Introduction
Spinal cord injury (SCI) is a serious and disabling
disease associated with a range of symptoms, including severe
movement dysfunction, muscle weakness, and sensory changes
(1). Based on pathological
patterns, SCI can be classified as a primary or secondary injury.
Although little is known regarding the pathocellular mechanisms of
SCI, secondary damage following primary SCI are considered to
involvea wide range of pathologies, including neural inflammation,
demyelination, axonal degeneration, and oligodendrocyte and
neuronal cell death (2,3). Great efforts have been made to
improve the functional outcomes of patients with SCI; however,
therapeutic advances have thus far been limited (4). Therefore, the specific objective of
the present study was to identify biomarkers and therapeutic
targets for SCI.
A large number of studies have implicated apoptosis,
autophagy, inflammation and endoplasmic reticulum stress as
important features of SCI; however, regulatory mechanisms based on
crosstalk between these factors remain to be delineated (5). A strong association between autophagy
and acute neurodegeneration caused by SCI has previously been
reported in the literature (6).
Autophagy is a highly conserved evolutionary phenomenon of
intracellular degradation that maintains the homeostasis of cells
by inducing rapid self-clearance or by degrading supramolecular
structures (7,8). Similar to cell division,
differentiation and death, autophagy dysfunctionis associated with
numerous diseases, including cancer, and cardiovascular and
neurodegenerative diseases (9,10).
Pott et al (11)
demonstrated that inadequate autophagy in intestinal epithelial
cells increases the induction of apoptosis and potentially impairs
barrier integrity due to inflammatory stimuli. A study conducted by
Papadakis et al (12) also
indicated that autophagy may have a protective role in an
oxygenglucose-deprivation neuronal and cerebral ischemia model.
However, recent evidence (10)
suggests that whether autophagy is protective or detrimental may be
based on the activation status of the cell, as well as other
factors. Notably, increased or decreased autophagy potentially
contributes to a variety of diseases and pathological conditions
(13).
Among the RNAs of different molecular ranges, the
sequences of long non-coding RNAs (lncRNAs) are the least
evolutionarily conserved. At the cellular level, epigenetic
modulation is one of the salient roles of lncRNAs, although lncRNAs
can also regulate gene expression through transcription,
alternative splicing, RNA translation and organization of important
structures for RNA processing. An abundance of chromatin
modification complexes can be targeted by lncRNAs to reconstruct
the structure and/or expression of their adjacent genes (14). Prior studies have noted the
critical roles of lncRNAs in the pathogenesis of various
neurological diseases, including SCI. For instance, there is
evidence that the lncRNA-XIST significantly aids in the recovery
from SCI by inhibiting apoptosis (15). Qiao et al (16) also suggested that the lncRNA MALAT1
has a neuroprotective role in spinal cord ischemic/reperfusion
injury, where it acts by regulating miR-204. Furthermore, Zhou
et al (17) confirmed that
MALAT1 can inhibit acute SCI by inhibiting the inflammatory
response of microglial cells.
Recent evidence has indicated that lncRNAs can
regulate the expression of messenger RNAs (mRNAs) by competitively
binding to microRNAs (miRNAs) and acting ascompeting endogenous
RNAs (ceRNAs), which can systematically functionalize non-coding
transcripts based on competitive sharing of miRNAs (18). It is considered that lncRNA
transcripts function as competing endogenous RNAs (ceRNAs) or
natural microRNA sponges that contain numerous miRNA binding sites.
Salmena et al (19)
proposed the ‘ceRNA hypothesis’, according to which the expression
of a specific miRNA can be temporarily reduced due to ceRNA. In
addition, based on the ceRNA hypothesis, scholars discovered a
large-scale regulatory network in the transcriptome based on miRNA
binding sites, which greatly expands the information availableon
human functional genetics and details a network that may serve a
critical role in cancer pathology (19,20).
In the present study, a global triple network was
generated based on the ceRNA hypothesis by mining data from the
Gene Expression Omnibus (GEO), which is curated by the National
Center for Biotechnology Information (NCBI). Based on the resulting
network, target lncRNAs associated with SCI were identified.
Furthermore, a subnetwork of node lncRNAs was obtained from the
ceRNA network, which facilitated enrichment and identification of
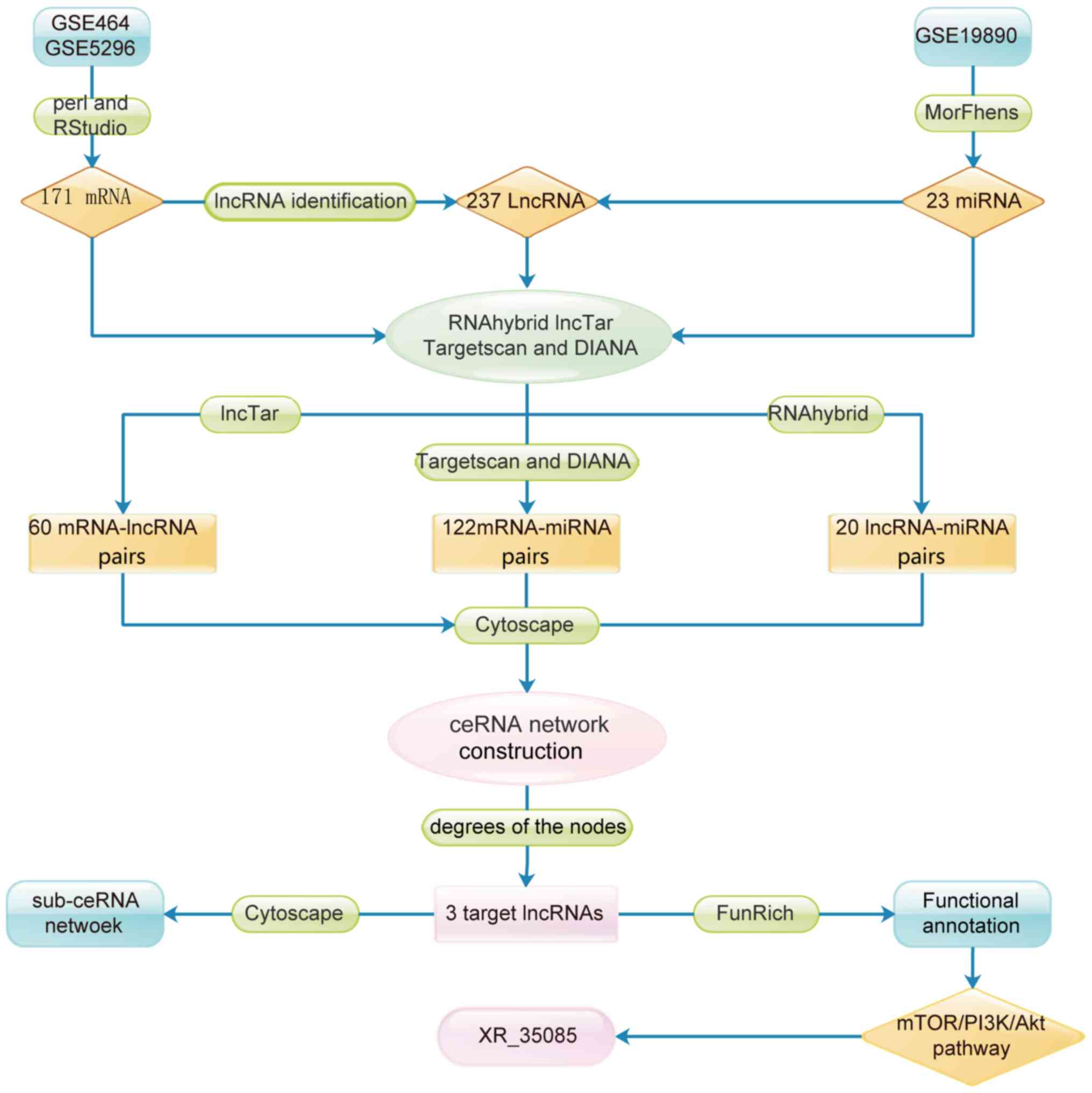
lncRNA pathways and functions. The flow chart for target lncRNA
selection is presented in Fig.
1.
Materials and methods
Data sources
GEO (www.ncbi.nlm.nih.gov/geo) is currently the largest
fully public gene expression resource data repository, from which
thousands of experiments and tens of millions of gene expression
profiles can be queried and downloaded. Data examined in the
present study were obtained from the NCBI GEO database. The raw
mRNA expression data were downloaded from the series GSE464
(www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE464) and
GSE5296 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE5296).
The lncRNA data were obtained from the aforementioned datasets by
repurposing the probes in the RG_U34A, RG_U34B, RG_U34C and
Mouse430_2 arrays of the Affymetrix annotation platform (www.thermofisher.com/cn/en/home/life-science/microarray-analysis/microarray-data-analysis/genechip-array-annotation-files.html)
(21). Data on miRNA were obtained
from GEO series GSE19890 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE19890).
Raw data analysis
Subsequent tologarithmic processing, the two mRNA
expression datasets were merged using Perl script (www.perl.org/), and batch effects were corrected using
the Combat method from the sva and limma packages (22–24).
Differential mRNA expression analysis between SCI and normal
samples was performed using the limma package (bioconductor.org/packages/release/bioc/html/limma.html)
(25). mRNAs with a P-value of
<0.05 and a log2fold-change (log2FC) of
>1.5 were considered to be differentially expressed.
Differential lncRNA and miRNA expression analysis between SCI and
normal samples was performed using the Morpheus platform
(https://software.broadinstitute.org/morpheus/) based
on the significance analysis of microarrays method, and the
threshold was also set tolog2FC>1.5 and
P<0.05.
Screening and pairwise matching of
lncRNAs, miRNAs and mRNAs
Pairwise matching of differentially expressed
lncRNAs, miRNAs and mRNAs was performed using RNAhybrid (bibiserv.cebitec.uni-bielefeld.de/rnahybrid),
LncTar (www.cuilab.cn/lnctar), TargetScan
Human version 7.1 (www.targetscan.org/vert_71/) and DIANA Tools
(diana.imis.athena-innovation.gr/DianaTools/index.php?r=microT_CDS/index).
The miRNA binding sites on lncRNAs were predicted using RNA hybrid
(26), which is a tool used to
calculate the minimum free energy hybridization of a long and short
RNA. LncTar (27) is a software
used for predicting putative interactions between mRNAs and lncRNAs
based on free energy minimization. In addition, TargetScan
(28) and DIANA (29,30)
were synchronously used to predict differentially expressed pairs
of miRNAs and mRNAs. TargetScan predicts the biological targets of
miRNAs by searching for the presence of conserved 8mer, 7mer and
6mer sites by matching the seed region of each miRNA (31). In mammals, predictions were ranked
based on the predicted efficacy of targeting as calculated using
the cumulative weighted context++ scores of the sites. In the
current study, the predicted mRNAs were set to a cumulative
weighted context++ score of ≤-0.4 in TargetScan (28). The arsenal of DIANA Tools included
the target prediction algorithms of microT v4 and microT-CDS, where
microT-CDS was used to predict the miRNA and mRNA pairs in the
present study. In order to avoid data omission, the two online
websites (TargetScan and DIANA) were used for prediction, and their
results were combined.
Construction of the ceRNA network
For a given lncRNA-mRNA pair, which included
negatively co-expressed mRNAs and lncRNAstargeted with a specified
common miRNA, co-expression competing triplets were identified
based on the ceRNA theory (32).
Using existing miRNA target online software (RNAhybrid, LncTar,
TargetScan and DIANA Tools), lncRNA-miRNA, lncRNA-mRNA and
miRNA-mRNA interactions were confirmed. Using an in-house Perl
script, the ceRNA associations were integrated (33). To elucidate the roles of lncRNAs,
miRNAs and mRNAs within the regulatory ceRNA network, their
interactive and visual mediated network was then created using
Cytoscape software, version 3.5.0 (34).
Molecular function analysis of the
ceRNA network
To elucidate the molecular functions within the
ceRNA network, mRNAs in the network were analyzed using FunRich
software (version 3.0; www.funrich.org/). The FunRich tool allows users to
assign the biological process, cellular component and molecular
function terms, as well as biological pathways, protein domains,
sites of expression, clinical phenotypes and transcription factors,
to enriched and depleted factors (35).
Reconstruction of a sub-ceRNA network
using node lncRNAs
Based on the established ceRNA network, network
analysis was subsequently performed to investigate the degree
distribution of the nodes (36).
Three node lncRNAs were screened based on the degrees of the nodes,
while miRNA and mRNA pairing with the lncRNAs was also examined.
Accordingly, three sub-ceRNA networks were established with the
Cytoscape software (version 3.5.0).
Functional annotation enrichment
analysis
Biological pathway functional enrichment analyses
were conducted using FunRich software, version 3.0. This software
was used to visualize and assess pathway interactions of the
lncRNA-targeted mRNAs. This tool also highlighted nodes that were
enriched in specific pathways and allowed for the creation of
subnetworks based on these highlighted nodes (35).
Results
Screening of differentially expressed
lncRNAs, miRNAs and mRNAs
Following logarithmic processing and batch
correction, the microarray data were analyzed by Morpheus and limma
packages. In total, 171 mRNAs and 237 lncRNAs that were
overexpressed in SCI were identified according to the threshold of
log2FC>1.5 and P<0.05. In addition, 23 miRNAs that
were downregulated in SCI were selected using Morpheus software
(Fig. 1).
Pairwise matching of lncRNAs, miRNAs
and mRNAs
Using RNAhybrid and LncTar, 20 mature miRNA-lncRNA
pairs and 60 lncRNA-mRNA pairs were respectively predicted.
Furthermore, a total of 122 unique mature miRNA-mRNA pairs,
predicted by both TargetScan and DIANA, were selected as reliable
interaction pairs.
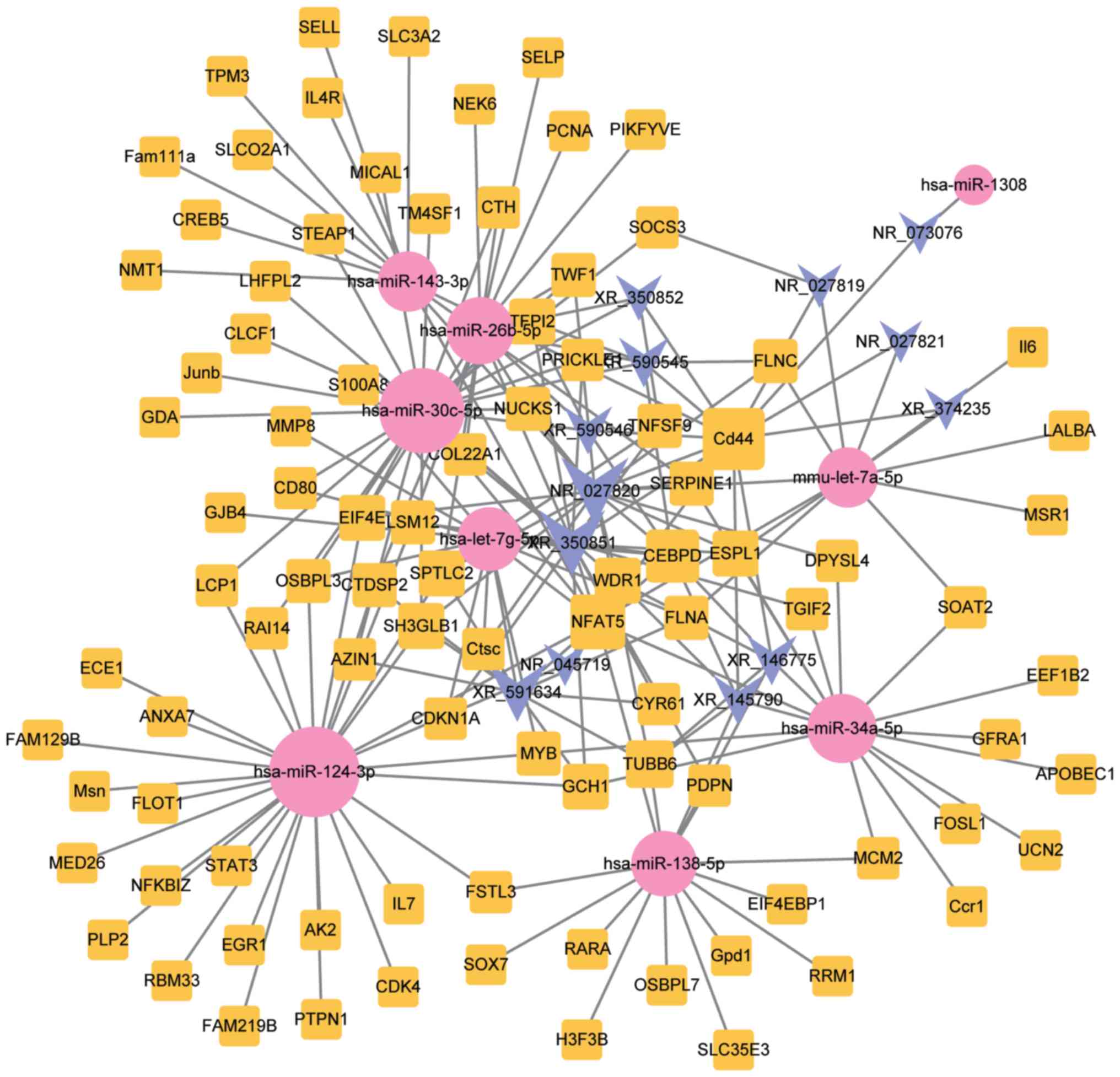
Construction of the lncRNA-miRNA-mRNA
ceRNA network
In order to elucidate the interactions between
lncRNAs, miRNAs and mRNAs in SCI, a ceRNA network was established.
As shown in Fig. 2, this network
consisted of 13 lncRNA, 93 mRNA and 9 miRNA nodes, with a total of
202 edges.
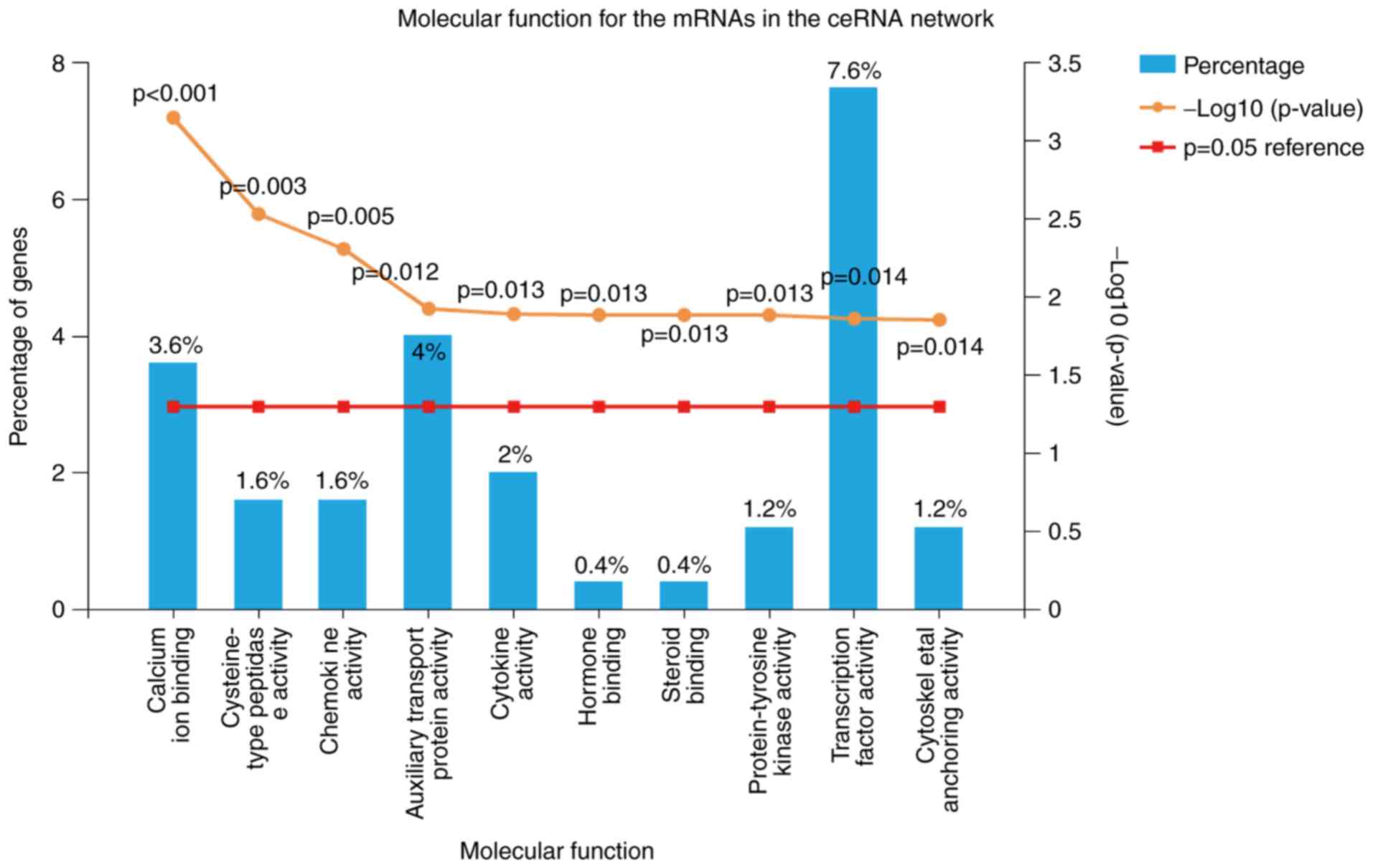
Molecular annotation of lncRNAs based
on the ceRNA network
To identify possible mechanisms associated with SCI,
75 molecular function modules were selected using FunRich 3.0
software. As displayed in Table I,
the primary modules included calcium ion-binding, cysteine-type
peptidase activity, chemokine activity, auxiliary transport protein
activity, cytokine activity, hormone binding, steroid binding,
protein-tyrosine kinase activity, transcription factor activity and
cytoskeletal anchoring activity. The top 10 critical molecular
functions of mRNAs in the ceRNA network are presented in Fig. 3. The percentage and P-values of
critical molecular functions are also presented; there was a higher
proportion of transcription factor activity (7.6%), auxiliary
transport protein activity (4%) and calcium ion-binding (3.6%;
Fig. 3).
 | Table I.A total of 75 molecular function
modules potentially involved in spinal cord injury. |
Table I.
A total of 75 molecular function
modules potentially involved in spinal cord injury.
| Molecular
function | Fold
enrichment | P-value |
|---|
| Calcium ion
binding | 3.760905 | 0.000706 |
| Cysteine-type
peptidase activity | 6.730617 | 0.002923 |
| Chemokine
activity | 5.841835 | 0.004888 |
| Auxiliary transport
protein activity | 2.321344 | 0.011811 |
| Cytokine
activity | 3.615564 | 0.012769 |
| Hormone
binding | 77.225850 | 0.012949 |
| Steroid
binding | 77.225850 | 0.012949 |
| Protein-tyrosine
kinase activity | 6.115491 | 0.012960 |
| Transcription
factor activity | 1.743523 | 0.013665 |
| Cytoskeletal
anchoring activity | 5.958724 | 0.013908 |
| Receptor
activity | 2.141300 | 0.019646 |
| Acyltransferase
activity | 3.870462 | 0.020167 |
|
Ubiquitin-like-protein-specific protease
activity | 38.805030 | 0.025730 |
| Translation
regulator activity | 3.065792 | 0.042320 |
| Metallopeptidase
activity | 3.065792 | 0.042320 |
| Complement
activity | 5.541734 | 0.050627 |
| Ion channel
activity | 3.521433 | 0.054121 |
| Intracellular
ligand-gated ion channel activity | 5.172408 | 0.057295 |
| Ligand-dependent
nuclear receptor activity | 4.433704 | 0.075174 |
| Lipid kinase
activity | 4.433704 | 0.075174 |
| Phosphoprotein
phosphatase activity | 12.978060 | 0.075229 |
| Guanylate cyclase
activity | 9.737592 | 0.099030 |
| Lipid transporter
activity | 8.656838 | 0.110701 |
| Transcription
factor binding | 7.792019 | 0.122222 |
| Hydrolase
activity | 1.905825 | 0.124300 |
| Lipid binding | 7.084297 | 0.133594 |
| Serine-type
peptidase activity | 2.371695 | 0.134282 |
| RNA
methyltransferase activity | 6.494431 | 0.144819 |
| Transmembrane
receptor activity | 2.984502 | 0.145688 |
| Cytoskeletal
protein binding | 1.774696 | 0.153605 |
| Oxidoreductase
activity | 1.923332 | 0.156614 |
| Water channel
activity | 5.567317 | 0.166836 |
| Kinase
activity | 5.567317 | 0.166836 |
| Receptor signaling
protein tyrosine phosphatase activity | 4.871837 | 0.188289 |
| Protein
binding | 1.701421 | 0.210739 |
| Deaminase
activity | 4.103004 | 0.219441 |
| Carboxy-lyase
activity | 3.897957 | 0.229558 |
| Phosphorylase
activity | 3.897957 | 0.229558 |
| Peroxidase
activity | 3.897957 | 0.229558 |
| Transaminase
activity | 3.389748 | 0.259133 |
| Kinase regulator
activity | 3.389748 | 0.259133 |
| Hormone
activity | 3.118677 | 0.278218 |
| Helicase
activity | 3.118677 | 0.278218 |
| Protein tyrosine
phosphatase activity | 3.118677 | 0.278218 |
| Transcription
regulator activity | 1.207568 | 0.282903 |
| Heat shock protein
activity | 2.887749 | 0.296813 |
| Protease inhibitor
activity | 1.651143 | 0.344255 |
| Extracellular
matrix structural constituent | 1.400216 | 0.364123 |
|
Galactosyltransferase activity | 2.052042 | 0.390878 |
| Extracellular
ligand-gated ion channel activity | 1.901929 | 0.414282 |
|
Ribonucleoprotein | 1.901929 | 0.414282 |
| Growth factor
activity | 1.410999 | 0.417894 |
| Ligase
activity | 1.385805 | 0.426817 |
| Isomerase
activity | 1.732906 | 0.444099 |
| Phospholipase
activity | 1.732906 | 0.444099 |
| Transporter
activity | 1.073903 | 0.470688 |
| DNA binding | 1.063906 | 0.474045 |
| Chaperone
activity | 1.231838 | 0.487156 |
| Cell adhesion
molecule activity | 1.086772 | 0.490374 |
| Transmembrane
receptor protein tyrosine kinase activity | 1.392575 | 0.518527 |
| Peptidase
activity | 1.344563 | 0.530951 |
| Structural
constituent of cytoskeleton | 1.132939 | 0.531709 |
| Defense/immunity
protein activity | 1.278448 | 0.548991 |
| Motor activity | 0.987193 | 0.643607 |
| RNA binding | 0.846085 | 0.700779 |
| Receptor signaling
complex scaffold activity | 0.721871 | 0.790031 |
| Receptor
binding | 0.604590 | 0.814912 |
| Voltage-gated ion
channel activity | 0.599939 | 0.817325 |
| Transferase
activity | 0.499956 | 0.870160 |
| Protein
serine/threonine kinase activity | 0.515677 | 0.903888 |
| Catalytic
activity | 0.582086 | 0.916616 |
| Ubiquitin-specific
protease activity | 0.411724 | 0.957707 |
| Structural molecule
activity | 0.289945 | 0.970716 |
| G-protein coupled
receptor activity | 0.105974 | 0.999944 |
| Molecular function
unknown | 0.557085 | 0.999999 |
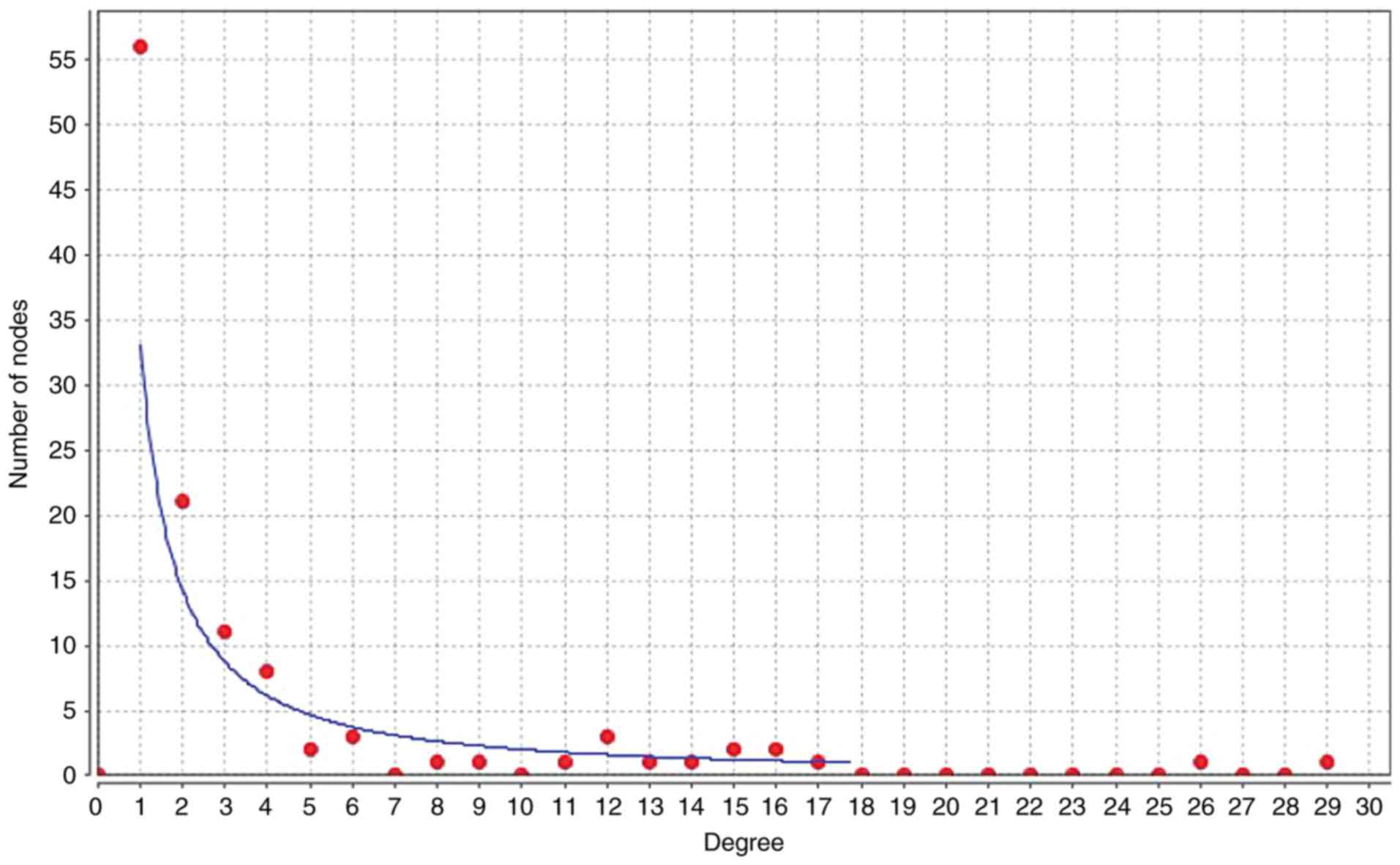
Target lncRNA-miRNA-mRNA
subnetwork
To illuminate the competitive mechanisms and
potential biological functions of lncRNAs in SCI, the degree
distribution of nodes in the ceRNA network were investigated using
Cytoscape software. The horizontal axis of Fig. 4 represents the degree of RNA in the
ceRNA network. As presented in Fig.
4 and Table II,
hsa-miR-124-3p had the highest degree of node, with a node value of
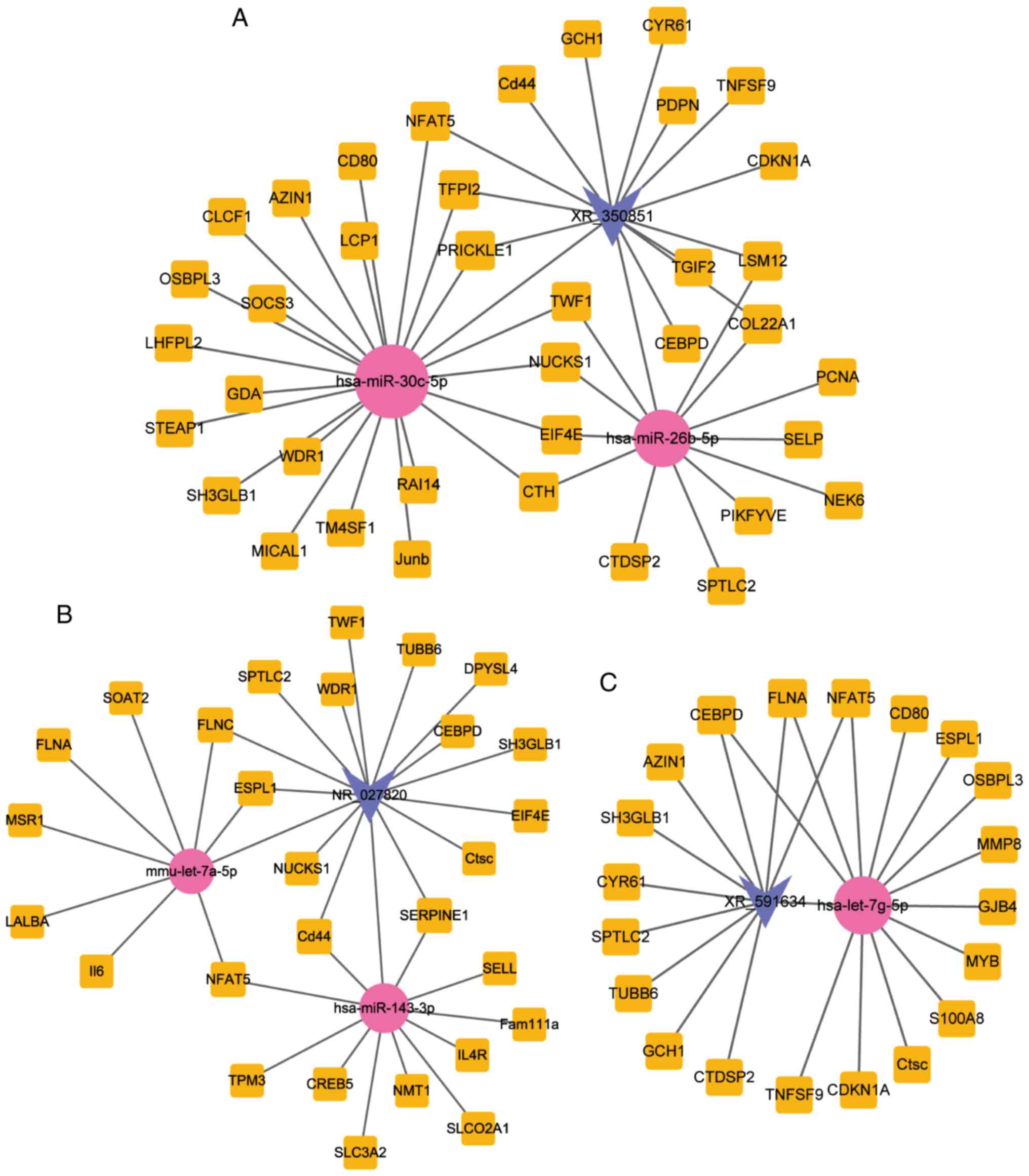
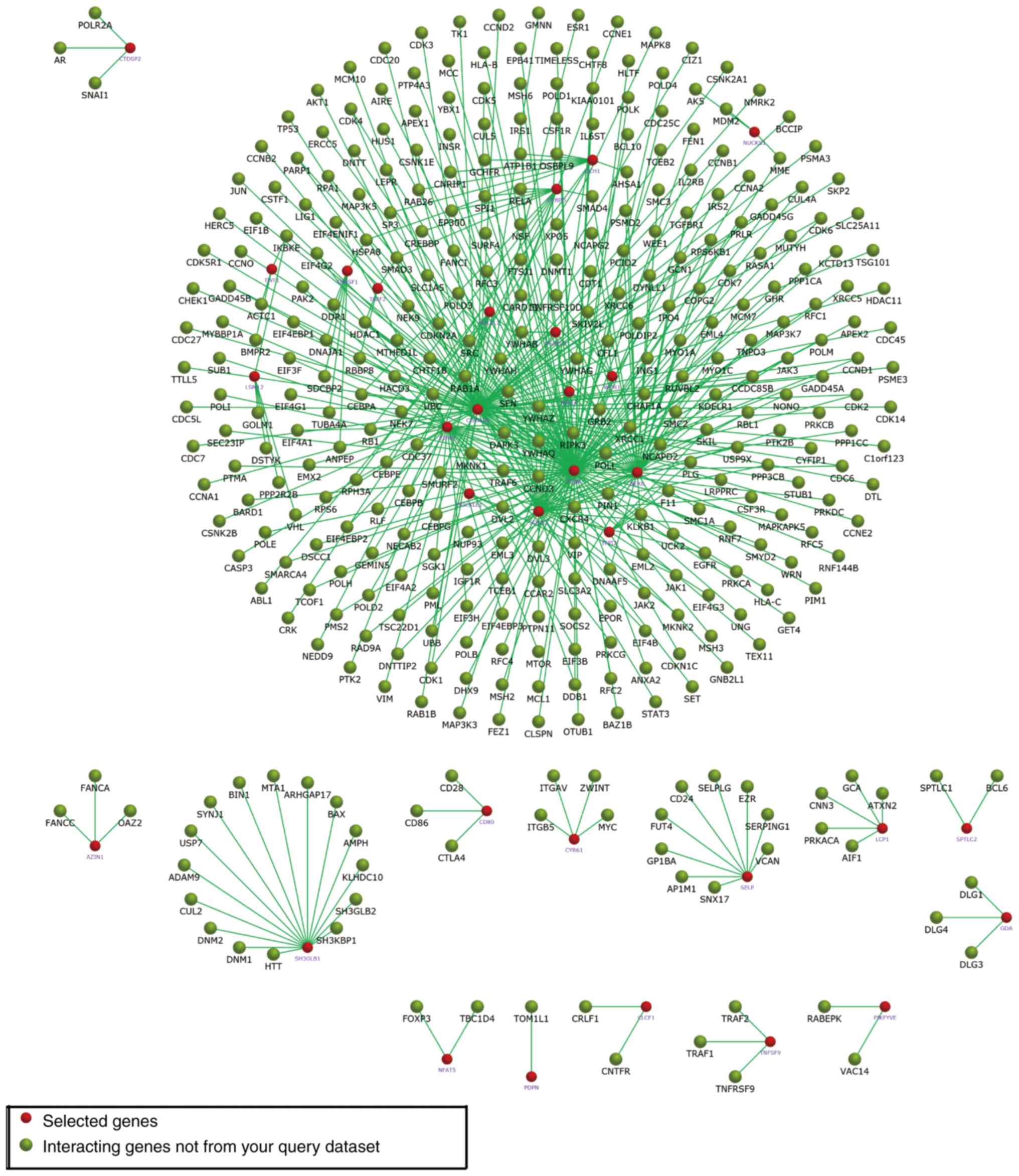
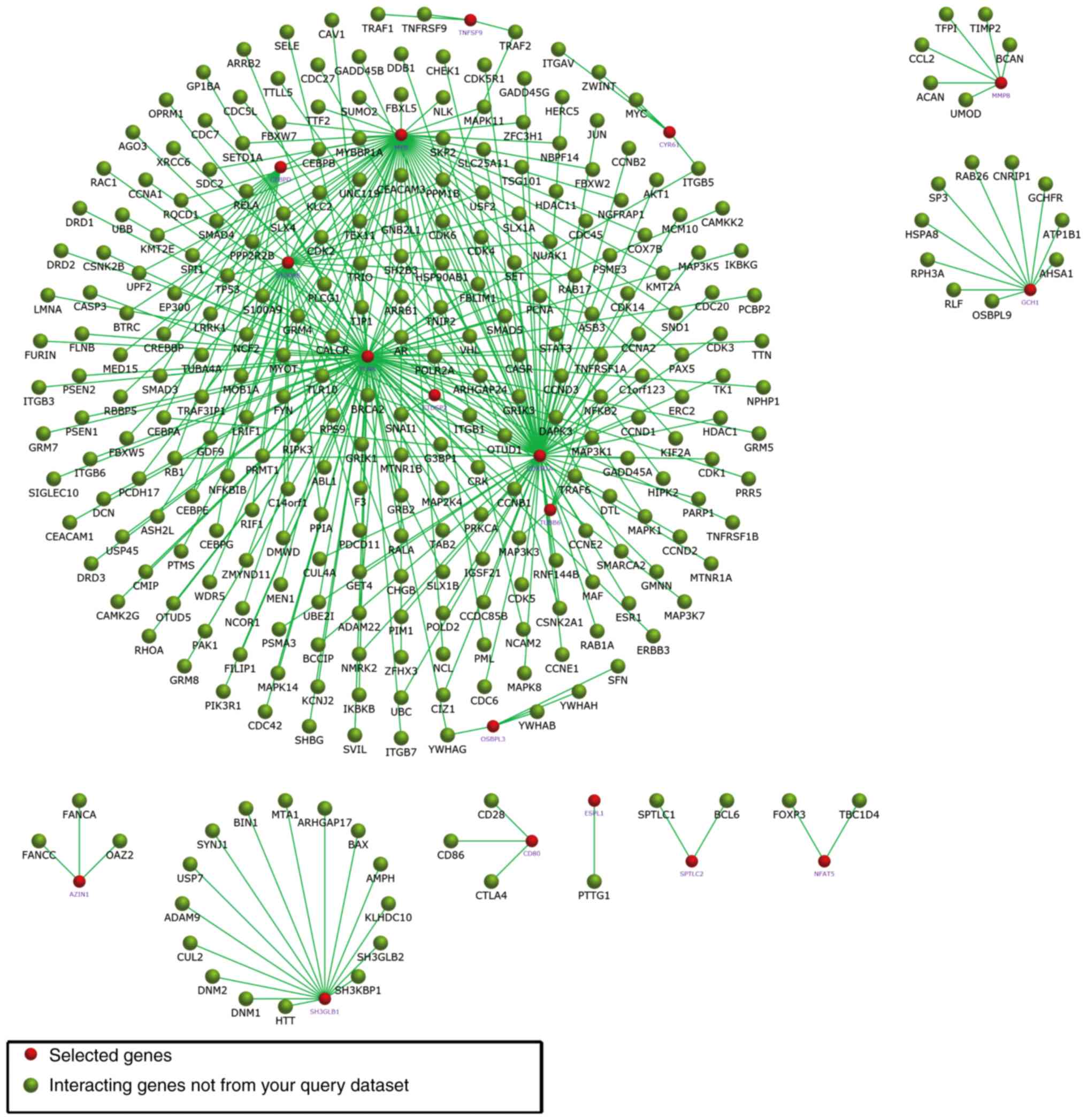
29. In Fig. 5, the subnetworks of
XR_350851, NR_027820, and XR_591634, respectively, are presented,
showing how the mRNA directly or indirectly is associated with the
lncRNA.
| Table II.Differentially expressed genes of the
competing endogenous RNAs (node degree ≥5). |
Table II.
Differentially expressed genes of the
competing endogenous RNAs (node degree ≥5).
| Number | RNA name | Degree |
|---|
| 1 | hsa-miR-124-3p | 29 |
| 2 | hsa-miR-30c-5p | 26 |
| 3 | hsa-miR-34a-5p | 17 |
| 4 | NR_027820 | 16 |
| 5 | hsa-miR-26b-5p | 16 |
| 6 | XR_350851 | 15 |
| 7 | hsa-miR-138-5p | 15 |
| 8 | hsa-let-7g-5p | 14 |
| 9 | Cd44 | 13 |
| 10 | hsa-miR-143-3p | 12 |
| 11 | mmu-let-7a-5p | 12 |
| 12 | XR_591634 | 11 |
| 13 | NFAT5 | 9 |
| 14 | CEBPD | 8 |
| 15 | XR_590545 | 6 |
| 16 | XR_145790 | 6 |
| 17 | XR_146775 | 6 |
| 18 | XR_590546 | 5 |
| 19 | TUBB6 | 5 |
Enrichment analysis of lncRNA-targeted
mRNAs
Biological pathway annotation analysis of
significantly differentially expressed mRNAs in the ceRNA
subnetwork revealed relevant pathways and molecular interactions
with associated genes. Based on FunRich software enrichment
analyses, 263, 139 and 174 biological pathways were identified to
be the components associate with the genes in the XR_350851,
NR_027820 and XR_591634 subnetworks, respectively. As shown in
Fig. 6, the critical biological
pathways in the XR_350851 subnetwork included the activator protein
1 (AP-1) transcription factor network, integrin-linked kinase
signaling, cell division cycle 42 (CDC42) signaling events,
sphingosine-1-phosphate receptor 1 (S1P1) pathway, ADP-ribosylation
factor 6 (Arf6) trafficking events, epidermal growth factor
receptor 1 downstream signaling, class I phosphoinositide 3-kinase
(PI3K) signaling events mediated by protein kinase B (Akt), insulin
pathway, mammalian target of rapamycin (mTOR) signaling pathway and
Arf6 downstream pathway. In addition, it was observed that the
significant biological pathways identified for the subnetwork of
NR_027820 were the following: Cell-extracellular matrix
interactions, platelet degranulation, response to elevated platelet
cytosolic Ca2+, cell junction organization, ceramide
biosynthesis, hemostasis, cell-cell communication, release of
eukaryotic translation initiation factor 4E, platelet activation,
signaling and aggregation, and glycoprotein Ib-IX–V activation
signaling (Fig. 7). Furthermore,
the top 10 biological pathways for the XR_591634 subnetwork were as
follows: C-MYB transcription factor network, AP-1 transcription
factor network, integrin-linked kinase signaling, CDC42 signaling
events, non-canonical Wnt signaling pathway, regulation of CDC42
activity, Wnt signaling network, glypican 3 network,
syndecan-4-mediated signaling events, and Glypican pathway
(Fig. 8).
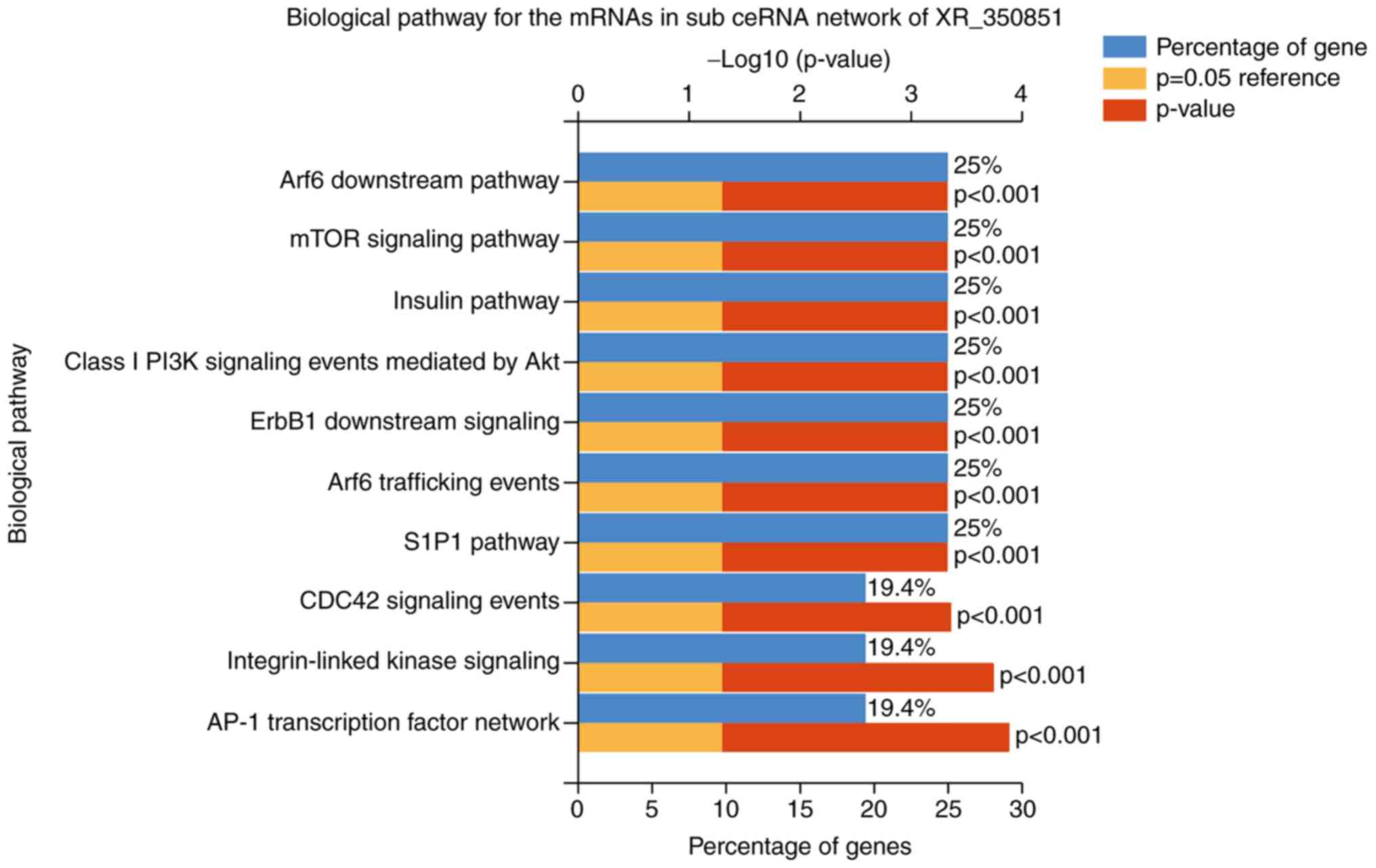 | Figure 6.Top 10 biological pathways of the
mRNAs in the sub-ceRNA network of the long noncoding RNA XR_350851.
The top identified pathways included the AP-1 transcription factor
network, integrin-linked kinase signaling, CDC42 signaling events,
S1P1 pathway, Arf6 trafficking events, ErbB1 downstream signaling,
class I PI3K signaling events mediated by Akt, insulin pathway,
mTOR signaling pathway and Arf6 downstream pathway. mRNA, messenger
RNA; ceRNA, competing endogenous RNA; AP-1, activator protein 1;
CDC42, cell division cycle 42; S1P1, sphingosine-1-phosphate
receptor 1; Arf6, ADP-ribosylation factor 6; ErbB1, epidermal
growth factor receptor 1; mTOR, mammalian target of rapamycin;
PI3K, phosphoinositide 3-kinase; Akt, protein kinase B. |
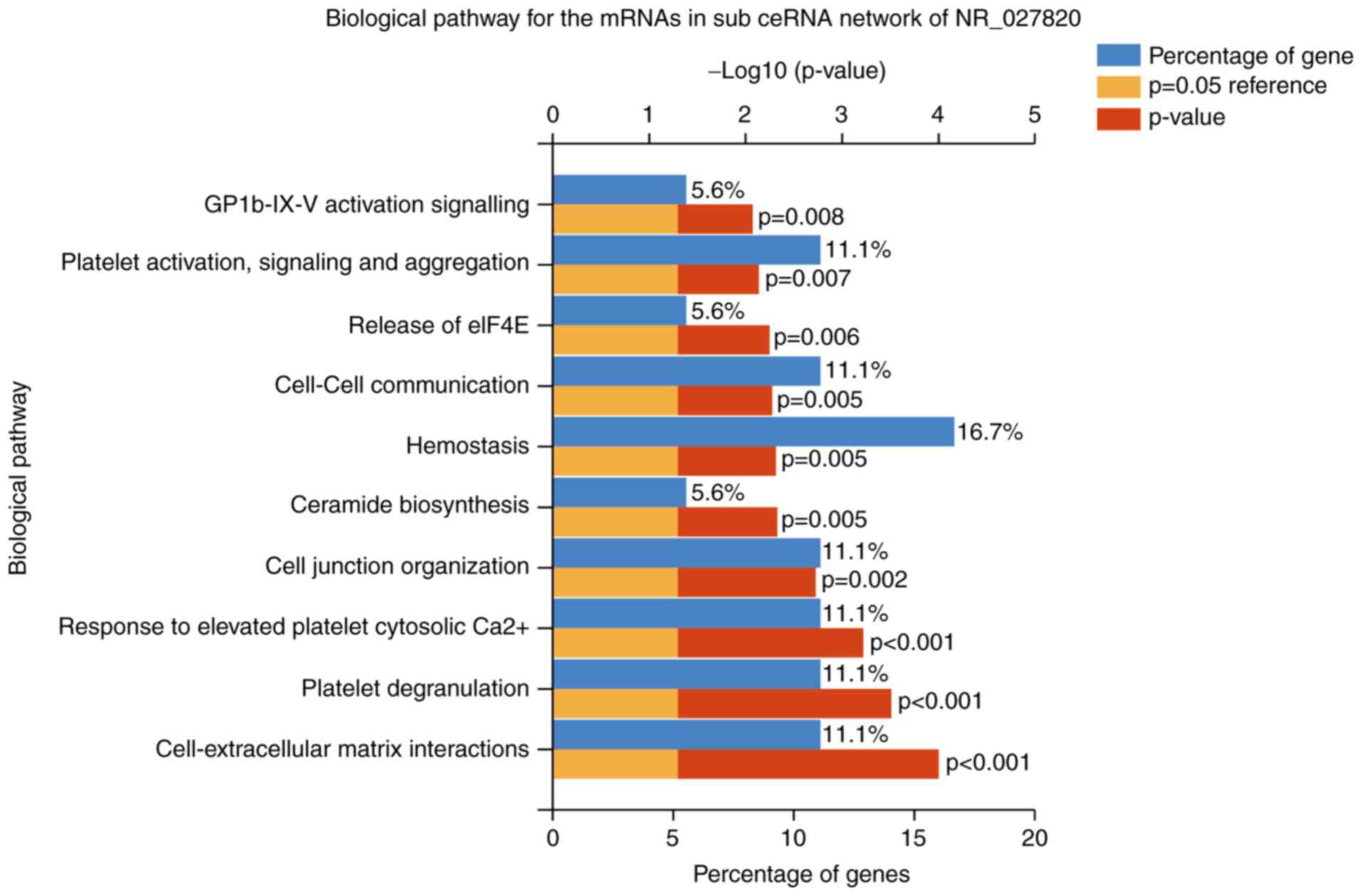 | Figure 7.Top 10 biological pathways of the
mRNAs in the sub-ceRNA network of the long noncoding RNA NR_027820.
These pathways included the cell-extracellular matrix interactions,
platelet degranulation, response to elevated platelet cytosolic
Ca2+, cell junction organization, ceramide biosynthesis,
hemostasis, cell-cell communication, release of eIF4E, platelet
activation, signaling and aggregation, GPIb-IX–V activation
signaling. mRNA, messenger RNA; ceRNA, competing endogenous RNA;
eIF4E, eukaryotic translation initiation factor 4E; GPIb-IX–V,
glycoprotein Ib-IX–V. |
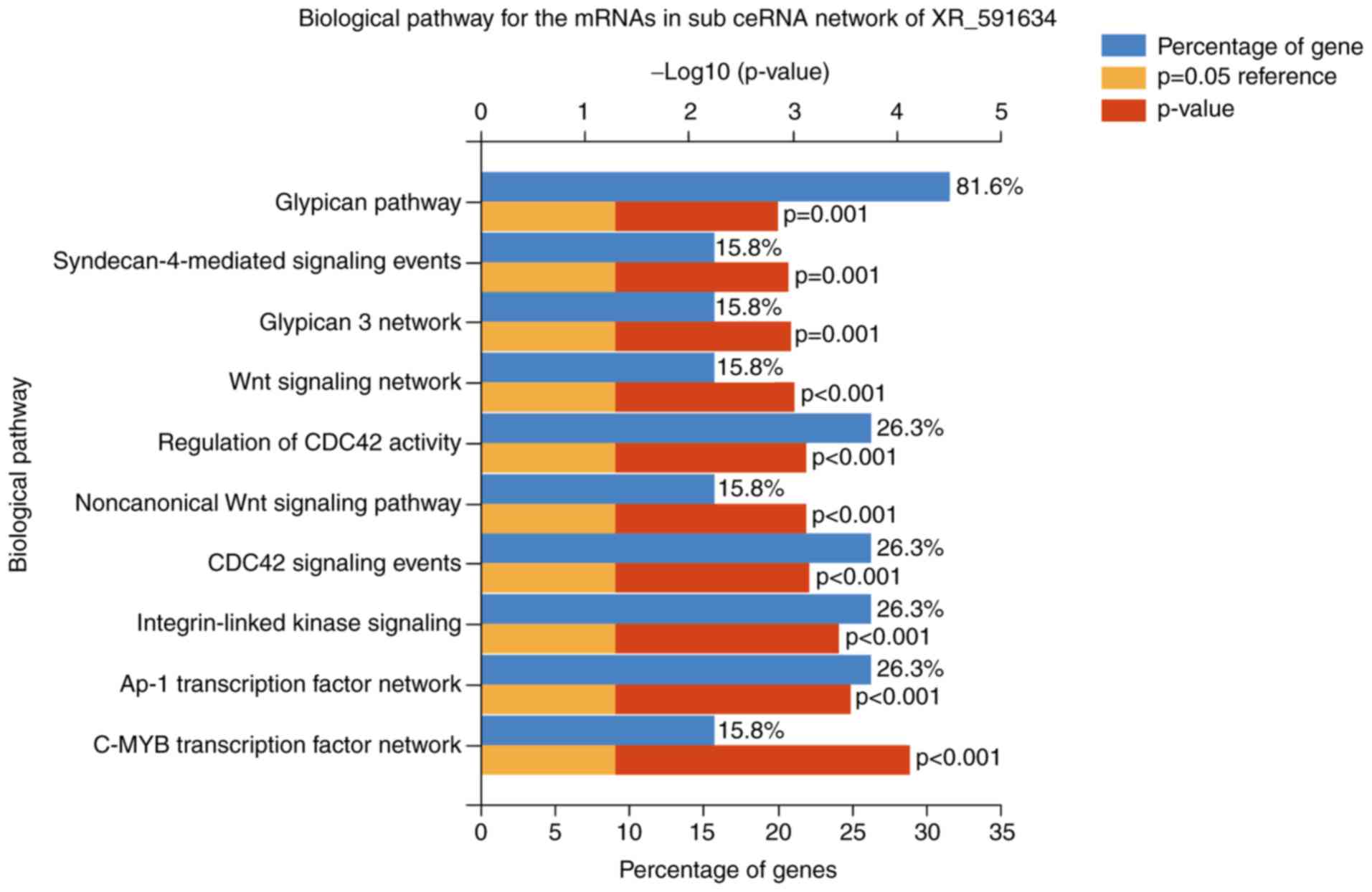 | Figure 8.Top 10 biological pathways of the
mRNAs in the sub-ceRNA network of the long noncoding RNA XR_591634.
The pathways included the C-MYB transcription factor network, AP-1
transcription factor network, integrin-linked kinase signaling,
CDC42 signaling events, noncanonical Wnt signaling pathway,
regulation of CDC42 activity, Wnt signaling network, glypican 3
network, syndecan-4-mediated signaling events and glypican pathway.
mRNA, messenger RNA; ceRNA, competing endogenous RNA; AP-1,
activator protein 1; CDC42, cell division cycle 42. |
Comparing the results of these three subnetworks,
XR_350851 was observed to be associated with the classic autophagy
pathway, namely the PI3K/Akt/mTOR signaling pathway. The
interactions of the three lncRNA-targeted mRNAs were also enriched
using FunRich software (Figs.
9–11).
Discussion
Approximately 130,000 people, predominantly young
adults, suffer from SCI paralysis annually worldwide, which has a
serious socioeconomic impact (37). The complex pathophysiology of SCI,
which includes primary and secondary mechanisms, involves a
complicated cascade of molecules and hampers the generation of
effective therapies (38). To
identify target lncRNAs that have potential as novel predictors in
clinical diagnoses and treatments, a network based on the ceRNA
theory was generated by mining lncRNA, miRNA and mRNA data from
NCBI GEO database. The resulting lncRNA-miRNA-mRNA regulatory
network consisted of 13 lncRNA, 93 mRNA and 9 miRNA nodes, with a
total of 202 edges.
Molecular function and biological pathway analyses
were used to assign the biological functions of differentially
expressed genes. The module analysis identified 75 molecular
function modules associated with SCI. The identified molecular
functions included calcium ion-binding, cysteine-type peptidase
activity, chemokine activity, auxiliary transport protein activity,
cytokine activity, hormone binding, steroid binding,
protein-tyrosine kinase activity, transcription factor activity and
cytoskeletal anchoring activity. In the current study, it was
observed that the node degrees of three lncRNAs, namely XR_350851,
NR_027820 and XR_591634, were significantly higher compared with
those of other lncRNAs. These three lncRNAs also exhibited an
increased number of lncRNA-miRNA and miRNA-mRNA pairs. Therefore,
it is suggested that these lncRNAs (XR_350851, NR_027820 and
XR_591634) may have profound implications in SCI andmay be
potential target lncRNAs. Subsequently, sub-ceRNA networks of the
three aforementioned node lncRNAs were reconstructed in the current
study.
Biological pathway analysis thenidentified that 263,
139 and 174 pathways were respectively enriched in the XR_350851,
NR_027820 and XR_591634 subnetworks. The predominant biological
pathways in the XR_350851 subnetwork were the AP-1 transcription
factor network, S1P1 pathway, class I PI3K signaling events
mediated by Akt, insulin pathway and mTOR signaling pathway. The
significant biological pathways in the NR_027820 subnetwork
included platelet degranulation, cell junction organization,
ceramide biosynthesis, hemostasis and platelet activation. In
addition, the AP-1 transcription factor network, integrin-linked
kinase signaling, regulation of CDC42 activity, Wnt signaling
network and glypican pathway were the critical biological pathways
in the XR_591634 subnetwork.
A previous study by Tsuboyama et al (39) determined that starvation conditions
or mTOR inhibition promoted VPS34-dependent ribophagic flux in
cells. In addition, a previous study reported that rapamycin may
aid in the restoration of motor function and act in a
neuroprotective manner by suppressing the mTOR pathway (40). As a central regulator of autophagy
and a serine/threonine kinase, mTOR is involved in cancer, andin
cardiovascular and neurological diseases. In addition, mTOR is the
catalytic subunit of two distinct signaling complexes, including
mTOR complex 1 (mTORC1) and mTORC2 (41,42),
which are significantly involved in the control of cell
proliferation. It is noteworthy that the growth factor/PI3K/Akt
signaling pathway is the upstream modulator of mTORC1 (43). Another important finding was that
mTORC2-ribosome binding can be improved by insulin stimulation of
the PI3K signaling pathway (44).
There is increasing evidence that inhibition of the
autophagy-associated mTOR pathway subsequent to SCI may have a
neuroprotective effect (39).
Furthermore, Bai et al (45) revealed that stimulating the
AMPK/mTOR signaling pathway of autophagy may facilitate functional
recovery from SCI. The PI3K/Akt/mTOR pathway has previously been
identified as a classic autophagy pathway (46). In addition, the PI3K/Akt/mTOR
pathway is important for XR_350851 associated mRNA. Therefore,
XR_350851 may be used as a target gene for neuroprotection
following SCI. However, whether the lncRNA XR_350851 regulates
autophagy in SCI has yet to be determined. Therefore, it is
suggested that future studies should focus on the role of XR_350851
in SCI.
In conclusion, using mined data and based on the
ceRNA theory, an SCI-associated lncRNA-miRNA-mRNA network was
constructed in the current study. According to the distribution of
the nodes in the ceRNA network, three lncRNAs (XR_350851, NR_027820
and XR_591634) were identified, and their sub-ceRNA networks
constructed. Furthermore, the functions of these three node lncRNAs
were predicted by enriching the pathways of their associated mRNAs
in the sub-ceRNA networks. The present study provided further
insight into the involvement of lncRNAs in the mechanism of SCI,
and the identified lncRNAs may serve as potential novel biomarkers
and therapeutic targets for SCI. Finally, it is speculated that the
lncRNA XR_350851 is closely associated with autophagy. However,
further research is required to determine the biological role of
XR_350851 in SCI.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZQ conceived and designed the present study. LW, JL
and BW collected, extracted and analyzed the data. LW wrote the
manuscript. LW and ZQ reviewed the final manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
SCI
|
spinal cord injury
|
|
lncRNAs
|
long non-coding RNAs
|
|
ceRNA
|
competing endogenous RNA
|
|
mRNAs
|
messenger RNAs
|
|
miRNAs
|
microRNAs
|
|
GEO
|
Gene Expression Omnibus
|
|
NCBI
|
National Center for Biotechnology
Information
|
|
AP-1
|
activator protein 1
|
|
CDC42
|
cell division cycle 42
|
|
S1P1
|
sphingosine-1-phosphate receptor 1
|
|
Arf6
|
ADP-ribosylation factor 6
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
Akt
|
protein kinase B
|
|
mTOR
|
mammalian target of rapamycin
|
|
mTORC1
|
mTOR complex 1
|
References
|
1
|
Ramer LM, Ramer MS and Bradbury EJ:
Restoring function after spinal cord injury: Towards clinical
translation of experimental strategies. Lancet Neurol.
13:1241–1256. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Su M, Guan H, Zhang F, Gao Y, Teng X and
Yang W: HDAC6 regulates the chaperone-mediated autophagy to prevent
oxidative damage in injured neurons after experimental spinal cord
injury. Oxid Med Cell Longev. 2016:72637362016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Walker CL, Walker MJ, Liu NK, Risberg EC,
Gao X, Chen J and Xu XM: Systemic bisperoxovanadium activates
Akt/mTOR, reduces autophagy and enhances recovery following
cervical spinal cord injury. PLoS One. 7:e300122012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thuret S, Moon LD and Gage FH: Therapeutic
interventions after spinal cord injury. Nat Rev Neurosci.
7:628–643. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li GL, Brodin G, Farooque M, Funa K, Holtz
A, Wang WL and Olsson Y: Apoptosis and expression of Bcl-2 after
compression trauma to rat spinal cord. J Neuropathol Exp Neurol.
55:280–289. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Galluzzi L, Bravo-San Pedro JM, Blomgren K
and Kroemer G: Autophagy in acute brain injury. Nat Rev Neurosci.
17:467–484. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ohsumi Y: Historical landmarks of
autophagy research. Cell Res. 24:9–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen ZH, Wang WT, Huang W, Fang K, Sun YM,
Liu SR, Luo XQ and Chen YQ: The lncRNA HOTAIRM1 regulates the
degradation of PML-RARA oncoprotein and myeloid cell
differentiation by enhancing the autophagy pathway. Cell Death
Differ. 24:212–224. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kroemer G: Autophagy: A druggable process
that is deregulated in aging and human disease. J Clin Invest.
125:1–4. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang L, Wang H, Shen Q, Feng L and Jin H:
Long non-coding RNAs involved in autophagy regulation. Cell Death
Dis. 8:e30732017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pott J, Kabat AM and Maloy KJ: Intestinal
epithelial cell autophagy is required to protect against
TNF-induced apoptosis during chronic colitis in mice. Cell Host
Microbe. 23:191–202.e4. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Papadakis M, Hadley G, Xilouri M, Hoyte
LC, Nagel S, McMenamin MM, Tsaknakis G, Watt SM, Drakesmith CW,
Chen R, et al: Tsc1 (hamartin) confers neuroprotection against
ischemia by inducing autophagy. Nat Med. 19:351–357. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu F, Wei X, Wu Y, Kong X, Hu A, Tong S,
Liu Y, Gong F, Xie L, Zhang J, et al: Chloroquine promotes the
recovery of acute spinal cord injury by inhibiting
autophagy-associated inflammation and endoplasmic reticulum stress.
J Neurotrauma. 35:1329–1344. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fritah S, Niclou SP and Azuaje F:
Databases for lncRNAs: A comparative evaluation of emerging tools.
RNA. 20:1655–1665. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gu S, Xie R, Liu X, Shou J, Gu W and Che
X: Long coding RNA XIST contributes to neuronal apoptosis through
the downregulation of AKT phosphorylation and is negatively
regulated by miR-494 in rat spinal cord injury. Int J Mol Sci.
18(pii): E7322017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qiao Y, Peng C, Li J, Wu D and Wang X:
LncRNA MALAT1 is neuroprotective in a rat model of spinal cord
ischemia-reperfusion injury through miR-204 regulation. Curr
Neurovasc Res. 15:211–219. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou HJ, Wang LQ, Wang DB, Yu JB, Zhu Y,
Xu QS, Zheng XJ and Zhan RY: Long noncoding RNA MALAT1 contributes
to inflammatory response of microglia following spinal cord injury
via the modulation of a miR-199b/IKKβ/NF-κB signaling pathway. Am J
Physiol Cell Physiol. 315:C52–C61. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tay Y, Rinn J and Pandolfi PP: The
multilayered complexity of ceRNA crosstalk and competition. Nature.
505:344–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu J, Li Y, Lu J, Pan T, Ding N, Wang Z,
Shao T, Zhang J, Wang L and Li X: The mRNA related ceRNA-ceRNA
landscape and significance across 20 major cancer types. Nucleic
Acids Res. 43:8169–8182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang X, Sun S, Pu JK, Tsang AC, Lee D,
Man VO, Lui WM, Wong ST and Leung GK: Long non-coding RNA
expression profiles predict clinical phenotypes in glioma.
Neurobiol Dis. 48:1–8. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Johnson WE, Li C and Rabinovic A:
Adjusting batch effects in microarray expression data using
empirical Bayes methods. Biostatistics. 8:118–127. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Leek JT, Scharpf RB, Bravo HC, Simcha D,
Langmead B, Johnson WE, Geman D, Baggerly K and Irizarry RA:
Tackling the widespread and critical impact of batch effects in
high-throughput data. Nat Rev Genet. 11:733–739. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Leek JT, Johnson WE, Parker HS, Jaffe AE
and Storey JD: The sva package for removing batch effects and other
unwanted variation in high-throughput experiments. Bioinformatics.
28:882–883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang X, Zhu S, Li L, Zhang L, Xian S, Wang
Y and Cheng Y: Identification of differentially expressed genes and
signaling pathways in ovarian cancer by integrated bioinformatics
analysis. Onco Targets Ther. 11:1457–1474. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rehmsmeier M, Steffen P, Hochsmann M and
Giegerich R: Fast and effective prediction of microRNA/target
duplexes. RNA. 10:1507–1517. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li J, Ma W, Zeng P, Wang J, Geng B, Yang J
and Cui Q: LncTar: A tool for predicting the RNA targets of long
noncoding RNAs. Brief Bioinform. 16:806–812. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. 4:2015. View Article : Google Scholar
|
|
29
|
Paraskevopoulou MD, Georgakilas G,
Kostoulas N, Vlachos IS, Vergoulis T, Reczko M, Filippidis C,
Dalamagas T and Hatzigeorgiou AG: DIANA-microT web server v5.0:
Service integration into miRNA functional analysis workflows.
Nucleic Acids Res. 41:(Web Server Issue). W169–W173. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reczko M, Maragkakis M, Alexiou P, Grosse
I and Hatzigeorgiou AG: Functional microRNA targets in protein
coding sequences. Bioinformatics. 28:771–776. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jiang H, Ma R, Zou S, Wang Y, Li Z and Li
W: Reconstruction and analysis of the lncRNA-miRNA-mRNA network
based on competitive endogenous RNA reveal functional lncRNAs in
rheumatoid arthritis. Mol Biosyst. 13:1182–1192. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sun J, Yan J, Yuan X, Yang R, Dan T, Wang
X, Kong G and Gao S: A computationally constructed ceRNA
interaction network based on a comparison of the SHEE and SHEEC
cell lines. Cell Mol Biol Lett. 21:212016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Morris JH, Wu A, Yamashita RA,
Marchler-Bauer A and Ferrin TE: cddApp: A Cytoscape app for
accessing the NCBI conserved domain database. Bioinformatics.
31:134–136. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pathan M, Keerthikumar S, Ang CS, Gangoda
L, Quek CY, Williamson NA, Mouradov D, Sieber OM, Simpson RJ, Salim
A, et al: FunRich: An open access standalone functional enrichment
and interaction network analysis tool. Proteomics. 15:2597–2601.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhou M, Diao Z, Yue X, Chen Y, Zhao H,
Cheng L and Sun J: Construction and analysis of dysregulated
lncRNA-associated ceRNA network identified novel lncRNA biomarkers
for early diagnosis of human pancreatic cancer. Oncotarget.
7:56383–56394. 2016.PubMed/NCBI
|
|
37
|
Jackson A and Zimmermann JB: Neural
interfaces for the brain and spinal cord-restoring motor function.
Nat Rev Neurol. 8:690–699. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Blesch A and Tuszynski MH: Spinal cord
injury: Plasticity, regeneration and the challenge of translational
drug development. Trends Neurosci. 32:41–47. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tsuboyama K, Koyama-Honda I, Sakamaki Y,
Koike M, Morishita H and Mizushima N: The ATG conjugation systems
are important for degradation of the inner autophagosomal membrane.
Science. 354:1036–1041. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sekiguchi A, Kanno H, Ozawa H, Yamaya S
and Itoi E: Rapamycin promotes autophagy and reduces neural tissue
damage and locomotor impairment after spinal cord injury in mice. J
Neurotrauma. 29:946–956. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ge D, Han L, Huang S, Peng N, Wang P,
Jiang Z, Zhao J, Su L, Zhang S, Zhang Y, et al: Identification of a
novel MTOR activator and discovery of a competing endogenous RNA
regulating autophagy in vascular endothelial cells. Autophagy.
10:957–971. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen JF, Wu P, Xia R, Yang J, Huo XY, Gu
DY, Tang CJ, De W and Yang F: STAT3-induced lncRNA HAGLROS
overexpression contributes to the malignant progression of gastric
cancer cells via mTOR signal-mediated inhibition of autophagy. Mol
Cancer. 17:62018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kim YC and Guan KL: mTOR: A pharmacologic
target for autophagy regulation. J Clin Invest. 125:25–32. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zinzalla V, Stracka D, Oppliger W and Hall
MN: Activation of mTORC2 by association with the ribosome. Cell.
144:757–768. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bai L, Mei X, Shen Z, Bi Y, Yuan Y, Guo Z,
Wang H, Zhao H, Zhou Z, Wang C, et al: Netrin-1 improves functional
recovery through autophagy regulation by activating the AMPK/mTOR
signaling pathway in rats with spinal cord injury. Sci Rep.
7:422882017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Saiki S, Sasazawa Y, Imamichi Y, Kawajiri
S, Fujimaki T, Tanida I, Kobayashi H, Sato F, Sato S, Ishikawa K,
et al: Caffeine induces apoptosis by enhancement of autophagy via
PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 7:176–187. 2011.
View Article : Google Scholar : PubMed/NCBI
|