Introduction
Polycystic kidney disease (PKD) is a monogenic
inherited disease characterized by bilateral polycystic kidneys and
their progressive enlargement, which affects renal structure and
function (1). According to the
inheritance pattern, PKD is divided into autosomal dominant PKD
(ADPKD, OMIM#173900) and autosomal recessive PKD (ARPKD,
OMIM#263200). ADPKD is a life-threatening disease, and is one of
the most common inherited diseases, with a morbidity of
1:500–1,000, affecting >12 million individuals worldwide
(2–4). In addition to the kidneys, other
organs may also be affected, including the liver, brain and
pancreas, resulting in end-stage kidney disease (ESRD) (5–7). The
manifestations of ADPKD usually appear in adulthood; the majority
of individuals with ADPKD have hypertension (8–10).
There are two primary genes in which mutations are reported to lead
to ADPKD: PKD1 (OMIM*601313, autosomal dominant) and PKD2
(OMIM*173910, autosomal dominant). PKD1 is located on chromosome
16p13 (11). The protein product
of PKD1, polycystin-1 (PC1), is an integral membrane protein with
11 membrane-spanning domains. PKD2 is located on chromosome 4q22.1,
and its protein product, polycystin-2 (PC2), also localizes to the
cell membrane, acting as a nonselective Ca2+ channel
(12). The C-termini of PC1 and
PC2 interact to regulate ion transportation; thus, mutations in
PKD1 and PKD2 may lead to disease (13–16).
Over 2,000 and 400 mutations in PKD1 and PKD2,
respectively, have been identified in association with PKD; ~85% of
patients with ADPKD carry mutations in PKD1 and its protein product
PC1, which comprises numerous domains (17). The PC1 C-terminal domains are the
most extensively studied, and include a coiled-coil domain (amino
acids 4,220–4,251) and a G-protein binding and activation sequence
(amino acids 4,111–4,184) (18).
The coiled-coil domain mediates PC2 binding in order to control
Ca2+ flow (19).
Providing the PC1/PC2 complex loses its function, intracellular
Ca2+ levels become too low to activate
phosphatidylinositol 3-kinase or Akt, which activate Camp-dependent
B-Raf (20,21). Thus, cyclic adenosine monophosphate
(cAMP) induces the activation of the mitogen-activated protein
kinase/extracellular signal-regulated kinase pathway via protein
kinase A/Ras/B-Raf signaling, stimulating cell proliferation and
promoting the expansion of cystic cavities (21–23).
Due to cAMP stimulation, cystic epithelial cells secrete
Cl− together with Na+ via the paracellular
pathway (21,24). Additionally, the C-terminal tail of
PC1 interacts with the α-subunit of
Na+/K+-adenosine 5′-triphosphatase,
increasing Na+ and K+ translocation (25). During osmotic action, fluid is
extracellularly secreted and accumulates in cystic cavities
(25); as a result, the kidneys
progressively engorge and fill with fluid. The G-protein-coupled
receptor (GPCR) phosphorylation site (GPS) motif within PC1 is part
of the GPCR-autoproteolysis inducing regulatory domain, which
mediates PC1 cis-autoproteolysis to generate an
unglycosylated N-terminus and a C-terminus that remains
non-covalently bound (19,26). The function of the observed
cleavage remains unknown; however, it may aid stable protein
folding and PC1 trafficking (27).
In the present study, whole exome sequencing of a
blood sample from a proband with ADPKD was performed to detect the
pathogenic variations present within their family. To identify the
pathogenic mutation site, nested polymerase chain reaction (PCR)
was conducted to amplify candidate regions present in the patient
and other unaffected family members. The PCR product was sequenced
via Sanger sequencing. As a result, the novel mutation was
determined to be the pathogenic factor. The mutation generates a
truncated PC1 without its C-terminal domains. Additionally, all the
pathogenic substitutions in PKD1 published from the dataset were
analyzed in order to evaluate the pathogenic domains of PC1,
representing the first report of this nature, to the best of our
knowledge. The present study provides a basis for assessing the
pathogenic properties of missense mutations, and may aid in the
genetic counseling of patients with ADPKD, and contribute to the
identification of asymptomatic individuals at risk of developing
ADPKD.
Materials and methods
Subjects and blood samples
The present study was approved by the Institutional
Research Board of Harbin Medical University (Harbin, China), and
all participants provided written informed consent. A
four-generation family was recruited on May, 2017; the pedigree of
the family included 29 individuals with 15 females and 14 males.
Clinical information and peripheral blood samples from the proband
and their daughters were collected into graded negative pressure
vacuum EDTA anticoagulant tubes.
Clinical evaluation
The family with ADPKD was identified at The Second
Affiliated Hospital of Harbin Medical University (Harbin, China).
The proband was a 56-year-old female who had been diagnosed with
PKD and polycystic liver disease one year prior to analysis.
Ultrasound examination for abdominal urology assessment revealed
numerous cysts of variable sizes in the patient's bilateral kidneys
and liver. The kidneys and liver were notably enlarged. The largest
cyst in the liver was ~72×58 mm. The size of the cholecyst was
normal; however, the gallbladder wall was thick and rough.
Additionally, the pancreas was normal. Liver puncture was performed
to extract the cystic liquid following local anesthetization with
2% lidocaine; ~430 ml of cystic liquid was extracted twice within
one week from 16 cysts.
Pathogenic gene detection
Whole exome sequencing of the blood sample from the
proband was performed by Novogene Co., Ltd. (Beijing, China). For
library generation, 1.0 µg of genomic DNA per sample was required,
which was randomly fragmented to 180–280 base pairs (bp) by a
sonicator (Covaris S220, Covaris, Woburn, MA, USA) using 450 W peak
incident power at 20 cycles per burst for 400 sec. The DNA
fragments were then end-repaired and phosphorylated (Illumina,
Inc., San Diego, CA, USA). Following determination of the size
distribution and concentration of the fragments, the DNA library
was sequenced on the Illumina HiSeq 4000 platform for paired-end
150 bp reads. DNA extraction was performed using the DNeasy Blood
and Tissue kit (Qiagen GmbH, Hilden, Germany) according to the
manufacturer's protocols. Briefly, a DNA collection spin tube was
employed to isolate genomic DNA from the blood sample. As the PKD1
gene is complex, long-range PCR (LR-PCR) was performed to amplify
PKD1 exons 22–26 (forward, 5′-TCCAGTCAAGTGGGCTCTC-3′, and reverse,
5′-CAATGAAGAGGAAAGCAGCAC-3′). In the LR-PCR system, 100 ng
template, 2 µl dNTP (4019; Takara Bio, Inc., Otsu, Japan), 10 µl
2XGC buffer (9155; Takara Bio, Inc.), 1 µl forward primer (10
pmol/l), 1 µl reverse primer (10 pmol/l), 0.2 µl LA Polymerase
(RR02MA; Takara Bio, Inc.) and water were used: 20 µl in total. The
thermocycling condition followed the LA Polymerase protocol:
Initialization at 94°C for 1 min, denaturation at 98°C for 10 sec,
annealing at 63°C for 30 sec, extension at 72°C for 3.5 min, a
final elongation at 72°C for 10 min and hold at 4°C, for 30 cycles.
The LR-PCR product was amplified as the template to obtain the
candidate region within the 26th exon (forward,
5′-GGCTGCCCACCCTGACTGACT-3′, and reverse,
5′-CCTGGGTGGGCTCGGCTCTATC-3′) and the 25th exon (forward,
5′-CGCCGGGCCCAGGAGGTC-3′, and reverse, 5′-CCCCGGCCCAATGAGCACAA-3′).
In the PCR system, 100 ng template, 3 µl dNTP (4019, Takara,
Japan), 3 µl 10XGC buffer (9151A; Takara Bio, Inc.), 1.5 µl forward
primer (10 pmol/l), 1.5 µl reverse primer (10 pmol/l), 0.3 µl Taq
Polymerase (R001A; Takara Bio, Inc.) and water were used: 30 µl in
total. The thermocycling condition followed the Taq Polymerase
protocol: Initialization at 98°C for 1 min, denaturation at 98°C
for 10 sec, annealing at 61°C for 30 sec, extension at 72°C for 45
sec, final elongation at 72°C for 10 min and hold at 4°C, for 30
cycles. The nest PCR product was ligated to the T linear vector
(Peasy-T1 Simple Cloning Kit, cat. no. CT111, Beijing Transgen
Biotech Co., Ltd., Beijing, China) according to the manufacturer's
protocol and sequenced by TsingKe Biological Technology (Beijing,
China) via Sanger sequencing.
Variation analysis
During whole exome sequencing, the reads that
aligned to exonic regions were collected for variant calling, and
the identification of single nucleotide polymorphisms (SNPs) and
indels using SAMtools mpileup and bcftools (28). The frequency of variants was
evaluated via the 1000 Genomes Project (http://browser.1000genomes.org) and the Exome
Aggregation Consortium (http://exac.broadinstitute.org). The significance of
mutation variants was assessed with the Online Mendelian
Inheritance in Man database (OMIM, http://omim.org;
clinical significance is assessed as pathogenic), the Human Gene
Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/all.php; clinical
significance is assessed as pathogenic), the SNP Database (dbSNP;
https://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?geneId=5310;
clinical significance is assessed as pathogenic) and the Autosomal
Dominant Polycystic Kidney Disease Database (PKDB, http://pkdb.pkdcure.org; clinical significance is
assessed as any pathogenic mutation), as well as the substitution
assessment tools SIFT (https://sift.bii.a-star.edu.sg; score ≤0.05),
Polyphen2_HVAR (http://genetics.bwh.harvard.edu/pph2; score ~1
assessed as probably pathogenic or possibly pathogenic),
MutationTaster (http://www.mutationtaster.org; small score assessed as
disease-causing automatic or disease-causing) and Mutation Assessor
(http://mutationassessor.org/r3/; large
score assessed as high or medium). The conserved properties of the
variants were analyzed using the University of California Santa
Cruz Genome Browser (http://genome.ucsc.edu). The division of variable
regions in PC1 was based on UniProt (http://www.uniprot.org).
Results
Studied family suffers from ADPKD
A family with ADPKD, including 29 individuals from
four generations, was studied. The family's medical history was
further investigated following diagnosis of the proband with PKD.
The proband was a 56-year-old female who was diagnosed with
bilateral PKD and polycystic liver disease at the age of 55. After
one year, the proband returned for a check-up and treatment.
According to the results of an abdominal ultrasound examination
(Table I), numerous cysts in the
liver were filled with cystic liquid; thus, liver puncture
treatment was required.
 | Table I.Abdominal ultrasound examination
findings. |
Table I.
Abdominal ultrasound examination
findings.
| Individuals | Age (years) | Sex | Findings |
|---|
| II4 | 56 | Female | Polycystic kidney
and polycystic liver with large liver, chronic cholecystitis |
|
III5 | 34 | Female | Non-apparent
abnormality |
|
III6 | 27 | Female | Non-apparent
abnormality |
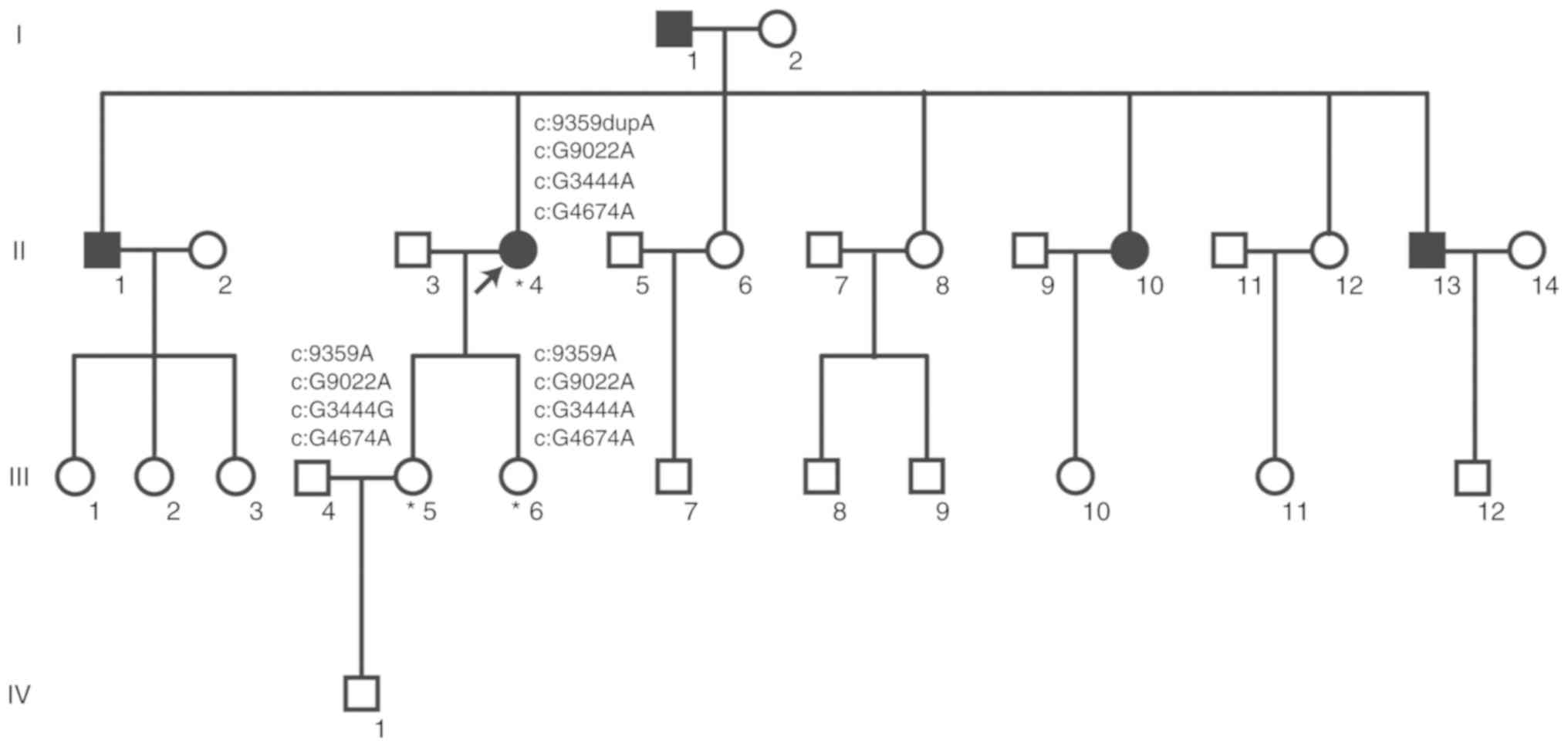
Based on the pedigree (Fig. 1), five patients with PKD were
evaluated in the family, including two females and three males. The
proband's father was diagnosed with PKD, while their mother
exhibited no symptoms. The remaining diagnosed patients in the
family were sisters or brothers of the proband; however, the two
daughters of the proband, who were 34 and 27 years old, exhibited
no manifestations of PKD. The other individuals in generations III
and IV were not examined or diagnosed. As the clinical symptoms of
the disease are not clear, it is difficult to determine whether
individuals suffer from the disease or not without ultrasound
examination. Almost half of the lineal consanguineous group (5 of
11 individuals) was diagnosed with PKD. Collectively, the results
indicated that the family analyzed suffers from ADPKD.
A novel duplication variant,
NM_001009944.2:c.9359dupA:p.Y3120_E3121delinsX, may be the
pathogenic mutation
To identify the pathogenic genes associated with
PKD, whole exome sequencing was performed on the proband. The
results revealed an adenine (A) duplication following chr16:2152099
(GRCh37/hg19, GCF_000001405.25), which resulted in a frameshift
mutation in PKD1 and generated a truncated PC1 protein. The
inserted A was located in the 26th exon of PKD1 in a heterozygous
form. The duplication variant was described as
c.9359dupA:p.Y3120_E3121delinsX in version NM_001009944.2.
Additionally, one missense site (NM_001009944.2:c.G9022A:p.V3008M)
and two synonymous sites (NM_001009944.2:c.G4674A:p.T1558T and
NM_001009944.2:c.G3444A:p.P1148P) were observed in PKD1 (Table II, Fig. S1).
| Table II.PKD1 variant information. |
Table II.
PKD1 variant information.
| Exon | Variant | Genotype | NM number | Amino acid
change | Domain | 1000 Ga | ExACb |
|---|
| 26 | Duplication | Heterozygote | NM_001009944.2 |
c.9359dupA:p.Y3120_E3121delinsX | PLAT | – | – |
| 25 | Missense | Heterozygote | NM_001009944.2 |
c.G9022A:p.V3008M | Near GPS | 0.001597 | 0.0005109 |
| 15 | Synonymous | Heterozygote | NM_001009944.2 |
c.G4674A:p.T1558T | PKD11 | 0.07109 | 0.07574 |
| 15 | Synonymous | Heterozygote | NM_001009944.2 |
c.G3444A:p.P1148P | PKD6 | 0.003395 | 0.003747 |
Notably, the duplication variant
(c.9359dupA:p.Y3120_E3121delinsX) identified in PKD1 is a novel
mutation that has not been reported in PKDB, OMIM, HGMD or other
published articles. To determine the mutations present in the
family, targeted DNA fragments from the proband and their two
normal daughters were sequenced via Sanger's method and analyzed.
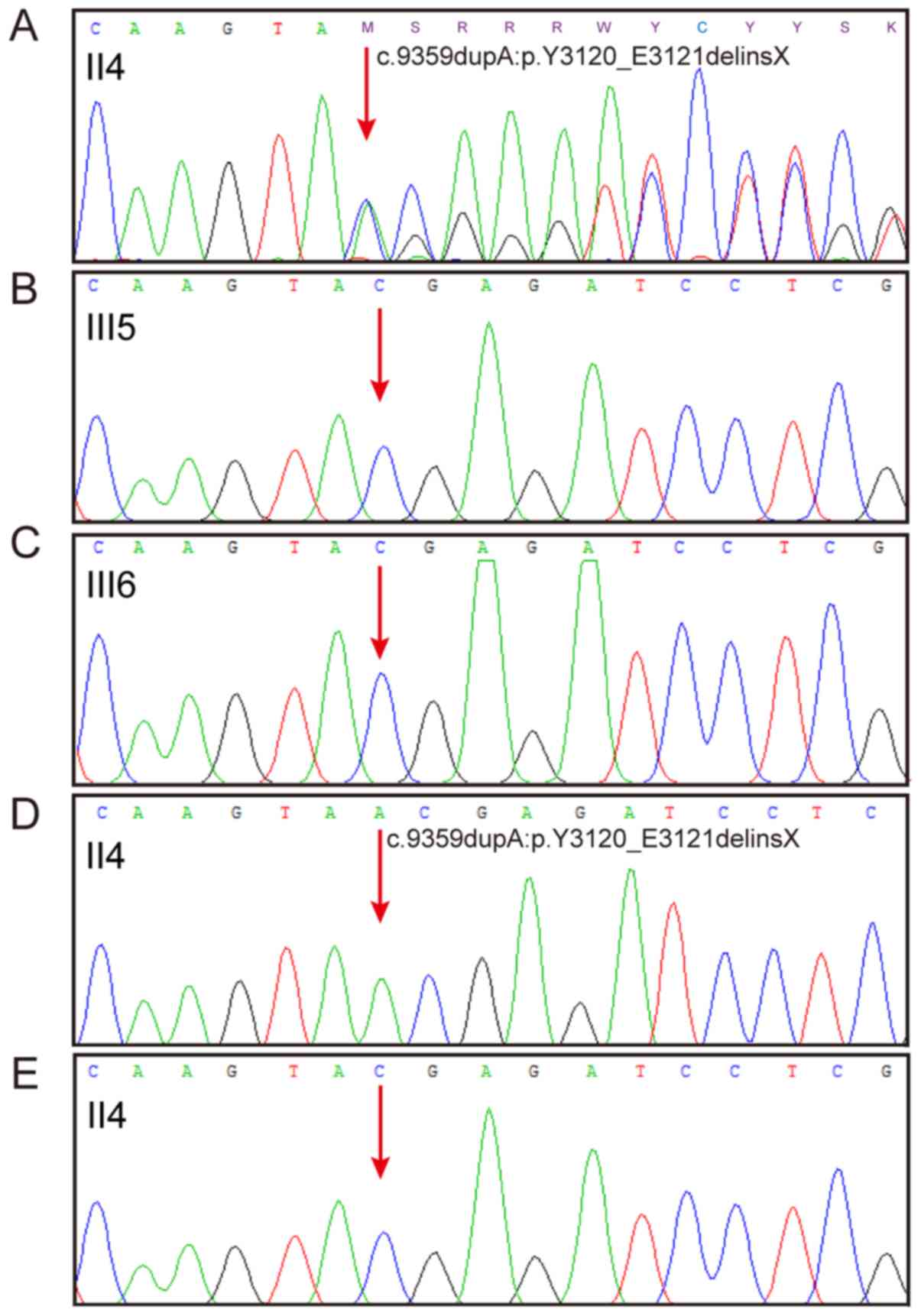
The results of Sanger sequencing indicated that the mother
possessed the duplication variant c.9359dupA:p.Y3120_E3121delinsX
in PKD1, while their two normal daughters did not (Fig. 2A and C). Further investigation of
the proband's sequence was performed via ligation of the nest PCR
product with the T linear vector, followed by sequencing. The
sequencing results revealed that the proband carried one mutated
sequence (Fig. 2D) and one normal
sequence (Fig. 2E), indicating
that the mutation exists in a heterozygous form.
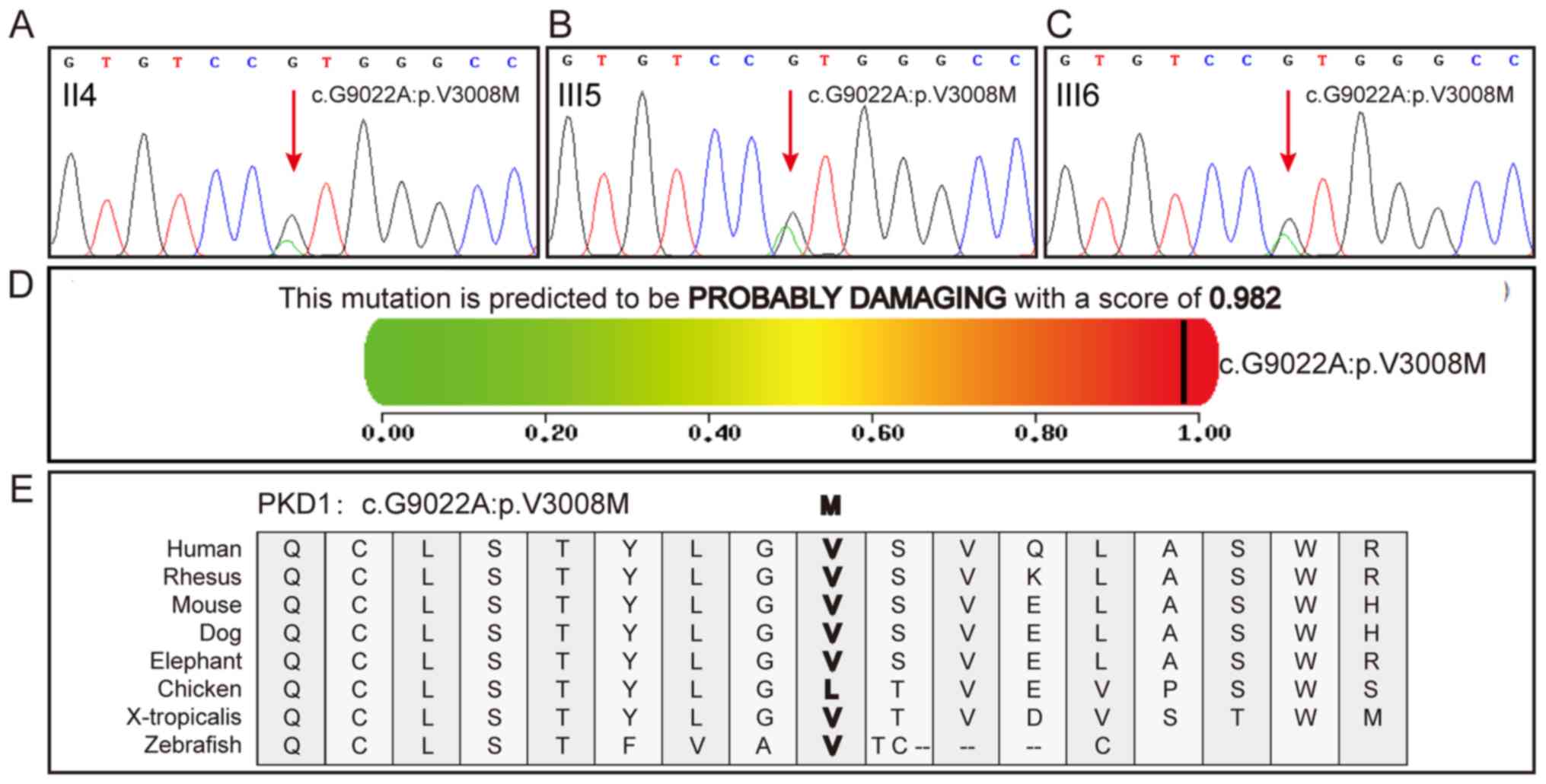
In addition, the proband and their daughters
presented the same c.G9022A:p.V3008M mutation in PKD1 (Fig. 3A-C). To further investigate the
importance of the missense site, the pathogenic and conservational
properties of the site were analyzed using various prediction
tools. Almost all the prediction results indicated that the
missense site was pathogenic (Table
III). The Polyphen2 prediction results for c.G9022A:p.V3008M
are presented in Fig. 3D.
Conservational property analysis revealed that human, mouse and
other animals, but not chicken, share the same amino acid (valine,
V) at a particular site, indicating high conservation (Fig. 3E). PC1 is an integral membrane
protein comprising 4,303 amino acids; >3,000 of the amino acids
in the first part of the protein are located extracellularly. A
diagram of PC1 is presented in Fig.
4. The position of c.9359dupA:p.Y3120_E3121delinsX is located
within the PC-1, lipoxygenase, α-toxin (PLAT) domain, and
c.G9022A:p.V3008M is located within an undefined region (amino
acids 2,834–3,011) near the GPS domain (Fig. 4). The first 23 amino acids in PC1
consist of a signal peptide that is cleaved during the
post-translational modification process (29). The remaining PC1 protein can be
separated into extracellular, transmembrane and cytoplasmic regions
that contain numerous domains.
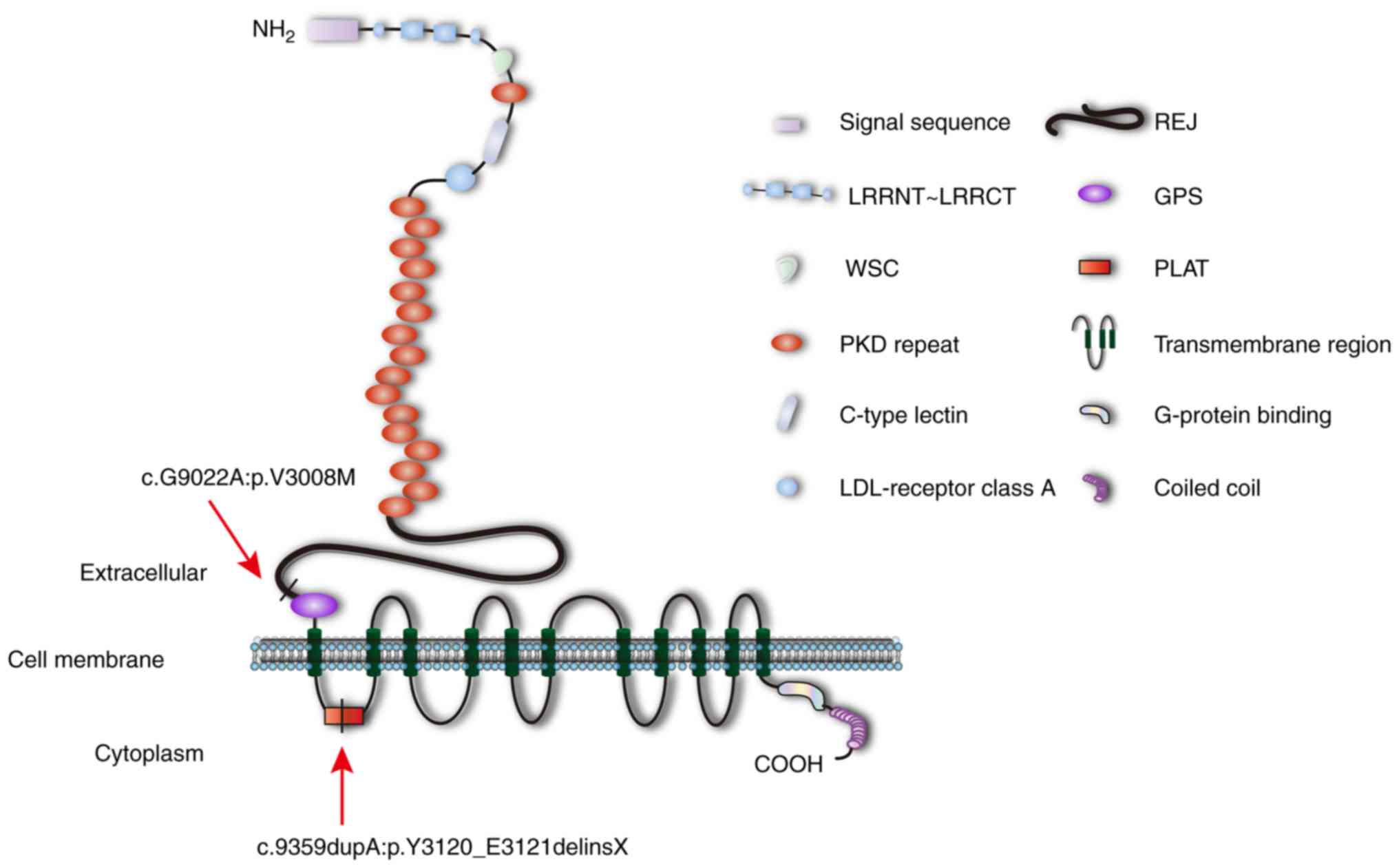 | Figure 4.Diagram of polycystin-1. Arrows
indicate the position of c.9359dupA:p.Y3120_E3121delinsX within the
PLAT domain and c.G9022A:p.V3008M within an undefined region (amino
acids 2,834–3,011). GPS, G protein-coupled receptor proteolysis
site; LDL, low-density lipoprotein; LRR, leucine-rich repeat;
LRRCT, C-terminal LRR domain; LRRNT, N-terminal LRR domain; PKD,
polycystic kidney disease; PLAT, polycystin-1, lipoxygenase,
α-toxin; REJ, receptor egg jelly; WSC, cell wall integrity and
stress-response component 1. |
| Table III.Assessment of the missense site in
PKD1. |
Table III.
Assessment of the missense site in
PKD1.
| Mutation | SIFTa |
Polyphen2_HVARb |
MutationTasterc |
MutationAssessord |
|---|
|
c.G9022A:p.V3008M | 0.005 | 0.982 | 21, D | 2.865, M |
Analysis of all the pathogenic sites
of PKD1 suggests that the missense c.G9022A:p.V3008M mutation is
likely pathogenic
The PKDB is a database that facilitates the
characterization of ADPKD variants in PKD1 and PKD2. In PKDB,
mutations in PKD1 and PKD2 are collected, and ADPKD mutation types
are classified as substitution, nonsense, synonymous,
3′-untranslated region (UTR), 5′-UTR, frameshift, insertion or
deletion, large deletion or duplication, splice, intervening
sequence (IVS) silent and IVS unknown mutations. The clinical
significance of each mutation is further characterized as
definitely pathogenic, highly likely pathogenic, likely pathogenic,
indeterminate, likely hypomorphic or likely neutral (30). According to the PKDB, it is
difficult to identify the clinical significance of substitution
mutations, and no substitutions have been classified as definitely
pathogenic.
To further investigate the pathogenic properties of
the single nucleotide variants in various regions, PKD1
substitutions listed in dbSNP and PKDB were studied. The single
nucleotide variants from the two websites were counted, and
repetitions were deleted. As the clinical significance of nonsense
mutations is assessed as definitely pathogenic, the single
nucleotide variant group in dbSNP was excluded. Substitutions
described as likely pathogenic and highly likely pathogenic in PKDB
were counted. In total, 4,805 single nucleotide variants, including
312 pathogenic variants, were observed in PC1 amino acids
24–4,303.
Provided that PC1 is an integral membrane protein,
in addition to its signal peptide, it can be divided into an
extracellular region (amino acids 24–3,074), a transmembrane region
(amino acids 3,075–4,106) and a cytoplasmic region (amino acids
4,107–4,303). The frequency of substitutions identified as highly
likely pathogenic or likely pathogenic was calculated within a
variety of regions with various numbers of single nucleotide
variants. Notably, the frequencies of highly likely pathogenic or
likely pathogenic substitutions in the extracellular (6.3%, 216 out
of 3,444 amino acids) and transmembrane (7.9%, 88 out of 1,111
amino acids) regions were similar, while that in the cytoplasmic
region was markedly lower (3.2%, 8 out of 250 amino acids)
(Table III). The results
suggested that substitutions occurring in the PC1 N-terminal region
were more likely to be pathogenic variants than substitutions in
the C-terminal region of the protein.
According to the domains and repeats divisions
listed in UniProt, the frequencies of likely pathogenic
substitutions were calculated in the various regions (Table IV). The frequencies in the
N-terminal leucine-rich repeat (LRR) domain (LRRNT, amino acids
24–67), LRR domain 1 (LRR1, amino acids 68–91), LRR domain 2 (LRR2,
amino acids 92–113), C-terminal LRR domain (LRRCT, amino acids
125–178), cell wall integrity and stress-response component 1 (WSC,
amino acids 177–271) and C-type lectin (amino acids 415–531)
motifs, which are located in the PC1 C-terminal region, were
notably higher. The GPS domain (amino acids 3,012–3,061) appeared
to exhibit a relatively high frequency (11.9%, 7 out of 59 amino
acids) compared with the other domains. The c.G9022A:p.V3008M
missense mutation is located in an undefined region within amino
acids 2,834–3,011. This region presented a frequency of 8.9% (21
out of 235 amino acids), which was higher than the majority of
other undefined regions, as well as the average frequency of 6.5%
(312 out of 4,805 amino acids).
| Table IV.Pathogenic substitutions and domains
in PKD1. |
Table IV.
Pathogenic substitutions and domains
in PKD1.
| Region | Domain | Amino acid
position | Amino acid length
(aa) | Nucleotide length
(bp) | Number of SNP | Number of
pathogenic SNPs | Pathogenic
SNPs/total SNPs (%) |
|---|
| Extracellular | LRRNT | 24-67 | 44 | 132 | 24 | 4 | 16.7 |
|
| LRR1 | 68-91 | 24 | 72 | 18 | 4 | 22.2 |
|
| LRR2 |
92-113 | 22 | 66 | 22 | 5 | 22.7 |
|
| UD | 114-124 | 11 | 33 | 8 | 1 | 12.5 |
|
| LRRCT | 125-178 | 54 | 162 | 26 | 4 | 15.4 |
|
| WSC | 177-271 | 95 | 285 | 63 | 9 | 14.3 |
|
| PKD1 | 272-359 | 88 | 264 | 73 | 4 |
5.5 |
|
| UD | 360-414 | 55 | 165 | 38 | 3 |
7.9 |
|
| C-type lectin | 415-531 | 117 | 351 | 95 | 18 | 18.9 |
|
| UD | 532-637 | 106 | 318 | 83 | 5 | 6 |
|
| LDL-receptor class
A | 638-671 | 34 | 102 | 37 | 1 |
2.7 |
|
| UD | 672-742 | 71 | 213 | 80 | 7 |
8.8 |
|
| PKD2 | 743-817 | 34 | 102 | 60 | 1 |
1.7 |
|
| UD | 818-854 | 37 | 111 | 43 | 1 |
2.3 |
|
| PKD3 | 855-928 | 75 | 225 | 83 | 0 | 0 |
|
| UD | 929-934 | 6 | 18 | 2 | 1 | 50 |
|
| PKD4 |
935-1020 | 74 | 222 | 80 | 4 | 5 |
|
| UD | 1021-1022 | 2 | 6 | 1 | 0 | 0 |
|
| PKD5 | 1023-1129 | 86 | 258 | 134 | 1 |
0.7 |
|
| PKD6 | 1127-1215 | 107 | 321 | 112 | 4 |
3.6 |
|
| PKD7 | 1213-1298 | 86 | 258 | 120 | 5 |
4.2 |
|
| PKD8 | 1294-1383 | 90 | 270 | 135 | 5 |
3.7 |
|
| PKD9 | 1382-1469 | 88 | 264 | 101 | 4 | 4 |
|
| PKD10 | 1468-1551 | 84 | 252 | 98 | 6 |
6.1 |
|
| PKD11 | 1550-1635 | 86 | 258 | 110 | 1 |
0.9 |
|
| PKD12 | 1634-1721 | 88 | 264 | 110 | 3 |
2.7 |
|
| PKD13 | 1719-1805 | 87 | 261 | 109 | 1 |
0.9 |
|
| UD | 1806 | 1 | 3 | 2 | 0 | 0 |
|
| PKD14 | 1807-1890 | 84 | 252 | 110 | 6 |
5.5 |
|
| PKD15 | 1889-1974 | 86 | 258 | 107 | 2 |
1.9 |
|
| UD | 1976 | 1 | 3 | 2 | 0 | 0 |
|
| PKD16 | 1977-2057 | 81 | 243 | 116 | 8 |
6.9 |
|
| UD | 2058-2059 | 2 | 6 | 9 | 0 | 0 |
|
| PKD17 | 2060-2148 | 89 | 267 | 103 | 3 |
2.9 |
|
| REJ | 2146-2833 | 688 | 2064 | 863 | 67 |
7.8 |
|
| UD | 2834-3011 | 178 | 534 | 235 | 21 |
8.9 |
|
| GPS | 3012-3061 | 50 | 150 | 59 | 7 | 11.9 |
|
| UD | 3062-3074 | 13 | 39 | 15 | 1 |
6.7 |
|
|
| 24-3074 | 3051 | 9153 | 3444 | 216 |
6.3 |
| Transmembrane | UD | 3075-3117 | 43 | 129 | 40 | 3 |
7.5 |
|
| PLAT | 3118-3233 | 116 | 348 | 113 | 5 |
4.4 |
|
| UD | 3234-4106 | 873 | 2619 | 958 | 80 |
8.4 |
|
|
| 3075-4106 | 1032 | 3096 | 1111 | 88 |
7.9 |
| Cytoplasmic | UD | 4107-4303 | 197 | 591 | 250 | 8 |
3.2 |
|
| UD | 4107-4303 | 197 | 591 | 250 | 8 |
3.2 |
| Total |
| 24-4303 | 4280 | 12840 | 4805 | 312 |
6.5 |
Collectively, the results indicated that the
c.G9022A:p.V3008M missense mutation was likely neutral in PKDB;
however, it may be pathogenic. This finding may explain emerging
concepts in PKD, including hypomorphic alleles and gene dosage.
Further investigations should be performed to identify the
potential underlying mechanism.
Discussion
In the present study, a four-generation ADPKD family
including 29 individuals was examined. According to the pedigree,
there were five patients with PKD distributed across two
generations, including two females (II4 and
II10) and three males (I1, II1,
and II13). A parent of the proband suffered from PKD;
however, neither of the daughters were affected. Of the individuals
assessed for PKD, almost half (5 patients among 11 individuals with
lineal consanguinity) were patients with PKD. Collectively, the
results allowed this family to be diagnosed with ADPKD.
Following whole exome sequencing and analysis, the
duplication variant NM_001009944.2:c.9359dupA:p.Y3120_E3121delinsX
and the missense mutation c.G9022A:p.V3008M in PKD1 can be
considered as candidate pathogenic factors accounting for the
family's disease. At present, >2,000 and >400 mutations in
PKD1 and PKD2, respectively, have been identified according to
PKDB; however, the association between phenotype and genotype
requires further study. Dedoussis et al (31) reported that the coexistence of PKD1
and PKD2 mutations in a single patient may induce a more severe
phenotype compared with individuals with PKD1 mutations. Numerous
reports have demonstrated that patients with PKD1 mutations usually
progress to ESRD at an earlier age, particularly those expressing
truncated proteins compared with individuals harboring PKD2
mutations (32–34). The novel duplication variant
c.9359dupA:p.Y3120_E3121delinsX in PKD1 may contribute to
understanding the association between phenotype and genotype in
future studies.
PKD1 is a long gene with 46 exons, among which exons
1–35 exhibit six highly similar repeats on chromosome 16 (35,36).
Considering the complexity of PKD1, nest PCR was employed to
amplify the candidate fragments. The results of Sanger sequencing
for the nest PCR product demonstrated that an A was inserted into
the coding sequence, resulting in a frameshift mutation. PKDB, OMIM
and HGMD were examined, and published articles were employed to
determine whether the mutation was novel. There are no previous
reports of this duplication variant; however, the missense mutation
c.G9361A:p.E3121K, representing the next amino acid substitution of
the duplication variant, has been reported to be likely pathogenic
(32). Following
post-translational modification, PC1 constitutes 4,279 amino acids,
with numerous functional domains. The
c.9359dupA:p.Y3120_E3121delinsX duplication variant altered a TAC
codon to TAA, which is a termination signal in protein translation.
As a result, protein translation was stopped at the 3,120th amino
acid (tyrosine, T), and a truncated PC1 protein was generated. The
truncated PC1 was terminated in the PLAT domain, which may have
affected cell signaling mediated by the protein and its roles as a
lipid/protein binding scaffold (19). The premature stop codon resulted in
an ~3,000 amino acid PC1 sequence, causing the protein to lose the
majority of its C-terminal domains.
A missense site (NM_001009944.2:c.G9022A:p.V3008M)
and two synonymous sites (NM_001009944.2:c.G4674A:p.T1558T and
NM_001009944.2: c.G3444A:p.P1148P) in PKD1 were also observed
according to the whole exome sequencing results from the proband.
All three substitutions were reported as likely neutral in
PKDB.
The missense site c.G9022A:p.V3008M is located in
the 25th exon of PKD1, which lies in an undefined region (amino
acids 2,834–3,011) near the GPS motif, upstream of
c.9359dupA:p.Y3120_E3121delinsX. The 9,022nd nucleotide (guanine,
G) in the consensus coding sequence containing 12,912 nucleotides
is changed to A, resulting in an amino acid substitution from V to
methionine (M). V is a nonpolar amino acid, while M is polar,
indicating that a structural alteration occurs in the PKD1 protein
PC1 following substitution. When pathogenic properties were
assessed with the prediction tools, the missense mutation
(c.G9022A:p.V3008M) was indicated as likely to lead to a diseased
state. The results of conservation analysis also supported the
pathogenic properties of the missense mutation. Additionally, the
frequency of pathogenic substitutions in various regions was
analyzed. The region (from amino acids 2,834–3,011) that contained
the missense mutation (c.G9022A:p.V3008M) exhibited a relatively
high frequency. Collectively, the results indicated that the
c.G9022A:p.V3008M missense mutation is likely to be pathogenic;
however, it was identified as likely neutral in PKDB.
The results regarding pathogenic substitutions in
PKD1 within various regions also revealed that the PC1 N-terminal
domain, including LRRNT, LRR1, LRR2, LRRCT, WSC and C-type lectin,
exhibited a relatively high frequency compared with the C-terminal
domain, suggesting that missense mutations in the N-terminus are
more likely to induce ADPKD. Additionally, the frequency of
pathogenic substitutions in the C-type lectin domain was notably
higher, indicating that it is a key functional domain.
To determine whether the missense mutation is a
disease-inducing factor along with the duplication variant, Sanger
sequencing was employed to detect the missense mutation in the
proband and their two normal daughters. The missense mutation was
observed in the sequencing results for all three individuals,
indicating that the missense mutation was not a pathogenic factor
in the family with ADPKD. It should be noted that the
c.G9022A:p.V3008M missense mutation may not be responsible for PKD
in the studied family; however, it may be involved in the process
of PKD development according to the gene dosage theory (20).
The present study identified a novel duplication
variant, NM_001009944.2:c.9359dupA:p.Y3120_E3121delinsX, within the
PLAT domain, and a missense mutation, c.G9022A:p.V3008M in PKD1,
within an undefined region (amino acids 2,834–3,011), in a family
that suffers from ADPKD. The duplication variant is considered to
be the pathogenic factor for ADPKD in this family; however, the
c.G9022A:p.V3008M missense mutation has an uncertain importance.
Numerous, but not all, classical methods were used in the study to
assess the pathogenic properties of missense mutations. Only three
individuals were sequenced, as informed consent was not obtained
from other individuals in the family; the assessment of additional
samples may improve understanding into the genetic basis of PKD.
Furthermore, recently reported novel substitutions were not
included in the calculation of pathogenic mutations within various
regions in PC1 (37–39). The pathogenic mechanism of the
variants, as well as the association with clinical manifestations,
require further investigation.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Xue Zhang
(Institute of Basic Medical Sciences of the Chinese Academy of
Medical Sciences, McKusick-Zhang Center for Genetic Medicine,
School of Basic Medicine, Harbin Medical University, Harbin, China)
for his advice and help. The authors also thank Dr Mengdi Cai
(Laboratory of Medical Genetics, Harbin Medical University, Harbin,
China) for the modification of Fig.
4.
Funding
The present study was supported by the National Key
Research and Development Program of China (grant no.
2016YFC1000504).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KD, XJ, JW, WS, JY, XZ and SF conceived and designed
the study; HM, HWu, JS, HWa and XZ collected the clinical
information; KD, HM, HWu, JS, WJ, HS, SZ and GJ performed the
experiments; KD, LX, XZ, GJ, RG and JB analyzed the data; and KD,
HWa, WS and SF prepared the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Research Board of Harbin Medical University (no. HMUIRB20180001,
Harbin, China), and all participants provided written informed
consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PKD
|
polycystic kidney disease
|
|
ADPKD
|
autosomal dominant polycystic kidney
disease
|
|
ARPKD
|
autosomal recessive polycystic kidney
disease
|
|
PC1
|
polycystin-1
|
|
PC2
|
polycystin-2
|
|
ESRD
|
end-stage renal disease
|
|
LR-PCR
|
long-range polymerase chain
reaction
|
References
|
1
|
Obeidova L, Elisakova V, Stekrova J,
Reiterova J, Merta M, Tesar V, Losan F and Kohoutova M: Novel
mutations of PKD genes in the Czech population with autosomal
dominant polycystic kidney disease. BMC Med Genet. 15:412014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Edrees BM, Athar M, Abduljaleel Z,
Al-Allaf FA, Taher MM, Khan W, Bouazzaoui A, Al-Harbi N, Safar R,
Al-Edressi H, et al: Functional alterations due to amino acid
changes and evolutionary comparative analysis of ARPKD and ADPKD
genes. Genom Data. 10:127–134. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Robinson C, Hiemstra TF, Spencer D, Waller
S, Daboo L, Karet Frankl FE and Sandford RN: Clinical utility of
PKD2 mutation testing in a polycystic kidney disease cohort
attending a specialist nephrology out-patient clinic. BMC Nephrol.
13:792012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Abdelwahed M, Hilbert P, Ahmed A, Mahfoudh
H, Bouomrani S, Dey M, Hachicha J, Kamoun H, Keskes-Ammar L and
Belguith N: Mutational analysis in patients with Autosomal Dominant
Polycystic Kidney Disease (ADPKD): Identification of five mutations
in the PKD1 gene. Gene. May 31–2018.(Epub ahead of print).
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yu G, Qian X, Wu Y, Li X, Chen J, Xu J and
Qi J: Analysis of gene mutations in PKD1/PKD2 by multiplex
ligation-dependent probe amplification: Some new findings. Ren
Fail. 37:366–371. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Litvinchuk T, Tao Y, Singh R and Vasylyeva
TL: A case of new familiar genetic variant of autosomal dominant
polycystic kidney disease-2: A case study. Front Pediatr. 3:822015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Casteleijn NF, Spithoven EM, Rookmaaker
MB, Vergouwen MD and Gansevoort RT: Bilateral cysts in the choroid
plexus in a patient with autosomal dominant polycystic kidney
disease. Nephrol Dial Transplant. 30:859–860. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu B, Chen SC, Yang YM, Yan K, Qian YQ,
Zhang JY, Hu YT, Dong MY, Jin F, Huang HF and Xu CM: Identification
of novel PKD1 and PKD2 mutations in a Chinese population with
autosomal dominant polycystic kidney disease. Sci Rep. 5:174682015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hafer AS and Conran RM: Autosomal
recessive polycystic kidney disease. Acad Pathol.
4:23742895177185602017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thomas C, Zühlsdorf A, Hörtnagel K,
Mulahasanovic L, Grauer OM, Kümpers P, Wiendl H and Meuth SG: A
novel PKD1 mutation associated with autosomal dominant kidney
disease and cerebral cavernous malformation. Front Neurol.
9:3832018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Somlo S, Wirth B, Germino GG,
Weinstat-Saslow D, Gillespie GA, Himmelbauer H, Steevens L, Coucke
P, Willems P, Bachner L, et al: Fine genetic localization of the
gene for autosomal dominant polycystic kidney disease (PKD1) with
respect to physically mapped markers. Genomics. 13:152–158. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gainullin VG, Hopp K, Ward CJ, Hommerding
CJ and Harris PC: Polycystin-1 maturation requires polycystin-2 in
a dose-dependent manner. J Clin Invest. 125:607–620. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim DY and Park JH: Genetic mechanisms of
ADPKD. Adv Exp Med Biol. 933:13–22. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shin YB and Park JH: Recent trends in
ADPKD research. Adv Exp Med Biol. 933:3–11. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang Y and Ehrlich BE: Structural studies
of the C-terminal tail of polycystin-2 (PC2) reveal insights into
the mechanisms used for the functional regulation of PC2. J
Physiol. 594:4141–4149. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Venugopal J and Blanco G: On the many
actions of ouabain: Pro-cystogenic effects in autosomal dominant
polycystic kidney disease. Molecules. 22:E7292017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rangan GK, Lopez-Vargas P, Nankivell BJ,
Tchan M, Tong A, Tunnicliffe DJ and Savige J: Autosomal dominant
polycystic kidney disease: A path forward. Semin Nephrol.
35:524–537. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Saigusa T and Bell PD: Molecular pathways
and therapies in autosomal-dominant polycystic kidney disease.
Physiology (Bethesda). 30:195–207. 2015.PubMed/NCBI
|
|
19
|
Ong AC and Harris PC: A polycystin-centric
view of cyst formation and disease: The polycystins revisited.
Kidney Int. 88:699–710. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Santoso NG, Cebotaru L and Guggino WB:
Polycystin-1, 2, and STIM1 interact with IP(3)R to modulate ER Ca
release through the PI3K/Akt pathway. Cell Physiol Biochem.
27:715–726. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yamaguchi T, Wallace DP, Magenheimer BS,
Hempson SJ, Grantham JJ and Calvet JP: Calcium restriction allows
cAMP activation of the B-Raf/ERK pathway, switching cells to a
cAMP-dependent growth-stimulated phenotype. J Biol Chem.
279:40419–40430. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yamaguchi T, Pelling JC, Ramaswamy NT,
Eppler JW, Wallace DP, Nagao S, Rome LA, Sullivan LP and Grantham
JJ: cAMP stimulates the in vitro proliferation of renal cyst
epithelial cells by activating the extracellular signal-regulated
kinase pathway. Kidney Int. 57:1460–1471. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rossetti S, Kubly VJ, Consugar MB, Hopp K,
Roy S, Horsley SW, Chauveau D, Rees L, Barratt TM, van't Hoff WG,
et al: Incompletely penetrant PKD1 alleles suggest a role for gene
dosage in cyst initiation in polycystic kidney disease. Kidney Int.
75:848–855. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nagao S, Nishii K, Yoshihara D, Kurahashi
H, Nagaoka K, Yamashita T, Takahashi H, Yamaguchi T, Calvet JP and
Wallace DP: Calcium channel inhibition accelerates polycystic
kidney disease progression in the Cy/+ rat. Kidney Int. 73:269–277.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ghata J and Cowley BD Jr: Polycystic
kidney disease. Compr Physiol. 7:945–975. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Trudel M, Yao Q and Qian F: The role of
G-protein-coupled receptor proteolysis site cleavage of
polycystin-1 in renal physiology and polycystic kidney disease.
Cells. 5:E32016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chapin HC, Rajendran V and Caplan MJ:
Polycystin-1 surface localization is stimulated by polycystin-2 and
cleavage at the G protein-coupled receptor proteolytic site. Mol
Biol Cell. 21:4338–4348. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R; 1000 Genome
Project Data Processing Subgroup, : The Sequence Alignment/Map
format and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Al-Bhalal L and Akhtar M: Molecular basis
of autosomal dominant polycystic kidney disease. Adv Anat Pathol.
12:126–133. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gout AM, Martin NC, Brown AF and Ravine D:
PKDB: Polycystic kidney disease mutation database-a gene variant
database for autosomal dominant polycystic kidney disease. Hum
Mutat. 28:654–659. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dedoussis GV, Luo Y, Starremans P,
Rossetti S, Ramos AJ, Cantiello HF, Katsareli E, Ziroyannis P,
Lamnissou K, Harris PC and Zhou J: Co-inheritance of a PKD1
mutation and homozygous PKD2 variant: A potential modifier in
autosomal dominant polycystic kidney disease. Eur J Clin Invest.
38:180–190. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cornec-Le Gall E, Audrézet MP, Chen JM,
Hourmant M, Morin MP, Perrichot R, Charasse C, Whebe B, Renaudineau
E, Jousset P, et al: Type of PKD1 mutation influences renal outcome
in ADPKD. J Am Soc Nephrol. 24:1006–1013. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Neumann HP, Bacher J, Nabulsi Z, Ortiz
Brüchle N, Hoffmann MM, Schaeffner E, Nürnberger J, Cybulla M,
Wilpert J, Riegler P, et al: Adult patients with sporadic
polycystic kidney disease: The importance of screening for
mutations in the PKD1 and PKD2 genes. Int Urol Nephrol.
44:1753–1762. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu C, Yang Y, Zou L, Hu Z, Li J, Liu Y, Ma
Y, Ma M, Su D and Zhang S: Identification of novel mutations in
Chinese Hans with autosomal dominant polycystic kidney disease. BMC
Med Genet. 12:1642011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kinoshita M, Higashihara E, Kawano H,
Higashiyama R, Koga D, Fukui T, Gondo N, Oka T, Kawahara K, Rigo K,
et al: Technical evaluation: Identification of pathogenic mutations
in PKD1 and PKD2 in patients with autosomal dominant polycystic
kidney disease by next-generation sequencing and use of a
comprehensive new classification system. PLoS One. 11:e01662882016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu J, Li L and Liu Q: Mutational analysis
of PKD1 gene in a Chinese family with autosomal dominant polycystic
kidney disease. Int J Clin Exp Pathol. 8:13289–13292.
2015.PubMed/NCBI
|
|
37
|
Carrera P, Calzavara S, Magistroni R, den
Dunnen JT, Rigo F, Stenirri S, Testa F, Messa P, Cerutti R, Scolari
F, et al: Deciphering variability of PKD1 and PKD2 in an italian
cohort of 643 patients with autosomal dominant polycystic kidney
disease (ADPKD). Sci Rep. 6:308502016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sha YK, Sha YW, Mei LB, Huang XJ, Wang X,
Lin SB, Li L and Li P: Use of targeted sequence capture and
high-throughput sequencing identifies a novel PKD1 mutation
involved in adult polycystic kidney disease. Gene. 634:1–4. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mallawaarachchi AC, Furlong TJ, Shine J,
Harris PC and Cowley MJ: Population data improves variant
interpretation in autosomal dominant polycystic kidney disease.
Genet Med. Oct 29–2018.(Epub ahead of print). doi:
10.1038/s41436-018-0324-x. View Article : Google Scholar : PubMed/NCBI
|