Introduction
Previous genomic studies identified several common
genetic variants that could play a role in large-vessel ischemic
stroke (LVIS) (1). Precise type of
their effect on brain ischemia remains unclear, and it is assumed
that common genetic variants could explain between 39–66% of
variation in ischemic stroke incidence (2). It has also been postulated that the
accumulated effect of the remaining uncommon or rare damaging
variants (called genetic burden) might explain a significant
portion of the genetic predisposition to many common diseases or
phenotypes (3,4). A small number of re-sequencing
reports of European patients with LVIS were published so far. These
studies have demonstrated that infrequent coding variants in
numerous genes might be linked to stroke (5–8).
Platelets have an important role in the pathogenesis
of LVIS based on their activation and adherence to the endothelium
within cerebral arteries, as well as progression of thrombus
(9,10). Most research strategies to date
revealed the effect of genetic variation on reactivity of platelets
and were obtained by analyzing common variants within candidate
genes and/or genome-wide association studies (GWAS), followed by
in vitro studies to assess platelet functions (11). Overall, previous studies on the
genetic background of platelet reactivity indicated that many
different genes contribute to platelet function. Thus far, the
potential contribution of genetic variants within genes encoding
proteins essential to thrombus formation in LVIS have been analyzed
in a small number of studies and the majority of them focused on
the common single nucleotide polymorphisms (SNPs) within a few
genes associated with glycoproteins on the platelet surface
(11). The results of these
studies suggested that additional loci associated with platelet
functions are yet to be found and that the known loci may contain
high effect rare risk variants that have thus far gone undetected
by GWAS.
Rare coding variants appear to be restricted to
small populations and that was shown in only one previous study,
which concentrated on the re-sequencing of the common variants in
the platelet genes associated with membrane function (12). In addition, another recent
investigation revealed that the rare variants in receptors commonly
associated with platelet functions (e.g. purinergic receptors)
could be associated with the occurrence of LVIS in the Polish
population (13).
Thus, the objective of our current study was to
explain the contribution played by the another set of uncommon and
damaging genetic variants within selected genes associated with
changes in platelet functions in LVIS. We have chosen 54 genes
associated with less known or investigated biochemical processes
associated with platelet functions as the focus for the
re-sequencing study (14–21).
Materials and methods
Clinical material
The study was permitted by both local ethics
committee of the Institute of Psychiatry and Neurology, Warsaw,
Poland, and Warsaw Medical University, Warsaw, Poland. The study
conduct conformed to the most recent version of the Declaration of
Helsinki. All participants in the study signed the informed
consent. Full description of the study cohort, including the
inclusion and exclusion criteria were published previously
(12,13,22–24).
Based on the Trial of Org 10172 in Acute Stroke Treatment (TOAST)
classification we included: i) All patients classified as having
ischemic stroke due to large-vessel atherosclerosis and ii) a
subset of patients classified as having ischemic stroke of unknown
etiology, provided they had at least 50% stenosis of the carotid
artery ipsilateral to the infarct side and no evidence or no
history of atrial fibrillation. Patient with the history of
hemorrhagic or embolic stroke were excluded from the study. As a
control group we used samples from age- and sex-matched 500
patients without history of stroke coming from the same
geographical area as patients with LVIS (13) and collected for unrelated studies
performed at Warsaw Medical University in Poland. Both cohorts of
patients (study and control) were white Caucasian of Polish
ethnicity and originated from central Poland. DNA extraction from
collected blood samples was done as described before (9).
Pooled sequencing
The list of 54 re-sequenced genes is shown in
Table I. These targets contain 846
exons and 10 adjoining bases beyond each exon on both sides. The
genetic loci were selected using the human database (H.
sapiens, hg19, GRCh37, February 2009). Pooled targeted
enrichment of DNA, from LVIS patients (five polls with 100 subjects
per pool) and 500 age-, sex-matched control patients, without
stroke history (five pools with 100 subjects per pool), was done as
described previously. Further explanation of re-sequencing and
analysis of data is provided in the Supplementary material
(Tables SI–SIV).
 | Table I.List of 54 platelet genes with exons
sequenced in the present study. |
Table I.
List of 54 platelet genes with exons
sequenced in the present study.
| Author, year | Gene | Protein
encoded | Chromosome and
regions | (Ref.) |
|---|
| Janicki et
al, 2017 | FCER1G | Fc fragment of IgE,
high affinity I, receptor for; γ polypeptide | 1q23.3region | (13) |
| Janicki et
al, 2017 | VAV3 | vav 3 guanine
nucleotide exchange factor | 1p13.3 | (13) |
| Jones et al,
2009 | RAF1 | v-raf-1 murine
leukemia viral oncogene homolog 1 | 3p25.1 | (14) |
| Jones et al,
2009 | MAPK14 | Mitogen-activated
protein kinase 14 | 6p21.31 | (14) |
| Jones et al,
2009 | JAK2 | Janus kinase 2 | 9p24.1 | (14) |
| Jones et al,
2009 | MAP2K4 | Mitogen-activated
protein kinase kinase 4 17. p12 | 17p12 | (14) |
| Jones et al,
2009 | AKT2 | v-akt murine
thymoma viral oncogene homolog 2 | 19q13.1-q13.2 | (14) |
| Jones et al,
2009 | MAP2K2 | Mitogen-activated
protein kinase kinase 2 | 19p13.3 | (14) |
| Jones et al,
2009 | GNAZ | Guanine nucleotide
binding protein (G protein), α z polypeptide | 22q11.22 | (14) |
| Jones et al,
2009; Goodall et al, 2010 | TRIM27 | Tripartite motif
containing 27 | 6p22.1 | (14,15) |
| Goodall et
al, 2010 | LRRFIP1 | Leucine rich repeat
(in FLII) interacting protein 1 | 2q37.3 | (15) |
| Goodall et
al, 2010 | COMMD7 | COMM domain
containing 7 | 20q11.21 | (15) |
| Postula et
al, 2013 | RGS7 | Regulator of
G-protein signaling 7 | 1q23.1 | (16) |
| Guerrero et
al, 2011 | LPAR1 | Lysophosphatidic
acid receptor 1 | 9q31.3 | (17) |
| Guerrero et
al, 2011 | MYO5B | Myosin VB | 18q21.1 | (17) |
| Mathias et
al, 2010 | LDHAL6A | Lactate
dehydrogenase A-like 6A | 11p15.1 | (18) |
| Mathias et
al, 2010 | ANKS1B | Ankyrin repeat and
sterile α motif domain containing 1B | 12q23.1 | (18) |
| Johnson et
al, 2010 | PIK3CG |
Phosphoinositide-3-kinase, catalytic, γ
polypeptide | 7q22.3 | (19) |
| Johnson et
al, 2010 | SHH | Sonic hedgehog
homolog | 7q36.3 | (19) |
| Johnson et
al, 2010 | JMJD1C | Jumonji domain
containing 1C | 10q21.2 | (19) |
| Johnson et
al, 2010 | MRVI1 | Murine retrovirus
integration site 1 homolog | 11p15.4 | (19) |
| Johnson et
al, 2010; Johnson, 2011 | RGS18 | Regulator of
G-protein signaling 18 | 1q31.2 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | ST3GAL3 | ST3 β-galactoside
α-2,3-sialyltransferase 3 | 1p34.1 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | UGT1A10 | UDP
glucuronosyltransferase 1 family, polypeptide A10 | 2q37.1 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | NUP210 | Nucleoporin 210
kDa | 3p25.1 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | RAPGEF2 | Rap guanine
nucleotide exchange factor (GEF) 2 | 4q32.1 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | ADAMTS2 | ADAM
metallopeptidase with thrombospondin type 1 motif, 2 | 5q35.3 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | FBXL7 | F-box and
leucine-rich repeat protein 7 | 5p15.1 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | KLHL31 | Kelch-like family
member 31 | 6p12.1 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | GMDS | GDP-mannose
4,6-dehydtase | 6p25.3 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | WBSCR17 | Williams-Beuren
syndrome chromosome region 17 | 7q11.22 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | ATP6V1F | ATPase,
H+ transporting, lysosomal 14 kDa, V1 subunit F | 7q32.1 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | SGCZ | Sarcoglycan, ζ | 8p22 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | STMN4 | Stathmin-like
4 | 8p21.2 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | PSKH2 | Protein serine
kinase H2 | 8q21.3 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | PIP5K1B |
Phosphatidylinositol-4-phosphate 5-kinase,
type I, β | 9q21.11 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | PTPRD | Protein tyrosine
phosphatase, receptor type, D | 9p24.1 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | MIPOL1 | Mirror-image
polydactyly 1 | 14q13.3-q21.1 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | SVIL | Supervillin | 10p11.23 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | CUBN | Cubilin (intrinsic
factor-cobalamin receptor) | 10p13 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | ST3GAL4 | ST3 β-galactoside
α-2,3-sialyltransferase 4 | 11q24.2 | (19,20) |
| Johnson, 2010
Johnson, 2011 | KCNQ1 | Potassium
voltage-gated channel, KQT-like subfamily, member 1 | 11p15.5-p15.4 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | HSD17B6 | Hydroxysteroid
(17-β) dehydrogenase 6 | 12q13.3 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | RAP1B | RAP1B, member of
RAS oncogene family | 12q15 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | PTPN11 | Protein tyrosine
phosphatase, non-receptor type 11 | 12q24 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | THSD4 | Thrombospondin,
type I, domain containing 4 | 15q23 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | TAOK1 | TAO kinase 1 | 17q11.2 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | SETBP1 | SET binding protein
1 | 18q12.3 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 |
KIAA0802 | SOGA family member
2 | 18p11.22 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | CTCFL | CCCTC-binding
factor (zinc finger protein)-like | 20q13.31 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | PCK1 | Phosphoenolpyruvate
carboxykinase 1 | 20q13.31 | (19,20) |
| Johnson et
al, 2010; Johnson, 2011 | PRNP | Prion protein | 20p 13 | (19,20) |
| Shiffman et
al, 2006*** | VAMP8 | Vesicle-associated
membrane protein 8 | 2p12-p11.2 | (21) |
| Lee et al,
2014 | GLIS3 | GLIS family zinc
finger 3 | 9p24.2 | (26) |
Verification of selected variants by
individual genotyping
Individual genotyping for selected markers in
individual DNA samples was performed using a custom Sequenom iPLEX
assay in conjunction with the Mass ARRAY platform (Sequenom Inc.,
La Jolla, CA, USA). Panels of SNP markers were designed using
Sequenom Assay Design 3.2 software (Sequenom Inc.), in a similar
fashion to the previously described methodology from our laboratory
(9,16,17).
Statistical analysis
A cumulative minor allele frequency (cMAF) was
utilized to show the allelic frequency of the investigated
variants, which encompasses all rare damaging variants individually
genotyped in the investigated cohorts, as well as within each of
the analyzed loci. Pearson Chi-square test was used in order to
analyze differences in cMAF for all individually genotyped variants
between the study and control cohorts (VassarStats: Website for
Statistical Computation on http://vassarstats.net/). The pooled minor allele test
(CMAT) (10,000 × permutations) was used for comparison of all
variants within investigated loci to estimate the statistical
significance of the observed differences in the accumulation of
variants. CMAT is a pooling method proposed by Zawistowski et
al (25) and works by
comparing weighted minor-allele counts (for cases and controls)
against the weighted major-allele counts (for cases and controls).
Although the CMAT test statistic is based on a chi-square
statistic, it does not follow a known distribution and its
significance has to be determined by a permutation procedure. The
calculations of CMAT were performed using AssotesR package (0.1-10)
from CRAN repository (cran.r-project.org/package=AssotesteR) and written by
Gaston Sanchez (gastonsanchez.com/) as documented at www.rdocumentation.org/packages/AssotesteR/versions/0.1-10.
The significance threshold was adjusted to the number of
re-sequenced loci, when needed (13,25).
Power and sample size
considerations
For the power calculations, instead of using
individual rare variants, we decided to use predicted cMAF for all
deleterious rare variants in the sequenced loci. We have followed a
self-sufficient, closed-form, maximum-likelihood estimator for
allele frequencies that accounts for errors associated with
sequencing, and a likelihood-ratio test statistic that provides a
simple means for evaluating the null hypothesis of monomorphism
(13,26,27).
Unbiased estimates of allele frequencies 10/N (where N is the
number of individuals sampled) appear to be achievable, and
near-certain identification of a single nucleotide polymorphism
(SNP) requires a cMAF of at least 0.01 (i.e., 10 variants per
cohort). In addition, because the power to detect significant
allele-frequency differences between the two populations is
limited, we set both the number of sampled individuals (500 in the
cohort) and depth of sequencing coverage in excess of 100.
Fluorescence-based (FLIPR) functional
assay for KCNQ1 variants
Chinese hamster ovaries cells (CHO) cultures stably
transfected with human muscarinic type 1 receptor (M1) were
purchased from cDNA Resource Center, Bloomberg, PA. The CHO-M1
cells were then transiently co-transfected with KCNQ1 cDNA
constructs. Wild-type KCNQ1 cDNA constructs (in pcDNA3.1) were
prepared by Watson Bio Sciences (Houston, TX, USA). The fidelity of
the mutations for each variant was confirmed by Sanger sequencing.
Details about the transfection techniques and cell culture
conditions used in our laboratory were published before (13).
The heterologous expressed potassium KCNQ1 channels
were inhibited by the M1 receptor agonist oxotremorine (OxoM, 10
nM), resulting in significant changes in membrane polarization
(fully antagonized by muscarinic receptor antagonist atropine at 1
µM-not shown).
The Fluorescence Imaging Plate Reader (FLIPR) on
Flex Station 3 (Molecular Devices, San Jose, CA, USA) was employed
to measure the fluorescence changes. After the cells were loaded
with membrane dye, the plate with the cells and plate containing
OxoM (10 nM) were inserted into the plate reader. The raw
fluorescence readings were converted and plotted as the change in
relative fluorescence before (F0 baseline=100%) and after OxoM
application F), according to the formula ((F/F0)*100%). For all
experimental conditions, minimum fluorescence (Fmin) and maximum
fluorescence (Fmax) were recorded in triplicates after
administration. The summary data are presented as summary trend
lines for WT and mutants, as well as averages (with standard
deviations) for Fmax and Fmin and for each variant and WT cells
used in FLIPR experiments. The final data represent all
measurements done in triplicates and in three independent
experiments (cell populations). Data and statistical analysis were
performed using analysis of variance, followed by the Student's
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Study design and sequencing
coverage
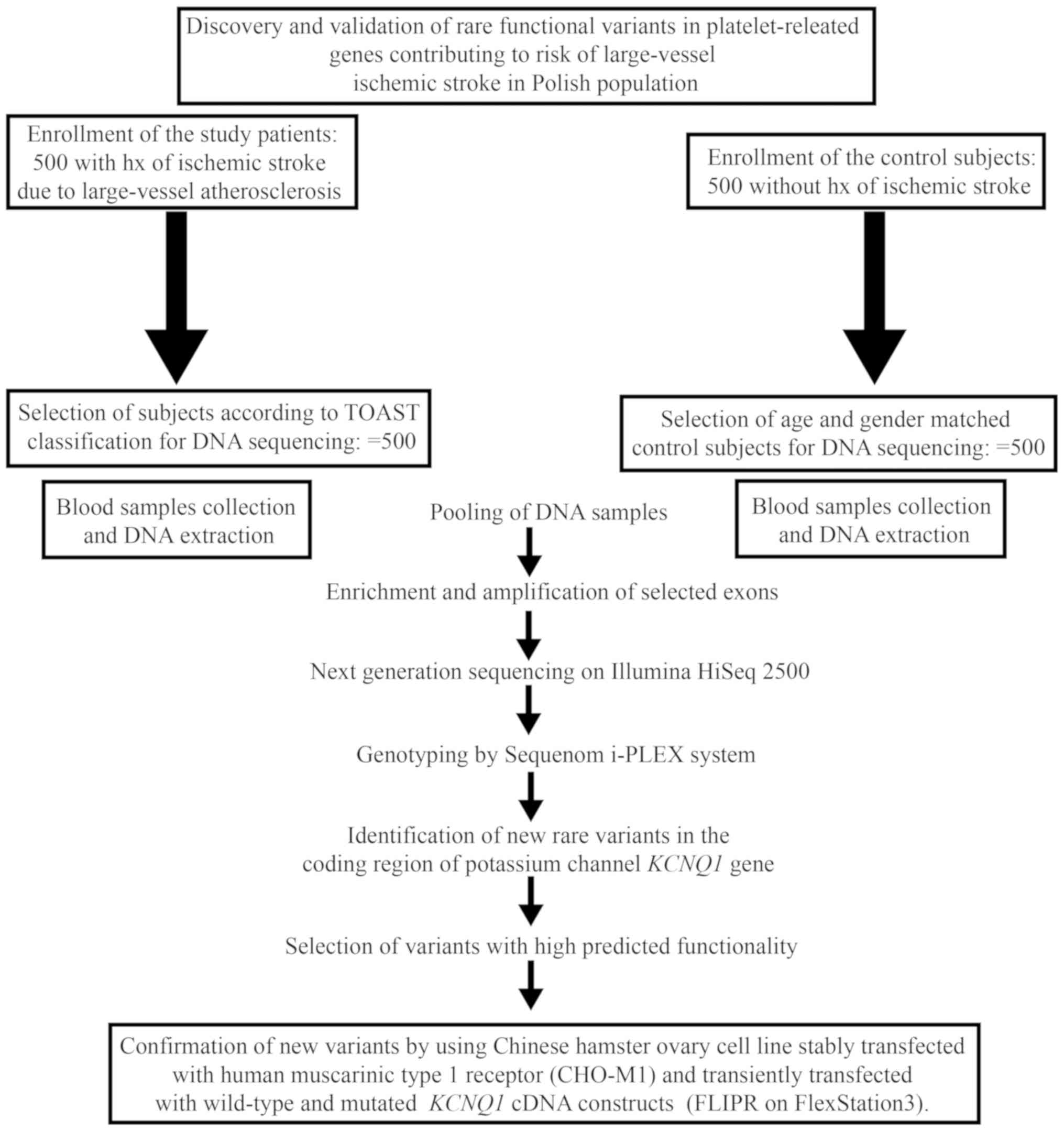
The design of the study is presented in Fig. 1. The enrichment of the target loci
resulted in coverage of 99.6% and produced 36.1 (22.7–45.9 range)
million reads which is 5.3 (3–7 range) Gbp per re-sequenced sample.
It relates to an average coverage of 12,000× per pool and an
average coverage of 120× per patient sample (range 21–369).
Selection of rare variants (based on
MAF<1%)
The step-wise analysis of the observed variants is
presented in Fig. 2. In total,
1018 unique variants, irrespective of MAF and with adequate quality
were observed in both investigated cohorts. The complete list of
all observed variants is provided in the Supplementary materials,
Tables SII for all non-coding
variants and SIII for all non-synonymous variants. Seventy two
percent of all variants were previously listed in the available
databases, and 28% were new. Out of all observed variants, 327
(32.1%) were located in the coding segments of the sequenced genes,
and the remainder was located in untranslated regions. Out of all
coding variants, 120 variants (Supplementary material, Table SIV-list of all rare non-synonymous
variants) were selected based on MAF <1% (i.e. rare variants)
(28), which consisted of 52 known
(by dbSNP149 November 2016) and 68 unlisted, novel variants.
Verification of selected variants by
individual genotyping
In total, 28 SNVs with the highest predicted
damaging score calculated by Combined Annotation Dependent
Depletion (CADD) score were chosen for individual genotyping. The
minimum CADD score of 10 served as a threshold for predicted
deleteriousness. The individual genotyping was performed in
patients from the original cohort and revealed that the identity of
all 28 initially selected variants could be confirmed by individual
genotyping, which indicates no sequencing errors for these
variants.
The initial statistical analysis utilized Pearsons
Chi-square test for the assessment of the differences in the
cumulative frequency of all 28 individually genotyped variants
between the investigated cohorts. There was a highly statistically
significant (P=0.00045) difference (cMAF control=1.2% vs. cMAF
stroke=3.6%) in cMAF for all damaging variants in the LVIS group
when compared with controls.
The statistical analysis of the number of variants
within the single gene loci was based on combined minor allele
association test (CMAT). The region-based, Bonferroni corrected,
significance threshold was P=0.00092 (nominal P=0.05/54 sequenced
genes). It demonstrated a statistically significant difference
(P=0.0009) between control and IS cohorts for the KCNQ1
location. The KCNQ1 exons locus contained 3 novel and 3
known rare and deleterious (CADD score range 13.22-39) coding
variants (Table II).
| Table II.cMAF for 28 rare and most damaging
variants observed in the individually genotyped patients from
control and LVIS cohorts. |
Table II.
cMAF for 28 rare and most damaging
variants observed in the individually genotyped patients from
control and LVIS cohorts.
| Gene locus | Number of carriers
in control cohort, n=500 | Number of carriers
in LVIS cohort, n=500 | P-value |
|---|
| ADAMT2 | 2 | 3 | 0.4 |
| CUBN | 0 | 4 | 0.06 |
| GLIS3 | 2 | 0 | 0.1 |
| KCNQ1 | 0 | 6 | 0.0009a |
| LDHAL6A | 3 | 2 | 0.4 |
| MAP2K2 | 0 | 2 | 0.2 |
| MIPOL1 | 0 | 2 | 0.8 |
| MTCL1 | 4 | 3 | 0.9 |
| MYO5B | 0 | 4 | 0.06 |
| PTPRJ | 0 | 2 | 0.1 |
| SHH | 0 | 2 | 0.1 |
| SVIL | 1 | 3 | 0.15 |
| UGT1A1 | 0 | 2 | 0.1 |
| Total number of
carriers and cMAF for all loci | 12 | 36 |
0.00045b |
| Odds ratio and 95%
confidence intervals (in brackets) |
| 3.07
(1.59–5.94) |
|
Functional analysis of selected rare
variants within KNCQ1 gene
To evaluate whether the observed variants exert a
damaging effect on KCNQ1 function, we selected 3 novel and
most damaging variants from the KCNQ1 locus to examine the
coupling between the heterologous expressed human M1 receptor and
KCNQ1 channels in CHO-M1 cells (Table III). Fig. 3 shows the summary of changes in
relative fluorescence for CHO-M1 cells expressing wild-type
KCNQ1, and variants KCNQ1 c. G855T p. K285N,
KCNQ1 c. G1545T p. K515N, and KCNQ1 c. 1637A p.
S546X. Following oxoM application (final concentration 10 nM), the
fluorescence decreased rapidly, indicating corresponding changes in
cell membrane potential. We expressed the changes in fluorescence
both as the differences between baseline fluorescence and its
changes (in % of baseline=Fo=100%) over 250 sec after
administration of oxoM (shown as trend line for all observations)
and average values of minimum (Fmin) and maximum (Fmax) of relative
fluorescence during the recording period (separately for WT and
variants). The provided trend lines represent summary values for
all observations (2–3 independent cell cultures and all
measurements in triplicates of plate wells). A significant decrease
in fluorescence signal for each variant was observed after
administration of oxoM when compared with the KCNQ1
wild-type-expressing CHO-M1 cells. Correspondingly, the
statistically significant decrease (P<0.05 ANOVA, followed by
t-test) in the average Fmin and Fmax was observed for all 3
investigated variants, indicating at least partial loss-of-function
characteristics for variant proteins, when compared to WT.
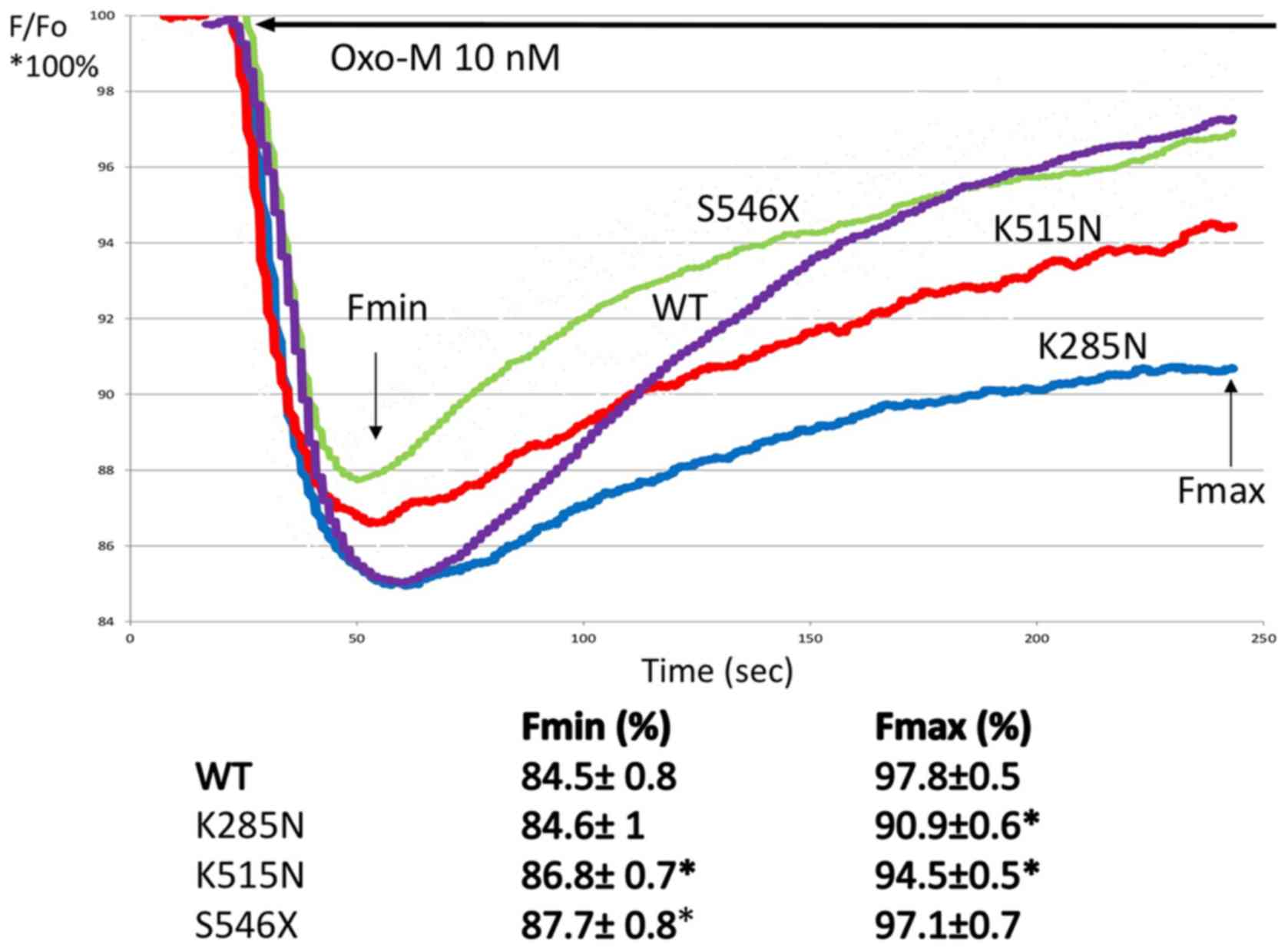 | Figure 3.Effect of Oxo-M on relative
fluorescence in CHO-M1 cells heterologously expressing KCNQ1
wild-type and KCNQ1 variant channels. Each panel shows % change
(from baseline Fo=100%) of F in CHO-M1 transfected with wild-type
K285N, K515N and S546X variants (where X is random amino acid
substituted). Fluorescence signals were acquired before and during
Oxo-M (10 nM, solid line) application. The data are presented trend
lines for all the experimental points (2–3 independent experiments,
triplicate measurements). For each variant and the calculated Fmin
and Fmax relative fluorescence change, including standard deviation
was calculated. Fmax and Fmin averages were compared with WT using
analysis of variance, followed by a t-test. *P<0.05. Oxo-M,
oxotremorine; CHO-M1, human muscarinic type 1 receptor; KCNQ1,
potassium voltage-gated channel subfamily Q member 1; F,
fluorescence; Fmin, minimum fluorescence; Fmax, maximum
fluorescence; Fo, baseline fluorescence; WT, wild-type. |
| Table III.Rare coding variants for KCNQ1
gene observed in the study patients, as identified by exome
sequencing and verified by individual genotyping. |
Table III.
Rare coding variants for KCNQ1
gene observed in the study patients, as identified by exome
sequencing and verified by individual genotyping.
| dbSNP | MAFctrl pooled | MAFstroke
pooled | MAF individual
genotyping in control | MAF in individual
genotyping in stroke | DNA change | Amino acid
change | CADD |
|---|
| rs12720457 | 0.80% | ND | 0.01% | ND | c.G1179T | p.K393N |
6.560 |
| novel | ND | 0.75% | ND | 0.1% | c.G4T | p.D2Y |
5.53 |
| rs199472712 | ND | 0.95% | ND | 0.1% | c.G724T | p.D242Y | 19.35 |
| Novel | ND | 0.74% | ND | 0.1% | c.G855T | p.K285N | 15.48 |
| Novel | ND | 0.65% | ND | 0.1% |
c.G1545T | p.K515N | 13.22 |
| rs199472793 | ND | 0.71% | ND | 0.1% | c.C1597T | p.R533W | 15.28 |
| Novel | ND | 0.85% | ND | 0.1% |
c.C1637A | p.S546X | 39 |
Discussion
We present the results of the analysis of the
genetic burden of the infrequent coding variants in 54 genes
associated with platelet function and its possible association with
LVIS. The assessment of MAF for the investigated uncommon coding
variants established that there was a significant accumulation of
those variants in the LVIS group when compared to the control
group. By grouping these variants by sequenced loci instead of
analyzing them individually, we were able to observe associations
which could be underpowered when applied to single variants, as
shown in previous studies of other traits (12,13).
In particular, we found an association between the increased
accumulation of rare variants in KCNQ1 locus and LVIS. It is
important to note that, with the exception of 3 already known
variants, the remaining 3 observed damaging variants within
KCNQ1 locus were novel. It is therefore likely that at least
some of the observed variants might be restricted to the Polish
cohort. So far detailed genotypes in the Polish population have
been rather poorly characterized in the available genomic
databases. This in turn might suggest that the verification of the
obtained results in the independent cohorts could be challenging.
For example, it was previously demonstrated that, at least in case
of rare damaging variants associated with ulcerative colitis, the
rare variants observed in the Dutch population could not be
replicated in a German cohort (29). Another study on inflammatory bowel
disease, that included several thousands of European individuals
and individuals of other ancestry, showed that although the
majority of the loci with MAF>5% were shared between different
ancestry groups (30), no such
similarities were observed for uncommon alleles. In fact, rare
variants were even more likely to be specific to a particular
population, as was confirmed by a recent sequencing study (31). What is more, rare variants might
differ significantly among even closely related populations
(32).
We were able to observe only few (out of several
hundreds) of previously listed rare coding variants in the
KCNQ1 locus, which might indicate either limited power of
the study or population-specific distribution of these variants.
Further studies in other populations will be helpful to verify if
the rare damaging variants in the KCNQ1 coding locus are
indeed associated with large-vessel IS, or also with other types of
stroke (small-vessel and embolic).
The KCNQ channels, members of the voltage-gated
(KV7.1) K+-selective channel subfamily, play
a major role in K+ ion transport. For instance, previous
studies have shown that both KV channels KCNA3 and G
protein-gated inwardly rectifying K+ channels (GIRK)
regulate platelet activation (33,34).
The presence of Kv7.1 channel in blood cells, including platelets,
suggests that they play a role in agonist-mediated regulation of
platelet-driven thrombotic pathways that is crucial to hemostasis
during IS (35,36).
Our in vitro results indicate that
oxoM-mediated changes in the membrane potential of cells expressing
M1 receptors and KCNQ1 channel variants were attenuated
(loss-of-function characteristics) when compared to cells
expressing the wild-type receptors. These findings suggest that the
signaling might be diminished in cells expressing the mutant KCNQ1
channels. The coding variants in KCNQ1 were previously
evaluated in different cardiovascular diseases and DM (37–41).
It has been also reported that obesity along with IS may modify
methylation of KCNQ1 gene and plasma KCNQ1 protein
concentration (38,39). The coding variants in KCNQ1 have
been reported to lead to congenital long QT syndrome (42), an autosomal dominant disorder.
Because of the demonstrated deleterious properties of the
investigated variants, it should be also considered that the stroke
patients in the study may have suffered from an ischemic stroke
secondary to a congenital disorder. Whether KCNQ1 variants are
disease-associated with ischemic stroke (possibly via platelet
function) or disease-causing for congenital QT syndrome (with
reportedly higher incidence of ischemic stroke is one of the
questions which should be clarified in the future investigations
(41).
Moreover, in one of the largest to date GWAS on
platelet function, KCNQ1 locus was discovered to contribute
to platelet function variability (19,20).
The exact mechanism of these interactions remains unknown, as
KCNQ1 is mostly co-assembled with the product of the
KCNE1 (minimal K+-channel protein) gene in the
heart to form a cardiac-delayed rectifier-like K+
current and the effect of KCNQ1 channels on platelet function has
not been not directly investigated so far. However, Gallego-Fabrega
et al (43) reported
recently that the methylation pattern of KCNQ1 locus might be
associated with vascular recurrence in aspirin-treated stroke
patients.
The study is limited by the absence of independent
verification of accumulation of deleterious KCNQ1 rare variants.
However, it was demonstrated previously that the occurrence of rare
variants, because of their private character, is often limited to
very restricted cohorts and has been difficult to repeat in other
cohorts, unless the confirmation cohorts are truly large (in this
case several tens of thousands of patients). The added drawback of
the presented research is that the direct effect of the observed
genomic variants on the platelet function (e.g. aggregation) was
not evaluated. This might raise an issue if the observed change in
the frequency of variants were entirely related to the platelet
function or, perhaps, some other mechanisms related to biochemical
pathways. Moreover, it should be noted that only a limited number
of all known genes related to platelet function were re-sequenced
in this study. We would like to stress that the results of
re-sequencing of the more frequently investigated 26 genes related
to platelet function were published by our group in the past
(13).
The outcome of this study indicates that the
increased accumulation of rare damaging variants in the exons of
the sequenced 54 platelet genes (and in particular for variants
located in the region of potassium channel KCNQ1 gene) could
be associated with LVIS. The mechanism of the interaction of these
variants with LVIS currently appears unclear and therefore requires
further investigations. It is also uncertain if our results could
be directly translated to other populations, as the variants
responsible for the observed associations appear to be limited to
the investigated cohort. Further studies in different, as well as
much larger cohorts, are required to address this problem.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Dr Michal Karlinski
(Institute of Psychiatry and Neurology, Warsaw, Poland) and Dr
Agnieszka Cudna (Center for Preclinical Research and Technology
CEPT, Warsaw, Poland) for preparing the samples and database for
further analysis.
Funding
Research subject was implemented with CEPT
infrastructure financed by the European Union-the European Regional
Development Fund within the Operational Program ‘Innovative
economy’ for 2007–2013. The study was supported financially as part
of the research grant from the National Science Center OPUS
research grant (grant no. 2013/11/B/NZ7/01541).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
PKJ and MP conceived the concept and design for the
study, were involved in data collection and analysis, and
supervised the work. CE and VRV contributed to the design of the
research, and were involved in data collection and analysis. SS and
YIK verified the analytical methods. JP, AC, IKJ and DMG were
involved in data collection and analysis. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All procedures followed were in accordance with the
ethical standards of the responsible committee on human
experimentation (institutional and national) and with the Helsinki
Declaration of 1975, as revised in 2000. Informed consent was
obtained from all patients for being included in the study.
Patient consent for publication
The consent for publication was obtained from all
patients included in the study.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
LVIS
|
large-vessel ischemic stroke
|
|
CHO-M1
|
human muscarinic type 1 receptor
|
|
FLIPR
|
fluorescence imaging plate reader
|
|
GWAS
|
genome wide association studies
|
|
SNP
|
single nucleotide polymorphism
|
|
CAD
|
coronary artery disease
|
|
cMAF
|
cumulative minor allele frequency
|
|
CMAT
|
combined minor allele test
|
|
SNVs
|
single nucleotide variants
|
|
Fmin
|
minimum fluorescence
|
|
Fmax
|
maximum fluorescence
|
|
CADD
|
Combined Annotation Dependent
Depletion
|
|
CHF
|
congestive heart failure
|
|
DM
|
diabetes mellitus
|
|
GIRK
|
G-protein-gated inwardly rectifying
K+ channels
|
References
|
1
|
Welter D, MacArthur J, Morales J, Burdett
T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L and
Parkinson H: The NHGRI GWAS Catalog, a curated resource of
SNP-trait associations. Nucleic Acids Res. 42:D1001–D1006. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Holliday EG, Maguire JM, Evans TJ, Koblar
SA, Jannes J, Sturm JW, Hankey GJ, Baker R, Golledge J, Parsons MW,
et al: Common variants at 6p21.1 are associated with large artery
atherosclerotic stroke. Nat Genet. 44:1147–1151. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bevan S, Traylor M, Adib-Samii P, Malik R,
Paul NL, Jackson C, Farrall M, Rothwell PM, Sudlow C, Dichgans M
and Markus HS: Genetic heritability of ischemic stroke and the
contribution of previously reported candidate gene and genomewide
associations. Stroke. 43:3161–3167. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gibson G: Rare and common variants: Twenty
arguments. Nat Rev Genet. 13:135–145. 2011. View Article : Google Scholar
|
|
5
|
Genome of the Netherlands Consortium, .
Whole-genome sequence variation, population structure and
demographic history of the Dutch population. Nat Genet. 46:818–25.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cheng YC, Cole JW, Kittner SJ and Mitchell
BD: Genetics of ischemic stroke in young adults. Circ Cardiovasc
Genet. 7:383–392. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Auer PL, Nalls M, Meschia JF, Worrall BB,
Longstreth WT Jr, Seshadri S, Kooperberg C, Burger KM, Carlson CS,
Carty CL, et al: Rare and coding region genetic variants associated
with risk of ischemic stroke: The NHLBI exome sequence project.
JAMA Neurol. 72:781–788. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lindgren A: Stroke genetics: A review and
update. J Stroke. 16:114–123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Anyanwu C, Hahn M, Nath M, Li J, Barone
FC, Rosenbaum DM and Zhou L: Platelets pleiotropic roles in
ischemic stroke. Austin J Cerebrovasc Dis Stroke. 3:10482016.
|
|
10
|
del Zoppo GJ: The role of platelets in
ischemic stroke. Neurology. 51 (Suppl 3):S9–S14. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Milanowski L, Pordzik J, Janicki PK and
Postula M: Common genetic variants in platelet surface receptors
and its association with ischemic stroke. Pharmacogenomics.
17:953–971. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Postula M, Janicki PK, Milanowski L,
Pordzik J, Eyileten C, Karlinski M, Wylezol P, Solarska M,
Czlonkowka A, Kurkowska-Jastrzebka I, et al: Association of
frequent genetic variants in platelet activation pathway genes with
large-vessel ischemic stroke in Polish population. Platelets.
28:66–73. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Janicki PK, Eyileten C, Ruiz-Velasco V,
Sedeek KA, Pordzik J, Czlonkowska A, Kurkowska-Jastrzebska I,
Sugino S, Imamura-Kawasawa Y, Mirowska-Guzel D and Postula M:
Population-specific associations of deleterious rare variants in
coding region of P2RY1-P2RY12 purinergic receptor genes in
large-vessel ischemic stroke patients. Int J Mol Sci. 18:E26782017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jones CI, Bray S, Garner SF, Stephens J,
de Bono B, Angenent WG, Bentley D, Burns P, Coffey A, Deloukas P,
et al: A functional genomics approach reveals novel quantitative
trait loci associated with platelet signaling pathways. Blood.
114:1405–1416. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Goodall AH, Burns P, Salles I, Macaulay
IC, Jones CI, Ardissino D, de Bono B, Bray SL, Deckmyn H, Dudbridge
F, et al: Transcription profiling in human platelets reveals
LRRFIP1 as a novel protein regulating platelet function. Blood.
116:4646–4656. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Postula M, Janicki PK, Rosiak M,
Kaplon-Cieslicka A, Trzepla E, Filipiak KJ, Kosior DA, Czlonkowski
A and Opolski G: New single nucleotide polymorphisms associated
with differences in platelets reactivity in patients with type 2
diabetes treated with acetylsalicylic acid: Genome-wide association
approach and pooled DNA strategy. J Thromb Thrombolysis. 36:65–73.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guerrero JA, Rivera J, Quiroga T,
Martinez-Perez A, Antón AI, Martínez C, Panes O, Vicente V,
Mezzano D, Soria JM and Corral J: Novel loci involved in platelet
function and platelet count identified by a genome-wide study
performed in children. Haematologica. 96:1335–1343. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mathias RA, Kim Y, Sung H, Yanek LR,
Mantese VJ, Hererra-Galeano JE, Ruczinski I, Wilson AF, Faraday N,
Becker LC and Becker DM: A combined genome-wide linkage and
association approach to find susceptibility loci for platelet
function phenotypes in European American and African American
families with coronary artery disease. BMC Med Genomics. 3:222010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Johnson AD, Yanek LR, Chen MH, Faraday N,
Larson MG, Tofler G, Lin SJ, Kraja AT, Province MA, Yang Q, et al:
Genome-wide meta-analyses identifies seven loci associated with
platelet aggregation in response to agonists. Nat Genet.
42:608–613. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Johnson AD: The genetics of common
variation affecting platelet development, function and
pharmaceutical targeting. J Thromb Haemost. 9 (Suppl 1):S246–S257.
2011. View Article : Google Scholar
|
|
21
|
Shiffman D, Rowland CM, Louie JZ, Luke MM,
Bare LA, Bolonick JI, Young BA, Catanese JJ, Stiggins CF, Pullinger
CR, et al: Gene variants of VAMP8 and HNRPUL1 are associated with
early-onset myocardial infarction. Arterioscler Thromb Vasc Biol.
26:1613–1618. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
No authors listed, . Stroke-1989.
Recommendations on stroke prevention, diagnosis, and therapy.
Report of the WHO task force on stroke and other cerebrovascular
disorders. Stroke. 20:1407–1431. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Foulkes MA, Wolf PA, Price TR, Mohr JP and
Hier DB: The stroke data bank: Design, methods, and baseline
characteristics. Stroke. 19:547–554. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Grabska K, Gromadzka G and Członkowska A:
Infections and ischemic stroke outcome. Neurol Res Int.
2011:6913482011.PubMed/NCBI
|
|
25
|
Zawistowski M, Gopalahrishnan S, Ding J,
Li Y, Grimm S and Zöllner S: Extending rare-variant testing
strategies: Analysis of noncoding sequence and imputed genotypes.
Am J Hum Genet. 87:604–617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee S, Abecasis GR, Boehnke M and Lin X:
Rare-variant association analysis: Study designs and statistical
tests. Am J Hum Genet. 95:5–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lynch M, Bost D, Wilson S, Maruki T and
Harrison S: Population-genetic inference from pooled-sequencing
data. Genome Biol Evol. 6:1210–1228. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kircher M, Witten DM, Jain P, O'Roak BJ,
Cooper GM and Shendure J: A general framework for estimating the
relative pathogenicity of human genetic variants. Nat Genet.
46:310–315. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Visschedijk MC, Alberts R, Mucha S, Deelen
P, de Jong DJ, Pierik M, Spekhorst LM, Imhann F, van der Meulen-de
Jong AE, van der Woude CJ, et al: Pooled resequencing of 122
ulcerative colitis genes in a large dutch cohort suggests
population-specific associations of rare variants in MUC2. PLoS
One. 11:e01596092016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu JZ, van Sommeren S, Huang H, Ng SC,
Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, et al:
Association analyses identify 38 susceptibility loci for
inflammatory bowel disease and highlight shared genetic risk across
populations. Nat Genet. 47:979–986. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hong KW, Shin MS, Ahn YB, Lee HJ and Kim
HD: Genomewide association study on chronic periodontitis in Korean
population: Results from the Yangpyeong health cohort. J Clin
Periodontol. 42:703–710. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Prescott NJ, Lehne B, Stone K, Lee JC,
Taylor K, Knight J, Papouli E, Mirza MM, Simpson MA, Spain SL, et
al: Pooled sequencing of 531 genes in inflammatory bowel disease
identifies an associated rare variant in BTNL2 and implicates other
immune related genes. PLoS Genet. 11:e10049552015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
McCloskey C, Jones S, Amisten S, Snowden
RT, Kaczmarek LK, Erlinge D, Goodall AH, Forsythe ID and
Mahaut-Smith MP: Kv1.3 is the exclusive voltage-gated K+
channel of platelets and megakaryocytes: Roles in membrane
potential, Ca2+ signalling and platelet count. J
Physiol. 588:1399–1406. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shankar H, Kahner BN, Prabhakar J, Lakhani
P, Kim S and Kunapuli SP: G-protein-gated inwardly rectifying
potassium channels regulate ADP-induced cPLA2 activity in platelets
through Src family kinases. Blood. 108:3027–3034. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kapural L and Fein A: Suppression of the
voltage-gated K+ current of human megakaryocytes by
thrombin and prostacyclin. Biochim Biophys Acta. 1355:331–342.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schmidt EM, Münzer P, Borst O, Kraemer BF,
Schmid E, Urban B, Lindemann S, Ruth P, Gawaz M and Lang F: Ion
channels in the regulation of platelet migration. Biochem Biophys
Res Commun. 415:54–60. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Al-Shammari MS, Al-Ali R, Al-Balawi N,
Al-Enazi MS, Al-Muraikhi AA, Busaleh FN, Al-Sahwan AS, Al-Elq A,
Al-Nafaie AN, Borgio JF, et al: Type 2 diabetes associated variants
of KCNQ1 strongly confer the risk of cardiovascular disease among
the Saudi Arabian population. Genet Mol Biol. 40:586–590. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gómez-Úriz AM, Milagro FI, Mansego ML,
Cordero P, Abete I, De Arce A, Goyenechea E, Blázquez V,
Martínez-Zabaleta M, Martínez JA, et al: Obesity and ischemic
stroke modulate the methylation levels of KCNQ1 in white blood
cells. Hum Mol Genet. 24:1432–1440. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Abete I, Gómez-Úriz AM, Mansego ML, De
Arce A, Goyenechea E, Blázquez V, Martínez-Zabaleta MT,
González-Muniesa P, López De Munain A, et al: Epigenetic changes in
the methylation patterns of KCNQ1 and WT1 after a weight loss
intervention program in obese stroke patients. Curr Neurovasc Res.
12:321–333. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Harmer SC, Mohal JS, Royal AA, McKenna WJ,
Lambiase PD and Tinker A: Cellular mechanisms underlying the
increased disease severity seen for patients with long QT syndrome
caused by compound mutations in KCNQ1. Biochem J. 462:133–142.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu J, Wang F, Wu Y, Huang X, Sheng L, Xu
J, Zha B, Ding H, Chen Z and Sun T: Meta-analysis of the effect of
KCNQ1 gene polymorphism on the risk of type 2 diabetes. Mol Biol
Rep. 40:3557–3567. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Henninger N, Haussen DC, Kakouros N, Selim
M, Searls DE, Kumar S, Schlaug G and Caplan LR: QTc-prolongation in
posterior circulation stroke. Neurocrit Care. 19:167–175. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gallego-Fabrega C, Carrera C, Reny JL,
Fontana P, Slowik A, Pera J, Pezzini A, Serrano-Heras G, Segura T,
Bin Dukhyil AA, et al: PPM1A methylation is associated with
vascular recurrence in aspirin-treated patients. Stroke.
47:1926–1929. 2016. View Article : Google Scholar : PubMed/NCBI
|