Introduction
Cardiac hypertrophy is characterized by an increase
in the volume of myocardial cells without cell division, resulting
in increased protein accumulation in cells and increased formation
of new sarcomeres and myofibrils. Cardiac hypertrophy induced by
prolonged stress can lead to heart failure, which may increase the
incidence and mortality rates of patients suffering from
cardiovascular diseases (1).
Therefore, understanding the mechanism of cardiac hypertrophy is
important in the field of cardiovascular diseases. Compensatory
hypertrophy after myocardial failure involves a series of complex
events at the cellular and molecular levels that may lead to
structural and functional changes in the myocardium (2). To the best of our knowledge,
treatment of advanced heart failure consists primarily of
palliative care. Therefore, understanding the signaling pathway
involved in cardiomyocyte growth may provide new therapeutic
targets for the treatment of cardiac hypertrophy.
Angiotensin II (Ang II) is an effector of the
renin-angiotensin system, and it can increase blood pressure by
inducing vasoconstriction via the activation of the angiotensin
receptor system (3). An increasing
number of studies have shown that Ang II serves an important role
in cardiac hypertrophy in vitro and in vivo (4–7). The
Ang II-mediated cardiomyocyte hypertrophy model has become an
increasingly popular model to investigate cardiac hypertrophy
(8,9). The H9c2 cell line, an established
cardiomyocyte cell line derived from embryonic rat ventricular
tissue, is an important model for studying hypertension-induced
cardiac hypertrophy (10).
Therefore, the present study constructed a model of cardiomyocyte
hypertrophy in H9c2 cells using Ang II treatment.
The transient receptor potential (TRP) channel gene
was discovered in the visual transmission system of
Drosophila (11). The
trp mutation in Drosophila, which prevents
Ca2+ signaling in the photoreceptors of
Drosophila, results in transient spikes but not sustained
spikes under continuous light stimulation (12,13).
According to amino acid sequence homology, the 28 mammalian TRP
channels are divided into seven subfamilies: TRP canonical (TRPC),
TRP ankyrin (TRPA), TRP melastatin (TRPM), TRP vanilloid (TRPV),
TRP polycystin (TRPP), TRP no mechanoreceptor potential C (TRPN)
and TRP mucolipin 7 (TRPML7) (14). The mammalian homologues that
display the greatest similarity to the Drosophila TRP
protein have been named TRP canonical channels (15). The TRPC subfamily consists of seven
subtypes (TRPC1-TRPC7), which are generally composed of
heteropolymers and are highly expressed in myocardial fibroblasts
and myocardial cells (16). TRPC
channels have six transmembrane domains, named S1-S6, and a
nonselective cation channel is formed between the S5 and S6
segments at the N-terminus, allowing cations such as calcium ions
to pass through the cell membrane (17). The N-termini of TRPC channels have
3 or 4 anchoring protein-like repeat structures, which can regulate
the release of calcium ions in the calcium pool by binding to the
anchoring protein (18). TRPC
channels are expressed in a number of organs, are important for
organogenesis, and their dysfunction may result in organ damage
(19). TRPC channel family members
are the molecular basis of receptor-operated Ca2+
channels (ROCs) and store-operated Ca2+ channels (SOCs)
on the cell membrane. TRPC3, TRPC6 and TRPC7 function as ROCs
(20), and TRPC1, TRPC4 and TRPC5
function as SOCs (21–23).
Ca2+ plays a crucial role in maintaining
cardiovascular physiological functions, such as cardiac
contractility, hemodynamic stretching, expansion and repair
(24). Malfunctions of TRPC
channels are closely associated with a number of cardiovascular
diseases (25,26). Therefore, TRPC channels have been
regarded as drug therapeutic targets for cardiac hypertrophy
(27). A number of previous
studies have demonstrated that the expression of TRPC1, TRPC5,
TRPC6 and TRPC7 are markedly upregulated in cardiac hypertrophy,
and accumulating evidence has demonstrated that TRPC channels are
related to cardiac hypertrophy (28–31).
Whether TRPC channels have a role in the development of
cardiomyocyte hypertrophy, and whether TRPC channels are involved
in the process of cardiomyocyte hypertrophy induced by Ang II
remain unclear. In addition, the potential roles of TRPC channels
in cardiomyocyte hypertrophy requires further investigation.
In the present study, the effects of three doses (1,
5 and 10 µM) of SKF-96365, a non-selective TRPC inhibitor, on Ang
II-induced cardiomyocyte hypertrophy were investigated in H9c2
cells, and its possible mechanisms were examined.
Materials and methods
Cell culture
H9c2 cardiomyocytes were obtained from Chi
Scientific, Inc., and cultured in complete high-glucose DMEM [cat.
no. 06-1055-57-1ACS; Biological Industries (BI)] with 10% FBS (cat.
no. 04-001-1ACS; BI) and 1% penicillin/streptomycin (cat. no.
03-031-1B; BI). The cells were incubated with 5% CO2 at
37°C.
Establishment of cardiomyocyte
hypertrophy
The cells were divided into four groups: i) The 0 µM
Ang II group (control); ii) the 0.01 µM Ang II group; iii) the 0.1
µM Ang II group; and iv) the 1 µM Ang II group. After treatment for
72 h, the cells were collected to detect the protein expression
levels of two factors associated with cardiomyocyte hypertrophy,
such as atrial natriuretic peptide (ANP) and α-actinin, by western
blot assay. The optimal concentration to induce cell hypertrophy in
subsequent experiments was selected as 0.1 µM because it induced
the highest expression of ANP and α-actinin compared with the other
concentrations.
Drug treatment
H9c2 cells were divided into five groups: i) The
control group; ii) the 0.1 µM Ang II group; iii) the 0.1 µM Ang
II+1 µM SKF-96365 group; iv) the 0.1 µM Ang II+5 µM SKF-96365
group; and v) the 0.1 µM Ang II+10 µM SKF-96365 group. The cells
were pretreated with SKF-96365 for 30 min and subsequently treated
with Ang II for an additional 72 h. SKF-96365 was purchased from
Selleck Chemicals (cat. no. S7999).
Measurement of cell surface area
Cells were digested into a cell suspension by 0.25%
trypsin for 30 sec at 37°C, and the degree of digestion was
controlled to avoid cell shrinkage. A total of five fields were
randomly selected for each group, and 20 cells were randomly
selected from each field for imaging. The cell surface area
(µm2) was measured using ImageJ 2× software (Rawak
Software, Inc.), and the mean value was calculated.
Detection of protein synthesis
rate
The protein synthesis rate of cells in all groups
was determined by a [3H] leucine incorporation assay
(8). The cultured cells were
treated with a [3H] leucine isotope marker (GE
Healthcare Life Sciences) 12 h before the test. The medium was
removed, and the cells were rinsed twice with PBS. The cells were
digested with 0.25% trypsin and repeatedly agitated for 4 min to
detach the cells. The cell suspension was added to a glass fiber
filter membrane to remove the liquid, dried and fixed with 5%
trichloroacetic acid for 30 min at 4°C, and the free isotope
markers were washed away. The membrane was dried and placed in a 5
ml flask with scintillation solution. The radioactive intensity was
detected by a liquid scintillation counter (MicroBeta; PerkinElmer,
Inc.). The data are expressed as counts/min.
Measurement of intracellular
Ca2+ concentration
Cells were cultured in special confocal dishes and
washed with PBS 2–3 times. Then, cells were treated with 100 µl of
a 5 µM Fluo-4/AM calcium fluorescent probe solution (Beyotime
Institute of Biotechnology), and were incubated at 37°C for 30 min.
After incubation, the cells were washed with PBS 2–3 times, and 1
ml PBS was added to the cells. The Fluo-4/AM positive cells were
detected by flow cytometry (Sysmex Partec GmbH) at an excitation
wavelength of 488 nm and an emission wavelength of 512–520 nm. The
fluorescence intensity of the cells was also quantitatively
analyzed using FlowMax version 2.8 flow cytometry software (Sysmex
Partec GmbH).
Reverse transcription-quantitative PCR
(RT-qPCR) assay
The expression of ANP, nuclear factor of activated
T-cells (NFAT), α-actinin, β-myosin heavy chain (MHC), TRPC3 and
TRPC6 at the mRNA level was determined by RT-qPCR assay. Total RNA
was obtained by TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). The RNA concentration was measured by a
Nanodrop 2000 (Thermo Fisher Scientific, Inc.). Then, cDNA was
synthesized from the total RNA with PrimeScript RT Mix Kit (Takara
Bio, Inc.) at 37°C for 15 min and then 85°C for 5 sec. The cDNA was
amplified with SYBR Green PCR Mix (Thermo Fisher Scientific, Inc.)
and an ABI 7300 system (Thermo Fisher Scientific, Inc.). The
following thermocycling conditions were used for the qPCR: Initial
denaturation, 95°C for 10 min; followed by 40 cycles of 95°C for 15
sec and 60°C for 60 sec. The expression levels were normalized to
that of GAPDH. The primers for qPCR are as follows: TRPC3-forward
(F), 5′-TGTGGTCTGAGTGCAAGGAG-3′ and TRPC3-reverse (R),
5′-ACCTCTGGTGGGAGTGTGAC-3′; TRPC6-F, 5′-TTTGCTGAAGGCAAGAGGTT-3′ and
TRPC6-R, 5′-TTGTTTCTGGCTGCATTCTG-3′; ANP-F,
5′-ATACAGTGCGGTGTCCAACA-3′ and ANP-R, 5′-CGAGAGCACCTCCATCTCTC-3′;
brain natriuretic peptide (BNP)-F, 5′-GGAAATGGCTCAGAGACAGC-3′ and
BNP-R, 5′-CGATCCGGTCTATCTTCTGC-3′; β-MHC-F,
5′-CCTCGCAATATCAAGGGAAA-3′ and β-MHC-R, 5′-TACAGGTGCATCAGCTCCAG-3′;
GAPDH-F, 5′-CTCATGACCACAGTCCATGC-3′ and GAPDH-R,
5′-TTCAGCTCTGGGATGACCTT-3′.
Western blotting assay
Protein was extracted with RIPA lysis buffer
(Beyotime Institute of Biotechnology). Total protein was quantified
using a Bicinchoninic Acid Assay kit (Beyotime Institute of
Biotechnology). A total of 40 µg of protein was used for 10%
SDS-PAGE and transferred to PVDF membranes. Subsequently, the
membranes were blocked with 5% skim milk at 4°C overnight. The
membranes were incubated with the following primary antibodies
overnight at 4°C: anti-ANP (1:1,000; cat. no. A14755; ABclonal
Biotech Co., Ltd.), anti-NFAT2 (1:1,000; cat. no. A1539; ABclonal
Biotech Co., Ltd.), anti-α-actinin (1:1,000; cat. no. sc-17829;
Santa Cruz Biotechnology, Inc.), anti-β-MHC (1:1,000; cat. no.
A7564; ABclonal Biotech Co., Ltd.), anti-TRPC3 (1:1,000; cat. no.
A7742; ABclonal Biotech Co., Ltd.), anti-TRPC6 (1:1,000; cat. no.
bs-2393R; BIOSS) and anti-GAPDH (1:2,000; cat. no. AC033; ABclonal
Biotech Co., Ltd.). Subsequently, the membranes were incubated with
a horseradish peroxidase (HRP) conjugated goat anti-rabbit
immunoglobulin (Ig)G secondary antibody (1:5,000; cat. no.
bs-0295G-HRP; BIOSS) or HRP-conjugated goat anti-mouse IgG
secondary antibody (1:5,000; cat. no. AS003; ABclonal Biotech Co.,
Ltd.) for 2 h at room temperature. The membranes were detected with
Immobilon Western HRP substrate (EMD Millipore). The bands were
semi-quantified by ImageJ 2× software (Rawak Software, Inc.).
Immunofluorescence assay
A total of 1×105 cells were seeded in
6-well plates. After treatment, the cells were fixed in 4%
paraformaldehyde (PFA) for 30 min at room temperature,
permeabilized with 0.2% Triton X-100 for 5 min at room temperature
and then blocked with 5% BSA for 30 min at room temperature. Then,
the cells were incubated with the α-actinin antibody (1:100) in
PBS, a FITC-conjugated goat anti-mouse IgG antibody (1:50; cat. no.
AS001; ABclonal Biotech Co., Ltd.) for 30 min at room temperature,
and DAPI (Beyotime Institute of Biotechnology) for 5 min at room
temperature. The cells from the different groups were observed by
fluorescence microscopy (magnification, ×200; Nikon
Corporation).
Statistical analysis
Data are presented as the mean ± SD. Statistical
analysis was determined by one-way ANOVA, followed by Tukey's
multiple comparisons test with GraphPad Prism software (version
5.0a; GraphPad Software, Inc.). P<0.05 was considered to
indicate a statistically significant difference.
Results
Cardiomyocyte hypertrophy markers ANP
and α-actinin are increased after Ang II treatment, and TRPC
channels are involved in this process
The effect of Ang II on the cardiomyocyte
hypertrophy markers ANP and α-actinin in H9c2 cells was detected by
western blot assay. As shown in Fig.
1A, Ang II (0.01 and 0.1 µM) significantly promoted the protein
expression levels of ANP and α-actinin. The semi-quantitative
results of the western blot assay identified that the expression
levels of ANP and α-actinin were the highest after treatment with
0.1 µM Ang II (Fig. 1B and C).
According to the results of the western blot analysis, the
expression levels of TRPC3 and TRPC6 were higher in the Ang
II-treated groups compared with the control group, and their
expression levels were higher in the 0.1 µM Ang II-treated group
than in the 0.01 µM Ang II-treated group. However, there was no
difference between the 0.1 µM Ang II-treated group and the 1 µM Ang
II-treated group (Fig. 1A, D and
E). The present results indicated that TRPC channels may be
associated with the process of Ang II-induced cardiomyocyte
hypertrophy in H9c2 cells. Moreover, 0.1 µM Ang II for 72 h was the
optimal concentration for the induction of cardiomyocyte
hypertrophy markers ANP and α-actinin in H9c2 cells.
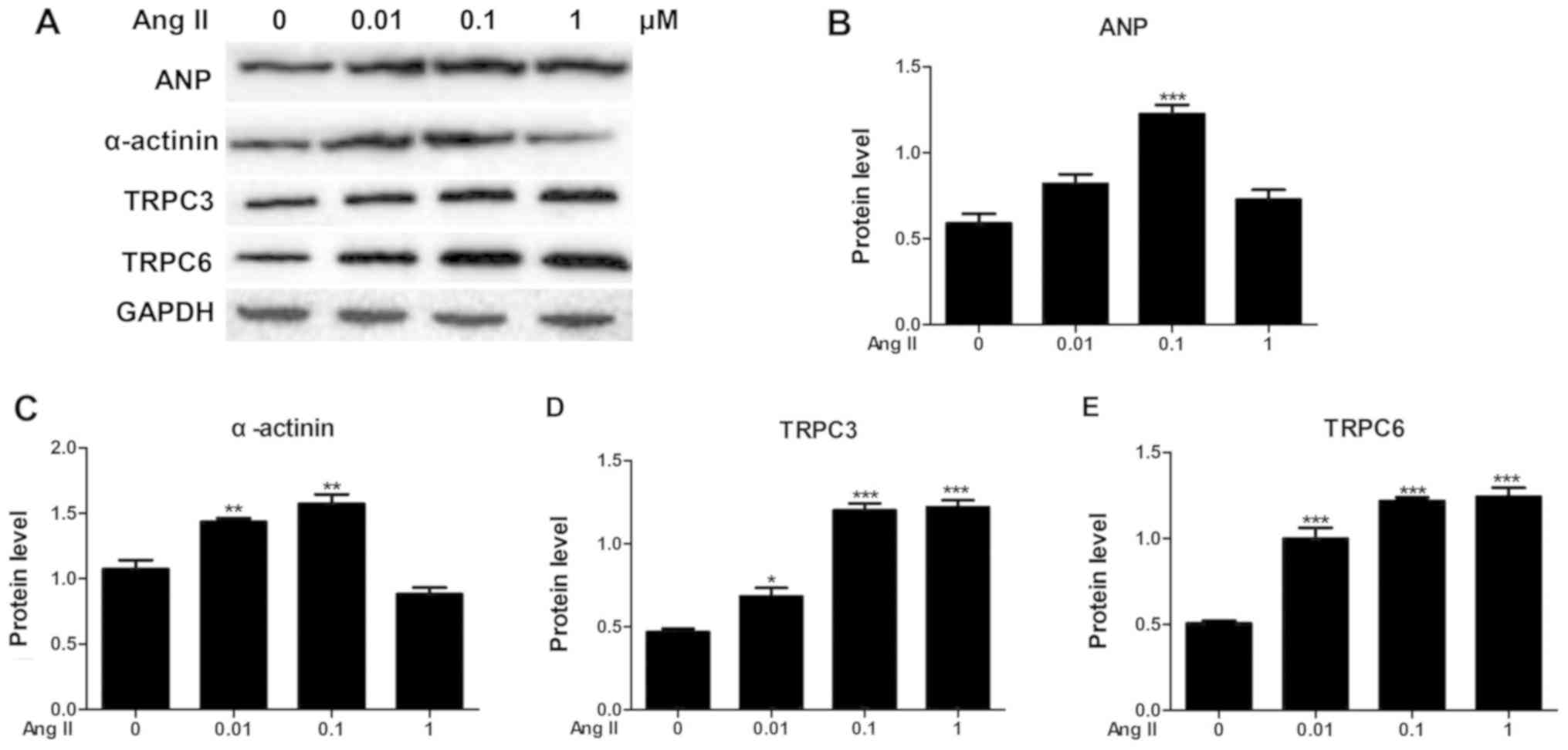 | Figure 1.Ang II induces the protein expression
levels of ANP, α-actinin, TRPC3 and TRPC6 in H9c2 cells. (A)
Protein levels of ANP, α-actinin, TRPC3 and TRPC6 in H9c2 cells
treated with different concentrations of Ang-II were detected by
western blotting. (B-E) Relative semi-quantitative analysis of (B)
ANP, (C) α-actinin, (D) TRPC3 and (E) TRPC6 protein expression in
all groups. *P<0.05, **P<0.01, ***P<0.001 vs. 0 µM group.
Ang II, angiotensin II; ANP, atrial natriuretic peptide; TRPC,
transient receptor potential canonical. |
SKF-96365 inhibits the expression
levels of TRPC3, TRPC6 and cardiomyocyte hypertrophy marker genes
in Ang-II-treated H9c2 cells
To determine the inhibitory role of the TRPC channel
inhibitor SKF-96365 on cardiomyocyte hypertrophy in H9c2 cells
treated with Ang-II, the expression levels of two genes encoding
for TRPC channels, TRPC3 and TRPC6, and the cardiomyocyte
hypertrophy markers ANP, BNP, and β-MHC by qPCR and western
blotting. As shown in Fig. 2A, the
present qPCR results showed that SKF-96365 significantly suppressed
the increased expression of TRPC3, TRPC6, ANP, BNP and β-MHC
induced by Ang II. The western blot results were consistent with
the qPCR results (Fig. 2B and C).
These results suggested that SKF-96365 suppressed the expression
levels of cardiomyocyte hypertrophy markers in Ang II-treated H9c2
cells.
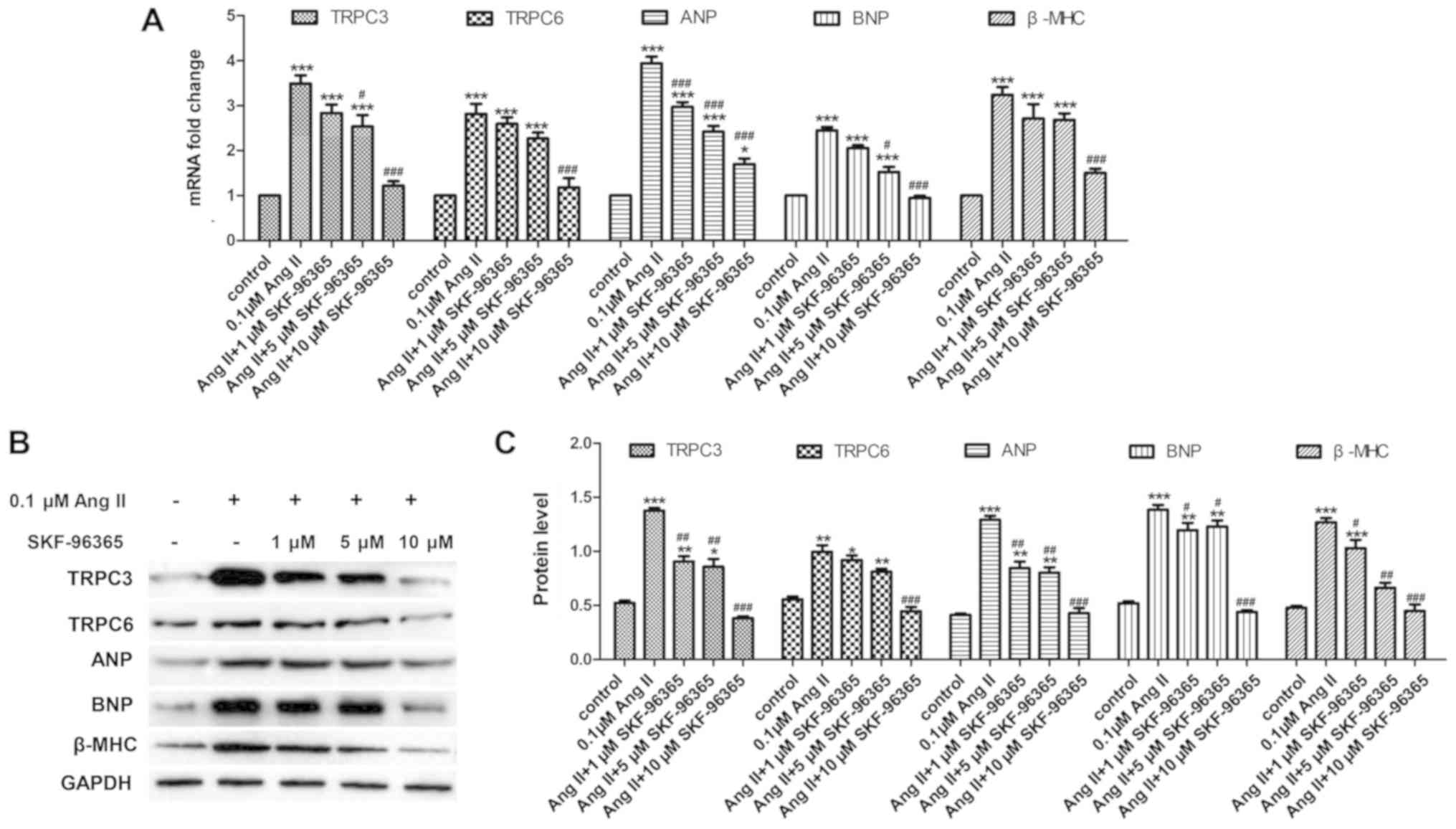 | Figure 2.SKF-96365 inhibits the increase in
expression levels of TRPC3, TRPC6, ANP, BNP and β-MHC induced by
Ang II treatment. TRPC3, TRPC6, ANP, BNP and β-MHC mRNA levels in
H9c2 cells treated with different concentrations of SKF-96365 were
detected by (A) quantitative PCR and (B) western blotting. (C)
Relative semi-quantitative analysis of TRPC3, TRPC6, ANP, BNP and
β-MHC protein expression in all groups. *P<0.05, **P<0.01,
***P<0.001 vs. control group; #P<0.05,
##P<0.01, ###P<0.001 vs. 0.1 µM Ang II
group. Ang II, Angiotensin II; ANP, atrial natriuretic peptide;
BNP, brain natriuretic peptide; β-MHC, β-myosin heavy chain; TRPC,
transient receptor potential canonical. |
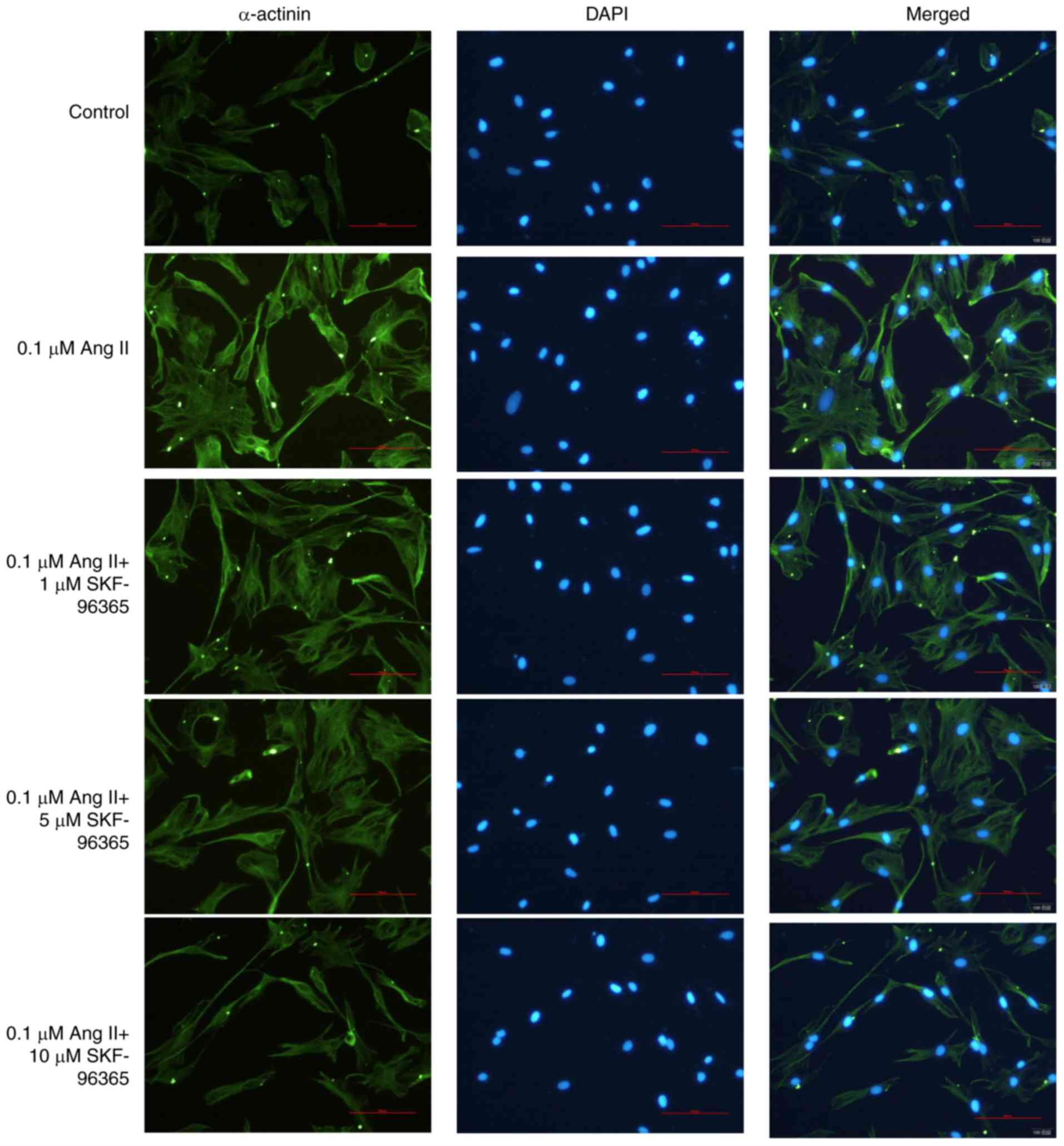
SKF-96365 inhibits Ang II-induced
cardiomyocyte hypertrophy
To demonstrate the potential inhibitory role of
SKF-96365 in cardiomyocyte hypertrophy induced by Ang II, the
protein level of α-actinin was examined by immunofluorescence. In
addition, the H9c2 cell surface area and the [3H]
leucine incorporation rate were investigated in all groups. As
shown in Fig. 3, H9c2
cardiomyocytes were attached and exhibited a triangular or
irregular shape, and the α-actinin protein was localized in the
cytoplasm. Ang II (0.1 µM) markedly increased the size of the
cardiomyocytes and the level of α-actinin. However, SKF-96365
markedly decreased the size of the cardiomyocytes and the level of
α-actinin in a dose-dependent manner (Figs. 3 and 4). These results suggested that the TRPC
channel inhibitor SKF-96365 may suppress Ang II-induced
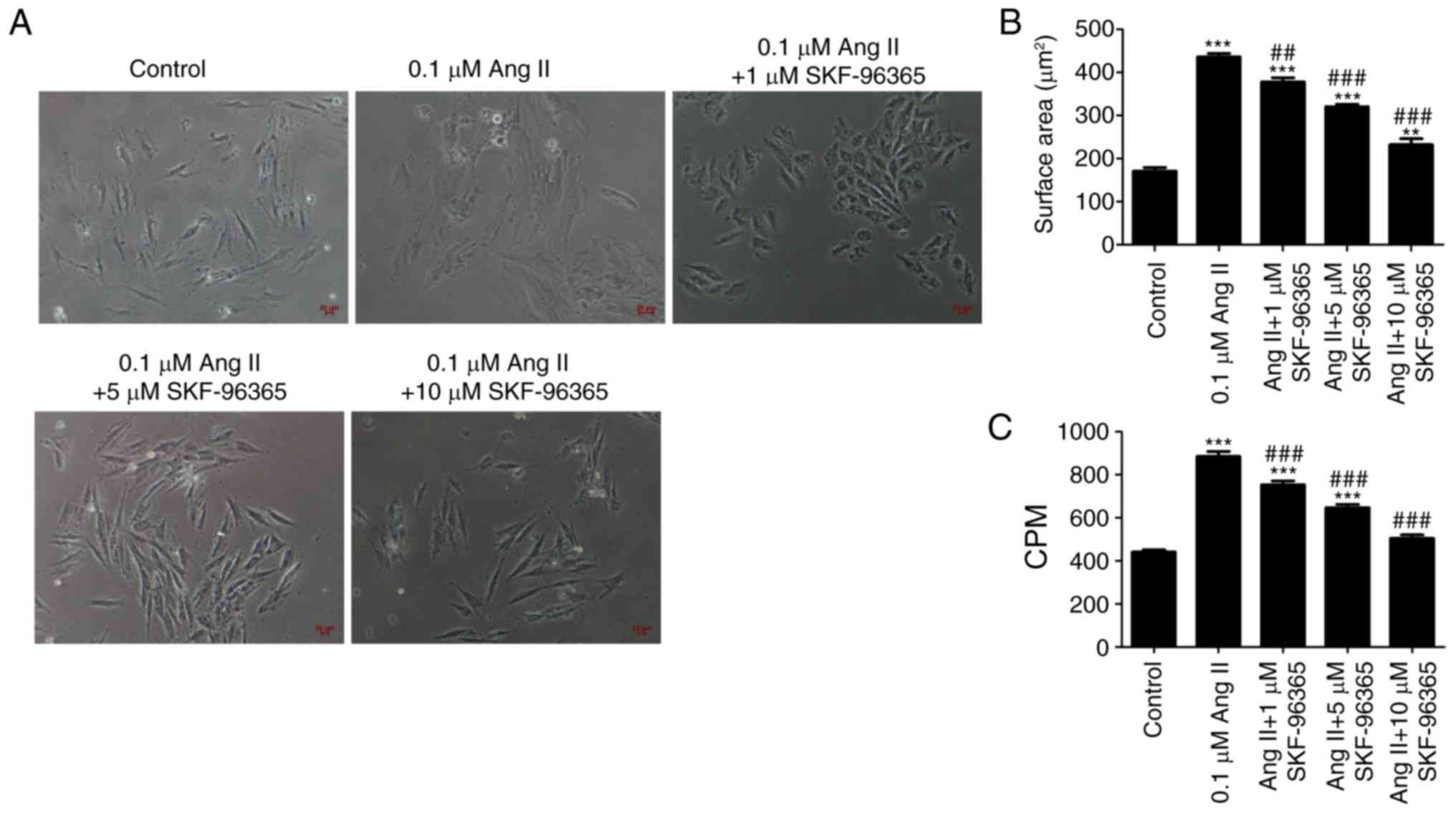
cardiomyocyte hypertrophy. As shown in Fig. 4A and B, Ang II (0.1 µM) increased
the surface area of H9c2 cells, and SKF-96365 significantly
attenuated the increased surface area of H9c2 cells induced by Ang
II. Ang II (0.1 µM) also induced an increase in the [3H]
leucine incorporation rate compared with the control group, but
SKF-96365 inhibited the increased [3H] leucine
incorporation rate induced by Ang II in a dose-dependent manner
(Fig. 4C). The present results
suggested that blocking TRPC channels inhibited cardiomyocyte
hypertrophy induced by Ang II.
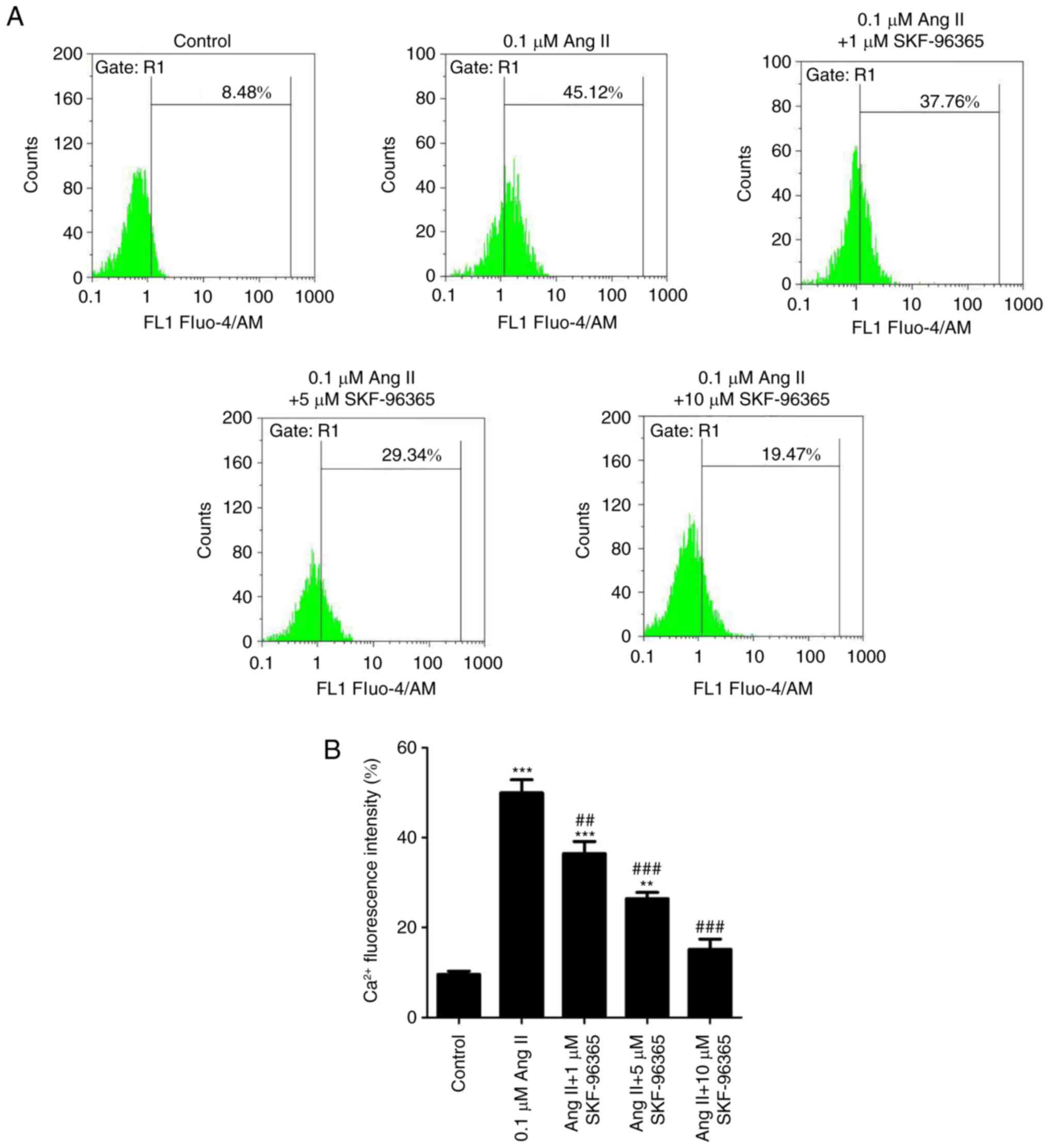
SKF-96365 inhibits Ang II-induced
intracellular Ca2+ rise
The effect of SKF-96365 on the Ang II-induced
Ca2+ increase was determined using the Ca2+
indicator Fluo-4/AM and flow cytometry. The Fluo-4 percentage is an
indicator of the intracellular Ca2+ concentration.
Ang-II (0.1 µM) significantly increased the intracellular
Ca2+ concentration compared with the control group, but
SKF-96365 inhibited the increased Ca2+ concentration
induced by Ang-II in a dose-dependent manner (Fig. 5A). In addition, the Ca2+
levels were quantified by flow cytometry (Fig. 5B).
Discussion
Pathological myocardial hypertrophy, defined as
myocardial cell enlargement and cardiac systolic dysfunction, is a
risk factor for cardiovascular diseases, and may cause severe
arrhythmia and heart failure (32,33).
However, the molecular mechanisms involved in cardiac hypertrophy
are still unclear. In the present study, the effects of SKF-96365,
a non-selective TRPC inhibitor, on Ang II-induced cardiomyocyte
hypertrophy were investigated in H9c2 cells.
The TRP family is a superfamily of nonselective
cation-permeable channels that have a crucial role in the
pathophysiological process of various diseases (34–36).
According to amino acid sequence homology, the mammalian TRP
channel superfamily has been divided into seven subfamilies: TRPC,
TRPM, TRPA, TRPV, TRPP, TRPML and TRPN (37). TRPC channels mediate
Ca2+ influx controlled by the calcium reservoir
(38). Previous studies have found
that the TRPC family is associated with the regulation of tumor
growth, invasion and metastasis (39,40),
is involved in the occurrence and development of central nervous
system diseases (41), and plays a
role in primary hypertension and myocardial cell apoptosis
(42). It has been reported that
miR-103 exerts an inhibitory effect on cardiac hypertrophy by
reducing cardiac autophagy through TRPV3 (43). TRPA1 inhibition can ameliorate
cardiac hypertrophy and fibrosis induced by increased blood
pressure in mice (44). In
addition, TRPC1 knockdown protects heart function and morphology in
mouse models of pressure overload (45). Moreover, the inhibition of TRPC6
has antihypertrophic results on the activity of the cardiac
ANP/BNP-GC-A pathway (46). In the
present study, the inhibition of TRPC channels suppressed the
hypertrophy of H9c2 cells induced by Ang II by decreasing the
concentration of Ca2+. Accumulating evidence has
indicated that TRPC channels have a pivotal function in the process
of cardiac hypertrophy (47,48).
SKF-96365 is a non-selective TRPC channel blocker
(49). It was first recommended as
an inhibitor of ionotropic receptor-mediated Ca2+ entry
(50). A previous study reported
that SKF-96365 suppresses voltage-gated sodium currents in rat
ventricular myocytes (51). The
present results show that SKF-96365 strongly reduces the
Ca2+ concentration.
Ang II serves an important role in promoting the
hypertrophy of myocardial cells, which can increase myocardial cell
volume and total protein content, without affecting the number of
cells (52,53). Previous studies have indicated that
Ang II directly regulates myocardial contractility and hypertrophic
growth (54). The present results
suggest that Ang II induced high levels of TRPC3 and TRPC6 in H9c2
cells. A previous study showed that TRPC3 and TRPC6 are crucial for
Ang II-induced cardiac hypertrophy (55), consistent with the present results.
Harada et al (56) found
that TRPC3 is highly expressed in freshly harvested rat cardiac
fibroblasts and that pyrazole-3, as a selective TRPC3 channel
blocker, inhibits Ang II-induced calcium ion flow, thus reducing
fibroblast proliferation. The present results indicated that
SKF-96365, a nonselective TRPC inhibitor, suppressed cardiomyocyte
hypertrophy markers and TRPC3 and TRPC6 expression. In a previous
study, Gao et al (57)
demonstrated that Nifedipine is more efficient than SKF-96365 at
blocking Ca2+ influx and cardiac hypertrophy. Because
this previous study focused on the source of hypertrophic
Ca2+, the Nifedipine (L-type Ca2+ channel
antagonist), SKF-96365 (TRP channel antagonist) and Nickel (T-type
Ca2+ channel antagonist) were chosen to reduce the
levels of Ca2+. The present study aimed to investigate
the role of a TRPC channel inhibitor in cardiomyocyte hypertrophy
induced by Ang II. Therefore, the TRPC channel inhibitor SKF-96365
was selected to block the TRPC channel pathway. SKF-96365 was
initially used as a Ca2+ blocker; however, it was later
used as a TRPC channel blocker (58). In the present study, the role of
SKF-96365 was investigated in myocardial hypertrophy, and the
present results provided a theoretical basis for the use of
SKF-96365 in clinical practice.
In the present study a cardiomyocyte hypertrophy
model was established using H9c2 cells treated with 0.1 µM Ang II.
After treatment with 1, 5 or 10 µM SKF-96365, the expression levels
of various cardiomyocyte hypertrophy markers were investigated,
including ANP, BNP, and β-MHC, and the TRPC channel-related genes
TRPC3 and TRPC6 were investigated. In addition, the fluorescence
intensity of α-actinin, the cell surface area, the protein
synthesis rate, and the intracellular Ca2+ concentration
were examined. SKF-96365 was found to decrease the expression
levels of ANP, BNP, β-MHC, TRPC3 and TRPC6 induced by Ang II in a
dose-dependent manner. SKF-96365 significantly suppressed the
increased cell surface area and the protein synthesis rate induced
by Ang II. Furthermore, the intracellular Ca2+
concentration was decreased by SKF-96365 treatment. The present
data suggested that SKF-96365 inhibited Ang II-induced
cardiomyocyte hypertrophy by decreasing the intracellular
Ca2+ concentration. Therefore, the non-selective TRPC
inhibitor SKF-96365 may be considered as a potential treatment for
myocardial hypertrophy.
Acknowledgements
Not applicable.
Funding
This work was supported by the Special Project of
Yunnan Science and Technology Department-Kunming Medical University
Applied Basic Research [grant no. 2017FE467(−095)] and the Open
Subject of Key Laboratory of Cancer Immune Prevention and Control
in Yunnan Province (grant no. 2017DG004-06).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HC, JL and YZ conceived and designed the study. QW,
XZ, YG, QY, NS and MH performed the experiments. HC, JL, XZ, QW and
QY analyzed the data. YZ, HC, XZ and YG wrote the manuscript. YZ
and HC reviewed and edited the manuscript. All authors read and
approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Messerli FH, Aristizabal D and Soria F:
Reduction of left ventricular hypertrophy: How beneficial. Am Heart
J. 125:1520–1524. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Oka T, Akazawa H, Naito AT and Komuro I:
Angiogenesis and cardiac hypertrophy: Maintenance of cardiac
function and causative roles in heart failure. Circ Res.
114:565–571. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ito M, Oliverio MI, Mannon PJ, Best CF,
Maeda N, Smithies O and Coffman TM: Regulation of blood pressure by
the type 1A angiotensin II receptor gene. Proc Natl Acad Sci USA.
92:3521–3525. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sadoshima J, Xu Y, Slayter HS and Izumo S:
Autocrine release of angiotensin II mediates stretch-induced
hypertrophy of cardiac myocytes in vitro. Cell. 75:977–984. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim S, Ohta K, Hamaguchi A, Yukimura T,
Miura K and Iwao H: Angiotensin II induces cardiac phenotypic
modulation and remodeling in vivo in rats. Hypertension.
25:1252–1259. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mollmann H, Schmidt-Schweda S, Nef H,
Möllmann S, Burstin JV, Klose S, Elsässer A and Holubarsch CJ:
Contractile effects of angiotensin and endothelin in failing and
non-failing human hearts. Int J Cardiol. 114:34–40. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fabris B, Candido R, Bortoletto M,
Zentilin L, Sandri M, Fior F, Toffoli B, Stebel M, Bardelli M,
Belgrado D, et al: Dose and time-dependent apoptotic effects by
angiotensin II infusion on left ventricular cardiomyocytes. J
Hypertens. 25:1481–1490. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guo H, Liu B, Hou L, The E, Li G, Wang D,
Jie Q, Che W and Wei Y: The role of mAKAPβ in the process of
cardiomyocyte hypertrophy induced by angiotensin II. Int J Mol Med.
35:1159–1168. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bai Y, Sun X, Chu Q, Li A, Qin Y, Li Y,
Yue E, Wang H, Li G, Zahra SM, et al: Caspase-1 regulates Ang
II-induced cardiomyocyte hypertrophy via up-regulation of IL-1β.
Biosci Rep. Apr 27–2018.(Epub ahead of print). View Article : Google Scholar
|
|
10
|
Kimes BW and Brandt BL: Properties of a
clonal muscle cell line from rat heart. Exp Cell Res. 98:367–381.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cosens DJ and Manning A: Abnormal
electroretinogram from a Drosophila mutant. Nature.
224:285–287. 1969. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pak WL, Grossfield J and Arnold KS:
Mutants of the visual pathway of Drosophila melanogaster.
Nature. 227:518–520. 1970. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hotta Y and Benzer S: Genetic dissection
of the Drosophila nervous system by means of mosaics. Proc
Natl Acad Sci USA. 67:1156–1163. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nilius B and Szallasi A: Transient
receptor potential channels as drug targets: From the science of
basic research to the art of medicine. Pharmacol Rev. 66:676–814.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Montell C, Birnbaumer L, Flockerzi V,
Bindels RJ, Bruford EA, Caterina MJ, Clapham DE, Harteneck C,
Heller S, Julius D, et al: A unified nomenclature for the
superfamily of TRP cation channels. Mol Cell. 9:229–231. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiang Y, Huang H, Liu P, Wei H, Zhao H,
Feng Y, Wang W and Niu W: Expression and localization of TRPC
proteins in rat ventricular myocytes at various developmental
stages. Cell Tissue Res. 355:201–212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mu YP, Lin DC, Yan FR, Jiao HX, Gui LX and
Lin MJ: Alterations in caveolin-1 expression and receptor-operated
Ca2+ entry in the aortas of rats with pulmonary hypertension. Cell
Physiol Biochem. 39:438–452. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huynh KW, Cohen MR, Chakrapani S, Holdaway
HA, Stewart PL and Moiseenkova-Bell VY: Structural insight into the
assembly of TRPV channels. Structure. 22:260–268. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tai Y, Yang S, Liu Y and Shao W: TRPC
Channels in Health and Disease. Adv Exp Med Biol. 976:35–45. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kwon J, An H, Sa M, Won J, Shin JI and Lee
CJ: Orai1 and orai3 in combination with stim1 mediate the majority
of store-operated calcium entry in astrocytes. Exp Neurobiol.
26:42–54. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li N, Si B, Ju JF, Zhu M, You F, Wang D,
Ren J, Ning YS, Zhang FQ, Dong K, et al: Nicotine induces
cardiomyocyte hypertrophy through trpc3-mediated Ca2+
NFAT signalling pathway. Can J Cardiol. 32:1260.e1–1260.e10. 2016.
View Article : Google Scholar
|
|
22
|
Antigny F, Sabourin J, Sauc S, Bernheim L,
Koenig S and Frieden M: TRPC1 and TRPC4 channels functionally
interact with STIM1L to promote myogenesis and maintain fast
repetitive Ca2+ release in human myotubes. Biochim
Biophys Acta Mol Cell Res. 1864:806–813. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ambudkar IS, de Souza LB and Ong HL:
TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell
Calcium. 63:33–39. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pecoraro M, Pinto A and Popolo A:
Mitochondria and cardiovascular disease: A Brief Account. Crit Rev
Eukaryot Gene Expr. 29:295–304. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yue Z, Xie J, Yu AS, Stock J, Du J and Yue
L: Role of TRP channels in the cardiovascular system. Am J Physiol
Heart Circ Physiol. 308:H157–182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eder P: Cardiac Remodeling and Disease:
SOCE and TRPC Signaling in Cardiac Pathology. Adv Exp Med Biol.
993:505–521. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Falcón D, Galeano-Otero I,
Calderón-Sánchez E, Del Toro R, Martín-Bórnez M, Rosado JA, Hmadcha
A and Smani T: TRP Channels: Current perspectives in the adverse
cardiac remodeling. Front Physiol. 10:1592019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ohba T, Watanabe H, Murakami M, Takahashi
Y, Iino K, Kuromitsu S, Mori Y, Ono K, Iijima T and Ito H:
Upregulation of TRPC1 in the development of cardiac hypertrophy. J
Mol Cell Cardiol. 42:498–507. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sunggip C, Shimoda K, Oda S, Tanaka T,
Nishiyama K, Mangmool S, Nishimura A, Numaga-Tomita T and Nishida
M: TRPC5-eNOS axis negatively regulates ATP-induced cardiomyocyte
hypertrophy. Front Pharmacol. 9:5232018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kuwahara K, Wang Y, McAnally J, Richardson
JA, Bassel-Duby R, Hill JA and Olson EN: TRPC6 fulfills a
calcineurin signaling circuit during pathologic cardiac remodeling.
J Clin Invest. 116:3114–3126. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Satoh S, Tanaka H, Ueda Y, Oyama J, Sugano
M, Sumimoto H, Mori Y and Makino N: Transient receptor potential
(TRP) protein 7 acts as a G protein-activated Ca2+ channel
mediating angiotensin II-induced myocardial apoptosis. Mol Cell
Biochem. 294:205–215. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lyon RC, Zanella F, Omens JH and Sheikh F:
Mechanotransduction in cardiac hypertrophy and failure. Circ Res.
116:1462–1476. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hou J and Kang YJ: Regression of
pathological cardiac hypertrophy: Signaling pathways and
therapeutic targets. Pharmacol Ther. 135:337–354. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li H: TRP Channel Classification. Adv Exp
Med Biol. 976:1–8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sawamura S, Shirakawa H, Nakagawa T, Mori
Y and Kaneko S: Frontiers in Neuroscience TRP Channels in the
Brain: What Are They There For? Neurobiology of TRP Channels. Emir
TLR: 2nd. CRC Press/Taylor & Francis; Boca Raton, FL: pp.
295–322. 2017, View Article : Google Scholar
|
|
36
|
Caterina MJ and Pang Z: TRP Channels in
Skin Biology and Pathophysiology. Pharmaceuticals (Basel). 9(pii):
E772016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pedersen SF, Owsianik G and Nilius B: TRP
channels: An overview. Cell Calcium. 38:233–252. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee HC, Yoon SY, Lykke-Hartmann K, Fissore
RA and Carvacho I: TRPV3 channels mediate Ca2+ influx
induced by 2-APB in mouse eggs. Cell Calcium. 59:21–31. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
El Boustany C, Bidaux G, Enfissi A,
Delcourt P, Prevarskaya N and Capiod T: Capacitative calcium entry
and transient receptor potential canonical 6 expression control
human hepatoma cell proliferation. Hepatology. 47:2068–2077. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Song MY and Yuan JX: Introduction to TRP
channels: Structure, function, and regulation. Adv Exp Med Biol.
661:99–108. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Greka A, Navarro B, Oancea E, Duggan A and
Clapham DE: TRPC5 is a regulator of hippocampal neurite length and
growth cone morphology. Nat Neurosci. 6:837–845. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lepage PK and Boulay G: Molecular
determinants of TRP channel assembly. Biochem Soc Trans. 35:81–83.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Qi H, Ren J, E M, Zhang Q, Cao Y, Ba L,
Song C, Shi P, Fu B and Sun H: MiR-103 inhibiting cardiac
hypertrophy through inactivation of myocardial cell autophagy via
targeting TRPV3 channel in rat hearts. J Cell Mol Med.
23:1926–1939. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang Z, Xu Y, Wang M, Ye J, Liu J, Jiang
H, Ye D and Wan J: TRPA1 inhibition ameliorates pressure
overload-induced cardiac hypertrophy and fibrosis in mice.
EBioMedicine. 36:54–62. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Seth M, Zhang ZS, Mao L, Graham V, Burch
J, Stiber J, Tsiokas L, Winn M, Abramowitz J, Rockman HA, et al:
TRPC1 channels are critical for hypertrophic signaling in the
heart. Circ Res. 105:1023–1030. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kinoshita H, Kuwahara K, Nishida M, Jian
Z, Rong X, Kiyonaka S, Kuwabara Y, Kurose H, Inoue R, Mori Y, et
al: Inhibition of TRPC6 channel activity contributes to the
antihypertrophic effects of natriuretic peptides-guanylyl cyclase-A
signaling in the heart. Circ Res. 106:1849–1860. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Watanabe H, Iino K, Ohba T and Ito H:
Possible involvement of TRP channels in cardiac hypertrophy and
arrhythmia. Curr Top Med Chem. 13:283–294. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Eder P and Molkentin JD: TRPC channels as
effectors of cardiac hypertrophy. Circ Res. 108:265–272. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ding J, Xiao Y, Lu D, Du YR, Cui XY and
Chen J: Effects of SKF-96365, a TRPC inhibitor, on melittin-induced
inward current and intracellular Ca2+ rise in primary sensory
cells. Neurosci Bull. 27:135–142. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Merritt JE, Armstrong WP, Benham CD,
Hallam TJ, Jacob R, Jaxa-Chamiec A, Leigh BK, McCarthy SA, Moores
KE and Rink TJ: SK&F 96365, a novel inhibitor of
receptor-mediated calcium entry. Biochem J. 271:515–522. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen KH, Liu H, Yang L, Jin MW and Li GR:
SKF-96365 strongly inhibits voltage-gated sodium current in rat
ventricular myocytes. Pflugers Arch. 467:1227–1236. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hartman JC: The role of bradykinin and
nitric oxide in the cardioprotective action of ACE inhibitors. Ann
Thorac Surg. 60:789–792. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Weber KT, Brilla CG and Janicki JS:
Myocardial fibrosis: Functional significance and regulatory
factors. Cardiovasc Res. 27:341–348. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rohini A, Agrawal N, Koyani CN and Singh
R: Molecular targets and regulators of cardiac hypertrophy.
Pharmacol Res. 61:269–280. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Onohara N, Nishida M, Inoue R, Kobayashi
H, Sumimoto H, Sato Y, Mori Y, Nagao T and Kurose H: TRPC3 and
TRPC6 are essential for angiotensin II-induced cardiac hypertrophy.
EMBO J. 25:5305–5316. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Harada M, Luo X, Qi XY, Tadevosyan A,
Maguy A, Ordog B, Ledoux J, Kato T, Naud P, Voigt N, et al:
Transient receptor potential canonical-3 channel-dependent
fibroblast regulation in atrial fibrillation. Circulation.
126:2051–2064. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gao H, Wang F, Wang W, Makarewich CA,
Zhang H, Kubo H, Berretta RM, Barr LA, Molkentin JD and Houser SR:
Ca(2+) influx through L-type Ca(2+) channels and transient receptor
potential channels activates pathological hypertrophy signaling. J
Mol Cell Cardiol. 53:657–667. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Singh A, Hildebrand ME, Garcia E and
Snutch TP: The transient receptor potential channel antagonist
SKF96365 is a potent blocker of low-voltage-activated T-type
calcium channels. Br J Pharmacol. 160:1464–1475. 2010. View Article : Google Scholar : PubMed/NCBI
|