Introduction
Alzheimer's disease (AD) is a common progressive
neurodegenerative disease associated with increasing age, which is
characterized by the abnormal deposition of senile plaques and
intracellular neurofibrillary tangles owing to the formation of
extracellular amyloid β (Aβ) protein and hyperphosphorylation of
tau protein in the brain (1).
Enhanced brain Aβ and amyloid precursor protein (APP) aggregation
is closely associated with the development of AD (2). Extracellular amyloid plaques
identified in AD, including Aβ1-40 and
Aβ1-42, are derived from the larger APP by β-secretase
and γ-secretase (3). An inhibitor
of γ-secretase, semagacestat, has been reported to cause
neurotoxicity, thus its clinical application is limited; therefore,
a safe and effective suppressor of β-secretase may be considered a
promising treatment approach (4,5).
Galangin is a natural flavonol, which is extracted
from Alpinia officinarum (6). It has been reported that galangin
exerts anti-inflammatory effects on mast cell-derived allergies and
collagen-induced arthritis via the suppression of receptor
activator of nuclear factor (NF)-κB ligand-induced activation of
Janus kinase, p38 and NF-κB pathways (7). In addition to these effects, galangin
has been considered as a potential drug candidate for AD therapy.
It may decrease levels of β-secretase and acetylated H3 in the
β-secretase promoter regions via upregulation of endogenous histone
deacetylase 1 (HDAC1)-mediated deacetylation in SH-SY5Y cells
(8).
It has been reported that there is a close
association between abnormal degradation of misfolded proteins and
AD pathogenesis (9);
overaccumulation of Aβ can lead to it autonomously aggregating into
oligomers, which block proteasome function and clog neuronal
processes (10).
Hyperphosphorylation of tau means it cannot bind to microtubules,
thus resulting in self-aggregation of tau into neurofibrillary
tangles, which injure axonal transport and normal neuronal function
(11). The effects of autophagy in
AD are contradictory, or the level of autophagy initiation varies
with disease progression. On the one hand, the autophagy-initiation
Beclin-1 protein is reduced in early AD (12). However, transcriptional data
indicate the factors of autophagy activation were upregulation in
AD brains (13). Further studies
are required to clarify the level of autophagic activity during the
different stages of AD. Okadaic acid (OA) has also been used to
study AD in numerous species. OA is able to inhibit protein
phosphatase 2A (PP2A) activity, thereby causing tau
hyperphosphorylation in the AD brain (14). The present study aimed to determine
the potential mechanisms underlying the effects of galangin on
autophagy in OA-induced PC12 cells.
Materials and methods
Materials and reagents
Galangin (lot no. 111699-200501) was purchased from
the National Institute of Control of Pharmaceutical and Biological
Products (Beijing, China). OA (cat. no. 78111-17-8) was purchased
from Shanghai Adamas Reagent Co. Ltd. (Shanghai, China).
3-methyladenine (3MA; cat. no. M9281) and rapamycin (cat. no.
V900930) were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany). Dulbecco's modified Eagle's medium (DMEM), PBS and fetal
bovine serum (FBS) were purchased from Gibco (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The ELISA kits for
phosphorylated (p)-tau (cat. no. P261FC), β-secretase enzyme (cat.
no. B096FC) and Aβ42 (cat. no. A227FC) were purchased
from Hermes Criterion Biotechnology (Elixir Canada Medicine Company
Ltd., Vancouver, Canada).
OA and galangin preparation
OA was diluted to 10 µg/ml with PBS containing
0.001% dimethyl sulfoxide (DMSO), and was stored in sterile
microcentrifuge tubes and maintained at 20°C. For use, it was
diluted to various concentrations with high glucose DMEM containing
10% FBS. Galangin was diluted to 1 µg/ml with PBS containing 0.001%
DMSO, and was stored in sterile microcentrifuge tubes and
maintained at −20°C. For use, it was diluted to various
concentrations with high glucose DMEM medium containing 10%
FBS.
Experimental design
Highly differentiated PC12 cells (Type Culture
Collection of the Chinese Academy of Sciences, Shanghai, China)
were grown in DMEM supplemented with 10% FBS. The cells were
incubated at 37°C, in an atmosphere containing 5% CO2
for 48 h; the media were replaced daily. Cells were divided into a
control group, AD model group, galangin groups (0.25, 0.50 or 1.00
µg/ml galangin), 3MA group (5 nM 3MA) and rapamycin group (100 nM
rapamycin). With the exception of the control group cells, PC12
cells in all other groups were treated with OA (175 nM) for 48 h,
following pretreatment for 0.5 h with galangin (0.25, 0.50 and 1.00
µg/ml), 3MA (5 nM) or rapamycin (100 nM).
Cell Counting Kit (CCK)-8 assay
Cell viability was determined using a CCK-8 kit
(Dojindo Molecular Technologies, Inc., Japan), according to the
manufacturer's protocol. The PC12 cell groups with various
concentrations of OA (0–250 nM) were seeded at a density of
1×105 cells/well in 96-well plates and were cultured at
37°C in an atmosphere containing 5% CO2 for 12, 24 and
48 h. The PC12 cell groups treated with 175 nM OA + galangin (0.25,
0.50, 0.75 and 1.00 µg/ml) were seeded at a density of
1×105 cells/well in 96-well plates and were cultured at
37°C in an atmosphere containing 5% CO2 for 48 h.
Subsequently, 10 µl CCK-8 (0.5 g/l) was added and the cells were
incubated for 2 h at 37°C in an atmosphere containing 5%
CO2. The optical density (OD) of each group was measured
at a wavelength of 450 nm; OD values were used to calculate cell
viability.
Analysis of p-tau, Aβ42 and
β-secretase
The cells were treated as aforementioned prior to
ELISA. Briefly, the media from treated PC12 cells were collected
and centrifuged at 10,000 × g for 10 min at 4°C. Subsequently, the
supernatant was collected, and p-tau, Aβ42 and
β-secretase expression was measured using ELISA kits, according to
the manufacturer's protocol (Hermes Criterion Biotechnology; Elixir
Canada Medicine Company Ltd.).
Western blot analysis of p-Akt,
p-GSK3β, p-mTOR and Beclin-1 expression
PC12 cells were harvested and lysed using
phenylmethanesulfonyl fluoride lysis buffer (Sigma-Aldrich; Merck
KGaA). The lysates were incubated for 30 min at 4°C, centrifuged at
13,000 × g for 15 min at 4°C, and total protein was extracted and
quantified using a bicinchoninic acid kit (Wuhan Boster Biological
Technology, Ltd., Wuhan, China). Subsequently, 40 µg protein was
separated by 10% SDS-PAGE and transferred onto polyvinylidene
difluoride membranes. The membranes were blocked in 5% bovine serum
albumin (BSA; cat. no. 810652, Merck KGaA) for 1 h at 4°C and were
incubated with primary antibodies against GAPDH (cat. no. ab8245;
Abcam), p-Akt (cat. no. 9275S; Cell Signaling Technology, Inc.,
Danvers, MA, USA), Akt (cat no. 4691S; Cell Signaling Technology,
Inc.), p-GSK3β (cat. no. ab75745; Abcam), GSK3β (cat. no. ab131356;
Abcam), p-mTOR (cat. no. ab109268; Abcam), mTOR (cat. no. ab134903;
Abcam) and Beclin-1 (cat. no. ab62557; Abcam) overnight at 4°C (all
1:1,000). Subsequently, the blots were washed and incubated with
their respective horseradish peroxidase (HRP)-conjugated
immunoglobulin G (anti-mouse and anti-rabbit) secondary antibodies
(1:2,000; cat nos. 7076S and 7074S, respectively; Cell Signaling
Technology, Inc.) at room temperature for 1 h. GAPDH was used as an
internal control. Bound secondary antibodies were visualized using
an enhanced chemiluminescence kit (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) using Image Lab 4.1 software (Bio-Rad
Laboratories, Inc.). Western blotting was repeated at least three
times for each condition. After development, the band intensities
were semi-quantified with Image-Pro Plus software version 6.0
(Media Cybernetics, Inc., Rockville, MD, USA).
Immunocytochemistry
The cells (1×106 cells/well) were treated
with cell culture medium (control group), 175 nM OA (model group),
3MA (5 nM), rapamycin (100 nM) or galangin (1 µg/ml) + 175 nM OA
for 48 h. Following fixation in 4% paraformaldehyde at 4°C for 60
min, the cells were washed with PBS and incubated with 300 µl BSA
for 20 min at room temperature. Subsequently, cells were incubated
with anti-Beclin-1 antibody (cat. no. ab62557; 1:50 dilution;
Abcam) for 1 h at 37°C. After washing with PBS, cells were
incubated with HRP-conjugated anti-rabbit immunoglobulin (Ig)G
(1:200; cat no. 7074S; Cell Signaling Technology, Inc.) for 20 min
at 37°C. The cells were washed again with PBS and amplified with
avidin biotin-peroxidase complex (ABC) labeling by adding 300 µl
streptavidin (S)ABC (cat no. SA1021; Wuhan Boster Biological
Technology, Ltd.) for 20 min overnight. Thereafter, the cells were
washed and were stained with 300 µl 3,3′-diaminobenzidine (cat no.
AR1022; Wuhan Boster Biological Technology, Ltd.) for 5 min at room
temperature. Subsequently, the nuclei were stained with hematoxylin
at room temperature for 5 min. Finally, images were obtained using
a light microscope (U-SPT; Olympus Corporation, Tokyo, Japan).
Brown staining indicated positive expression, and the darker the
color, the more expression of Beclin-1. Data were analyzed using
ImageJ software v1.48 (National Institutes of Health, Bethesda, MD,
USA); the integrated optical density (IOD) of the positive neurons
and the area of image were used to calculate the average optical
density (AOD), according the following formula:
AOD=IODArea×100
Immunofluorescence
The cells (1×106 cells/well) were treated
with cell culture medium (control), 175 nM OA (model), 3MA (5 nM),
rapamycin (100 nM) or galangin (1 µg/ml) + 175 nM OA for 48 h.
After fixation in 4% paraformaldehyde for 60 min at 4°C, cells were
washed with PBS and 300 µl BSA was added for 20 min at room
temperature. Subsequently, the cells were incubated with rabbit
anti-Beclin-1 antibody (1:50 dilution; Abcam) for 1 h at 37°C.
After washing with PBS, cells were incubated with Alexa
488-conjugated anti-rabbit IgG (1:200; cat no. 4412S; Cell
Signaling Technology, Inc.) for 30 min at 37°C. The cells were
washed again with PBS and 300 µl SABC-fluorescein isothiocyanate
was added for 30 min at 37°C in the dark. Thereafter, the cells
were washed and 300 µl DAPI was added for 5 min at room temperature
to stain the nuclei (Wuhan Boster Biological Engineering, Wuhan,
China). Finally, images of the cells were captured using a light
microscope (U-SPT; Olympus, Tokyo, Japan). Data were analyzed using
ImageJ software v1.48 (National Institutes of Health); the AOD was
calculated as aforementioned.
Statistical analysis
Data are expressed as the means ± standard
deviation, and significant differences among different groups were
determined by one-way analysis of variance followed by a Bonferroni
post hoc test for multiple comparisons. The experiments were
repeated at least three times. Correlations among p-tau,
Aβ42, β-secretase, p-Akt, p-GSK3β, p-mTOR and Beclin-1
expression were determined using Pearson's correlation analysis.
All statistical analyses were performed using SPSS statistical
software version 13.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
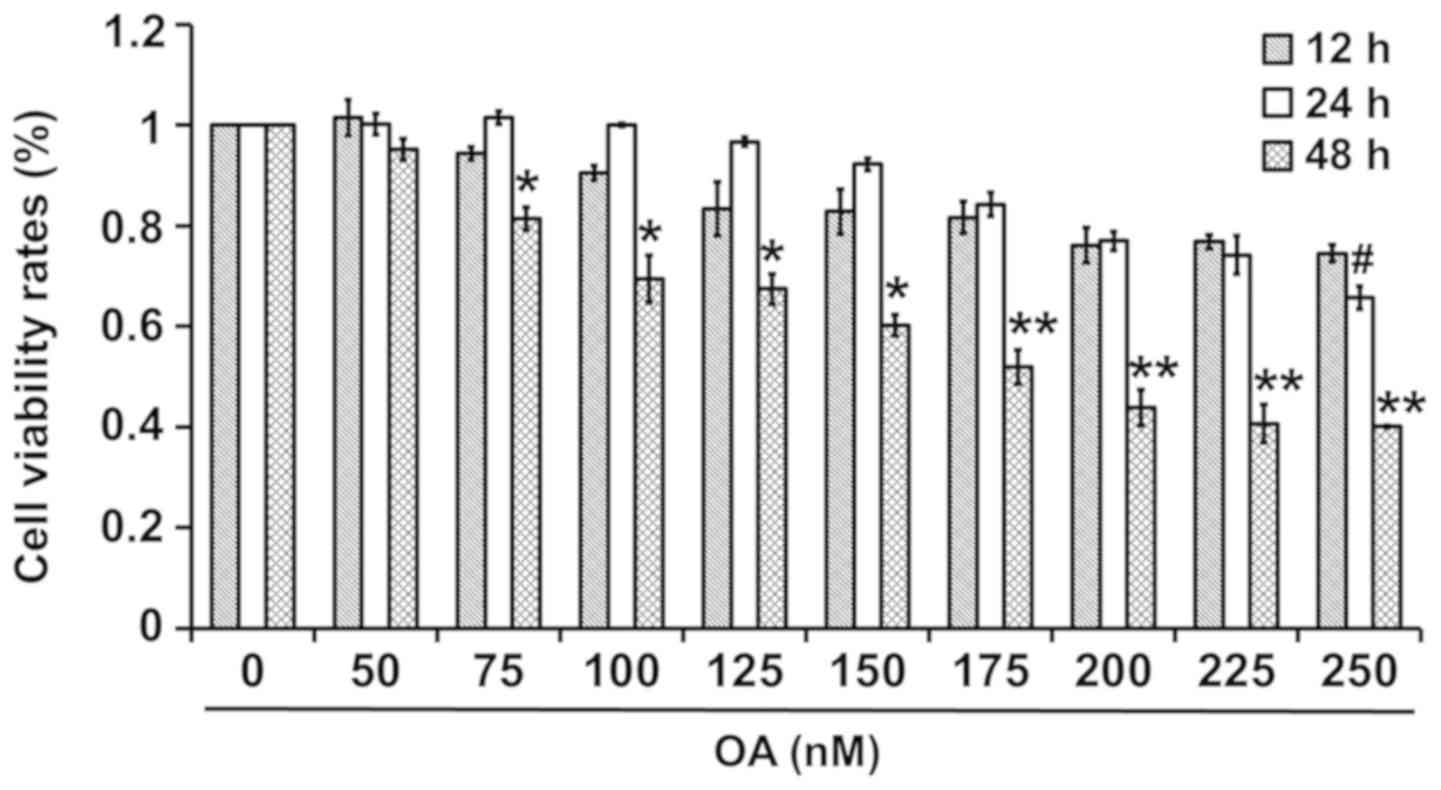
Effects of OA on the viability rates
of PC12 cells
The effects of various concentrations of OA on the
viability rates of PC12 cells are shown in Fig. 1. Cell viability rates were similar
at 12 and 24 h, but were reduced at 48 h following exposure to 50
and 175–225 nM OA. In addition, the cell viability rates were
highest at 24 h, followed by at 12 and 48 h following treatment
with 75–150 nM OA, and were highest at 12 h, followed by at 24 and
48 h following treatment with 225–250 nM OA. Therefore, treatment
with 175 nM OA for 48 h was selected in this experiment, as it
reduced viability by 50%.
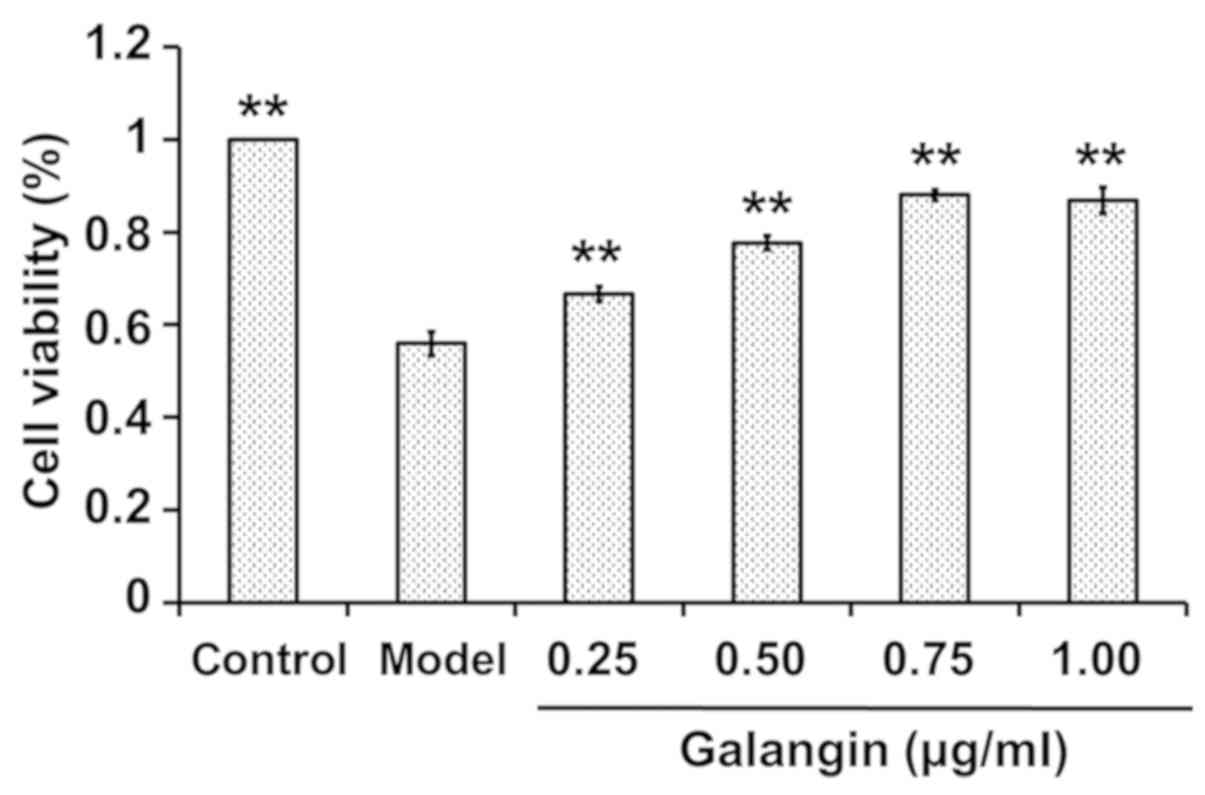
Effects of various concentrations of
galangin on OA-induced cytotoxicity in PC12 cells
PC12 cells were incubated with various
concentrations of galangin (0.25, 0.50, 0.75 and 1.00 µg/ml) in the
presence of 175 nM OA for 48 h, and CCK-8 assays were used to
detect the effect of galangin. The results demonstrated that,
compared with in the control group, OA decreased cell viability,
whereas galangin significantly increased the viability of PC12
cells in a concentration-dependent manner (P<0.01; Fig. 2). These results indicated that
galangin may alleviate cellular damage caused by OA, thus
suggesting that galangin has a neuroprotective effect.
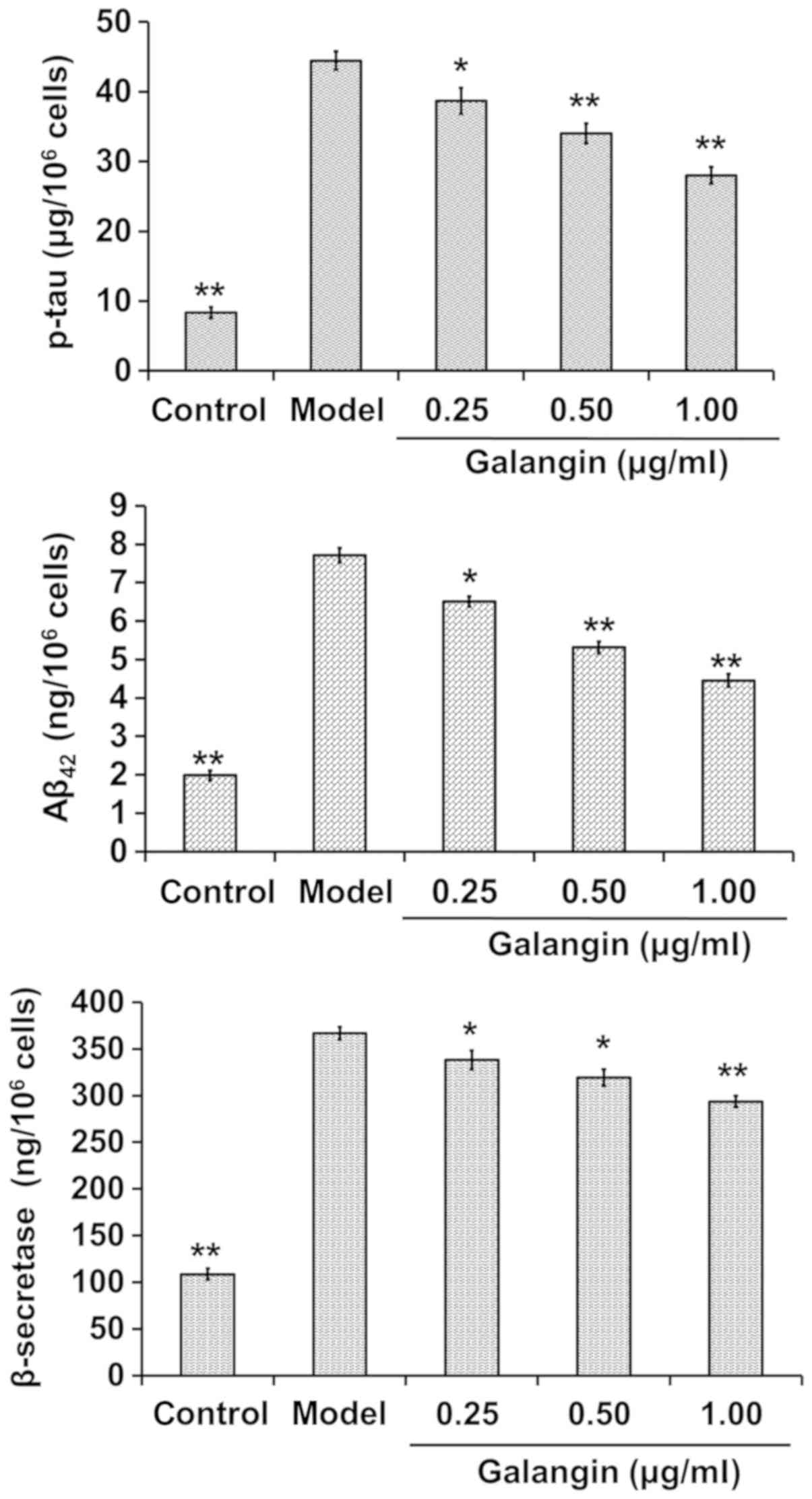
Galangin decreases β-secretase, p-tau
and Aβ42 levels
The cells were treated as aforementioned and media
were collected for p-tau, Aβ42 and β-secretase
measurement. p-tau, Aβ42 and β-secretase levels were
increased in the model group compared with in the control group
(P<0.01). Conversely, p-tau, Aβ42 and β-secretase
levels were significantly decreased in the galangin-treated groups
compared with in the model group (P<0.05). The findings
indicated that p-tau, Aβ42 and β-secretase levels were
reduced with increases in the dose of galangin (Fig. 3).
Effects of galangin on the expression
levels of p-Akt, p-GSK3β, p-mTOR and Beclin-1 in PC12 cells treated
with OA
The cells were treated as aforementioned, with
untreated cells used as a control. The expression levels of p-Akt
and p-mTOR were decreased (P<0.01), whereas the expression
levels of p-GSK3β and Beclin-1 were increased (P<0.05), in the
model group compared with in the control group. In addition,
galangin evidently increased p-Akt and p-mTOR expression
(P<0.01), and significantly attenuated p-GSK3β and Beclin-1
expression (P<0.01), compared with in the model group. In
addition, pretreatment with the autophagy inhibitor 3MA
significantly increased p-Akt and p-mTOR expression, but decreased
p-GSK3β and Beclin-1 expression, compared with in the model group
(P<0.01). Conversely, pretreatment with the autophagy activator
rapamycin significantly reduced p-Akt and p-mTOR expression, but
increased p-GSK3β and Beclin-1 expression compared with in the
model group (P<0.05; Fig.
4).
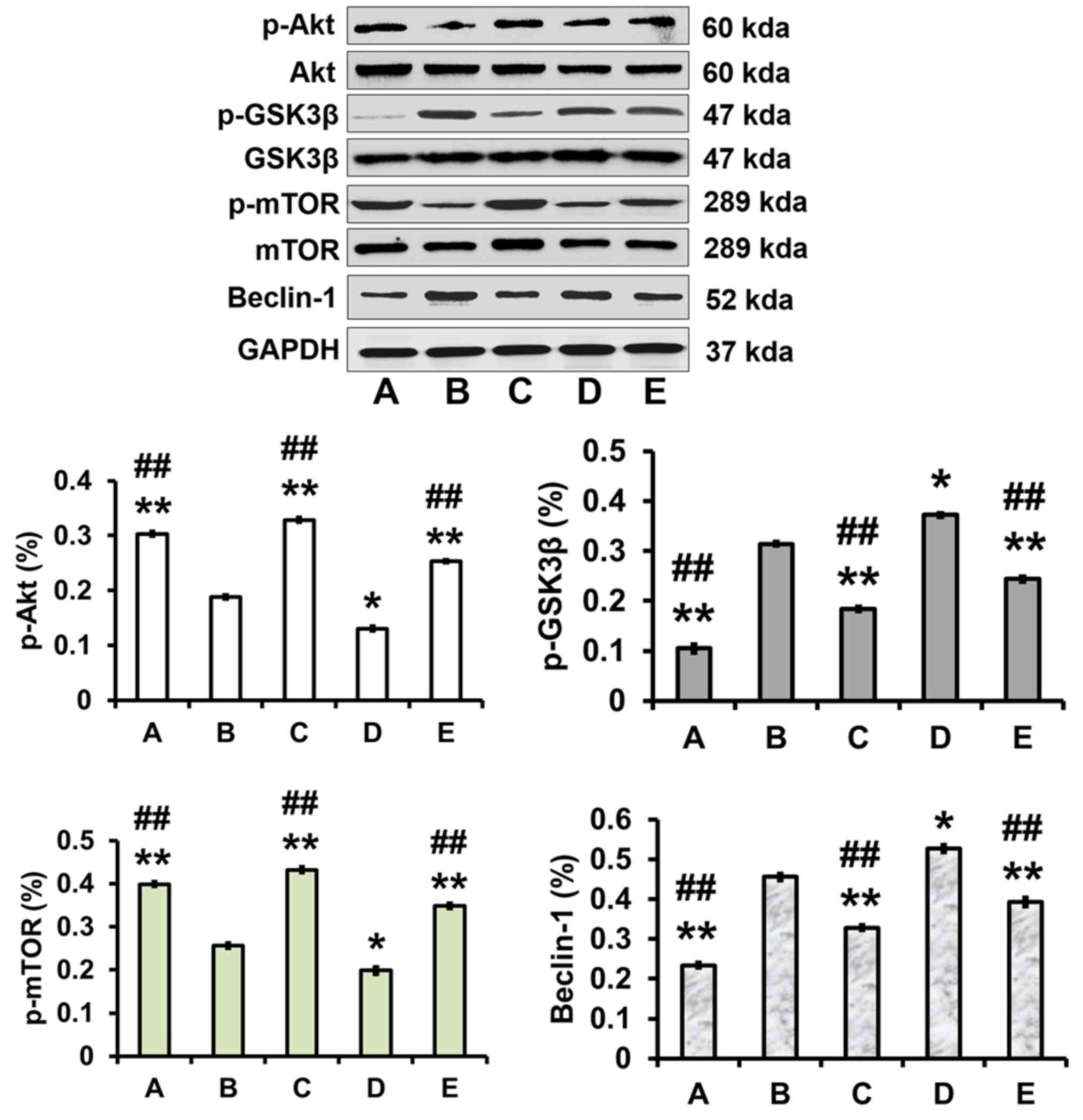 | Figure 4.Effects of galangin on the protein
expression levels of p-Akt, p-GSK3β, p-mTOR and Beclin-1. PC12
cells were pretreated with 3MA (5 nM), rapamycin (100 nM) and
galangin (1.00 µg/ml) for 0.5 h prior to exposure to OA (175 nM)
for 48 h. Protein expression levels of p-Akt and p-mTOR were
significantly increased; however, p-GSK3β and Beclin-1 protein
expression was clearly decreased in response to galangin.
Representative western blot images are shown. A, control group; B,
model group; C, 3-methyladenine group; D, rapamycin group; E,
galangin group. Data are expressed as the means ± standard
deviation (n=3). *P<0.05, **P<0.01 vs. the model group;
##P<0.01 vs. the rapamycin group. Akt, protein kinase
B; GSK, glycogen synthase kinase; mTOR, mechanistic target of
rapamycin; OA, okadaic acid; p, phosphorylated. |
Galangin inhibits OA-induced autophagy
in PC12 cells
The detailed mechanism underlying the suppressive
effects of galangin was investigated. OA significantly increased
Beclin-1 expression compared with in the control group (P<0.05).
Conversely, pretreatment with galangin prior to exposure to OA
significantly suppressed Beclin-1 expression (P<0.01). In
addition, pretreatment with the autophagy activator rapamycin
promoted activation of Beclin-1 expression (P<0.05), whereas
pretreatment with the autophagy inhibitor 3MA suppressed Beclin-1
expression (P<0.01; Fig.
5).
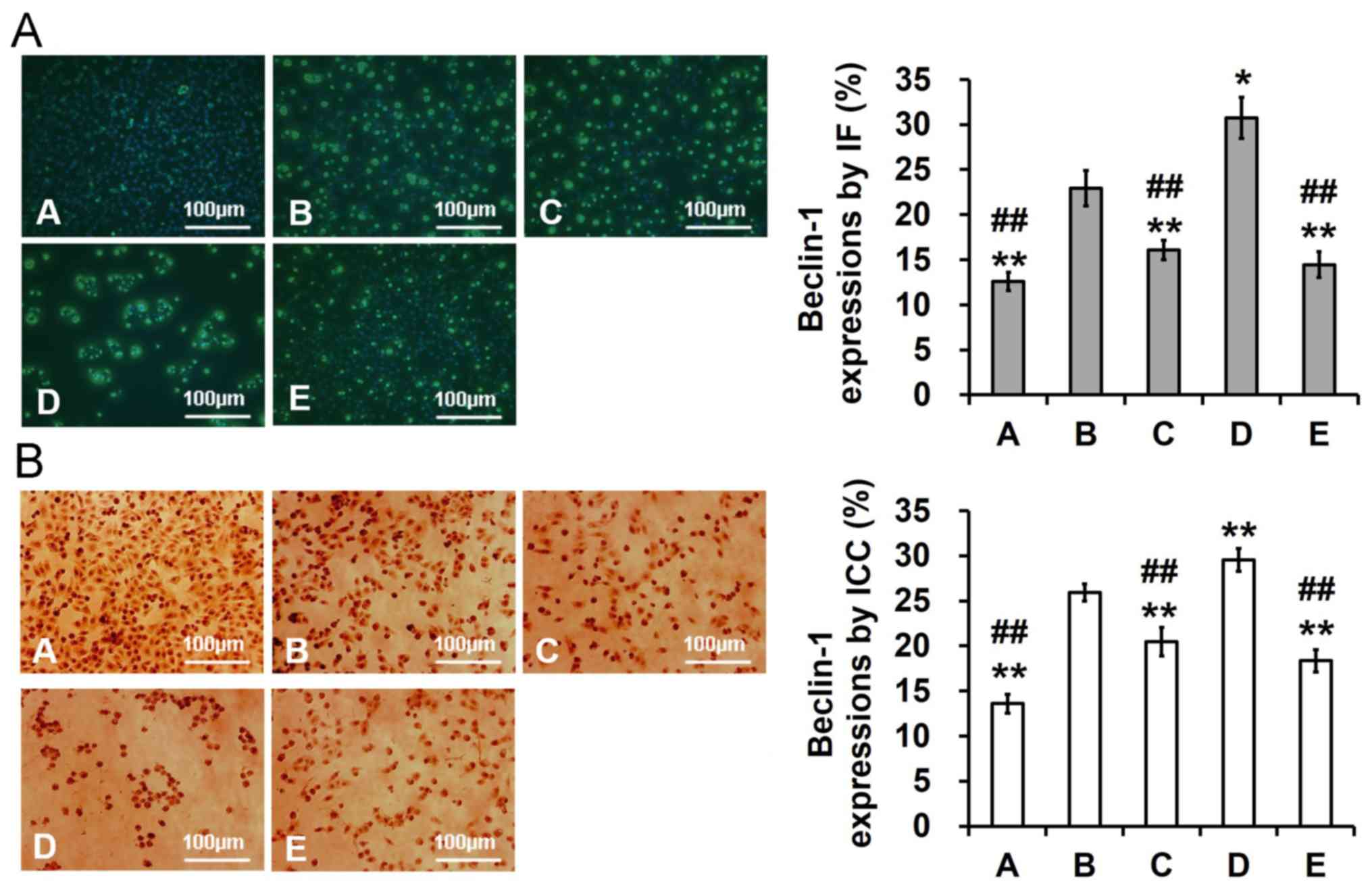 | Figure 5.Effects of galangin on Beclin-1
expression in OA-stimulated PC12 cells (magnification, ×200). Cells
were pretreated (0.5 h) with 5 nM 3MA, 100 nM rapamycin and 1.00
µg/ml galangin and were then exposed to OA (175 nM) for 48 h. (A)
Beclin-1 expression, as determined by IF and AOD analysis. (B)
Beclin-1 expression, as determined by immunocytochemistry and AOD
analysis. Data are expressed as the means ± standard deviation.
*P<0.05, **P<0.01 vs. the model group; ##P<0.01
vs. the rapamycin group. A, control group; AOD, average optical
density; B, model group; C, 3-methyladenine group; D, rapamycin
group; E, galangin group; IF, immunofluorescence; OA, okadaic
acid. |
Correlation analysis
Finally, the associations between p-tau,
Aβ42, β-secretase, p-Akt, p-GSK3β, p-mTOR and Beclin-1
expression levels were investigated using Pearson correlation
analysis (Table I). Pearson
analysis demonstrated that there were significant positive
correlations between p-tau and Aβ42/β-secretase/p-GSK3β
(0.839/0.733/0.789, respectively; P<0.01), Aβ42 and
β-secretase/p-GSK3β/Beclin-1 (0.978/0.507/0.618, respectively;
P<0.01 or P<0.05), β-secretase and Beclin-1 (0.701;
P<0.05), and p-Akt and p-mTOR (0.826; P<0.01). In addition,
there were significant negative correlations between β-secretase
and p-Akt/p-mTOR (−0.692/-0.715; P<0.05), p-Akt and Beclin-1
(−0.990; P<0.01), and p-mTOR and p-GSK3β/Beclin-1
(−0.761/-0.918; P<0.01).
 | Table I.Correlations between p-tau,
Aβ42, β-secretase, p-Akt, p-GSK3β, p-mTOR and Beclin-1
in the OA model group. |
Table I.
Correlations between p-tau,
Aβ42, β-secretase, p-Akt, p-GSK3β, p-mTOR and Beclin-1
in the OA model group.
|
| Gene |
|---|
|
|
|
|---|
| Gene | p-tau |
Aβ42 | β-secretase | p-Akt | p-GSK3β | p-mTOR | Beclin-1 |
|---|
| p-tau | 1.000 | 0.839b | 0.733b | −0.231 | 0.789b | −0.282 | 0.240 |
|
Aβ42 |
| 1.000 | 0.978b | −0.622a | 0.507a | −0.688a | 0.618a |
| β-secretase |
|
| 1.000 | −0.692a | 0.434 | −0.715a | 0.701a |
| p-Akt |
|
|
| 1.000 | −0.171 | 0.826b | −0.990b |
|
p-GSK3β |
|
|
|
| 1.000 | −0.761b | 0.242 |
| p-mTOR |
|
|
|
|
| 1.000 | −0.918b |
| Beclin-1 |
|
|
|
|
|
| 1.000 |
Discussion
Autophagy is an essential degradation pathway
involved in the clearance of abnormal protein aggregates and
protein homeostasis in neuronal and non-neuronal cells (15). A previous study suggested that
autophagy initiation by Beclin-1 may be involved in the
pathogenesis of AD (16).
Targeting Aβ, tau and β-secretase has long been believed to be a
viable strategy for drug discovery against AD (13). Aβ has been reported to be
associated with autophagy, and is able to induce reactive oxygen
species (ROS) accumulation and apoptotic cell death (17). Excessive ROS production has been
suggested to serve a role in the initiation of autophagic cell
death (18). In addition,
Aβ1-42 can induce autophagic cell death in human glioma
and human neuroblastoma cell lines (19). The Aβ-loading of autophagosomes
could lead not only of the extracellular secretion of Aβ, but of
its lysosomal degradation, in which APP, β-secretase (BACE1) and
γ-secretase reside. Studies have confirmed that autophagy is
essential for Aβ secretion effectively, since loss of the Atg7 gene
in APP transgenic mice results in reduced extracellular Aβ plaques
and promoted Aβ accumulation (20,21).
Research revealed that autophagy plays dual roles in the release of
Aβ to the extracellular space. Exposing the neurons to rapamycin,
autophagy enhanced and Aβ secretion decreased. Conversely,
inhibition of autophagy by spautin-1, which decreased Aβ secretion
(22). According to one study,
age-induced reduction of autophagy-related gene expression is
associated with onset of AD and tau seems to mediate the neuronal
toxicity of Aβ (23). Pimozide
reduces toxic forms of tau in tauC3 mice via AMP-activated protein
kinase-mediated autophagy (24).
AD is the most common neurodegenerative disorder in the elderly,
and it has previously been confirmed that BACE1 at the mRNA and
protein level is decreased in SH-SY5Y cells treated with galangin
(8). However, to the best of our
knowledge, the effects and mechanism of galangin on autophagy in
the OA-induced PC12 cell model has not been reported.
In vivo and in vitro, OA leads to Aβ
deposition and subsequent neuronal degeneration, synaptic loss,
memory impairment and tau hyperphosphorylation, all of which
resemble AD pathology (25,26).
OA also induces neurotoxicity of the SH-SY5Y cell line and cultured
neuronal cells, resulting in the development of pathological
conditions similar to AD (27).
Neurotoxicity induced by OA, which is associated with abnormally
phosphorylated tau protein, promotes AD-like pathology (28). The results of the present study
indicated that there was evident neurotoxicity in PC12 cells
treated with OA (75–250 nM) for 48 h. In addition, galangin exerted
dose-dependent neuroprotective effects on PC12 cells treated with
175 nM OA.
Aβ is generated through serial cleavage of APP.
β-secretase is a key enzyme for Aβ production, and is a promising
target for AD therapy (29).
OA-induced PP2A gene regulation and kinase remodeling serve a
crucial role in the generation of abnormal p-tau, which results in
neurotoxicity and an AD-like pathology (30). To investigate the neuroprotective
effects of galangin on OA-induced PC12 cell injury, β-secretase,
p-tau and Aβ42 levels were detected following treatment
with galangin (0.5 h) and OA (48 h). A recent study indicated that
galangin reduces β-secretase at the mRNA and protein level, and Aβ
levels in SH-SY5Y cells through the upregulation of endogenous
HDAC1-mediated deacetylation (8).
The results of the present study also demonstrated that galangin
increased cell viability and significantly decreased β-secretase,
p-tau and Aβ42 levels. These results indicated that
pretreatment with galangin protected PC12 cells from OA-induced
cytotoxicity.
The mechanism underlying the effects of galangin on
OA-induced PC12 cell autophagy was also examined. Autophagy serves
an important role in clearing Aβ aggregates and preserving neuronal
function in AD (31). Beclin-1 is
an autophagy-associated protein, which is involved in the
initiation of autophagy (32). It
has previously been reported that Beclin-1 deficiency increases the
expression of APP and APP-like proteins, accelerates Aβ
accumulation and promotes neurodegeneration in mice, indicating
that Beclin-1 may be involved in the pathogenesis of AD (33); the present study results in
OA-induced PC12 cells are consistent with this finding. The present
results demonstrated that OA could activate autophagy and increase
the expression of Beclin-1. Conversely, galangin significantly
decreased Beclin-1 expression, thus suggesting that galangin may
suppress OA-induced autophagy. In other words, galangin has the
pharmacological effect of alleviating AD: the autophagy of the
model group is up-regulated, and the autophagy is down-regulated
after treatment with galangin. It is concluded that galangin has a
certain effect of autophagy inhibition to OA induced PC12 cells.
Furthermore, Beclin-1 expression was significantly reduced in
response to pretreatment with the autophagy inhibitor 3MA; however,
Beclin-1 expression was significantly enhanced following
pretreatment with the autophagy activator rapamycin.
To elucidate the precise molecular mechanism
underlying the effects of galangin on autophagy in OA-treated PC12
cells, the present study focused on the Akt/mTOR and Akt/GSK-3β
pathways. Akt has a key role in regulating cell signals that are
important for cell death and survival (34). mTOR, which functions downstream of
Akt, is mainly mediated by phosphoinositide 3-kinase/Akt signaling
transduction and suppresses autophagy (35); therefore, the present study used
the classic mTOR inhibitor rapamycin to investigate the effects of
mTOR on autophagy. GSK3, as an integrator of Akt and Wnt signals,
also serves a central role in the regulation of mTOR during
synaptic plasticity (36). GSK3β
has also been identified as serving a prominent role in the
formation of abnormal protein accumulation, which is important in
AD (37). The present study
revealed that p-Akt and p-mTOR levels were decreased, whereas
p-GSK3β was increased in the OA treatment group compared with in
the normal control group. However, galangin increased levels of
p-Akt and p-mTOR, but reduced levels of p-GSK3β, compared in with
the OA treatment group. These results indicated that galangin
attenuated autophagy through modulation of the Akt/GSK3β/mTOR
pathway.
Finally, significant positive correlations were
detected in the present study, including between Beclin-1 and
β-secretase, and Aβ42 and p-GSK3β/p-tau, whereas there
were significant negative correlations between Beclin-1 and
p-Akt/p-mTOR, β-secretase and p-Akt/p-mTOR, and Aβ42 and
p-Akt/p-mTOR. These findings indicated that galangin decreased
β-secretase, Aβ42, p-tau and Beclin-1 levels, which may
interact with the Akt/GSK3β/mTOR pathway in PC12 cells following OA
treatment.
In conclusion, galangin attenuated OA-induced
autophagy via the Akt/GSK3β/mTOR pathway. Galangin promoted Akt and
mTOR phosphorylation, but suppressed GSK3β phosphorylation, which
served to inhibit autophagy. The findings of the present study
indicated that galangin may be a potential preventive drug for
AD.
Acknowledgements
Not applicable.
Funding
The present study was supported by The Lingnan
Normal University-Level Talent Project (grant no. ZL1801), The
Natural Science Foundation of Guangdong Province of China (grant
nos. 2018A030307037, 2016A030310362 and 2017A030310604), The Hainan
Natural Science Foundation of China (grant nos. 20168266 and
817140), The Program of Hainan Association for Science and
Technology Plans to Youth R&D Innovation (grant no.
HAST201635), The Scientific Research Cultivating Fund of Hainan
Medical University (grant nos. HY2015-01 and HY2016-02), The
Scientific Research Project of Guangdong Provincial Administration
of Traditional Chinese Medicine (grant nos. 20181114) and The
National Natural Science Foundation of China (grant no.
201705071).
Availability of data and materials
The data sets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
LH and MD conceived the study, designed the
experiments, analyzed the data and prepared the manuscript. LH, ML,
XZ, HY and MD selected the subjects and obtained samples for the
present study. XZ, MD and LH performed the experiments. All authors
have read and approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interest.
References
|
1
|
Swomley AM, Förster S, Keeney JT, Triplett
J, Zhang Z, Sultana R and Butterfield DA: Abeta, oxidative stress
in Alzheimer disease: Evidence based on proteomics studies. Biochim
Biophys Acta 1842. 1248–1257. 2014.
|
|
2
|
Guerreiro RJ, Gustafson DR and Hardy J:
The genetic architecture of Alzheimer's disease: Beyond APP, PSENs
and APOE. Neurobiol Aging. 33:437–456. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yen CH, Wang CH, Wu WT and Chen HL:
Fructo-oligosaccharide improved brain β-amyloid, β-secretase,
cognitive function, and plasma antioxidant levels in
D-galactose-treated Balb/cJ mice. Nutr Neurosci. 20:228–237. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wolfe MS: Processive proteolysis by
γ-secretase and the mechanism of Alzheimer's disease. Biol Chem.
393:899–905. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Imbimbo BP and Giardina GA: γ-secretase
inhibitors and modulators for the treatment of Alzheimer's disease:
Disappointments and hopes. Curr Top Med Chem. 11:1555–1570. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang X, Gong G, Yang W, Li Y, Jiang M and
Li L: Antifibrotic activity of galangin, a novel function evaluated
in animal liver fibrosis model. Environ Toxicol Pharmacol.
36:288–295. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huh JE, Jung IT, Choi J, Baek YH, Lee JD,
Park DS and Choi DY: The natural flavonoid galangin inhibits
osteoclastic bone destruction and osteoclastogenesis by suppressing
NF-κB in collagen-induced arthritis and bone marrow-derived
macrophages. Eur J Pharmacol. 698:57–66. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zeng H, Huang P, Wang X, Wu J, Wu M and
Huang J: Galangin-induced down-regulation of BACE1 by epigenetic
mechanisms in SH-SY5Y cells. Neuroscience. 294:172–181. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Penke B, Bogár F and Fülöp L: Protein
folding and misfolding, endoplasmic reticulum stress in
neurodegenerative diseases: In trace of novel drug targets. Curr
Protein Pept Sci. 17:169–182. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Viola KL and Klein WL: Amyloid β oligomers
in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta
Neuropathol. 129:183–206. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guo JL and Lee VM: Seeding of normal Tau
by pathological Tau conformers drives pathogenesis of
Alzheimer-like tangles. J Biol Chem. 286:15317–15331. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pickford F, Masliah E, Britschgi M, Lucin
K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E,
Levine B and Wyss-Coray T: The autophagy-related protein beclin 1
shows reduced expression in early Alzheimer disease and regulates
amyloid beta accumulation in mice. J Clin Invest. 118:2190–2199.
2008.PubMed/NCBI
|
|
13
|
Lipinski MM, Zheng B, Lu T, Yan Z, Py BF,
Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR and Yuan J:
Genome-wide analysis reveals mechanisms modulating autophagy in
normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci
USA. 107:14164–14169. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kamat PK and Nath C: Okadaic acid: A tool
to study regulatory mechanisms for neurodegeneration and
regeneration in Alzheimer's disease. Neural Regen Res. 10:365–367.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Q, Liu Y and Sun M: Autophagy and
Alzheimer's disease. Cell Mol Neurobiol. 37:377–388. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Salminen A, Kaarniranta K, Kauppinen A,
Ojala J, Haapasalo A, Soininen H and Hiltunen M: Impaired autophagy
and APP processing in Alzheimer's disease: The potential role of
Beclin 1 interactome. Prog Neurobiol. 106-107:33–54. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li RC, Pouranfar F, Lee SK, Morris MW,
Wang Y and Gozal D: Neuroglobin protects PC12 cells against
beta-amyloid-induced cell injury. Neurobiol Aging. 29:1815–1822.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen Y, McMillan-Ward E, Kong J, Israels
SJ and Gibson SB: Oxidative stress induces autophagic cell death
independent of apoptosis in transformed and cancer cells. Cell
Death Differ. 15:171–182. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang H, Ma J, Tan Y, Wang Z, Sheng C, Chen
S and Ding J: Amyloid-beta1-42 induces reactive oxygen
species-mediated autophagic cell death in U87 and SH-SY5Y cells. J
Alzheimers Dis. 21:597–610. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cataldo AM, Petanceska S, Terio NB,
Peterhoff CM, Durham R, Mercken M, Mehta PD, Buxbaum J, Haroutunian
V and Nixon RA: Abeta localization in abnormal endosomes:
Association with earliest Abeta elevations in AD and Down syndrome.
Neurobiol Aging. 25:1263–1272. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
LaFerla FM, Green KN and Oddo S:
Intracellular amyloid-beta in Alzheimer's disease. Nat Rev
Neurosci. 8:499–509. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Choy RW, Cheng Z and Schekman R: Amyloid
precursor protein (APP) traffics from the cell surface via
endosomes for amyloid β (Aβ) production in the trans-Golgi network.
Proc Natl Acad Sci USA. 109:E2077–E2082. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Omata Y, Lim YM, Akao Y and Tsuda L:
Age-induced reduction of autophagy-related gene expression is
associated with onset of Alzheimer's disease. Am J Neurodegener
Dis. 3:134–142. 2014.PubMed/NCBI
|
|
24
|
Kim YD, Jeong EI, Nah J, Yoo SM, Lee WJ,
Kim Y, Moon S, Hong SH and Jung YK: Pimozide reduces toxic forms of
tau in TauC3 mice via 5′ adenosine monophosphate-activated protein
kinase-mediated autophagy. J Neurochem. 142:734–746. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cakir M, Duzova H, Tekin S, Taslıdere E,
Kaya GB, Cigremis Y, Ozgocer T and Yologlu S: ACA, an inhibitor
phospholipases A2 and transient receptor potential melastatin-2
channels, attenuates okadaic acid induced neurodegeneration in
rats. Life Sci. 176:10–20. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu X, Kosaraju J and Tam KY: SLM, a novel
carbazole-based fluorophore attenuates okadaic acid-induced tau
hyperphosphorylation via down-regulating GSK-3β activity in SH-SY5Y
cells. Eur J Pharm Sci. 110:101–108. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang J, Cheng Y and Zhang JT: Protective
effect of (−) clausenamide against neurotoxicity induced by okadaic
acid and beta-amyloid peptide25-3. Yao Xue Xue Bao. 42:935–942.
2007.(In Chinese). PubMed/NCBI
|
|
28
|
Kamat PK, Rai S and Nath C: Okadaic acid
induced neurotoxicity: An emerging tool to study Alzheimer's
disease pathology. Neurotoxicology. 37:163–172. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao XJ, Gong DM, Jiang YR, Guo D, Zhu Y
and Deng YC: Multipotent AChE and BACE-1 inhibitors for the
treatment of Alzheimer's disease: Design, synthesis and
bio-analysis of 7-amino-1,4-dihydro-2H-isoquilin-3-one derivates.
Eur J Med Chem. 138:738–747. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang CC, Kuai XX, Gao WB, Yu JC, Wang Q,
Li L and Zhang L: Morroniside-induced PP2A activation antagonizes
Tau hyperphosphorylation in a cellular model of neurodegeneration.
J Alzheimers Dis. 51:33–44. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aminyavari S, Zahmatkesh M, Farahmandfar
M, Khodagholi F, Dargahi L and Zarrindast MR: Protective role of
Apelin-13 on amyloid β25-35-induced memory deficit; Involvement of
autophagy and apoptosis process. Prog Neuropsychopharmacol Biol
Psychiatry. 89:322–334. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yu J, Lan L, Lewin SJ, Rogers SA, Roy A,
Wu X, Gao P, Karanicolas J, Aubé J, Sun B and Xu L: Identification
of novel small molecule Beclin 1 mimetics activating autophagy.
Oncotarget. 8:51355–51369. 2017.PubMed/NCBI
|
|
33
|
Jaeger PA, Pickford F, Sun CH, Lucin KM,
Masliah E and Wyss-Coray T: Regulation of amyloid precursor protein
processing by the Beclin 1 complex. PLoS One. 5:e111022010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hotfilder M, Sondermann P, Senss A, van
Valen F, Jürgens H and Vormoor J: PI3K/AKT is involved in mediating
survival signals that rescue Ewing tumour cells from fibroblast
growth factor 2-induced cell death. Br J Cancer. 92:705–710. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Du C, Zhang T, Xiao X, Shi Y, Duan H and
Ren Y: Protease-Activated Receptor-2 promotes kidney tubular
epithelial inflammation by inhibiting autophagy via the
PI3K/Akt/mTOR signalling pathway. Biochem J. 474:2733–2747. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ma T, Tzavaras N, Tsokas P, Landau EM and
Blitzer RD: Synaptic stimulation of mTOR is mediated by Wnt
signaling and regulation of glycogen synthetase kinase-3. J
Neurosci. 31:17537–17546. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang S, Chen Z, Cao M, Li R, Wang Z and
Zhang M: Pioglitazone ameliorates Aβ42 deposition in rats with
diet-induced insulin resistance associated with AKT/GSK3β
activation. Mol Med Rep. 15:2588–2594. 2017. View Article : Google Scholar : PubMed/NCBI
|