Introduction
Recent advancements in neonatal intensive care units
have led to an increase in the survival rates of the majority of
early gestation premature infants; however, some of these develop
bronchopulmonary dysplasia (BPD). BPD is one of the most common
consequences of premature birth, affecting ~30% of infants with
birth weights of <1,500 g (1).
With an increased survival rate in infants with extremely low birth
weights, BPD has increased in premature infants with a gestational
age of <28 weeks (2). Infants
exhibiting signs of BPD may require respiratory support following
hospital discharge and may experience chronic respiratory morbidity
throughout their whole lives; they are at higher risk of
neurodevelopment problems and mortality (3). Infants and children with a history of
BPD may require re-admission to hospital repeatedly in their early
years, with some developing severe lung function abnormalities,
including asthma, chest deformities and adolescent pulmonary
hypertension. Delayed neurodevelopment can cause cognitive and
motor function defects, such as cerebral palsy, which is common in
BPD infants (4). Currently, there
are a lack of effective treatments options for BPD. BPD is
characterized by the arrest of alveolar development and is
secondary to myofibroblast-mediated extracellular matrix deposition
and fibrotic lung formation (5).
Type II alveolar epithelial cells (AECIIs) serve an important role
in the development of BPD. Hyperoxia can induce AECIIs to
differentiate into fibroblasts and affect alveolar development
through the epithelial-to-mesenchymal (EMT) transition (6).
EMT is the process in which epithelial cells lose
their polarity and cell junctions, and acquire the characteristics
of mesenchymal cells (7). During
BPD, AECII cells undergo EMT, which promotes the progression of
pulmonary fibrosis (8).
SMAD-dependent (SMAD2 and SMAD3) and non-dependent cellular
signaling pathways [PI3K/AKT, RhoA, partitioning defective 6
homolog and mitogen-activated protein kinase (MAPK)] have been
reported to be associated with EMT, and it has been previously
demonstrated that bleomycin-induced lung fibrosis in mice can be
alleviated by the p38 MAPK protein inhibitorFR-167653 (9). Epithelial cell (E)-cadherin, an
important member of the cadherin family, is widely distributed in a
variety of epithelial cells. E-cadherin is important for epithelial
cell adhesion, the maintenance of epithelial tissue integrity and
polarity. The downregulation of E-cadherin is associated with the
development of EMT (10). The loss
of E-cadherin expression in the epithelia is considered a hallmark
of EMT (11). Zinc-finger E-box
binding homeobox 2 (ZEB2) has also been demonstrated to be an
important regulatory molecule in EMT; the upregulation of ZEB2 can
inhibit E-cadherin transcription and induce EMT, which may lead to
cell invasion and increased malignancy (12).
Vascular endothelial growth factors (VEGFs) are a
family of essential angiogenic mediators that serve important roles
in the regulation of angiogenesis. Placental growth factor (PLGF)
and VEGFA are two important molecules in this family (13). PLGF was first identified in
placental tissue, and subsequent studies have demonstrated that it
is expressed in numerous tissues types, including heart, lung and
thyroid (14–17). In breast cancer cells, PLGF has
been indicated to promote the progression of EMT (18). In cervical cancer cells, PLGF has
been reported to be associated with the process of EMT, allowing
cancer cells to leave the primary tumor, invade surrounding tissues
and spread to the distal organ (19). Our previous study reported that
PLGF expression is increased in lungs exposed to hyperoxia
(20). It was therefore
hypothesized that PLGF may be associated with lung injury through
the promotion of EMT. To address this hypothesis, PLGF gene
silencing was used in neonatal rat lung tissue to investigate the
underlying mechanisms by which this may occur, which provided
valuable information regarding disease pathogenesis and potential
therapeutic approaches for BPD.
Materials and methods
Materials
A total of 32 14-day old Sprague-Dawley (SD) rats
(female:male, 17:15; 25.2–30.3 g) were provided by The Experimental
Animal Center of China Medical University. The animals were housed
at a temperature of 25–27°C, with a humidity of 50–70% and a 12 h
light/dark cycle with ad libitum access to food and water.
Xylene, absolute ethanol, eosin Y and hydrogen peroxide were
purchased from Wuhan USCN Business Co., Ltd. Hematoxylin, eosin and
goat serum (cat. no. SL038) were purchased from Beijing Solarbio
Science& Technology Co., Ltd. PLGF mouse monoclonal antibody
(cat. no. sc-518003) and E-cadherin mouse monoclonal antibody (cat.
no. sc-71007) were purchased from Santa Cruz Biotechnology, Inc.
Anti-phosphorylated (p)-p38 rabbit polyclonal antibody (cat. no.
bs-2210R) was purchased from (BIOSS). Anti-p38 rabbit monoclonal
(cat. no. M00176), anti-β-actin goat polyclonal (cat. no. BM0627)
and anti-ZEB2 rabbit polyclonal (cat. no. PA1959) antibodies were
purchased from Boster Biological Technology. Biotin-labeled goat
anti-mouse or goat anti-rabbit IgG and HRP-labeled streptavidin
(cat. nos. A0286; A0277; A0303, respectively) were purchased from
Beyotime Institute of Biotechnology. RIPA lysis buffer was
purchased from Tiangen Biotech Co., Ltd. BCA Protein Assay Reagent
kit was obtained from Pierce (Thermo Fisher Scientific, Inc.).
TRIzol was obtained from Thermo Fisher Scientific, Inc.
Animal experiments
Neonatal SD rats, delivered prematurely at 21 days
gestation from the rats detailed above, were placed in an oxygen
chamber with their mothers. Oxygen was continuously supplied to
maintain fraction of inspired oxygen (FiO2)=75%
(hyperoxic condition; ProOx110O2 Controller; BioSpherix,
Ltd.), a CO2 concentration of <0.5% (sodium lime
absorption CO2), a temperature of 22–27°C and a humidity
of 50–70%. Normoxic conditions were identical to hyperoxia except
FiO2=21%. Chambers were opened for 30 min every day to
add water, feed and replace the litter. Mother rats were alternated
between hyperoxia and normoxia conditions to prevent oxygen
toxicity and to provide equal nutrition. The neonatal rats
(8/group) were randomly divided into: i) Normoxia control group
(FiO2=21%); ii) hyperoxia group (FiO2=75%);
iii) hyperoxia + negative control (NC) lentivirus group (hyperoxia
+ shRNA-NC); and iv) hyperoxia + short hairpin (sh)RNA-PLGF
lentivirus group (hyperoxia + shRNA-PLGF). After being anesthetized
by intraperitoneal injection with pentobarbital (30–40 mg/kg)
(Tianjin Kemiou Chemical Reagent Co., Ltd.), rat pups were
sacrificed after 14 days of exposure (8/group).
Lentiviral PLGF interference plasmid
injection
The 72 bp oligonucleotide short hairpin (sh)RNAs
specific for PLGF were obtained from Shanghai GenePharma Co., Ltd.
The sequences of PLGF shRNAs were as follows:
5′-GCGCTAAAGACAGCCAACA-3′. Non-targeting shRNA, with a sequence of
5′-TTCTCCGAACGTGTCACGT-3′, was used as a negative control. The PLGF
shRNAs were sub-cloned into a lentiviral vector (GV248; Shanghai
GeneChem Co., Ltd.). Following this, the lentiviral particles with
the shRNA-PLGF were obtained from Shanghai GeneChem Co., Ltd and
were directly injected into the neonates via tail-intravenous
injection at a concentration of 3×108 TU/kg, every day
for 3 consecutive days. PLGF expression in the lung tissue was
determined using western blot analysis.
Immunohistochemistry
Lung tissues were harvested from the neonates, fixed
in 10% formalin at 37°C for 48 h and imbedded in paraffin. The
specimens were then cut into 5 µm sections, which were
deparaffinized with xylene and rehydrated in a descending ethanol
series (95, 85 and 75%), underwent antigen retrieval with 1% sodium
citrate buffer at 100°C for 20 min, followed by incubation with 3%
hydrogen peroxide and blocking with 10% goat serum for 30 min, both
at room temperature. Sections were incubated overnight at 4°C with
primary antibodies against PLGF (1:200) and E-cadherin (1:50),
followed by incubation a corresponding biotin-labelled secondary
antibody (1:200) at 37°C for 30 min with. Sections were then
incubated with DAB and counterstained with hematoxylin. Slides were
examined using a light microscope and images were captured at ×400
magnification. Cells exhibiting brown-yellow particles in the
cytoplasm were regarded as positive-stained cells.
Hematoxylin and eosin (H&E)
staining
The lung tissues sections embedded in paraffin were
cut into 4 µm thick sections. After deparaffinization and
rehydration, the sections were stained using H&E at room
temperature for 15 min. Images were captured at ×200 magnification
using a light microscope and changes in lung tissue morphology were
recorded.
Western blot analysis
Lung tissue (~1 cm3) was disrupted using
sonication (50 kHz at 4°C for 15 min), liquid nitrogen grinding and
RIPA lysis buffer protein extraction. Total protein was quantified
using the BCA method. Equal amounts of protein (20 µg) were
separated using 8% SDS-PAGE and transferred to PVDF membranes. The
membranes were blocked at room temperature for 2 h using 5% skim
milk powder. After blocking, the membranes were incubated overnight
at 4°C with primary antibodies (all 1:500) against PLGF,
E-cadherin, ZEB2, p38, phosphorylated (p)-p38, and subsequently
incubated with HRP-conjugated secondary antibodies (1:1,000) at
room temperature for 1 h and developed using ECL Plus western
blotting detection reagents. ImageJ 1.8.0 (National Institutes of
Health) was used to analyze band density. β-actin (1:5,000) was
used as a loading control and to normalize protein expression.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from 1 cm3 lung
tissue using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol, and RT
was performed using 1 µg total RNA using the PrimeScript™ RT Master
Mix (Takara Bio, Inc.). The protocol was: At 25°C for 10 min
followed by incubation at 42°C for 50 min. The reaction was
inactivated by heating at 70°C for 15 min.
qPCR was performed using an ABI PRISM 7500HT System
(Applied Biosystems; Thermo Fisher Scientific, Inc.) and the TB
Green Premix Ex Taq II kit (Takara Bio, Inc.) according to
the manufacturer's protocol. The relative gene expression was
calculated using the 2−ΔΔCq method (21), normalized to the housekeeping gene
GAPDH. The primers used were as follows: PLGF, forward
5′-CCCACCTGGATGCTGTT-3′, reverse 5′-ATAGAGGGTAGGTACCAGCA-3′;
E-cadherin, forward 5′-ACTTTGGTGTGGGTCTGGAG-3′, reverse
5′-TCTGTGGCAATGATGAGAGC-3′; ZEB2, forward
5′-TGATTGAGAACCACAGCATACC-3′, reverse 5′-GTTCATCAGAGTTGGGTTCCAT-3′;
GAPDH, forward 5′-GCACCGTCAAGGCTGAGAAC-3′, reverse
5′-TGGTGAAGACGCCAGTGGA-3′.
Statistical analysis
GraphPad Prism version 8.0 (GraphPad Software, Inc.)
was used to analyze the data. Pairwise comparisons were performed
using the Student's t-test method. Two-way ANOVA followed by
Student-Newman-Keuls pot hoc test was used to compare multiple
groups. All experiments were repeated at least three times. Data
are presented as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Results
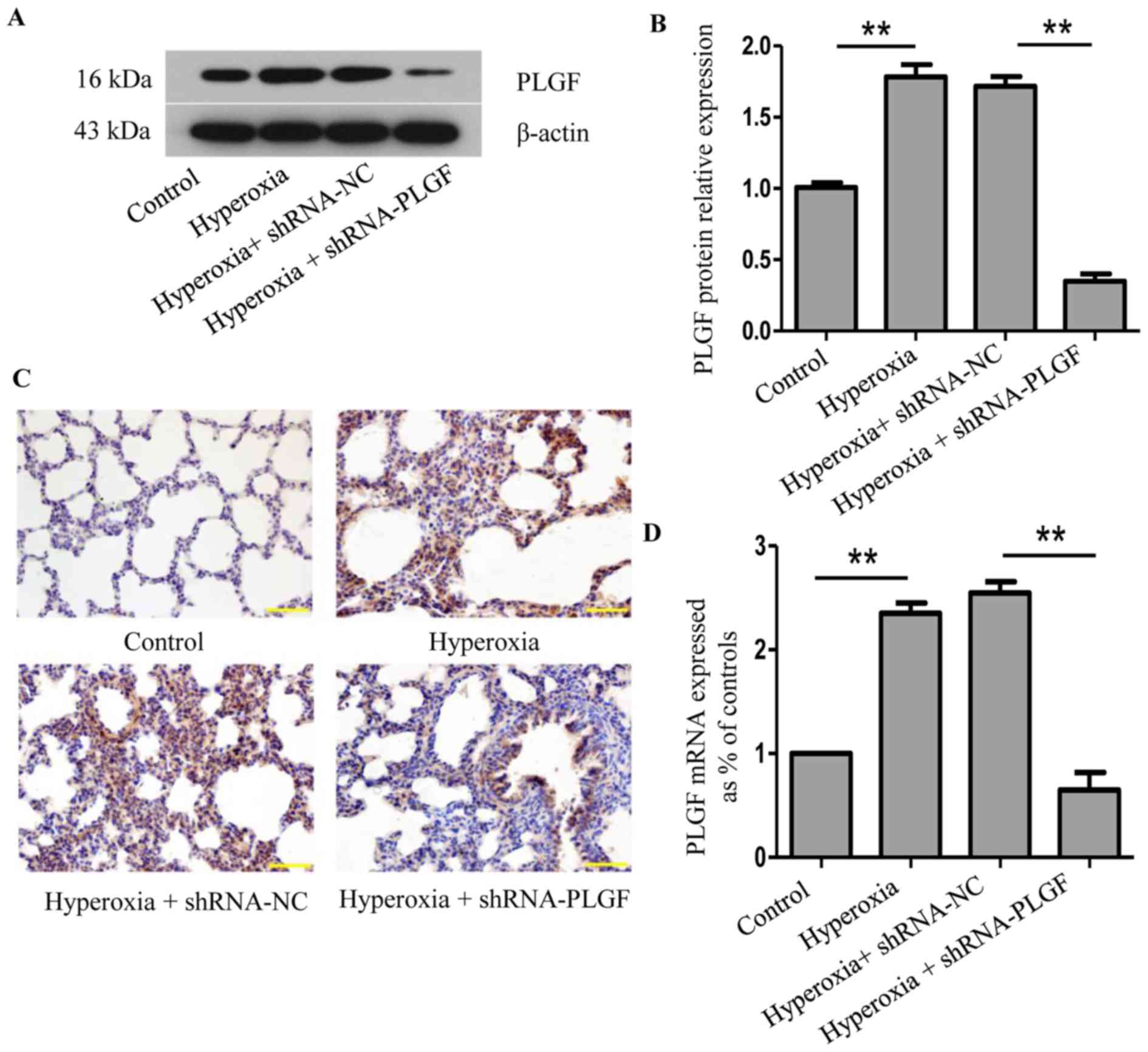
PLGF expression in hyperoxic lung
tissue after PLGF gene silencing
Following lentiviral plasmid injection, PLGF
expression was determined using western blot analysis and
immunohistochemistry 14 days after the first injection (Fig. 1). Compared with the normoxia
control group, PLGF protein expression levels in rat lung tissue
were significantly increased following exposure to hyperoxia
(Fig. 1A and B). A similar change
was detected at the mRNA level (Fig.
1D). Immunohistochemical analysis revealed a large number of
PLGF-positive cells in the hyperoxia group compared with the
normoxia control group (Fig. 1C).
In the hyperoxia + shRNA-NC group, western blotting analysis
revealed that the PLGF protein expression level in the lung tissue
was also significantly increased compared with the normoxia group,
and similar results were observed in the immunohistochemistry
experiments. In the hyperoxia + shRNA-PLGF group, lung tissue PLGF
protein expression levels were significantly lower compared with
the hyperoxia + shRNA-NC group, and the number of positively
stained cells decreased, as determined using immunohistochemistry
(Fig. S1). These data
demonstrated that hypoxia induced an increase in the expression of
PLGF in lung tissues and that PLGF gene expression was successfully
silenced following lentiviral interference plasmid injection in
rats.
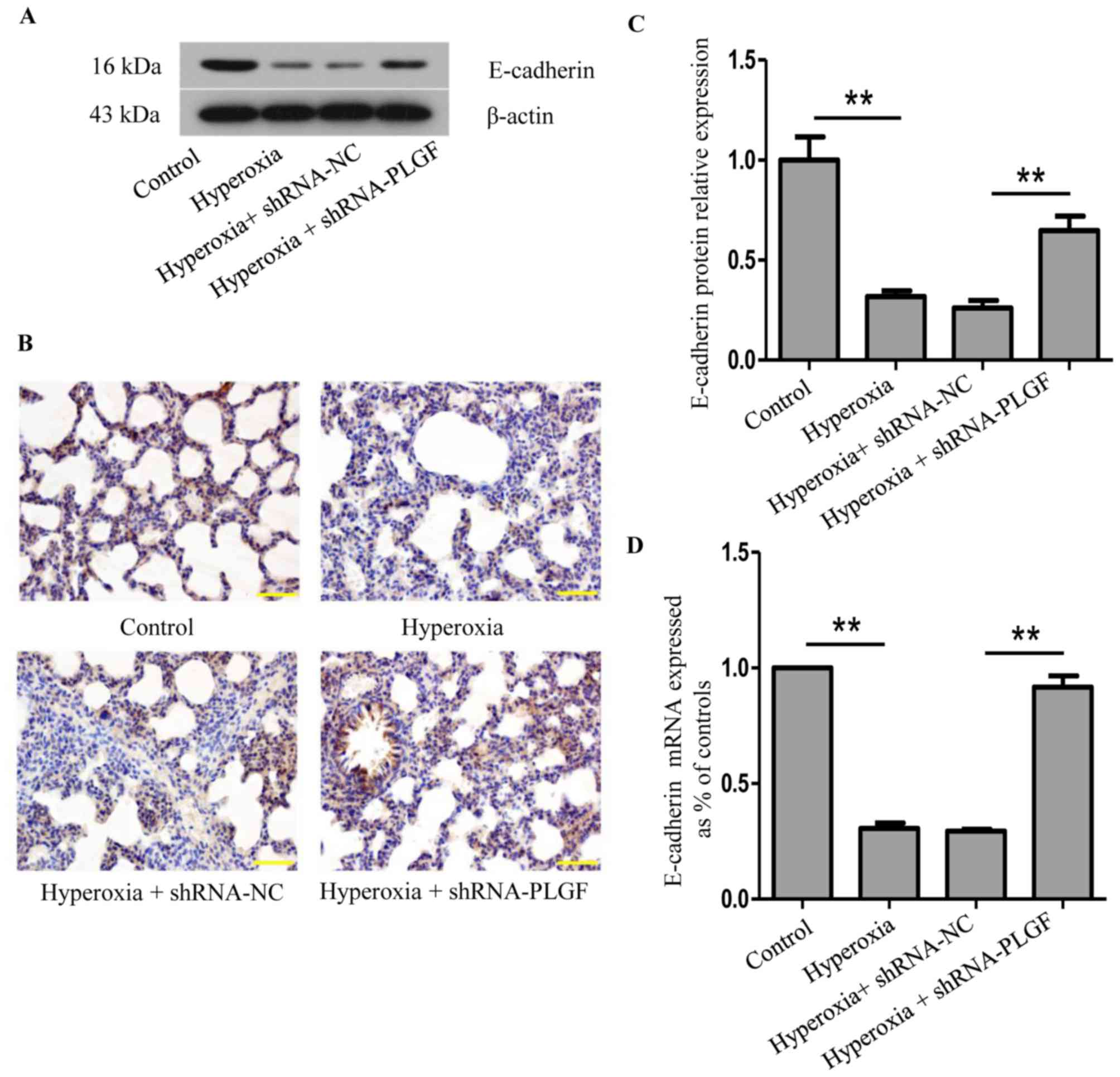
E-cadherin expression in hyperoxic
lung tissue after PLGF gene silencing
The effects of PLGF gene silencing on E-cadherin
protein expression in the lung tissue of hyperoxia-exposed rats was
determined using western blot, RT-qPCR and immunohistochemistry
(Fig. 2). Compared with the
normoxia control group, E-cadherin protein expression in lung
tissue following hyperoxia treatment was significantly decreased
(Fig. 2A and B). In the hyperoxia
+ shRNA-NC group, E-cadherin expression also significantly
decreased compared with the normoxia group; no significant
difference was identified in comparison with the hyperoxia group.
In hyperoxia + shRNA-PLGF rats, E-cadherin protein expression in
lung tissue was significantly increased compared with the hyperoxia
+ shRNA-NC group (Fig. 2A and B).
The number of positively stained cells was also increased in the
hyperoxia + shRNA-PLGF group compared with the hyperoxia + shRNA-NC
group, as determined using immunohistochemistry (Fig. 2C). These results indicated that
hyperoxia may induce EMT, as determined by the decreased E-cadherin
expression, and that shRNA-PLGF injection may delay this EMT in the
lung tissue of rats exposed to hyperoxia.
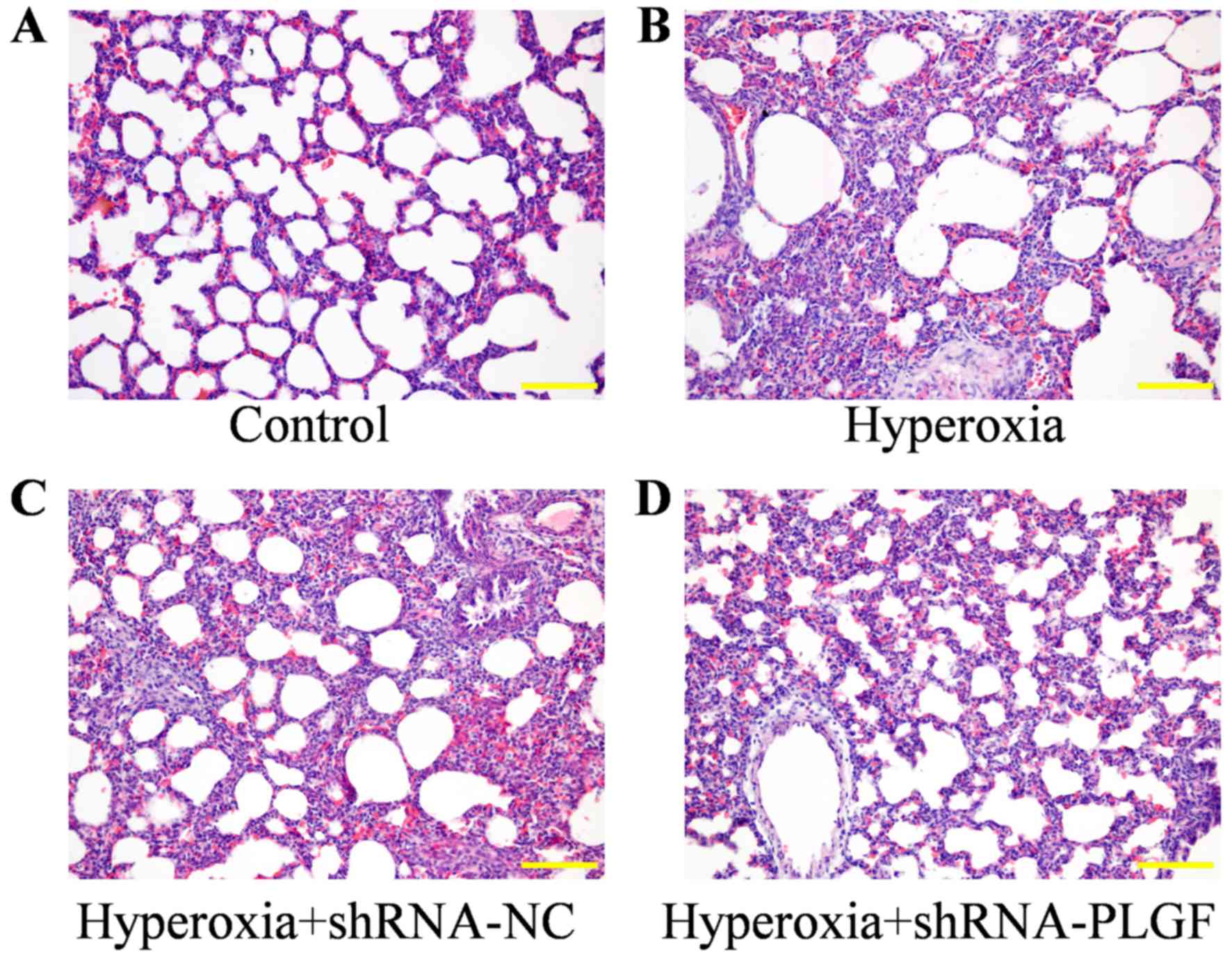
Pathological effects of PLGF gene
silencing on lung tissue in rats with hyperoxia-induced lung
injury
The pathological effect of PLGF gene silencing on
hyperoxia-induced lung injury rats was observed using H&E
staining (Fig. 3). Compared with
the control group, the alveolar epithelium was swollen in the
hyperoxia group, large amounts of exudate were present in the
alveolar space and alveolar structure was simplified. The lung
tissue of rats in the hypoxia + shRNA-NC group exhibited similar
results to the hyperoxia group, with the alveolar epithelium being
highly swollen and structurally simplified. In hyperoxia rats
injected with the shRNA-PLGF, the abnormal epithelial structure was
lessened and the lungs exhibited features, including a reduction in
alveolar exudate, a clear alveolar structure and alleviated
interstitial edema.
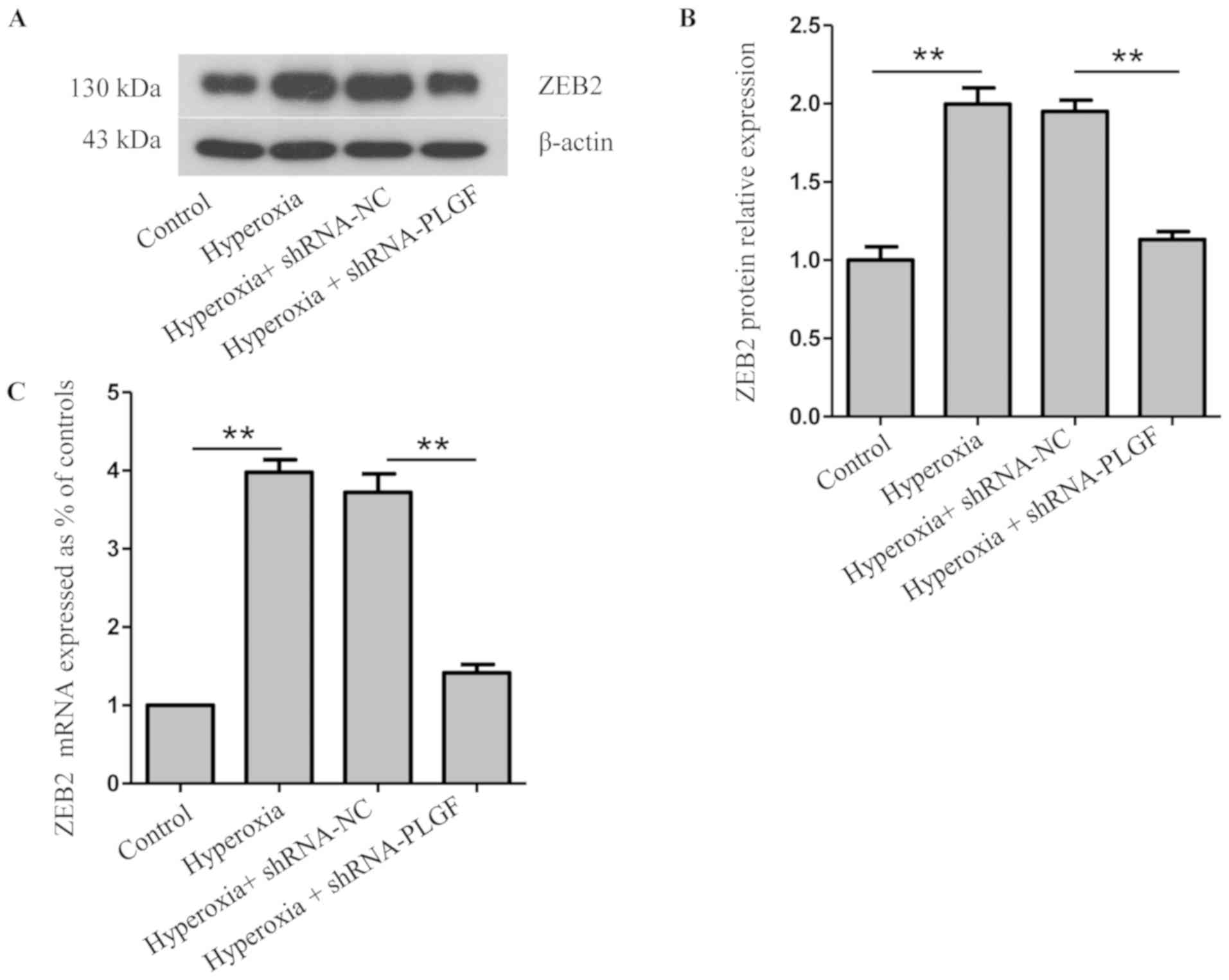
Expression of ZEB2 and
p-38MAPK/p-p38MAPK after PLGF gene silencing
ZEB2 is closely related to, and is a specific
antagonist of, E-cadherin; both serve important roles in EMT
(22). Compared with the normoxia
control group, ZEB2 protein and mRNA expression levels in rat lung
tissue was significantly increased in the hyperoxia group (Fig. 4A-C). ZEB2 expression in the lung
tissue of hyperoxia + shRNA-NC rats was also significantly
increased compared with the normoxia group. Compared with the
hyperoxia + shRNA-NC group, ZEB2 protein and mRNA expression levels
in the lung tissue of hyperoxia + shRNA-PLGF rats was significantly
decreased (Fig. 4A-C). These data
suggested that hyperoxia may cause EMT in rat lung tissue, whereas
PLGF gene silencing can reduce this effect (Fig. 4).
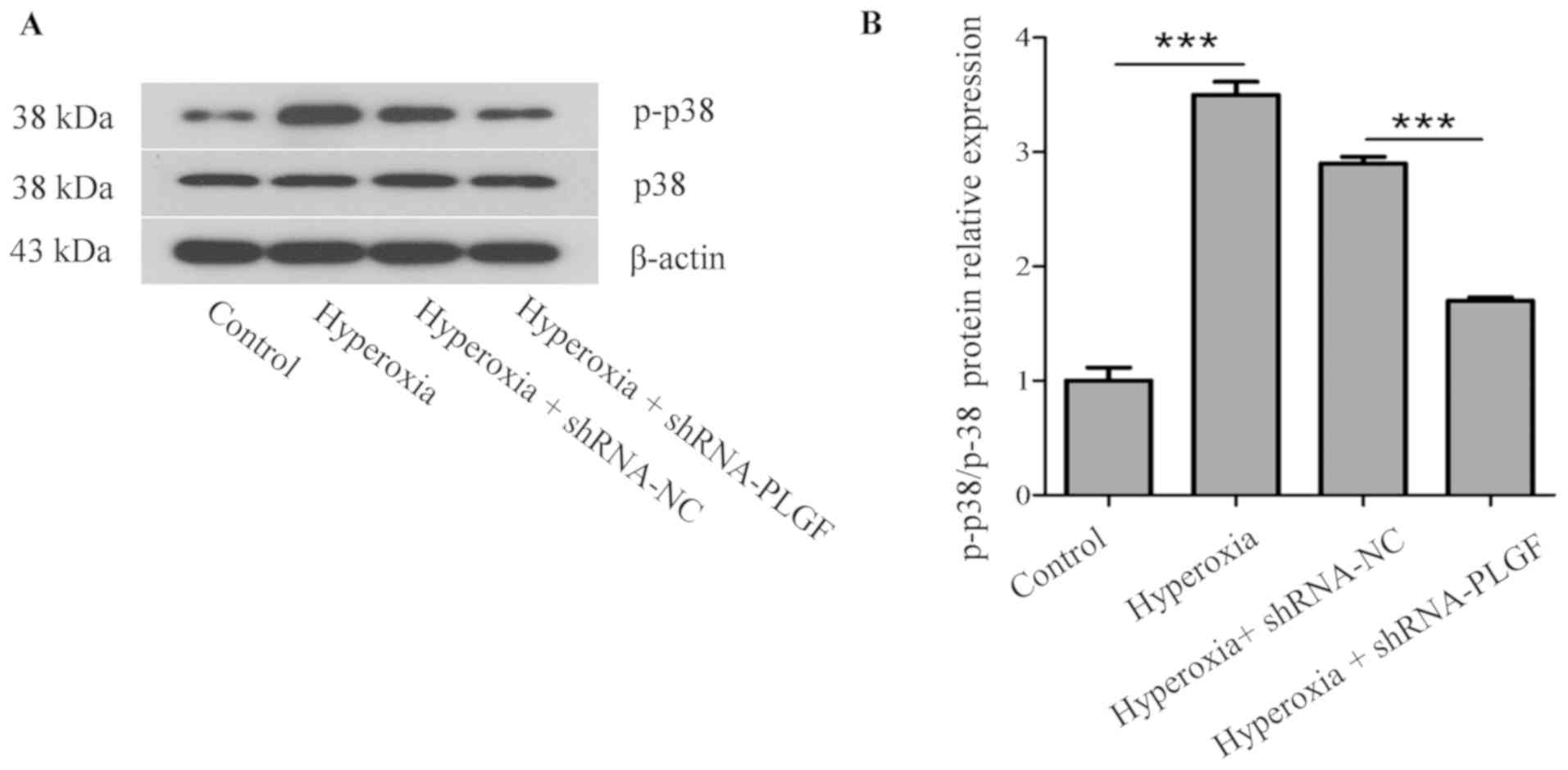
p38 protein kinase is a key factor in the MAPK
signaling pathway and is closely associated with the activation of
NF-κB pathways, which can induce EMT in the lungs (23). Compared with the normoxia control
group, p-p38 expression in the lung tissue of hyperoxia rats was
significantly increased (Fig. 5).
In the hyperoxia + shRNA-NC rats, p-p38 expression was also
significantly increased compared with the control group. p-p38
expression in lung tissue of hyperoxia + shRNA-PLGF rats was
significantly decreased compared with the hyperoxia + shRNA-NC
group. Although the treatment groups were different, the p38
content did not change significantly.
Discussion
In the present study, evidence of the role of PLGF
in the acute stage of hyperoxia-induced lung injury was presented
in the neonatal rat. The present study aimed to determine the
pathogenesis and the molecular mechanisms behind BPD with severe
lung function abnormalities. A previous study indicated the
molecular basis of lung injury using cultured lung cells in
vitro (24). In the present
study, the role of PLGF in hyperoxia-induced pulmonary dysplasia
in vivo was investigated using the neonatal rat disease
model of hyperoxia-induced lung injury.
Previous studies have demonstrated that PLGF can
induce EMT in a variety of disease, including cervical cancer and
breast cancer (18,19), and it has also been reported that
hyperoxia can promote EMT in alveolar cells (25). In concurrence with these previous
findings, results from the present study demonstrated that PLGF and
ZEB2 expression levels increased, whereas E-cadherin expression
decreased in hyperoxia exposed neonatal rats. To confirm these
results, a PLGF-silencing lentiviral plasmid was used to reduce
PLGF expression in hyperoxia-exposed neonatal rat lungs; the
results demonstrated that E-cadherin expression was increased and
ZEB2 expression was decreased. These results indicated that PLGF
may serve a role in regulating EMT in neonatal rat lung tissue
during exposure to hyperoxia.
The p38 MAPK signal transduction pathway is present
in the majority of cells. This pathway transduces extracellular
stimuli into cells and their nuclei, and causes the induction of
cellular biological reactions including cell proliferation,
differentiation, transformation and apoptosis (26). The activation of the p38 MAPK
signaling pathway promotes NF-κB inhibitor α phosphorylation and
degradation and activates the NF-κB pathway. This suggests that p38
MAPK may induce pulmonary fibrosis by activating NF-κB (27). In our previous study, it was
demonstrated that NF-κB increased during hyperoxia (28). To determine if p38 MAPK served a
role in the process of EMT, p-p38MAPK expression was determined in
neonatal rat lungs in the present study. The results indicated that
PLGF and p-p38MAPK expression increased in neonatal rat lungs
during hyperoxia. PLGF silencing resulted in the reduced expression
of p-p38MAPK. These results suggested that the p38 MAPK pathway may
be associated with the regulation of the EMT process in neonatal
rat lung tissue during hyperoxia. Previous studies have suggested
that ERK activation in lung cells has a protective effect in
response to hyperoxia (29,30),
through stimulation of DNA repair and antioxidant mechanisms, and
prolonged cell survival. Conversely, JNK1/2 and p38 kinase have
been most frequently reported to have roles in induction of
apoptotic responses (31). The
present study indicated that PLGF may be able to regulate
hyperoxia-induced lung injury in rats via the p38 MAPK pathway.
It had been reported that PLGF promoted migration
through regulating EMT-related protein expression in cervical
cancer (19). In the presented
study, the activation of p38 in hyperoxia-induced lung tissue was
demonstrated, which may have harmful effects on the lung cells. It
is possible that the different types of cells have the different
ability of resistance to injury. Further investigation will be done
using the alveolar epithelial cell lines such as RLE-6TN to better
dissect the role of the signaling axis during hyperoxia-induced
lung injury.
A previous study has revealed that PLGF expression
increased in hyperoxia-exposed primary AECIIs (24), which contributed to
hyperoxia-induced lung injury through the promotion of apoptosis
and EMT. These data suggested that PLGF may be a potential
therapeutic target for hyperoxia-induced lung injury. In the
present study, the role of PLGF in hyperoxia-induced pulmonary
dysplasia was examined using a neonatal SD rat model, with a focus
on the alterations of lung tissue in vivo. To further
investigate the role of PLGF in hyperoxia-induced pulmonary injury,
lentiviral plasmids were used to silence the PLGF gene and the
effects on lung injury were examined in vivo. These results
indicated that PLGF knockdown in vivo may attenuate lung
tissue injury under hyperoxia, which was predominantly due to the
depletion of PLGF inhibiting p38 MAPK-mediated EMT. Additionally,
treating cultured lung cells with an ERK inhibitor, such as
PD98059, will be necessary to delineate the signaling axis in
future studies.
In summary, the results of the present in
vivo study further supported previous in vitro findings,
which suggested that the reduction of PLGF, using RNA
interference-based gene silencing, maybe a potential method to use
to reduce hyperoxia-induced lung injury.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Science Foundation of Liaoning (grant no. 20180530094).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
SZ and LZ designed the study. SZ and GL performed
the experiments. HMW and LZ were involved in the statistical
analyses. SZ and LZ wrote and revised the manuscript. All authors
read and approved the final manuscript, and all authors confirm its
accuracy.
Ethics approval and consent to
participate
Ethical approval for the present study was provided
by China Medical University Ethics Committee (Shenyang, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jobe AH: The new bronchopulmonary
dysplasia. Curr Opin Pediatr. 23:167–172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stoll BJ, Hansen NI, Bell EF, Walsh MC,
Carlo WA, Shankaran S, Laptook AR, Sánchez PJ, Van Meurs KP,
Wyckoff M, et al: Trends in care practices, morbidity, and
mortality of extremely preterm neonates, 1993–2012. JAMA.
314:1039–1051. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Greenough A: Long term respiratory
outcomes of very premature birth (<32 weeks). Semin Fetal
Neonatal Med. 17:73–76. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kinsella JP, Greenough A and Abman SH:
Bronchopulmonary dysplasia. Lancet. 367:1421–1431. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li J, Li Y, He H, Liu C, Li W, Xie L and
Zhang Y: Csk/Src/EGFR signaling regulates migration of
myofibroblasts and alveolarization. Am J Physiol Lung Cell Mol
Physiol. 310:L562–L571. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang H, Fu J, Xue X, Yao L, Qiao L, Hou A,
Jin L and Xing Y: Epithelial-mesenchymal transitions in
bronchopulmonary dysplasia of newborn rats. Pediatr Pulmonol.
49:1112–1123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song JS, Kang CM, Park CK, Yoon HK, Lee
SY, Ahn JH and Moon HS: Inhibitory effect of receptor for advanced
glycation end products (RAGE) on the TGF-β-induced alveolar
epithelial to mesenchymal transition. Exp Mol Med. 43:517–524.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matsuoka H, Arai T, Mori M, Goya S, Kida
H, Morishita H, Fujiwara H, Tachibana I, Osaki T and Hayashi S: A
p38 MAPK inhibitor, FR-167653, ameliorates murine bleomycin-induced
pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol.
283:L103–L112. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Feldkoren B, Hutchinson R, Rapoport Y,
Mahajan A and Margulis V: Integrin signaling potentiates
transforming growth factor-beta 1 (TGF-β1) dependent
down-regulation of E-Cadherin expression-important implications for
epithelial to mesenchymal transition (EMT) in renal cell carcinoma.
Exp Cell Res. 355:57–66. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Serrano-Gomez SJ, Maziveyi M and Alahari
SK: Regulation of epithelial-mesenchymal transition through
epigenetic and post-translational modifications. Mol Cancer.
15:182016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vandewalle C, Van Roy F and Berx G: The
role of the ZEB family of transcription factors in development and
disease. Cell Mol Life Sci. 66:773–787. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Andraweera PH, Dekker GA and Roberts CT:
The vascular endothelial growth factor family in adverse pregnancy
outcomes. Hum Reprod Update. 18:436–457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hayes Ryan D, McCarthy FP, O'Donoghue K
and Kenny LC: Placental growth factor: A review of literature and
future applications. Pregnancy Hypertens. 14:260–264. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Iwasaki H, Kawamoto A, Tjwa M, Horii M,
Hayashi S, Oyamada A, Matsumoto T, Suehiro S, Carmeliet P and
Asahara T: PlGF repairs myocardial ischemia through mechanisms of
angiogenesis, cardioprotection and recruitment of myo-angiogenic
competent marrow progenitors. PLoS One. 6:e248722011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tsao PN, Li H, Wei SC, Ko ML, Chou HC,
Hsieh WS and Hsieh FJ: Expression of angiogenic factors and their
receptors in postnatal mouse developing lung. J Formos Med Assoc.
103:137–143. 2004.PubMed/NCBI
|
|
17
|
Korevaar TI, Steegers EA, de Rijke YB,
Visser WE, Jaddoe VW, Visser TJ, Medici M and Peeters RP: Placental
angiogenic factors are associated with maternal thyroid function
and modify hCG-mediated FT4 stimulation. J Clin Endocrinol Metab.
100:E1328–E1334. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ning Q, Liu C, Hou L, Meng M, Zhang X, Luo
M, Shao S, Zuo X and Zhao X: Vascular endothelial growth factor
receptor-1 activation promotes migration and invasion of breast
cancer cells through epithelial-mesenchymal transition. PLoS One.
8:e652172013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang W, Zhu S, Liu Q, Li C and Li L:
Placenta growth factor promotes migration through regulating
epithelial-mesenchymal transition-related protein expression in
cervical cancer. Int J Clin Exp Pathol. 7:8506–8519.
2014.PubMed/NCBI
|
|
20
|
Zhang L, Zhao S, Yuan L, Wu H, Jiang H and
Luo G: Placenta growth factor contributes to cell apoptosis and
epithelial-to-mesenchymal transition in the hyperoxia-induced acute
lung injury. Life Sci. 156:30–37. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu GJ, Song PP, Zhou H, Shen XH, Wang JG,
Ma XF, Gu YJ, Liu DD, Feng AN, Qian XY and Gao X: Role of
epithelial-mesenchymal transition markers E-cadherin, N-cadherin,
β-catenin and ZEB2 in laryngeal squamous cell carcinoma. Oncol
Lett. 15:3472–3481. 2018.PubMed/NCBI
|
|
23
|
Cuadrado A and Nebreda AR: Mechanisms and
functions of p38 MAPK signalling. Biochem J. 429:403–417. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang L, Zhao S, Yuan L, Wu H, Jiang H and
Luo G: Placental growth factor triggers epithelial-to-mesenchymal
transition-like changes in rat type II alveolar epithelial cells:
Activation of nuclear factor κB signalling pathway. Basic Clin
Pharmacol Toxicol. 119:498–504. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mourani PM and Abman SH: Pulmonary
hypertension and vascular abnormalities in bronchopulmonary
dysplasia. Clin Perinatol. 42:839–855. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sun Y, Liu WZ, Liu T, Feng X, Yang N and
Zhou HF: Signaling pathway of MAPK/ERK in cell proliferation,
differentiation, migration, senescence and apoptosis. J Recept
Signal Transduct Res. 35:600–604. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hu X, Shen H, Wang Y and Zhao M: Liver X
receptor agonist TO901317 attenuates paraquat-induced acute lung
injury through inhibition of NF-κB and JNK/p38 MAPK signal
pathways. Biomed Res Int. 2017:46526952017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang L and Zhao S, Yuan L, Wu H, Jiang H,
Luo G and Zhao S: Knockdown of placental growth factor (PLGF)
mitigates hyperoxia-induced acute lung injury in neonatal rats:
Suppressive effects on NFkappaB signaling pathway. Int
Immunopharmacol. 38:167–174. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nie M, Wang Y, Lu Y, Yuan Y, Liu Y and Li
X: Protective effects of fucoidan against hyperoxic lung injury via
the ERK signaling pathway. Mol Med Rep. 17:1813–1818.
2018.PubMed/NCBI
|
|
30
|
Bao XC, Fang YQ, You P, Zhang S and Ma J:
Protective role of peroxisome proliferator-activated receptor
beta/delta in acute lung injury induced by prolonged hyperbaric
hyperoxia in rats. Respir Physiol Neurobiol. 199:9–18. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Porzionato A, Sfriso MM, Mazzatenta A,
Macchi V, De Caro R and Di Giulio C: Effects of hyperoxic exposure
on signal transduction pathways in the lung. Respir Physiol
Neurobiol. 209:106–114. 2015. View Article : Google Scholar : PubMed/NCBI
|