Introduction
A cataract is the loss or decrease of vision due to
opacification of the lens. At present, cataracts, which account for
50% of cases of blindness in low- and middle-income countries, are
the most common ophthalmic diseases (1,2).
Congenital cataract (CC) refers to a lens opacity present at birth,
responsible for 10–30% of all vision loss in infants worldwide
(3). In industrialized countries,
the occurrence of CC is 1–6 cases per 10,000 births, whereas in
China it is ~5 per 10,000 births (4).
CC with or without other systemic abnormalities is
clinically and genetically heterogeneous. At present, at least 50
genes and loci have been demonstrated to be associated with
inherited cataract, including crystalline proteins, lens
cytoskeletal proteins, membrane junction proteins [including lens
fiber major intrinsic protein (MIP)], trans-membrane proteins
[including LEM domain-containing protein 2 (LEMD2)], transcription
factors [including paired like homeodomain 3 (PITX3), forkhead box
protein E3 (FOXE3), transcription factor Maf (MAF), paired box
protein Pax-6 (PAX6) and eyes absent homolog 1 (EYA1)] and other
functionally associated genes [including FYVE and coiled-coil
domain-containing protein 1 (FYCO1), wolframin and transient
receptor potential cation channel subfamily M member 3] (5). Mutations in PITX3 have been
demonstrated to be associated with isolated CC and CC with anterior
segment dysgenesis (ASD) (6).
In the present study, whole exome sequencing (WES)
was performed to identify the molecular defects of a
four-generation Chinese family with CC. A PITX3 variant (c.608delC)
was identified to lead to the development of congenital posterior
subcapsular cataract, which was confirmed by slit lamp exam on the
proband in family 10003. The CC caused by PITX3 mutations in an
additional 194 Chinese families with CC were also investigated. A
second PITX3 mutation (c.640_656del) identified in family 10094 and
10178 was confirmed to follow an autosomal dominant inheritance
pattern, which was in contrast to the autosomal recessive pattern
initially described when it was identified in Saudi Arabia in 2011
(7). Furthermore, the in
vitro functional studies of these two PITX3 mutations performed
in the present study demonstrated comparable molecular consequences
for these and other PITX3 mutations, and the results are closely
coincided with the hypothesis that these mutations may affect
transactivation of PITX3.
Materials and methods
Patients and clinical data
To search for a new locus for CC, 195 CC families
originating from 15 different provinces throughout China (Hunan,
Jilin, Guangdong, Guangxi Zhuang Autonomous Region, Hebei,
Shanghai, Shanxi, Sichuan, Anhui, Hubei, Liaoning, Jiangxi,
Jiangsu, Zhejiang and Beijing) were recruited in the present study.
Informed consent was gained directly from the participants, and the
study was approved by the Institutional Review Board of The Tongji
Eye Institute of Tongji University School of Medicine (Shanghai,
China) and adhered to the tenets of the Declaration of Helsinki.
The clinical data of the patients were gathered using slit lamp
examination. Total genomic DNA was extracted and isolated from
peripheral blood (5 ml) using DNA extraction kits (Tiangen Biotech
Co., Ltd., Beijing, China).
WES and bioinformatic analysis
In Family 10003 (laboratory reference number),
genomic DNA from 2 patients (IV:2 and IV:5) and a selected control
(III:3) were analyzed using WES by Genesky Bio-Tech Co., Ltd.,
(Shanghai, China). Whole-exome trapping was performed using the
Agilent SureSelect Human All Exon kit V6 (57Mb; Agilent
Technologies, Inc., Santa Clara, CA, USA). The entire protocol,
including construction of a shotgun library, in-solution
hybridization, washing and capture, was performed according to the
manufacturer's protocol. The captured DNA library was then
sequenced via Hiseq 2000 platform (2X150 bp) (Illumina, Inc., San
Diego, CA, USA), where each sample was provided an average coverage
depth of ~150 reads. Data were aligned to the human genome
reference assembly (UCSC Genome Browser hg19) (8) with the Burroughs-Wheeler Aligner.
Databases including 1000 Genomes Project, dbSNP137, 1000G_ASN and
esp6500si_all were used to filter the variants. The analyses of
single-nucleotide variants and indels were performed using the
Genome Analysis Toolkit (version 2.4–9 of GATK) The WES data of
structural variants and copy-number variations were also assessed.
Integrative Genomics Viewer (9)
and CoNIFER (version 0.2.2; http://conifer.sourceforge.net/index.html) were used
to analyze the bioinformatic prediction based on the BAM files.
Co-segregation analysis and mutation
detection
To confirm whether the disease phenotype was
co-segregated with the candidate gene in the family 10003 and to
screen for PITX3 mutations in probands of an additional 194 Chinese
CC families in the exon 4 of PITX3, DNA samples from the members of
the four-generation family and 194 CC families were amplified using
polymerase chain reaction (PCR). The following primers were used to
screen for the PITX3 mutation: PITX3_EXON2 Forward (F),
AGAGAACCTCTCAGCATGCAC; PITX3_EXON2 Reverse (R),
AAGCCAGCGCATATTCTCC; PITX3_EXON3 F, GGTGCAGGACATAACAGCTTC;
PITX3_EXON3 R, GGACAGTAGGATGGGGTTGAG; PITX3_EXON4 F,
CGTCTCTAGCCACCTCATCTC; PITX3_EXON4 R, TCCCTGTTCCTGGCTTTAGTC. Each
reaction mixture (25 µl) contained 40 ng genomic DNA, 2X Taq Master
Mix (Tiangen Biotech Co., Ltd.), 0.5 µM forward primer and 0.5 µM
reverse primer. The PCR thermocycler conditions were as follows:
95°C for 3 min; followed by 15 cycles of 95°C for 30 sec, 64–57°C
for 30 sec (annealing temperature decreased 0.5°C each cycle) and
72°C for 1 min, then 25 cycles of 95°C for 30 sec, 57°C for 30 sec
and 72°C for 1 min, followed by a final extension at 72°C for 10
min. Finally, the PCR products were sequenced using an ABI3730
Automated Sequencer (Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and compared with the reference sequence in
the National Center for Biotechnology Information database
(http://www.ncbi.nlm.nih.gov/).
Functional characterization of PITX3
mutations
Plasmid constructs and cell
culture
The cDNA of the PITX3 wild-type (WT) was synthesized
by Sangon Biotech Co., Ltd., (Shanghai, China), and the following
primers were used to construct PITX3 WT and mutant plasmids:
PTIX3-WT F, GCGAAGCTTATGGAGTTCGGCCTGCTCAG; PTIX3-WT R,
CGCGGATCCCTACGGGCGGGGCCGCTCATA; PITX3-c.608delC overlap F primer,
CTCCGCCGCGGCTGCCCCGGGCACCGT; PITX3-c.608delC overlap R primer,
GGGCAGCCGCGGCGGAGGGCACCATGGAGGC; PITX3-c.640_656del overlap F
primer, ACCGTGCCAGGGCCTGGGCGGGGGC; PITX3-c.640_656del overlap R
primer, CCCCGCCCAGGCCCTGGCACGGT. Then, PCR products from the cDNA
were inserted into the NheI-high fidelity (HF)- and BamH
I-HF-digested pcdna3.1-N-3×flag vector (Invitrogen; Thermo Fisher
Scientific, Inc.) to produce the pcdna3.1-N-3×flag-PITX3-WT,
pcdna3.1-N-3×flag-PITX3-c.640_656del. and
pcdna3.1-N-3×flag-PITX3-c.608delC expression plasmids. The
recombinant plasmids included an N-terminal three tandem FLAG-tag
insertion followed by the PITX3 WT or mutant sequences. The
luciferase reporter plasmids pGL3-MIP (+58/-598), pGL3-FOXE3
(−2988/-3722) and pGL3-LEMD2 (−77/-985) were constructed by
inserting 3 non-coding sequences containing bicoid binding sites
from MIP (a 656-bp fragment containing 597-bp of upstream and 59-bp
of downstream sequence from human MIP transcriptional start site),
FOXE3 (a 735-bp fragment derived from −2988 to −3722 of human FOXE3
promoter) and LEMD2 (a 908-bp fragment region from-77 to-985 of
human LEMD2 promoter) into the XhoI-HF- and
HindIII-HF-digested pGL3-Basic vector (Promega Corporation,
Madison, WI, USA) separately. Primers designed for luciferase
reporter gene plasmid construction were as follows: MIP_+58 to −598
F, GCACTCGAGAGCCAGACGCAGCAGAACTAT; MIP_+58 to −598 R,
GCAAAGCTTGCTGATCGCAGTTCCCACAT; FOXE3_-2988 to −3722 F,
GCACTCGAGCCCTACCCCATGTTCCTTGC; FOXE3_-2988 to −3722 R,
GCAAAGCTTGCGCCATGATGGGAAGGTCG; LEMD2_-77 to −985 F,
GCACTCGAGCTGCAAAGATGTGGGTTAAG; LEMD2_-77 to −985 R,
GCAAAGCTTGCGCCATGATGGGAAGGTCG. Finally, all the constructs were
verified by Sanger sequencing. HeLa and 293T cells (American Type
Culture Collection, Manassas, VA, USA) were cultured in Dulbecco's
modified Eagle medium (Invitrogen; Thermo Fisher Scientific, Inc.),
supplemented with 10% fetal bovine serum (FBS; Invitrogen; Thermo
Fisher Scientific, Inc.) and cultured in humidified air containing
5% CO2 at 37°C.
Western blot analysis
Following 36 h transfection with the PITX3 wild-type
and mutants plasmids 293T cells were lysed by
radioimmunoprecipitation assay lysis buffer (P0013B; Beyotime
Institute of Biotechnology, Haimen, China) including a proteinase
inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA, USA) on
ice. Then, cells were lysed for 2 h and centrifugated to obtain the
supernatant. For western blotting, total protein was quantified
using a bicinchoninic acid assay (cat. no. P0010S, Beyotime,
Shanghai, China) and 20 µg protein/lane was analyzed by means of
12% SDS-PAGE followed by transfer onto a polyvinylidene fluoride
membrane. To avoid non-specific binding, the membranes were blocked
with 5% non-fat milk at 25°C for 1 h. Following blocking, the
PITX3-FLAG recombinant proteins were detected with an anti-FLAG
mouse monoclonal antibody (1:1,000; cat. no. F1804; Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) at 25°C for 2 h. The secondary
antibody used was Goat Anti-Mouse IgG, HRP-Conjugated (1:5,000;
cat. no. CW0102; Beijing CWBio, Beijing, China) at 25°C for 1 h.
Membranes were visualized by electro chemiluminescent Western
Blotting Substrate (cat. no. 10026691, Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) with Tanon Imaging System (Tanon-5200; Tanon
Science & Technology Co. Ltd., Shanghai, China).
Immunofluorescence
HeLa cells were plated onto coverslips in 12-well
plates and seeded at 2×104 cells per well in DMEM with
10% FBS for 24 h. Cells were transfected with 1.5 µg of PITX3
wild-type and mutants plasmids respectively using transfection
reagent Vigofect (Vigorous Biotechnology, Inc., Beijing, China) and
harvested 24 h post-transfection. According to the manufacturer's
protocol. The cells were washed with PBS 3 times, fixed with purity
methanol (100% methanol) at −20°C for 10 min, permeabilized with
0.25% Triton X-100 at 25°C for 10 min, blocked with 1% bovine serum
albumin (Sangon Biotech Co., Ltd., Shanghai, China) at 25°C for 1
h, and incubated with a primary antibody PITX3 (N-20) (cat. no.
sc-19307; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) at a
1:1,000 dilution at 25°C for 1 h. Following washing 3 times with
PBS, the cells were subsequently incubated with secondary antibody
Alexa Fluor 594 Donkey anti-Goat IgG at a 1:200 dilution (cat. no.
A11058; Thermo Fisher Scientific, Inc.) for 1 h at 25°C. Nuclei
were counterstained with DAPI dye (0.5 µg/m1) for 1 min at 25°C.
Finally, the location of PITX3 wild-type and mutants protein was
detected by confocal fluorescent microscopy at two different
magnifications: ×20 and ×40.
Luciferase assays
293T cells were plated in 24-well plates and
transfected using transfection reagent Vigofect with 75 ng reporter
plasmid and luciferase reporter vectors MIP-pGL3, FOXE3-pGL3 and
LEMD2-pGL3 (a candidate target gene of PITX3) Each co-transfection
included 62.5 ng effector plasmid (PITX3 WT and mutants expression
constructs, the total DNA amount was kept the same in all
transfections by adding empty pcDNA3.1 vector when required) and 75
ng β-galactosidase vector, which was used as an internal control
for efficiency of transfection. Cells were harvested after 26 h and
luciferase assays were performed as previously described (10).
Statistical analysis
Data represents as means ± standard error of the
mean from 3 experiments performed in quadruplicate. Statistical
significance was determined using one-way analysis of variance and
Šidák's multiple comparisons test for the comparison of multiple
groups using GraphPad Prism v6 software (GraphPad Software, Inc.,
La Jolla, CA, USA).
Results
Clinical data
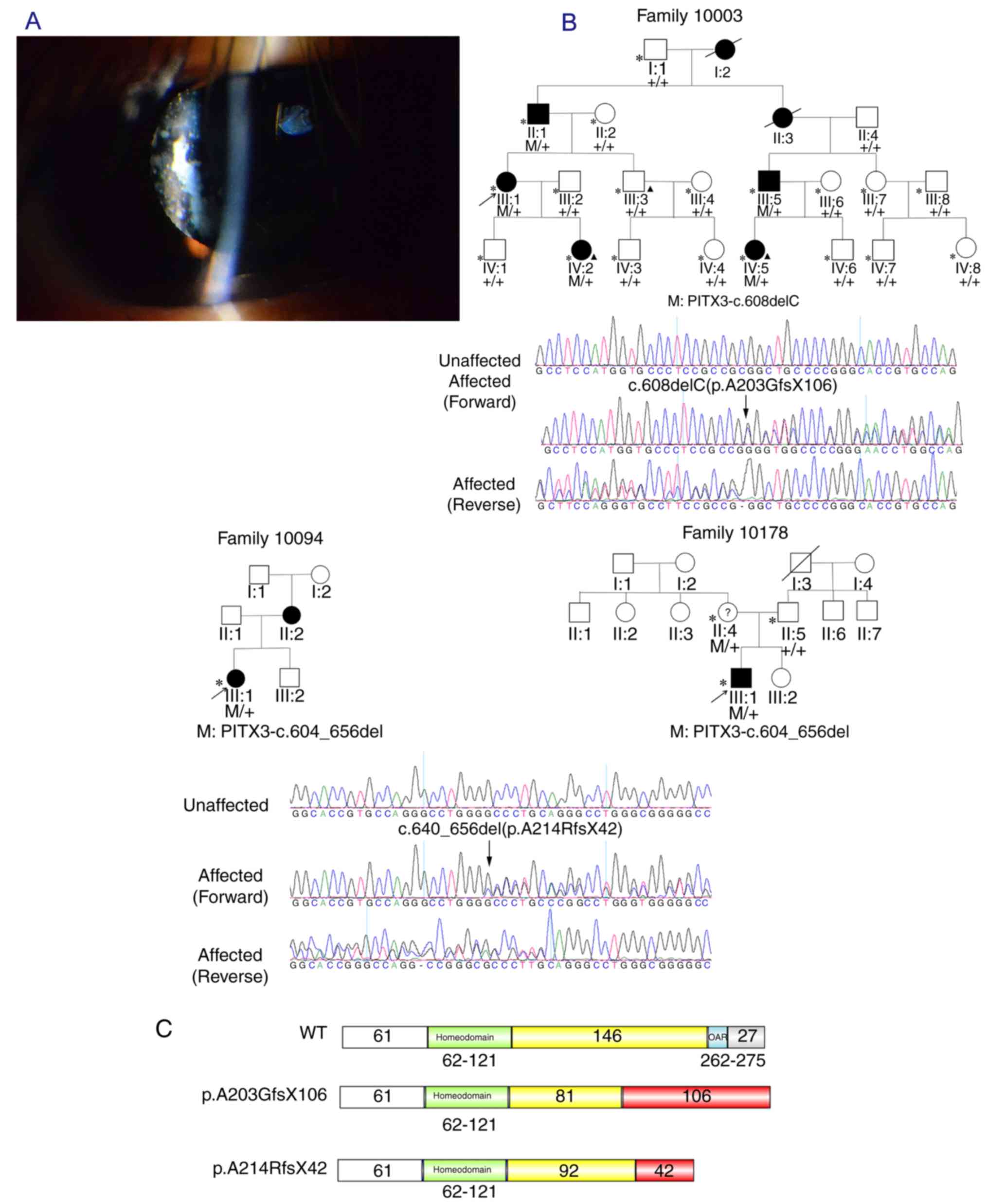
Family 10003 is a large four-generation family with
7 patients with CC (2 of them are deceased) demonstrating an
autosomal dominant pattern of inheritance (Fig. 1A). In the present study, WES was
conducted to map the causative gene for this family. Following
identification of the mutation and informing the family of the
result of the diagnosis, it was identified that all patients in
this family had a primary diagnosis of congenital subcapsular
cataract based on clinical descriptions. Therefore, it was
considered necessary to confirm the type of cataract via careful
ophthalmic examination. Following numerous contacts with members of
this family, permission was granted from a patient who had not
undergone cataract surgery, and the clinical images were finally
obtained (Fig. 1A. In family
10094, the transmission of the CC pathogenic gene from the affected
mother to her daughter in an autosomal dominant manner was
observed, while in the family 10178, it was verified that the
mutation that caused the CC of the proband was transmitted from the
asymptomatic mother, which may indicate that the mode of
inheritance is autosomal dominant with decreased penetrance
(Fig. 1B). As all patients in
these two families had undergone cataract surgery, the types of
cataracts present were not able to be identified.
Mutation analysis and co-segregation
analysis
In family 10003, WES was performed on 3 individuals
(III: 3, IV:2 and IV:5) and a PITX3 variant c.608delC
(p.A203GfsX106) in exon 4 was identified, which lead to the
substitution of alanine into glycine at residue 203. This deletion
mutation resulted in a frameshift in codon 203 and produced an
aberrant protein with 106 erroneous residues (Fig. 1C). In the probands of family 10094
and family 10178, Sanger sequencing was performed to investigate
the coding mutations in the exon 4 of PITX3 gene: An additional
PITX3 variant c.640_656del was identified, which resulted in an
alanine to arginine substitution at residue 214 (A214RfsX42) and
led to a truncation of the PITX3 protein (Fig. 1C). The co-segregation of these two
mutations with diseases was demonstrated in the corresponding
families with available DNA (Fig.
1A).
Subcellular localization
The PITX3 WT and mutant proteins in the present
study were overexpressed in HeLa cells and detected by
immunofluorescence (Fig. 2A). The
intracellular localization of PITX3 mutants c.608delC
(p.A203GfsX106) and c.640_656del (p.A214RfsX42) in the present
study were identified to be targeted to the nucleus, which was
similar to the PITX3 WT.
 | Figure 2.Functional analysis of the expression
of PITX3 WT and mutants including. Subcellular localization and
transactivation activity. (A) Subcellular localization of the PITX3
WT and mutant proteins transfected in HeLa cells. Cells were
stained with PITX3 (N-20) primary antibody and Alexa Fluor 568
donkey anti-mouse IgG as a secondary antibody (red); DAPI was used
as a nuclear counterstain (blue). Red fluorescence was not observed
in HeLa cells without exogenous gene introduction. For cells
transfected with PITX3 wild-type and mutants plasmids, the red
fluorescence was localized predominantly in the nucleus. Western
blot analysis indicated that the protein expression of PITX3
mutants was not affected. (B) Luciferase assay results for PITX3 WT
and mutants co-transfected with the (C) pGL3-MIP (+58/-598), (D)
pGL3-FOXE3 (−2988/-3722) or (E) pGL3-LEMD2 (−77/-985) reporters in
293T cells. All luciferase activities were normalized to
β-galactosidase activity. In comparison with the empty vector
pcDNA3.1, the values are indicated as fold changes of luciferase
activity. *P≤0.05, **P≤0.01 and ***P≤0.001. PITX3, paired like
homeodomain 3; WT, wild-type; MIP, lens fiber major intrinsic
protein; FOXE3, forkhead box protein E3; LEMD2, LEM
domain-containing protein 2; ns, not significant. |
Transactivation activity
To investigate the transcriptional activity of PITX3
WT and mutants in 293T cells, co-transfection assays were performed
using the pGL3-MIP and pGL3-FOXE3 reporter constructs and the PITX3
WT and mutant expression plasmids. To isolate additional genes that
are directly regulated by PITX3, a search for genomic sequences
that contained evolutionarily-conserved bicoid/PITX3 binding sites
in promoter regions and were located in the known
cataract-associated genes was also performed. This identified the
LEMD2 gene, which contains 2 tandem ‘TAATCC’ (bicoid binding site)
repeats. Then, the pGL3-LEMD2 reporter gene plasmid was constructed
to observe the expression change of pGL3-LEMD2 when co-transfected
with PITX3 WT or mutant expression plasmids. The result of the
western blot analysis indicated that the amount of protein of PITX3
WT and mutants were not different under visual observation
(Fig. 2B. When PITX3 WT was
co-transfected with the pGL3-MIP plasmid, a ~2-fold increase in
reporter gene activity was consistently observed compared with the
empty vector (Fig. 2C). However,
PITX3 mutants A214Rfs and A203Gfs co-transfected with the same
reporter exhibited decreases of 38 and 31% in luciferase activity
compared with the WT, respectively. A similar decrease in
luciferase activity was observed when using the FOXE3-pGL3
reporter. As a ~1.4-fold increase in luciferase activity was
detected when overexpressing PITX3 WT, the luciferase activity of
the mutants A214Rfs and A203Gfs decreased by 40 and 26%,
respectively (Fig. 2D). By
contrast, overexpression of PITX3 WT did not have any activation
effect on LEMD2-pGL3. However, the A214Rfs and A203Gfs mutants
exhibited increased transcriptional activity compared with the
empty vector and PITX3 WT (Fig.
2E).
Discussion
CC is a clinically heterogeneous disease. It may
occur in a variety of morphologic configurations: Total; nuclear;
cortical; anterior subcapsular; and posterior subcapsular (11). Compared with other types of
cataracts, posterior subcapsular cataract (PSC) is one of most
common forms of cataract in clinical surgical series (12), but as a type of CC, it is unusual.
At present, the PSC-associated genes include: Gap junction protein
alpha 8; Ras related GTP binding A; PITX3; abhydrolase domain
containing 12; charged multivesicular body protein 4B; crystallin
beta B2; Beta-1,4-galactosyltransferase 7; ornithine
aminotransferase; MAF; Unc-45 myosin chaperone B; and FYCO1
(13). PITX3 is one of PSC-causing
genes, and there are 2 PITX3 mutations associated with PSC. Along
with PITX1 and PITX2, PITX3 is the third gene in the PITX homeobox
family and it is essential to the formation of the lens during eye
development. There have been 9 PITX3 mutations demonstrated to be
associated with several different types of CC in several
populations (7,14–23)
(Table I). However, in China,
there have been few studies on PITX3 mutations. In the present
study, a wide-scale PITX3 mutation screening was implemented in 195
CC families from 15 different provinces of China. Among them, 2
PITX3 mutations were identified in 3 CC families: One was a
four-generation family with PITX3-c.608delC mutation causing PSC. A
second mutation, (PITX3-c.640_656del) identified in two other
families, resulted in a dominant and incompletely dominant
inheritance pattern in these two families, respectively, which is
different from the autosomal recessive inheritance pattern observed
when the mutation was initially identified in a family from Saudi
Arabia with ASD and severe congenital microphthalmia (7). The PITX3 protein has 2 domains: The
conserved homeodomain is required for DNA binding (a lysine at
residue 50 of the homeodomain interacts with base pairs 5 and 6 of
the hexanucleotide consensus 5′-TAATCC-3′) and the homeobox protein
orthopedia, Aristaless related homeobox and Retinal homeobox
protein Rx (OAR) domain is associated with target specificity and
transactivation of PITX3 (14,24).
However, as the majority of PITX3 mutated sites identified are
distributed in exon 4 of this gene, the N-terminal region of the
OAR domain is clearly a frequently mutated region. PITX3 deficiency
results in a range of phenotypes, from isolated cataracts to
microphthalmia in humans, and lens degeneration in mice (10). Although identification of
downstream targets of PITX3 is vital for understanding the
mechanisms of normal ocular development and human disease, these
targets remain largely unknown. As a demonstrated target gene of
PITX3, the expression of MIP is restricted to the lens. MIP is
abundant in the lens fibers and distributed throughout the plasma
membrane of the lens fiber cells, but it not present in the basal
or lateral plasma membrane of the lens epithelial cells (25,26),
making it difficult to detect endogenous levels of MIP mRNA in
other cell types. FOXE3 is an additional target gene of PITX3 in
mice (27) and as it is conserved
across mammalian species, direct PITX3 interaction with the
consensus bicoid-binding site located upstream of FOXE3 gene may be
additionally examined in humans. LEMD2 is a candidate target gene
of PITX3 as it has 2 tandem bicoid binding sites in the promoter
and is associated with cataract disease (28). LEMD2 is involved in the regulation
of several signaling pathways including mitogen-activated protein
(MAP) kinase and protein kinase B, and serves a role in cell
signaling and differentiation (29). The expression of LEMD2 has been
demonstrated in the human whole lens (28). Previous study (30) has indicated that downregulation of
mouse LEMD2 by RNA interference in myoblast cultures resulted in
increased phosphorylation of MAP kinases extracellular
signal-regulated kinase 1/2 (ERK1/2) and c-Jun N-terminal kinase
(JNK). ERK activation is required for lens fiber differentiation,
and the upregulation of MIP at the transcriptional level was
simultaneous with the activation of the fibroblast growth factor
downstream signaling components, ERK1/2 and JNK (31). Therefore, PITX3 may negatively
regulate LEMD2 to maintain fiber cell differentiation. Finally, a
luciferase reporter assay was performed in the present study to
examine the biological significance of PITX3 binding to the
upstream bicoid sites of MIP, FOXE3 and LEMD2. As hypothesized,
when PITX3 mutants were co-transfected with MIP-pGL3, a decrease in
luciferase activity was observed compared with the PITX3 WT. The
luciferase activity of the PITX3 mutants all decreased
significantly in comparison with the PITX3 WT when co-transfected
with FOXE3-pGL3. As a head-to-tail arrangement (5′-TAATCC…
TAATCC-3′) of bicoid elements is preferred over a head-to-head
arrangement (5′-TAATCC… GGATTA-3′) of bicoid elements (27,32),
the PITX3 WT and mutant sequences presented low levels of relative
luciferase activity compared with pcDNA3.1, as the reverse
complementary bicoid site GGATTA of FOXE3 may interact weakly with
PITX3. However, when PITX3 WT and mutants were co-transfected with
LEMD2-pGL3, the opposite effect was observed: When PITX3 WT was
co-transfected with LEMD2-pGL3, almost no activation was observed.
Conversely, the 2 PITX3 mutants exhibited an increased luciferase
activity compared with the WT. The data is consistent with the
hypothesis that PITX3 may negatively regulate LEMD2 in lens
epithelial cells, promoting the activation of ERK, which is
required for the differentiation of lens fiber cells. The
alterations in the OAR motif may affect the target specificity of
the homeobox-containing transcription factors that resulted in a
decrease in the specific binding to its target genes and
potentially enhanced the non-specific binding with other. Besides,
the reason for the differences in luciferase activity between
different PITX3 mutants may due to the fact that the destruction of
OAR would not lead to a complete loss of PITX3 specificity, but a
decrease in the specificity. In addition, the different affinity
between PITX3 and its target genes may also lead to a range of
luciferase activity levels when PITX3 WT and mutant plasmids are
co-transfected with the promoter of different target genes. The
nuclear localization of PITX3 WT and mutants may be attributed to
the probable nuclear localization signal (RRAKWRK), which is
located in the third helix of the PITX3 homeodomain and has been
identified in several other homeodomain proteins (33–37).
The pathogenic mechanism of CC from PITX3 mutations remains
complicated, and it may be as follows: On one hand, the alteration
of the OAR domain leads to a decrease in specific binding and
targeting to target genes, which will affect the regular
development of lens; conversely, by non-specific binding with other
genes, certain genes should have been inhibited by PITX3, yet may
be inactivated by interacting with PITX3 mutants and other genes,
in particular genes containing bicoid binding sites, which may
potentially also bind to PITX3 mutants. This may disrupt the
development of other tissue development processes in the lens and
lead to other phenotypes, including ASD.
 | Table I.Summary of 9 different mutations in
PITX3 associated with cataract. |
Table I.
Summary of 9 different mutations in
PITX3 associated with cataract.
| Authors | Nucleotide
change | Amino acid
change | Inheritance | Origin | Type of
cataract | Complication | (Refs.) |
|---|
| Semina EV, et
al 1998 | c.38G>A | p.S13N | AD | USA | Total cataract | – |
(15) |
|
|
c.640_656dup17bp | p.G220PfsX95 | AD | USA | Anterior
cortical | ASMD |
|
| Berry V, et
al 2004 |
c.640_656dup17bp | p.G220PfsX95 | AD | Two separate UK
families | Posterior
polar | ASMD | (18) |
|
|
c.640_656dup17bp | p.G220PfsX95 | AD | China | Posterior
polar | – |
|
|
| c.650delG | p.G217AfsX91 | AD | UK | Posterior
polar | – |
|
|
|
c.640_656dup17bp | p.G220PfsX95 | AD | Spain | Posterior
polar | – |
|
| Finzi S, et
al 2005 |
c.640_656dup17bp | p.G220PfsX95 | AD | USA | Posterior
polar | – | (19) |
| Burdon KP, et
al 2006 |
c.640_656dup17bp | p.G220PfsX95 | AD | Australia | Posterior
polar | – | (20) |
| Summers KM et
al 2008 |
c.640_656dup17bp | p.G220PfsX95 | AD | Australia | PSC | ASMD | (21) |
| Berry V, et
al 2011 | c.542delC | p.P181LfsX127 | AD | UK | Posterior
polar | – | (16) |
| Aldahmesh MA, et
al 2011 | c.640_656del | p.A214RfsX42 | AR | Saudi Arabia | – | ASMD | (7) |
| Verdin H, et
al 2014 | c.573delC | p.S192AfsX117 | AD | Belgo-Romanian
family | Cataract | ASMD | (14) |
|
|
c.640_656dup17bp | p.G220PfsX95 | AD | Two separate
Belgian families | PSC | ASMD |
|
|
|
c.640_656dup17bp | p.G220PfsX95 | AD | Two separate
Belgian families | Posterior
polar | ASMD |
|
| Bidinost C, et
al 2006 | c.650delG | p.G217AfsX91 | AD | Lebanese
family | Posterior
polar | – | (22) |
| Liu, H., et
al 2017 | c.608delC | p.A203GfsX106 | AD | China | Cataract |
| (17) |
|
| c.669delC | p.L225WfsX84 | AD | Iraq | Cataract | – |
|
| Zazo Seco C, et
al 2018 | c.582delC | p.I194MfsX115 |
| North Ireland | Cataract | – | (23) |
|
| c.38G>A | p.S13N | AD | France | – | – |
|
| Present study | c.608delC | p.A203GfsX106 | AD | Chinese family | PSC | ASMD |
|
|
| c.640_656del | p.A214RfsX42 | AD | Two separate
Chinese families | Cataract | – |
|
In conclusion, the present study extended the
mutation spectrum of PITX3 mutations in Chinese family with CC. In
the first family, the disease-causing gene was identified by WES,
but also detailed clinical images were obtained. The inheritance
and phenotype caused by mutation p.A214RfsX42 observed in the
present study was quite different from previous data. The
functional analysis of these 2 PITX3 mutations in the in
vitro functional studies is an important complement and
extension, which provides a potential interpretation for the
pathogenesis and molecular mechanism of PITX3 mutations associated
with CC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Key
Basic Research Program of China (973 Program; grant no.
2015CB964601), National Natural Science Foundation of China (grant
nos. 81371062 and 31501014) and Thousand Youth Talents Program of
China to Jianjun Chen.
Availability of data and materials
All data analyzed during the present study are
included in this article, and the datasets are available from the
corresponding author on reasonable request.
Authors' contributions
DM, BL, ZZho and JC were responsible for study
design. CF, HZ, ZZho and JC collected the samples. ZW, JLi, XZ, SL,
HZ and JLin performed the experiments. ZW, DM, HZ, ZZha, BL, ZZho
and JC conducted data interpretation and analysis. ZW, DM, ZZho and
JC were responsible for writing the manuscript. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all
participants including patients and their healthy family members,
and the study was approved by the Institutional Review Board of the
Tongji Eye Institute of Tongji University School of Medicine
(Shanghai, China).
Patient consent for publication
Written informed consent was obtained from all
patients and healthy volunteers.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
WHO. Visual impairment and blindness, .
2014.http://www.who.int/mediacentre/factsheets/fs282/en/May
14–2016
|
|
2
|
Liu YC, Wilkins M, Kim T, Malyugin B and
Mehta JS: Cataracts. Lancet. 390:6002017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li FF, Zhu SQ, Wang SZ, Gao C, Huang SZ,
Zhang M and Ma X: Nonsense mutation in the CRYBB2 gene causing
autosomal dominant progressive polymorphic congenital coronary
cataracts. Mol Vis. 14:750–755. 2008.PubMed/NCBI
|
|
4
|
Zhong Z, Wu Z, Han L and Chen J: Novel
mutations in CRYGC are associated with congenital cataracts in
Chinese families. Sci Rep. 7:1892017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shiels A and Hejtmancik JF: Molecular
genetics of cataract. John Wiley & Sons, Ltd. 37:672014.
|
|
6
|
Brémond-Gignac D, Bitoun P, Reis LM, Copin
H, Murray JC and Semina EV: Identification of dominant FOXE3 and
PAX6 mutations in patients with congenital cataract and aniridia.
Mol Vis. 16:1705–1711. 2010.PubMed/NCBI
|
|
7
|
Aldahmesh MA, Khan AO, Mohamed J and
Alkuraya FS: Novel recessive BFSP2 and PITX3 mutations: Insights
into mutational mechanisms from consanguineous populations. Genet
Med. 13:978–981. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kent WJ, Sugnet CW, Furey TS, Roskin KM,
Pringle TH, Zahler AM and Haussler D: The human genome browser at
UCSC. Genome Res. 12:996–1006. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Robinson JT, Thorvaldsdóttir H, Winckler
W, Guttman M, Lander ES, Getz G and Mesirov JP: Integrative
genomics viewer. Nat Biotechnol. 29:24–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sorokina EA, Muheisen S, Mlodik N and
Semina EV: MIP/Aquaporin 0 represents a direct transcriptional
target of PITX3 in the developing lens. PLoS One. 6:e211222011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Deng H and Yuan L: Molecular genetics of
congenital nuclear cataract. Eur J Med Genet. 57:113–122. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Adamsons I, Muñoz B, Enger C and Taylor
HR: Prevalence of lens opacities in surgical and general
populations. Arch Ophthalmol. 109:993–997. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shiels A, Bennett TM and Hejtmancik JF:
Cat-Map: Putting cataract on the map. Mol Vis. 16:2007–2015.
2010.PubMed/NCBI
|
|
14
|
Verdin H, Sorokina EA, Meire F, Casteels
I, de Ravel T, Semina EV and De Baere E: Novel and recurrent PITX3
mutations in Belgian families with autosomal dominant congenital
cataract and anterior segment dysgenesis have similar phenotypic
and functional characteristics. Orphanet J Rare Dis. 9:262014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Semina EV, Ferrell RE, Mintz-Hittner HA,
Bitoun P, Alward WL, Reiter RS, Funkhauser C, Daack-Hirsch S and
Murray JC: A novel homeobox gene PITX3 is mutated in families with
autosomal-dominant cataracts and ASMD. Nat Genet. 19:167–170. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Berry V, Francis PJ, Prescott Q, Waseem
NH, Moore AT and Bhattacharya SS: A novel 1-bp deletion in PITX3
causing congenital posterior polar cataract. Mol Vis. 17:1249–1253.
2011.PubMed/NCBI
|
|
17
|
Liu H, Liu H, Tang J, Lin Q, Sun Y, Wang
C, Yang H, Khan MR, Peerbux MW, Ahmad S, et al: Whole exome
sequencing identifies a novel mutation in the PITX3 gene, causing
autosomal dominant congenital cataracts in a Chinese family. Ann
Clin Lab Sci. 47:92–95. 2017.PubMed/NCBI
|
|
18
|
Berry V, Yang Z, Addison PK, Francis PJ,
Ionides A, Karan G, Jiang L, Lin W, Hu J, Yang R, et al: Recurrent
17 bp duplication in PITX3 is primarily associated with posterior
polar cataract (CPP4). J Med Genet. 41:e1092004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Finzi S, Li Y, Mitchell TN, Farr A,
Maumenee IH, Sallum JM and Sundin O: Posterior polar cataract:
Genetic analysis of a large family. Ophthalmic Genet. 26:125–130.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Burdon KP, McKay JD, Wirth MG,
Russell-Eggit IM, Bhatti S, Ruddle JB, Dimasi D, Mackey DA and
Craig JE: The PITX3 gene in posterior polar congenital cataract in
Australia. Mol Vis. 12:367–371. 2006.PubMed/NCBI
|
|
21
|
Summers KM, Withers SJ, Gole GA, Piras S
and Taylor PJ: Anterior segment mesenchymal dysgenesis in a large
Australian family is associated with the recurrent 17 bp
duplication in PITX3. Mol Vis. 14:2010–2015. 2008.PubMed/NCBI
|
|
22
|
Bidinost C, Matsumoto M, Chung D, Salem N,
Zhang K, Stockton DW, Khoury A, Megarbane A, Bejjani BA and
Traboulsi EI: Heterozygous and homozygous mutations in PITX3 in a
large Lebanese family with posterior polar cataracts and
neurodevelopmental abnormalities. Invest Ophthalmol Vis Sci.
47:1274–1280. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zazo Seco C, Plaisancié J, Lupasco T,
Michot C, Pechmeja J, Delanne J, Cottereau E, Ayuso C, Corton M,
Calvas P, et al: Identification of PITX3 mutations in individuals
with various ocular developmental defects. Ophthalmic Genet.
39:314–320. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sakazume S, Sorokina E, Iwamoto Y and
Semina EV: Functional analysis of human mutations in homeodomain
transcription factor PITX3. BMC Mol Biol. 8:842007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Takata K, Matsuzaki T and Tajika Y:
Aquaporins: Water channel proteins of the cell membrane. Prog
Histochem Cytochem. 39:1–83. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gorin MB, Yancey SB, Cline J, Revel JP and
Horwitz J: The major intrinsic protein (MIP) of the bovine lens
fiber membrane: Characterization and structure based on cDNA
cloning. Cell. 39:49–59. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ahmad N, Aslam M, Muenster D, Horsch M,
Khan MA, Carlsson P, Beckers J and Graw J: Pitx3 directly regulates
Foxe3 during early lens development. Int J Dev Biol. 57:741–751.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Boone PM, Yuan B, Gu S, Ma Z, Gambin T,
Gonzaga-Jauregui C, Jain M, Murdock TJ, White JJ, Jhangiani SN, et
al: Hutterite-type cataract maps to chromosome 6p21.32-p21.31,
cosegregates with a homozygous mutation in LEMD2 and is associated
with sudden cardiac death. Mol Genet Genomic Med. 4:77–94. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huber MD, Guan T and Gerace L: Overlapping
functions of nuclear envelope proteins NET25 (Lem2) and emerin in
regulation of extracellular signal-regulated kinase signaling in
myoblast differentiation. Mol Cell Biol. 29:5718–5728. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tapia O, Fong LG, Huber MD, Young SG and
Gerace L: Nuclear envelope protein Lem2 is required for mouse
development and regulates MAP and AKT kinases. PLoS One.
10:e01161962015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Golestaneh N, Fan J, Fariss RN, Lo WK,
Zelenka PS and Chepelinsky AB: Lens major intrinsic protein
(MIP)/aquaporin 0 expression in rat lens epithelia explants
requires fibroblast growth factor-induced ERK and JNK signaling. J
Biol Chem. 279:31813–31822. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Saadi I, Kuburas A, Engle JJ and Russo AF:
Dominant negative dimerization of a mutant homeodomain protein in
Axenfeld-Rieger syndrome. Mol Cell Biol. 23:1968–1982. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moede T, Leibiger B, Pour HG, Berggren P
and Leibiger IB: Identification of a nuclear localization signal,
RRMKWKK, in the homeodomain transcription factor PDX-1. FEBS Lett.
461:229–234. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hessabi B, Ziegler P, Schmidt I, Hessabi C
and Walther R: The nuclear localization signal (NLS) of PDX-1 is
part of the homeodomain and represents a novel type of NLS. Eur J
Biochem. 263:170–177. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kozlowski K and Walter MA: Variation in
residual PITX2 activity underlies the phenotypic spectrum of
anterior segment developmental disorders. Hum Mol Genet.
9:2131–2139. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sabherwal N, Schneider KU, Blaschke RJ,
Marchini A and Rappold G: Impairment of SHOX nuclear localization
as a cause for Léri-Weill syndrome. J Cell Sci. 117:3041–3048.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sabherwal N, Blaschke RJ, Marchini A,
Heine-Suner D, Rosell J, Ferragut J, Blum WF and Rappold G: A novel
point mutation A170P in the SHOX gene defines impaired nuclear
translocation as a molecular cause for Léri-Weill dyschondrosteosis
and Langer dysplasia. J Med Genet. 41:e832004. View Article : Google Scholar : PubMed/NCBI
|