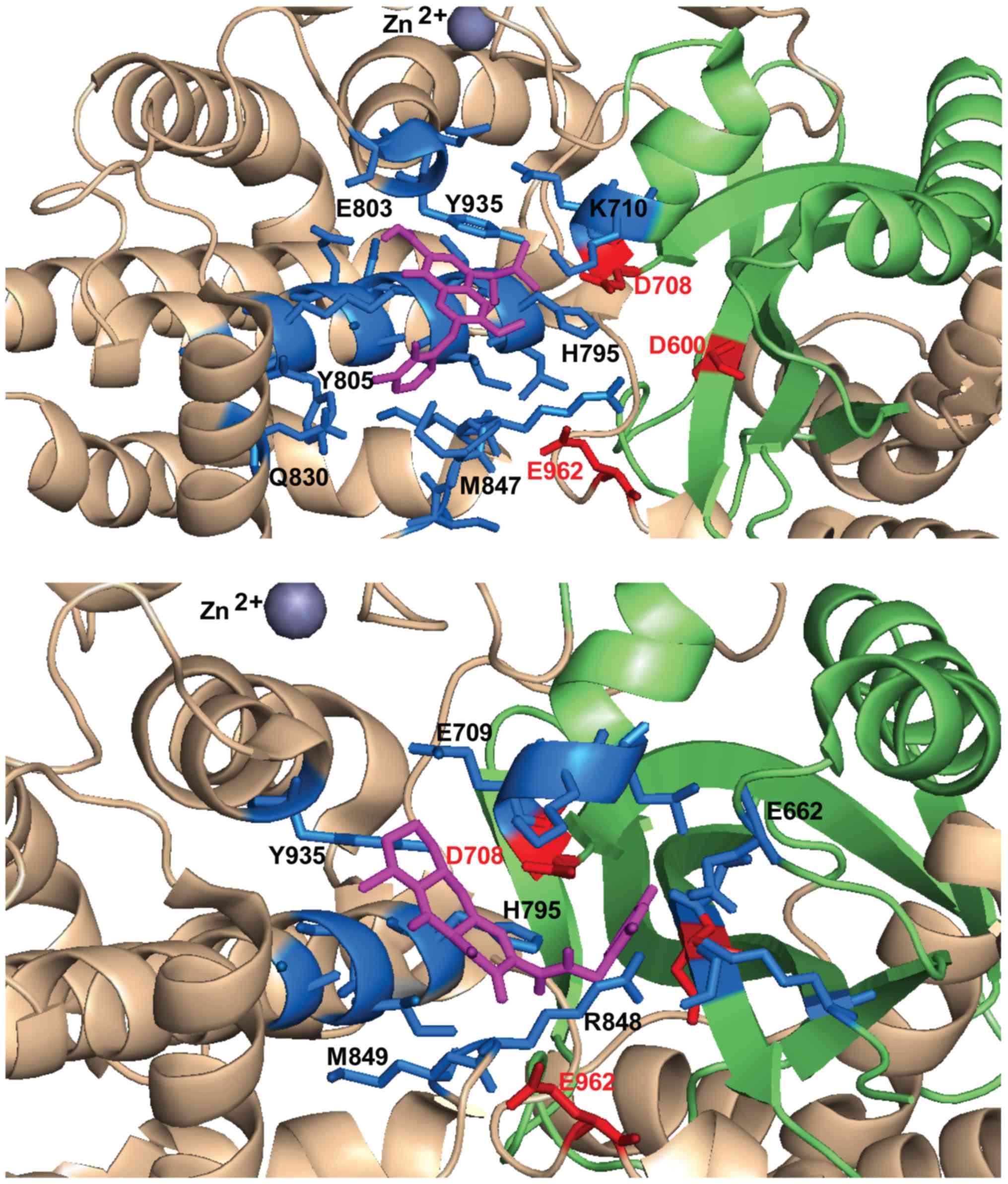
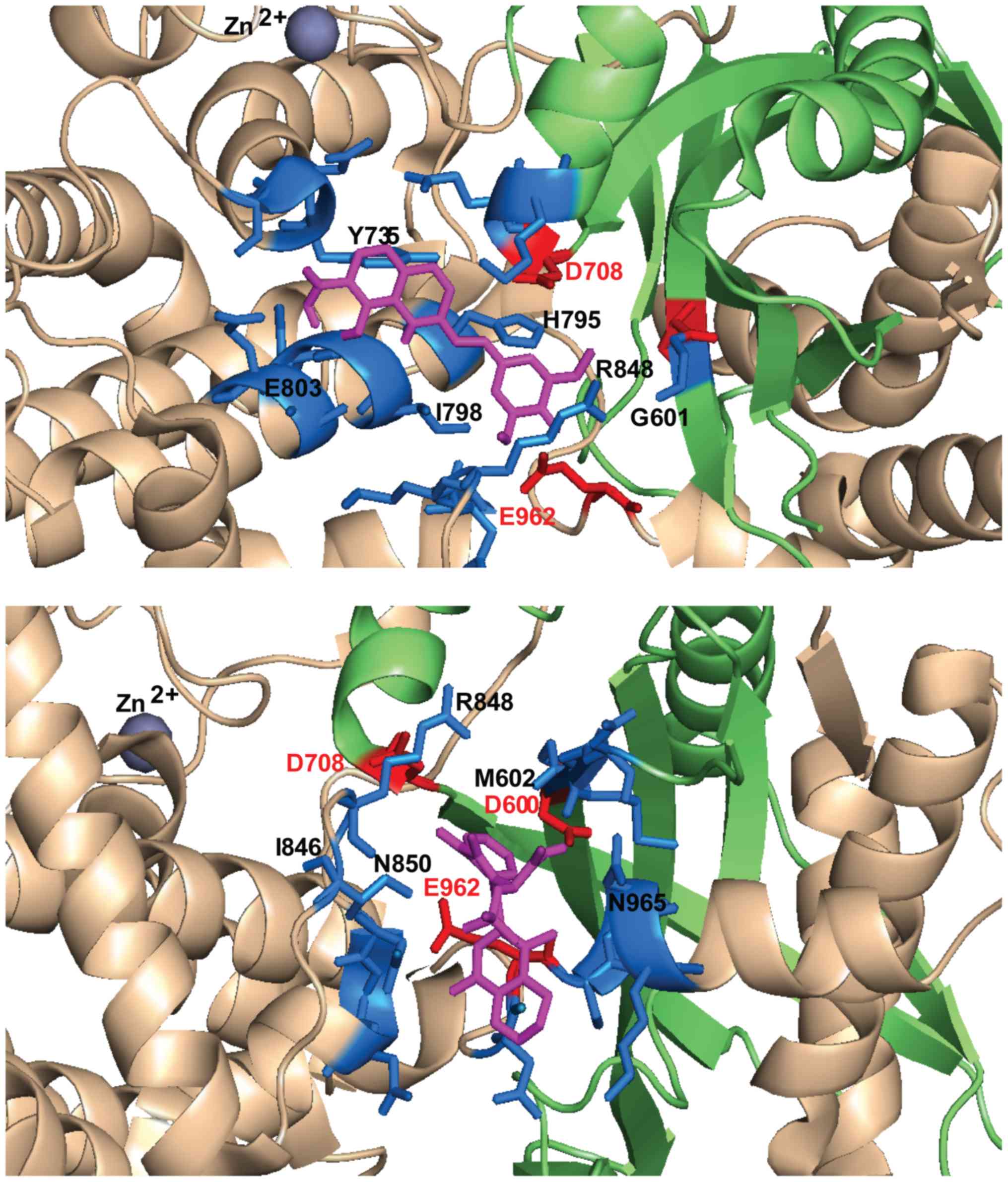
933–935) and on the loop around E962 (961–969) (Table I).
 | Table I.Residues which define the binding
pocket from docking simulations within RAG1 catalytic core
domain. |
Table I.
Residues which define the binding
pocket from docking simulations within RAG1 catalytic core
domain.
| Compound | Catalytic core
pocket residues within 5 Å of compound | Contacting residues
(H-bonding, stacking, electrostatic or hydrophobic
interactions) |
|---|
| Raltegravir | i) D600,
G601, M602, G603, D604, K618, R621, E662, D708, K710, L711,
L794, H795, I798, R848, M849, N858, S958, E959, E962,
S963 | G601, K618, E662,
D708, H795, I798, R848, S958, E962 |
|
| ii) D600,
G601, E662, D708, E709, K710, H795, I798, G799, M800, A802,
E803, R848, M849, T933, N934, Y935, E962 | G601, D708,
H795, G799, M849, T933, E962 |
| Elvitegravir | D708, E709,
K710, L794, H795, L796, I798, G799, N800, A802, E803, Y805, K806,
Q809, Q830, I846, M847, R848, M849, T933, N934, Y935 | D708, K710,
H795, E803, Y805, K806, N809, Q830, M847, Y935 |
| Dolutegravir | C599, D600,
G601, K618, E662, D708, E709, K710, L711, H795, I798, G799,
A802, R848, M849, Y935, E962 | G601, E662,
D708, E709, H795, R848, Y935 |
| L-708,906 | D708, E709,
H795, I798, G799, N800, A802, E803, Y805, K806, I846, M847, R848,
M849, T933, N934, Y935, E962 | D708, H795,
Y805, M847, R848, Y935 |
| FZ41 | G601, D708,
E709, K710, H795, I798, G799, N800, A802, E803, R848, M849, N850,
T933, N934, Y935, E962 | G601, D708,
H795, I798, G799, E803, R848, Y935, E962 |
| GSK1264 | D600, G601,
M602, G603, D604, I846, R848, N850, G851, N852, N961, E962,
S963, G964, N965, K966 | M602, D604, R848,
N850, N861, E962, N965, K966 |
| GS-A | D600, G601,
M602, G603, D604, N850, N852, N961, E962, S963, G964, N965,
K966, R969 | N961, E962,
S963, N965 |
| BI224436 | D600, G601,
M602, G603, D604, I846, M847, R848, M849, N850, G851, N852, N961,
E962, N965, R969 | D600, G601,
M602, M847, R848, N850, E962, N965 |
RAL emerged as the compound with the highest
frequency of docking in the described area (83%), followed by DTG
(57%), L-708,906 (43%), EVG and FZ41 (41%) (Table II). The highest calculated
affinity belonged to docking configurations in this pocket for DTG
and RAL (−8.2 kcal/mol and −8.1 kcal/mol, respectively), followed
by FZ41 (−7.8 kcal/mol), EVG (−7.5 kcal/mol) and L-708,906 (−7.2
kcal/mol). ALLINIs had the lowest frequency of docking in the
selected pocket, between 26 and 31% and with a corresponding low
affinity, between −7.1 kcal/mol and −6.6 kcal/mol (Table II). This is not a surprising
result taking into consideration that diketo acid derivatives and
styrylquinolines were designed to bind in the vicinity of the DDE
catalytic domain of IN, while ALLINIs have an affinity for the
protein-protein interface. RAL docking configurations were
categorized into two clusters (Tables
I and II). Cluster no. 1
contains configurations which protrude more deeply in the space
between the DDE residues (Fig. 5).
Representative conformations for ALLINIs are oriented differently
compared to diketo acids (Figs. 5
and 6) and FZ41, closer to the
D600 and E962, particularly oriented towards the loop containing
E962 and the helix that follows it (Fig. 7). Diketo acid derivatives and FZ41
all establish possible contacts with D708, while ALLINIs
configurations allow contacts preferentially with E962 (Table I). In the selected docking
conformations, D708 forms hydrogen bonds with RAL (3.2 Å), EVG (5
Å), DTG (4.3 Å), L-708,906 (3.1 Å), FZ41 (4.5 Å). E962 forms
hydrogen bonds or electrostatic interactions with RAL (3.6 Å), FZ41
(3.9 Å), GSK1264 (3.3 Å), GS-A (2.7 Å) and BI224436 (4.1 Å). A
possible hydrogen bond with D600 was observed only for BI224436
conformation (4.9 Å).
| Table II.Maximum apparent affinities and
frequency of docking conformations within 5 Å of the RAG1 DDE
motif. |
Table II.
Maximum apparent affinities and
frequency of docking conformations within 5 Å of the RAG1 DDE
motif.
| Compound | Maximum apparent
affinity (kcal/mol) for the selected conformations | % of total
conformations |
|---|
| Raltegravir | (1) −8.1 | 29 |
|
| (2) −7.9 | 54 |
| Elvitegravir | −7.5 | 41 |
| Dolutegravir | −8.2 | 57 |
| L-708,906 | −7.2 | 43 |
| FZ41 | −7.8 | 41 |
| GSK1264 | −7.1 | 31 |
| GS-A | −6.6 | 26 |
| BI224436 | −7.0 | 31 |
The compounds dock in an area critical for RSS
binding. K608, H609, G610, S611, G851, N852, R855, L794, S963 and
E959 are important residues for positioning the heptamer (21). N934 and T933 are part of the loop
that binds the first 5 nucleotides of the coding DNA (21). Notably, R848 is a common contact
for most of the compounds interacting through possible hydrogen
bonds with RAL (4.3 Å), DTG (4.2 Å), FZ41 (3.4 Å), L-708,906 (4.6
Å), GSK1264 (4 Å), BI224436 (3.6 Å). R848 is a key residue on the
848–855 loop of Zn finger domain and it is important for heptamer
CAC recognition. Upon hairpin formation, R848 modifies its position
and helps in substrate orientation by forming π-cation interactions
with the activated nucleotide and electrostatic interactions with
the scissile phosphate (21). This
residue is also involved in suppressing RAG-mediated transposition
(27). H795 establishes possible
hydrogen bonds or stacking contacts with RAL (3.6 Å), EVG (2.5 Å),
DTG (3.9 Å), L-708,906 (3.1 Å) and FZ41 (3.6 Å), but not with
ALLINIs. Mutations of this residue have been previously shown to
alter RAG catalytic activity in both 3′nicking and hairpin
formation steps, but not DNA binding (77).
HIV-1 IN inhibitors interfere with various
polynucleotidyl transferases
During the development of IN inhibitors,
transposases were considered as a surrogate model for HIV-1 based
on the structural similarity of the catalytic RNase H-like fold
domain. Among the first well characterized transposase-DNA
complexes was Tn5 (78), which
made it a suitable model enzyme for the identification of potential
anti-HIV-1 IN candidates. This strategy was employed for the
screening of 16,000 compounds and the subsequent identification of
20 Tn5 inhibitors, of which 6 also inhibited HIV-1 IN 3′P, namely
coumarin dimers, cinnamoyl derivatives and a chlorinated bithionol
sulfoxide, with IC50 values between 9 and 32 µM. Most compounds
inhibited Tn5 more potently than HIV-1 IN (79). Furthermore, six diketo acid
derivatives are able to interfere with paired complex formation and
with both donor DNA cleavage and ST steps of the reaction. It has
been suggested that the compounds bind at or near the active site,
independently of Mg2+ in the case of three of them
(80). Of note, L-708,906 does not
interfere with Tn10 transposase activity, which indicates that
despite of the similar RNaseH fold like catalytic core, not all
transposases are susceptible to inhibition by diketo acid compounds
(70).
RAL is able to inhibit other retroviral INs and
transposases (81). It has been
demonstrated that RAL is able to play a part in triggering and
exacerbating autoimmune disease in mice, by interacting with
endogenous retroelement INs, thus leading to accumulation of
pre-integration cDNA (82).
Certain autoimmune diseases, such as systemic lupus erythematosus
have been associated with the accumulation of cDNA in the cytoplasm
and the activation of the type I interferon response (83). The co-crystal structure of RAL in
complex with mariner transposase Mos1 (PDB accession code 4MDB)
revealed the compound's versatility in interacting with the
catalytic core pocket of the apo enzyme, by adopting a very
distinct, more compact conformation, than the extended one seen in
the PFV intasome (PDB accession code 3OYA). Diffraction data have
revealed how RAL's three coplanar oxygen atoms compete with the
enzyme's DDD motif (D156, D249 and D284) for the binding of the two
divalent metal cations. RAL also inhibited Mos1 enzymatic activity
in vitro, with an estimated IC50 value between 60 and 70 µM
on the first cleavage step and a significantly higher potency on ST
(IC50 ~2 µM). This suggests that RAL binds more efficiently to the
transpososome and can adopt yet a different and more efficient
spatial configuration than in the absence of DNA. By contrast, EVG
does not bind to Mos1 and it does not interfere with its activity
(84).
Mos1 shares 48.4% sequence similarity to the SETMAR
transposase catalytic domain, also a member of the Tc1/mariner
family. SETMAR, also known as metnase, is another example of
transposase domestication, a protein present only in anthropoid
lineage, resulting from the fusion of a SET histone methylase
domain and a Hsmar1 mobile element domain. Metnase performs a
variety of functions linked to DNA repair mechanisms through the
NHEJ complex and exogenous DNA integration (3). The SET domain methylates histones in
the vicinity of DSBs, thus stabilizing components of NHEJ
machinery, enhances DNA repair and suppresses chromosomal
translocations. The transposase domain is able to bind TIRs,
performs 5′nicking and has a modified DDN catalytic motif (D483,
D575 and N610), which does not allow double-strand cleavage and
mediates transposition with very low frequency. Metnase enhances
topoisomerase IIα activity and promotes the restart of stalled
replication forks. Metnase overexpression has been associated with
an increase in HIV-1 cDNA integration [reviewed in (85)]. Due to its genomic stabilizing and
repair properties, metnase overexpression is linked to resistance
to chemotherapy in cancer cells (metnase mediates resistance to
topoisomerase II inhibitors in breast cancer cells; metnase
mediates chromosome decatenation in acute leukemia cells). Both RAL
and EVG have been identified as active enzymatic inhibitors of
metnase in vitro 5′ cleavage at 2 µM (86).
Human cytomegalovirus [(HMCV), belonging to the
herpesvirus family] terminase is a 2-subunit protein with a
N-terminal U56 ATPase domain and C-terminal UL89 nuclease domain
and is responsible for cutting long genomic DNA head-to-tail
concatemers into individual units of genomic DNA to be singly
packed into viral capsids. The structure of the herpesvirus
packaging terminase UL89 nuclease C-terminal subunit (UL89C)
revealed a RNase H-like fold responsible for DNA cleavage, with a
DED motif (D463, E534 and D651), which coordinates two metal
divalent cations. In the presence of Mn2+ UL89C leads to
full DNA degradation, while in the presence of Mg2+ the
single strand nicking step is predominant. Of note, RAL inhibits
UL89C double-strand cleavage activity in vitro more
efficiently than nicking activity, beginning from 1 µM. By
contrast, EVG does not show any interference with UL89C under
similar conditions (16). HMCV
infection can be life-threatening for immunocompromised patients
and can cause serious birth defects. The versatility of INSTI metal
ion chelation mechanism is a source of inspiration and opens the
way for the design of novel DNA packaging inhibitors.
Human T-lymphotropic virus 1 (HTLV-1) belongs to the
same Orthoretrovirinae subfamily as HIV-1. The HTLV-1 infection of
CD4+ lymphocytes (to a lesser degree CD8+) is associated
with clonal expansion and adult T-cell leukemia/lymphoma, a very
aggressive and often treatment refractory form of cancer or
tropical spastic paraparesis/HTLV-1-associated myelopathy
(TSP/HAM). Both have a very poor disease prognosis and efficient
targeted therapy still remains a challenge (87). Given the similarities between the
two viruses, it was hypothesized that HIV-1 IN inhibitors can also
function as HTLV-1 IN inhibitors. Indeed, representatives from both
diketo acid INSTI and SQLs class have been demonstrated to
interfere in vitro with ST reaction catalyzed by HTLV-1 IN.
While L-731,988 and L-839,616 diketo acid derivatives maintain a
similar potency as observed on HIV-1 IN (0.051 and 0.069 µM), four
SQLs (KH161, KH211, FZ41, FZ149) displayed IC50s in micromolar
range (minimum 4.9 µM and maximum 7.4 µM), almost three times less
potent than on HIV-1 IN. The results were confirmed ex vivo
only for L-731,988 by assessing the number of integration events
which occur in infected cells in culture (88). RAL and the diketo acid derivative,
MK-2048, were later investigated in ex vivo in cell-free and
cell-to-cell HTLV-1 infectivity models. Under these conditions,
both compounds significantly reduced virus transmission, with IC50
values of 35 and 1 nM, respectively (15). In 2013, a phase I clinical trial
investigating the effects of RAL on HTLV-1 proviral load in
patients with HTLV-1 TSP/HAM was initiated (ClinicalTrials.gov Identifier: NCT01867320). EVG is
also active on a range of retroviral infections. EVG and L-870,810
potently inhibited murine leukemia virus and simian virus
replication in cell culture with EC50 values in nanomolar range
(89).
Conclusions
Metal-dependent catalytic amino acid triad assembled
by a RNAse H-like fold is a highly efficient and versatile feature
of polynucleotidyl transferases, relating enzymes with very
different cellular functions, present in all organisms, from
viruses to humans. On one hand, these structural and mechanistic
similarities between retroviral INs, ancient transposons and RAG1
recombinase have revealed a complex process of domestication during
species evolution. On the other hand, it has prompted more detailed
investigations into compounds acting as inhibitors within the
DDE(D) center.
Docking simulations have shown that IN inhibitors
designed to interfere with HIV-1 IN DDE motif also display affinity
towards the RAG1 catalytic pocket. The selected docking
conformations revealed interactions with key residues, such as
D708, E962, R848 and H795 which can explain compound interference
with RAG-mediated DNA cleavage demonstrated by previously reported
in vitro and ex vivo experiments (14,70)
However, given that these simulations were performed on RAG1 dimer
complex without DNA, it is possible that the identified binding
pockets for the compounds are not accessible upon synaptic complex
formation. This argument is supported by the fact that L-708,906
inhibits RAG cleavage more potently when added to the protein
before substrate (70). As
indicated herein, the styrylquinoline derivative FZ41 displays a
similar affinity to diketo acid INSTIs, and tBQA allosteric
inhibitors have a significantly lower affinity towards the
catalytic center.
Diketo acid INSTIs are able to bind and inhibit the
activity of various polynucleotide transferases, despite the
different architecture of their enzyme-DNA complex. While their
potency and selectivity for certain enzymes differ, it is suggested
that they all interact within the common RNase H-like fold domain,
harboring the catalytic DDE(D) motif. IN inhibitors interaction
with domesticated transposases, such as RAG1 and metnase prompts
researchers to be watchful on possible immunological adverse events
in patients, but also opens possibilities for the identification of
novel adjuvant therapies for treatment refractory cancers.
Moreover, the inhibition of related retroviral INs, such as HLTV-1
and other nucleases depending on a RNase H-like fold, such as HMCV
terminase, can expand INSTIs disease spectrum and offer lead
compounds for the development of more efficient novel targeted
drugs.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
MGM performed the literature research, participated
in writing the manuscript and performed the computational analysis.
MS assisted in the computational analysis and critical review of
the literature research. GMN and DM were involved in the design of
the review, participated in the writing of the manuscript and the
critical selection of the literature. DAS and AT were involved in
the design of the review. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
DAS is the Editor-in-Chief for the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this article.
The other authors declare that they have no competing
interests.
References
|
1
|
Pace JK II and Feschotte C: The
evolutionary history of human DNA transposons: Evidence for intense
activity in the primate lineage. Genome Res. 17:422–432. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jangam D, Feschotte C and Betrán E:
Transposable Element Domestication As an Adaptation to Evolutionary
Conflicts. Trends Genet. 33:817–831. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee SH, Oshige M, Durant ST, Rasila KK,
Williamson EA, Ramsey H, Kwan L, Nickoloff JA and Hromas R: The SET
domain protein Metnase mediates foreign DNA integration and links
integration to nonhomologous end-joining repair. Proc Natl Acad Sci
USA. 102:18075–18080. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hickman AB and Dyda F: DNA Transposition
at Work. Chem Rev. 116:12758–12784. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McCLINTOCK B: The origin and behavior of
mutable loci in maize. Proc Natl Acad Sci USA. 36:344–355. 1950.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chandler M, de la Cruz F, Dyda F, Hickman
AB, Moncalian G and Ton-Hoang B: Breaking and joining
single-stranded DNA: The HUH endonuclease superfamily. Nat Rev
Microbiol. 11:525–538. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wicker T, Sabot F, Hua-Van A, Bennetzen
JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O,
et al: A unified classification system for eukaryotic transposable
elements. Nat Rev Genet. 8:973–982. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yuan YW and Wessler SR: The catalytic
domain of all eukaryotic cut-and-paste transposase superfamilies.
Proc Natl Acad Sci USA. 108:7884–7889. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lacroix C, Giovannini D, Combe A, Bargieri
DY, Späth S, Panchal D, Tawk L, Thiberge S, Carvalho TG, Barale JC,
et al: FLP/FRT-mediated conditional mutagenesis in pre-erythrocytic
stages of Plasmodium berghei. Nat Protoc. 6:1412–1428. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ye J, Hong J and Ye F: Reprogramming rat
embryonic fibroblasts into induced pluripotent stem cells using
transposon vectors and their chondrogenic differentiation in
vitro. Mol Med Rep. 11:989–994. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rice PA and Baker TA: Comparative
architecture of transposase and integrase complexes. Nat Struct
Biol. 8:302–307. 2001. View
Article : Google Scholar
|
|
12
|
Schatz DG and Ji Y: Recombination centres
and the orchestration of V(D)J recombination. Nat Rev Immunol.
11:251–263. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dai Y, Wong B, Yen Y-M, Oettinger MA, Kwon
J and Johnson RC: Determinants of HMGB proteins required to promote
RAG1/2-recombination signal sequence complex assembly and catalysis
during V(D)J recombination. Mol Cell Biol. 25:4413–4425. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nishana M, Nilavar NM, Kumari R, Pandey M
and Raghavan SC: HIV integrase inhibitor, Elvitegravir, impairs RAG
functions and inhibits V(D)J recombination. Cell Death Dis.
8:e28522017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Seegulam ME and Ratner L: Integrase
inhibitors effective against human T-cell leukemia virus type 1.
Antimicrob Agents Chemother. 55:2011–2017. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nadal M, Mas PJ, Blanco AG, Arnan C, Solà
M, Hart DJ and Coll M: Structure and inhibition of herpesvirus DNA
packaging terminase nuclease domain. Proc Natl Acad Sci USA.
107:16078–16083. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Trott O and Olson AJ: AutoDock Vina:
Improving the speed and accuracy of docking with a new scoring
function, efficient optimization, and multithreading. J Comput
Chem. 31:455–461. 2010.PubMed/NCBI
|
|
18
|
Kim MS, Lapkouski M, Yang W and Gellert M:
Crystal structure of the V(D)J recombinase RAG1-RAG2. Nature.
518:507–511. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Avogadro, . Avogadro: an open-source
molecular builder and visualization tool. Version 1.0.3. http://AvogadroOpenmoleculesNet/2012
|
|
20
|
DeLano WL: The PyMOL Molecular Graphics
System, Version 1.8Schrödinger LLC; New York, NY: 2002
|
|
21
|
Kim MS, Chuenchor W, Chen X, Cui Y, Zhang
X, Zhou ZH, Gellert M and Yang W: Cracking the DNA Code for V(D)J
Recombination. Mol Cell. 70:358–370.e4. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ma Y, Pannicke U, Schwarz K and Lieber MR:
Hairpin opening and overhang processing by an Artemis/DNA-dependent
protein kinase complex in nonhomologous end joining and V(D)J
recombination. Cell. 108:781–794. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ru H, Chambers MG, Fu TM, Tong AB, Liao M
and Wu H: Molecular Mechanism of V(D)J Recombination from Synaptic
RAG1-RAG2 Complex Structures. Cell. 163:1138–1152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Grazini U, Zanardi F, Citterio E, Casola
S, Goding CR and McBlane F: The RING domain of RAG1 ubiquitylates
histone H3: A novel activity in chromatin-mediated regulation of
V(D)J joining. Mol Cell. 37:282–293. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matthews AGW, Kuo AJ, Ramón-Maiques S, Han
S, Champagne KS, Ivanov D, Gallardo M, Carney D, Cheung P, Ciccone
DN, et al: RAG2 PHD finger couples histone H3 lysine 4
trimethylation with V(D)J recombination. Nature. 450:1106–1110.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang S, Tao X, Yuan S, Zhang Y, Li P,
Beilinson HA, Zhang Y, Yu W, Pontarotti P, Escriva H, et al:
Discovery of an Active RAG Transposon Illuminates the Origins of
V(D)J Recombination. Cell. 166:102–114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang Y, Cheng TC, Huang G, Lu Q, Surleac
MD, Mandell JD, Pontarotti P, Petrescu AJ, Xu A, Xiong Y, et al:
Transposon molecular domestication and the evolution of the RAG
recombinase. Nature. 569:79–84. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kang YH, Son CY, Lee CH and Ryu CJ:
Aberrant V(D)J cleavages in T cell receptor β enhancer- and
p53-deficient lymphoma cells. Oncol Rep. 23:1463–1468.
2010.PubMed/NCBI
|
|
29
|
Lewis SM, Agard E, Suh S and Czyzyk L:
Cryptic signals and the fidelity of V(D)J joining. Mol Cell Biol.
17:3125–3136. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Papaemmanuil E, Rapado I, Li Y, Potter NE,
Wedge DC, Tubio J, Alexandrov LB, Van Loo P, Cooke SL, Marshall J,
et al: RAG-mediated recombination is the predominant driver of
oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia.
Nat Genet. 46:116–125. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Messier TL, O'Neill JP, Hou SM, Nicklas JA
and Finette BA: In vivo transposition mediated by V(D)J recombinase
in human T lymphocytes. EMBO J. 22:1381–1388. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Reddy YVR, Perkins EJ and Ramsden DA:
Genomic instability due to V(D)J recombination-associated
transposition. Genes Dev. 20:1575–1582. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li Z, Wu S, Wang J, Li W, Lin Y, Ji C, Xue
J and Chen J: Evaluation of the interactions of HIV-1 integrase
with small ubiquitin-like modifiers and their conjugation enzyme
Ubc9. Int J Mol Med. 30:1053–1060. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gupta K, Turkki V, Sherrill-Mix S, Hwang
Y, Eilers G, Taylor L, McDanal C, Wang P, Temelkoff D, Nolte RT, et
al: Structural Basis for Inhibitor-Induced Aggregation of HIV
Integrase. PLoS Biol. 14:e10025842016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lusic M and Siliciano RF: Nuclear
landscape of HIV-1 infection and integration. Nat Rev Microbiol.
15:69–82. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen JC-H, Krucinski J, Miercke LJW,
Finer-Moore JS, Tang AH, Leavitt AD and Stroud RM: Crystal
structure of the HIV-1 integrase catalytic core and C-terminal
domains: A model for viral DNA binding. Proc Natl Acad Sci USA.
97:8233–8238. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang W, Hendrickson WA, Crouch RJ and
Satow Y: Structure of ribonuclease H phased at 2 A resolution by
MAD analysis of the selenomethionyl protein. Science.
249:1398–1405. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Venanzi Rullo E, Ceccarelli M, Condorelli
F, Facciolà A, Visalli G, D'Aleo F, Paolucci I, Cacopardo B,
Pinzone MR, Di Rosa M, et al: Investigational drugs in HIV: Pros
and cons of entry and fusion inhibitors (Review). Mol Med Rep.
19:1987–1995. 2019.PubMed/NCBI
|
|
39
|
Wai JS, Egbertson MS, Payne LS, Fisher TE,
Embrey MW, Tran LO, Melamed JY, Langford HM, Guare JP Jr, Zhuang L,
et al: 4-Aryl-2,4-dioxobutanoic acid inhibitors of HIV-1 integrase
and viral replication in cells. J Med Chem. 43:4923–4926. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hazuda DJ, Felock P, Witmer M, Wolfe A,
Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau
C and Miller MD: Inhibitors of strand transfer that prevent
integration and inhibit HIV-1 replication in cells. Science.
287:646–650. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Summa V, Petrocchi A, Bonelli F, Crescenzi
B, Donghi M, Ferrara M, Fiore F, Gardelli C, Gonzalez Paz O, Hazuda
DJ, et al: Discovery of raltegravir, a potent, selective orally
bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS
infection. J Med Chem. 51:5843–5855. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
U.S. Food & Drug Administration, . HIV
Timeline and History of Approvals. https://www.fda.gov/patients/hivaids/hiv-timeline-and-history-approvalsAugust
1–2018
|
|
43
|
Di Santo R: Inhibiting the HIV integration
process: Past, present, and the future. J Med Chem. 57:539–566.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sato M, Motomura T, Aramaki H, Matsuda T,
Yamashita M, Ito Y, Kawakami H, Matsuzaki Y, Watanabe W, Yamataka
K, et al: Novel HIV-1 integrase inhibitors derived from quinolone
antibiotics. J Med Chem. 49:1506–1508. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lee JSF, Calmy A, Andrieux-Meyer I and
Ford N: Review of the safety, efficacy, and pharmacokinetics of
elvitegravir with an emphasis on resource-limited settings. HIV
AIDS (Auckl). 4:5–15. 2012.PubMed/NCBI
|
|
46
|
Barnhart M and Shelton JD: ARVs: The next
generation. Going boldly together to new frontiers of HIV
treatment. Glob Health Sci Pract. 3:1–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Johns BA, Kawasuji T, Weatherhead JG,
Taishi T, Temelkoff DP, Yoshida H, Akiyama T, Taoda Y, Murai H,
Kiyama R, et al: Carbamoyl pyridone HIV-1 integrase inhibitors 3. A
diastereomeric approach to chiral nonracemic tricyclic ring systems
and the discovery of dolutegravir (S/GSK1349572) and
(S/GSK1265744). J Med Chem. 56:5901–5916. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hare S, Gupta SS, Valkov E, Engelman A and
Cherepanov P: Retroviral intasome assembly and inhibition of DNA
strand transfer. Nature. 464:232–236. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hare S, Smith SJ, Métifiot M, Jaxa-Chamiec
A, Pommier Y, Hughes SH and Cherepanov P: Structural and functional
analyses of the second-generation integrase strand transfer
inhibitor dolutegravir (S/GSK1349572). Mol Pharmacol. 80:565–572.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hightower KE, Wang R, Deanda F, Johns BA,
Weaver K, Shen Y, Tomberlin GH, Carter HL III, Broderick T, Sigethy
S, et al: Dolutegravir (S/GSK1349572) exhibits significantly slower
dissociation than raltegravir and elvitegravir from wild-type and
integrase inhibitor-resistant HIV-1 integrase-DNA complexes.
Antimicrob Agents Chemother. 55:4552–4559. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yoshinaga T, Kobayashi M, Seki T, Miki S,
Wakasa-Morimoto C, Suyama-Kagitani A, Kawauchi-Miki S, Taishi T,
Kawasuji T, Johns BA, et al: Antiviral characteristics of
GSK1265744, an HIV integrase inhibitor dosed orally or by
long-acting injection. Antimicrob Agents Chemother. 59:397–406.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tsiang M, Jones GS, Goldsmith J, Mulato A,
Hansen D, Kan E, Tsai L, Bam RA, Stepan G, Stray KM, et al:
Antiviral activity of bictegravir (GS-9883), a novel potent HIV-1
integrase strand transfer inhibitor with an improved resistance
profile. Antimicrob Agents Chemother. 60:7086–7097. 2016.PubMed/NCBI
|
|
53
|
Mekouar K, Mouscadet JF, Desmaële D, Subra
F, Leh H, Savouré D, Auclair C and d'Angelo J: Styrylquinoline
derivatives: A new class of potent HIV-1 integrase inhibitors that
block HIV-1 replication in CEM cells. J Med Chem. 41:2846–2857.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Deprez E, Barbe S, Kolaski M, Leh H,
Zouhiri F, Auclair C, Brochon JC, Le Bret M and Mouscadet JF:
Mechanism of HIV-1 integrase inhibition by styrylquinoline
derivatives in vitro. Mol Pharmacol. 65:85–98. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Han Y-S, Xiao W-L, Quashie PK, Mesplède T,
Xu H, Deprez E, Delelis O, Pu JX, Sun HD and Wainberg MA:
Development of a fluorescence-based HIV-1 integrase DNA binding
assay for identification of novel HIV-1 integrase inhibitors.
Antiviral Res. 98:441–448. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bonnenfant S, Thomas CM, Vita C, Subra F,
Deprez E, Zouhiri F, Desmaële D, D'Angelo J, Mouscadet JF and Leh
H: Styrylquinolines, integrase inhibitors acting prior to
integration: A new mechanism of action for anti-integrase agents. J
Virol. 78:5728–5736. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Passos DO, Li M, Yang R, Rebensburg SV,
Ghirlando R, Jeon Y, Shkriabai N, Kvaratskhelia M, Craigie R and
Lyumkis D: Cryo-EM structures and atomic model of the HIV-1 strand
transfer complex intasome. Science. 355:89–92. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Quashie PK, Han YS, Hassounah S, Mesplède
T and Wainberg MA: Structural studies of the HIV-1 integrase
protein: Compound screening and characterization of a DNA-binding
inhibitor. PLoS One. 10:e01283102015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Shkriabai N, Patil SS, Hess S, Budihas SR,
Craigie R, Burke TR Jr, Le Grice SF and Kvaratskhelia M:
Identification of an inhibitor-binding site to HIV-1 integrase with
affinity acetylation and mass spectrometry. Proc Natl Acad Sci USA.
101:6894–6899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Du L, Zhao YX, Yang LM, Zheng YT, Tang Y,
Shen X and Jiang HL: Symmetrical 1-pyrrolidineacetamide showing
anti-HIV activity through a new binding site on HIV-1 integrase.
Acta Pharmacol Sin. 29:1261–1267. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ge H, Si Y and Roeder RG: Isolation of
cDNAs encoding novel transcription coactivators p52 and p75 reveals
an alternate regulatory mechanism of transcriptional activation.
EMBO J. 17:6723–6729. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Maertens G, Cherepanov P, Pluymers W,
Busschots K, De Clercq E, Debyser Z and Engelborghs Y: LEDGF/p75 is
essential for nuclear and chromosomal targeting of HIV-1 integrase
in human cells. J Biol Chem. 278:33528–33539. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Llano M, Delgado S, Vanegas M and Poeschla
EM: Lens epithelium-derived growth factor/p75 prevents proteasomal
degradation of HIV-1 integrase. J Biol Chem. 279:55570–55577. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
De Rijck J, Vandekerckhove L, Gijsbers R,
Hombrouck A, Hendrix J, Vercammen J, Engelborghs Y, Christ F and
Debyser Z: Overexpression of the lens epithelium-derived growth
factor/p75 integrase binding domain inhibits human immunodeficiency
virus replication. J Virol. 80:11498–11509. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cherepanov P, Ambrosio ALB, Rahman S,
Ellenberger T and Engelman A: Structural basis for the recognition
between HIV-1 integrase and transcriptional coactivator p75. Proc
Natl Acad Sci USA. 102:17308–17313. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Du L, Zhao Y, Chen J, Yang L, Zheng Y,
Tang Y, Shen X and Jiang H: D77, one benzoic acid derivative,
functions as a novel anti-HIV-1 inhibitor targeting the interaction
between integrase and cellular LEDGF/p75. Biochem Biophys Res
Commun. 375:139–144. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Christ F, Voet A, Marchand A, Nicolet S,
Desimmie BA, Marchand D, Bardiot D, Van der Veken NJ, Van Remoortel
B, Strelkov SV, et al: Rational design of small-molecule inhibitors
of the LEDGF/p75-integrase interaction and HIV replication. Nat
Chem Biol. 6:442–448. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Fader LD, Malenfant E, Parisien M, Carson
R, Bilodeau F, Landry S, Pesant M, Brochu C, Morin S, Chabot C, et
al: Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor
(NCINI) of HIV-1. ACS Med Chem Lett. 5:422–427. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Tsiang M, Jones GS, Niedziela-Majka A, Kan
E, Lansdon EB, Huang W, Hung M, Samuel D, Novikov N, Xu Y, et al:
New class of HIV-1 integrase (IN) inhibitors with a dual mode of
action. J Biol Chem. 287:21189–21203. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Melek M, Jones JM, O'Dea MH, Pais G, Burke
TR Jr, Pommier Y, Neamati N and Gellert M: Effect of HIV integrase
inhibitors on the RAG1/2 recombinase. Proc Natl Acad Sci USA.
99:134–137. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Goldgur Y, Craigie R, Cohen GH, Fujiwara
T, Yoshinaga T, Fujishita T, Sugimoto H, Endo T, Murai H and Davies
DR: Structure of the HIV-1 integrase catalytic domain complexed
with an inhibitor: A platform for antiviral drug design. Proc Natl
Acad Sci USA. 96:13040–13043. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
de Miguel R, Montejano R, Stella-Ascariz N
and Arribas JR: A safety evaluation of raltegravir for the
treatment of HIV. Expert Opin Drug Saf. 17:217–223. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Huhn GD, Badri S, Vibhakar S, Tverdek F,
Crank C, Lubelchek R, Max B, Simon D, Sha B, Adeyemi O, et al:
Early development of non-hodgkin lymphoma following initiation of
newer class antiretroviral therapy among HIV-infected patients -
implications for immune reconstitution. AIDS Res Ther. 7:442010.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Steigbigel RT, Cooper DA, Kumar PN, Eron
JE, Schechter M, Markowitz M, Loutfy MR, Lennox JL, Gatell JM,
Rockstroh JK, et al BENCHMRK Study Teams, : Raltegravir with
optimized background therapy for resistant HIV-1 infection. N Engl
J Med. 359:339–354. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Barbaro G and Barbarini G: HIV infection
and cancer in the era of highly active antiretroviral therapy
(Review). Oncol Rep. 17:1121–1126. 2007.PubMed/NCBI
|
|
76
|
Gillead Sciences: sNDA 207561/S-014.
Genvoya (elvitegravir/cobicistat/emtricitabine/tenofovir
alafenamide). Clinical and Cross-Discipline Team Leader Review.
|
|
77
|
Huye LE, Purugganan MM, Jiang M-M and Roth
DB: Mutational analysis of all conserved basic amino acids in RAG-1
reveals catalytic, step arrest, and joining-deficient mutants in
the V(D)J recombinase. Mol Cell Biol. 22:3460–3473. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Davies DR, Goryshin IY, Reznikoff WS and
Rayment I: Three-dimensional structure of the Tn5 synaptic complex
transposition intermediate. Science. 289:77–85. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ason B, Knauss DJ, Balke AM, Merkel G,
Skalka AM and Reznikoff WS: Targeting Tn5 transposase identifies
human immunodeficiency virus type 1 inhibitors. Antimicrob Agents
Chemother. 49:2035–2043. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Czyz A, Stillmock KA, Hazuda DJ and
Reznikoff WS: Dissecting Tn5 transposition using HIV-1 integrase
diketoacid inhibitors. Biochemistry. 46:10776–10789. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Koh Y, Matreyek KA and Engelman A:
Differential sensitivities of retroviruses to integrase strand
transfer inhibitors. J Virol. 85:3677–3682. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Beck-Engeser GB, Eilat D, Harrer T, Jäck
HM and Wabl M: Early onset of autoimmune disease by the retroviral
integrase inhibitor raltegravir. Proc Natl Acad Sci USA.
106:20865–20870. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Stetson DB, Ko JS, Heidmann T and
Medzhitov R: Trex1 prevents cell-intrinsic initiation of
autoimmunity. Cell. 134:587–598. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wolkowicz UM, Morris ER, Robson M,
Trubitsyna M and Richardson JM: Structural basis of Mos1
transposase inhibition by the anti-retroviral drug Raltegravir. ACS
Chem Biol. 9:743–751. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Shaheen M, Williamson E, Nickoloff J, Lee
SH and Hromas R: Metnase/SETMAR: A domesticated primate transposase
that enhances DNA repair, replication, and decatenation. Genetica.
138:559–566. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Williamson EA, Damiani L, Leitao A, Hu C,
Hathaway H, Oprea T, Sklar L, Shaheen M, Bauman J, Wang W, et al:
Targeting the transposase domain of the DNA repair component
Metnase to enhance chemotherapy. Cancer Res. 72:6200–6208. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Marino-Merlo F, Mastino A, Grelli S,
Hermine O, Bazarbachi A and Macchi B: Future Perspectives on Drug
Targeting in Adult T Cell Leukemia-Lymphoma. Front Microbiol.
9:9252018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Rabaaoui S, Zouhiri F, Lançon A, Leh H,
d'Angelo J and Wattel E: Inhibitors of strand transfer that prevent
integration and inhibit human T-cell leukemia virus type 1 early
replication. Antimicrob Agents Chemother. 52:3532–3541. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Shimura K, Kodama E, Sakagami Y, Matsuzaki
Y, Watanabe W, Yamataka K, Watanabe Y, Ohata Y, Doi S, Sato M, et
al: Broad antiretroviral activity and resistance profile of the
novel human immunodeficiency virus integrase inhibitor elvitegravir
(JTK-303/GS-9137). J Virol. 82:764–774. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Passos DO, Li M, Yang R, Rebensburg SV,
Ghirlando R, Jeon Y, Shkriabai N, Kvaratskhelia M, Craigie R and
Lyumkis D: Cryo-EM structures and atomic model of the HIV-1 strand
transfer complex intasome. Science. 355:89–92. 2017. View Article : Google Scholar : PubMed/NCBI
|