Introduction
Basal cell carcinoma (BCC) is a common type of skin
cancer that arises from the innermost layer of the epidermis or
from the outer root sheath of the hair follicle (1). According to the American Cancer
Society, non-melanoma skin cancer accounts for >95% of all skin
malignancies, and primarily includes squamous cell carcinoma (SCC)
and BCC (2). BCC occurs more
frequently in fair-skinned individuals; in the USA alone, there are
~800,000 new cases each year (3).
BCC incidence is the highest in Australia, reaching 726/10,000
individuals each year, whereas in Africa, BCC incidence is the
lowest, accounting for <1/10,000 individuals each year (4,5).
A recent study showed that somatic mutations in
normal skin cells were surprisingly high (6). Additionally, >25% of sun-exposed
skin cells carry cancer-causing mutations in genes that help
maintain the normal functions of the epidermis, thus indicating a
precancerous state and the promotion of skin cancer development
(including BCC, SCC and melanoma) due to sun exposure (7). UV light and genetic susceptibility
are important risk factors in BCC development (3), which contribute to a multistep
process in BCC development via the accumulation of genetic
mutations (3). Previous studies
(8–10) have shown that the alteration of
Hedgehog (HH) signaling and its synergistic signaling pathway are
associated with BCC tumorigenesis, as the HH signaling pathway
plays a critical role in the maintenance of skin stem cells during
early embryo and hair follicle development (11). The mammalian HH family of proteins
includes three members: Sonic HH, Indian HH and desert HH (12). Under normal circumstances, patched
homologue (PTCH), a tumor-suppressor protein, interacts with
smoothened protein (SMO) (5).
Binding of HH to PTCH permits SMO release, which then activates GLI
proteins to promote cell growth and proliferation, and leads to the
development of BCC (13–15). Indeed, loss of function of PTCH1 is
the most frequent mutation in 30–40% of sporadic BCC cases
(16–18), and mutations of genes such as p53,
SMO and suppressor of fused protein also occur in BCC (5).
SHANK-associated RH domain-interacting protein
(SHARPIN) is a component of the linear ubiquitin chain assembly
complex (LUBAC) and enhances tumor necrosis factor (TNF)-induced
NF-κB activity (19). The
C-terminal of SHARPIN protein contains a ubiquitin-like domain and
a Nuclear protein localization protein 4 zinc finger (NZF), with a
significant sequence homology to the longer isoform of
heme-oxidized iron-regulatory protein 2 ubiquitin ligase-1
(HOIL-1L), while the N-terminal contains a coiled-coil region that
can mediate the polymerization (20). SHARPIN, HOIL-1 and HOIL-1L
interacting protein (HOIP) together comprise the LUBAC, catalyzing
the formation of linear ubiquitin chains, and regulating the
activation of the NF-κB and JNK signaling pathways (21). The ubiquitin-like domain of SHARPIN
specifically binds to the ubiquitin-associated domain of HOIP to
associate with and activate HOIP; furthermore, SHARPIN and HOIL-1L
can separately or synergistically facilitate the E2 loading of HOIP
for its activation (22). Fujita
et al (23) demonstrated
that inhibition of LUBAC-tethering motifs-mediated HOIL-1L/SHARPIN
dimerization profoundly attenuates the function of LUBAC. Shimizu
et al (24) showed that the
binding of K63-linked ubiquitin chains to the NZF domain of
SHARPIN, but not HOIL-1L, appears to be involved in the recruitment
of LUBAC. Thus, selective recognition of ubiquitin chains by NZFs
in LUBAC underlies the regulation of LUBAC function (24). Loss of function of SHARPIN in mice
leads to the development of an idiopathic hypereosinophilic
syndrome with eosinophilic dermatitis (25). However, the geographical
heterogeneity of SHARPIN in various areas of the skin has not been
investigated. Previous studies showed that SHARPIN is a
cancer-associated gene. For example, Jung et al (26) demonstrated that SHARPIN was
upregulated in clear cell adenoma, hepatocellular carcinoma and
papillary serous adenocarcinoma. Additionally, several studies
showed that SHARPIN participated in the development of non-small
cell lung cancer, melanoma, mycosis fungoides, breast cancer,
prostate cancer and osteosarcoma (19,27–31).
Previous studies showed that the activation of the NF-κB pathway
induced Sonic HH expression (32–34),
and that UV light could also induce the activation of the NF-κB
pathway during BCC development (35–37).
Based on the BCC pathogenesis and the biological
functions of SHAPRIN in tumorigenesis, the present study
investigated the potential role of SHARPIN in skin BCC and the
underlying molecular mechanisms.
Materials and methods
Tissue samples
Tissues were fixed with 10% formalin for ~4 h at
room temperature. Then, formalin-fixed paraffin-embedded blocks of
26 BCC samples, five normal skin samples and one breast cancer
sample were collected from The Affiliated Hospitals of Southern
Medical University from July 2016 to December 2018. BCC was
diagnosed by experienced dermatologists and confirmed via
histopathological analysis. Breast cancer was diagnosed by a
pathologist. This study was approved by The Ethics Committee of
Shenzhen Hospital, Southern Medical University, and all
participants signed written informed consent forms.
Immunohistochemistry
Paraffin-embedded sections (thickness, 4 µm) were
prepared and deparaffinized in xylene, and then rehydrated in a
series of diluted ethanol solutions (100–70%). The endogenous
peroxidase activity was blocked for 30 min by incubation in 1%
methanolic hydrogen peroxide solution at room temperature. This was
followed by incubation with 20% goat serum (cat. no. C0265;
Beyotime Institute of Biotechnology) to minimize non-specific
binding of the secondary antibody (ready-to-use peroxidase
anti-Mouse/Rabbit IgG; cat. no. PV-9000; OriGene Technologies,
Inc.) and subsequently with rabbit anti-SHARPIN (1:100; cat. no.
sc-98127; Santa Cruz Biotechnology, Inc.), anti-cyclin D1 (1:200;
cat. no. WL01435a; Wanleibio Co., Ltd.), anti-cyclin-dependent
kinase 4 (CDK4; 1:5,000; cat. no. 12790; Cell Signaling Technology,
Inc.), antic-JUN (1:1,000; cat. no. 9165; Cell Signaling
Technology, Inc.) and anti-GLI family zinc finger 2 (GLI2; 1:200;
cat. no. sc-28674; Santa Cruz Biotechnology, Inc.) at 4°C
overnight. On the next day, the sections were subjected to a
two-step plusPoly-horseradish peroxidase anti-Mouse/Rabbit IgG
Detection System (cat. no. PV-9000; OriGene Technologies, Inc.) at
room for 1 h temperature and 3,3-diaminobenzidine Detection kit
(Enhanced Polymer; cat. no. PV-9000-D; OriGene Technologies, Inc.).
The immunostaining results were analyzed using the cross-product H
score, where the staining intensity was graded on a four-point
scale: i) 0, no staining; ii) 1+, weak; iii) 2+, moderate; and iv)
3+, strong staining (38). The H
score=tumor cell staining percentage × staining intensity for
SHARPIN expression, according to a previous study (39). In addition, the expression levels
of cyclin D1, CDK4, c-JUN and GLI2 were searched in the Human
Protein Atlas website (HPA; https://www.proteinatlas.org/; Version 16.1).
Cell lines and culture
The human BCC TE354.T cell line (40,41)
was purchased from the American Tissue Culture Collection (cat. no.
CRL-7762™). HaCaT cells, an immortalized non-tumorigenic
human keratinocyte cell line (42)
was obtained from Nanjing KeyGen Biotech. Co., Ltd. (cat. no.
KG300). HaCaT cells have been authenticated using STR profiling.
Both TE354.T and HaCaT cell lines were cultured in DMEM (cat. no.
C11995500BT; Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% FBS (cat. no. 10270; Gibco; Thermo Fisher Scientific,
Inc.) and 1% penicillin-streptomycin (cat. no. SV30010; GE
Healthcare Life Sciences) in a humidified incubator with 5%
CO2 at 37°C.
Western blotting
Cells were lysed in RIPA buffer (cat. no. 20-188;
EMD Millipore) containing a protease inhibitor (cat. no. CW2200S;
Beijing CoWin Biotech Co., Ltd.). The protein concentration was
measured using a bicinchoninic acid assay kit (cat. no. E112,
Vazyme Biotech Co., Ltd.). Then, 10 µl protein lysates were
subjected to 12% SDS-PAGE and transferred onto PVDF membranes (EMD
Millipore). For western blotting, the membranes were blocked in 5%
non-fat milk for 1 h at room temperature and then incubated with
antibodies against SHARPIN (1:2,000; cat. no. ab197853; Abcam),
cyclin D1 (1:800; cat. no. WL01435a; Wanleibio Co., Ltd.), CDK4
(1:1,000; cat. no. 12790; Cell Signaling Technology, Inc.), c-JUN
(1:2,000; cat. no. 9165; Cell Signaling Technology, Inc.),
phosphorylated (p)-c-JUN (1:1,000; cat. no. 2361; Cell Signaling
Technology, Inc.), GLI1 (1:1,000; cat. no. 8358; Cell Signaling
Technology, Inc.), GLI2 (1:200; cat. no. sc-28674; Santa Cruz
Biotechnology, Inc.), PTCH1 (1:1,000; cat. no. 2468; Cell Signaling
Technology, Inc.), PTCH2 (1:1,000; cat. no. 2470; Cell Signaling
Technology, Inc.) and β-actin (1:2,000; cat. no. AC001-R; Beijing
Dingguo Changsheng Biotechnology Co., Ltd.) at 4°C overnight. On
the next day, the membranes were incubated for 1 h with horseradish
peroxidase-conjugated immunoglobulin (1:2,500; cat. no.
bs-0295G-HRP; Bioss Antibodies) in room temperature and then
detected using a High-Sensitive ECL Chemiluminescence Detection kit
(cat. no. E411; Vazyme Biotech Co., Ltd.). The membranes were
exposed using an imaging system (Canon 5200; Canon Inc.). The
protein expression level was quantified using Image Pro Plus
(43).
Generation of a SHARPIN-deficient
stable cell line
Lentiviral vectors carrying SHARPIN short hairpin
RNA [shRNA; sh-SHARPIN;
pLV(shRNA)-EGFP:T2A:Puro-U6>hSHARPIN(shRNA2); Vector ID:
VB150923-10005] and the negative control [sh-Ctrl;
pLV(shRNA)-EGFP:T2A:Puro-U6>Scramble shRNA; Vector ID:
VB151023-10034] were designed and synthesized by Cyagen
Biosciences, Inc. The SHARPIN shRNA sequence was
5′-CCCTGGAGTCAGTTTCCTACA-3′, and the sh-Ctrl sequence was
5′-CCTAAGGTTAAGTCGCCCTCG-3′. These lentivirus vectors, including an
enhanced green fluorescent protein and an antibiotic resistance
gene against puromycin, were transfected into 293T cells (cat. no.
3111C0001CCC000091; National Infrastructure of Cell Line Resource)
for lentivirus production. 293T cells were cultured in DMEM
supplemented with 10% FBS and 1% penicillin-streptomycin in a
humidified incubator with 5% CO2 at 37°C. The
lentiviruses (Transducing units, 1×108 TU/ml) were
transfected into TE354.T cells (3–5×105/well in 6-well)
with 5 µg/ml polybrene according to the manufacturer's protocol;
polybrene is a transfection reagent with a positive charge, so it
can bind to the anion on the cell surface to facilitate and
significantly increase the transfection efficiency. After 72 h of
cell transfection, green fluorescence was observed under a
fluorescence microscope at ×200 magnification (Olympus IX71;
Olympus Corporation). Then, the cells were cultured in DMEM
supplemented with 10% FBS, 1% penicillin-streptomycin and 0.5 µg/ml
puromycin to obtain stable cell lines.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total cellular RNA was isolated from the
SHARPIN-shRNA lentivirus infected cells using TRIzol®
reagent (cat. no. 15596026; Invitrogen; Thermo Fisher Scientific,
Inc.). Then, 1 µg RNA was added with 4 µl 5X HiScript II qRT
SuperMix II (HiScript® II Reverse Transcriptase; cat.
no. R223; Vazyme Biotech Co., Ltd.) to form a 20 µl mixture, which
was placed in an ABI3130 automated sequencer (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The following RT conditions were
used: Reacted at 25°C for 10 min, 50°C for 30 min and 85°C for 5
min, for RT into cDNA using HiScript II Reverse Transcriptase (cat.
no. R223; Vazyme Biotech Co., Ltd.). qPCR was then performed using
an ABI 7500 RT PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.) with ChamQ SYBR qPCR Master mix (cat. no. Q331;
Vazyme Biotech Co., Ltd.). The qPCR conditions were as follows:
Initial denaturation at 95°C for 5 min; followed by 40 cycles of
95°C for 30, 72°C for 30 sec and 58°C for 30 sec; and a final
extension at 72°C for 10 min. The level of β-actin mRNA was used as
the endogenous control. Primer sequences used were as follows:
SHARPIN forward, 5′-TGTTCTCAGAGCTCGGTTT-3′ and reverse,
5′-AAGTTCCCCGTCCATCTT-3′; and β-actin forward,
5′-GGATGCAGAAGGAGATCACTG-3′ and reverse,
5′-CGATCCACACGGAGTACTTG-3′. All reactions were performed in
triplicate and repeated ≥1 time. The relative level of SHARPIN mRNA
was calculated using the 2−ΔΔCq method (44).
Cell counting Kit-8 (CCK-8) and
5-ethynyl-2′-deoxyuridine (EdU)-incorporation assays
A CCK-8 (cat. no. CK04; Dojindo Molecular
Technologies, Inc.) and EdU labeling kit, containing EdU labeling
media, Apollo reaction buffer, Apollo catalyst solution, Apollo
fluorescent dye solution and Apollo buffer additive (cat. no.
C10310-3; Guangzhou RiboBio Co., Ltd.), were used to investigate
cell viability and proliferation in SHARPIN knockdown BCC cells.
Stable TE354.T cells were seeded into 96-well plates at a density
of 5×103/well in 6-wells in each group for a final
volume of 100 µl and cultured for 3 days. At the end of 24, 48 and
72 h, 10 µl of the CCK-8 solution was added into each well and the
cells were incubated for 45 min in a humidified incubator with 5%
CO2 at 37°C, according to the manufacturer's
instructions. The optical absorbance rate of the cell solution was
measured using a Multimode Plate reader (PerkinElmer, Inc.) at 450
nm.
Stable sh-SHARPIN TE354.T cells and negative
controls were grown at a density of 8×103/well in
96-well plates for 48 h. Then, 50 µM EdU labeling media was added
into each well of the 96-well plates and incubated in a humidified
incubator with 5% CO2 at 37°C for 4 h. The cells were
fixed with 4% paraformaldehyde for 30 min at room temperature and
treated with 0.05% Triton X-100 solution for 10 min at room
temperature. Next, the cells were immunostained for 30 min in the
dark at room temperature with Apollo working solution containing 25
µl of Apollo reaction buffer, 5 µl of Apollo catalyst solution, 1.5
µl of Apollo fluorescent dye solution, 5 mg of Apollo buffer
additive and 469 µl of deionized water. Then, the cell nuclei were
stained with DAPI solution at room temperature in the dark for 15
min according to the manufacturer's instructions (cat. no. DA0001;
Beijing Leagene Biotechnology Co., Ltd.). The stained cells were
observed under a fluorescence microscope at ×200 magnification, and
five fields were randomly photographed for each group. The
EdU-positive cells and DAPI cells in each field were counted. The
proliferation rate was calculated using the following formula:
Proliferation rate=the green fluorescence-positive cells
(proliferating cells)/the blue fluorescence cells (total cells)
×100%.
Cell cycle analysis using flow
cytometric assays
TE354.T cells were plated into 6-well plates at a
density of 4×105 cells/well and cultured for 48 h. Then,
the cells were washed twice with ice-cold PBS, fixed with 75%
ethanol at −20°C overnight and stained with 400 µl propidium iodide
(cat. no. KGA512L; Nanjing KeyGen Biotech Co., Ltd.) at 4°C in the
dark for 30 min. Cell distribution was measured using a flow
cytometer (FACSCalibur; BD Biosciences; FlowJo, Version 10) at in
488 nm.
Annexin V assays
Apoptosis was detected using a Annexin
V-allophycocyanin (APC) kit (cat. no. 88-8007) and
7-aminoactinomycin D (7-ADD) solution (cat. no. 00-6993; both
eBioscience; Thermo Fisher Scientific, Inc.). An Annexin
V-APC/7-ADD apoptosis assay was used to assess tumor cell apoptosis
after sh-SHARPIN shRNA and sh-Ctrl transfection into BCC cells,
according to the manufacturer's instructions. TE354.T cells
(2×105) were plated into 6-well plates and cultured for
48 h. Cells were washed with PBS and then in 1X binding buffer
(cat. no. 00-0055; eBioscience; Thermo Fisher Scientific, Inc.),
and resuspended in 1X binding buffer at 3×105/ml. Then,
5 µl of fluorochrome-conjugated Annexin V was added to 100 µl of
the cell suspension and incubated for 10–15 min at the room
temperature. Cells were washed again in 1X binding buffer and
resuspended in 200 µl of 1X binding buffer. Then, 5 µl of 7-AAD
viability staining solution was added into the mixture and analyzed
within 4 h at 2–8°C in the dark. The cells were analyzed with a BD
FACSCalibur flow cytometer.
Transwell assay
Transwell units with 8-µm pore size membranes were
obtained from Falcon (cat. no. 353097; Corning Inc.). TE354.T cells
were harvested and suspended in serum-free DMEM medium and seeded
(3×104 cells) in Transwell upper chambers, which were
precoated with or without Matrigel (cat. no. 354234; Corning Inc.).
DMEM with 10% FBS was added into the lower chambers. The cells were
cultured for 16 h in a humidified incubator with 5% CO2
at 37°C, and the cells in the upper chamber were removed using a
cotton swab, while the cells that migrated or invaded into the
lower chambers were fixed with 4% paraformaldehyde for 30 min at
room temperature and stained with 0.5% crystal violet solution at
room temperature for 20 min. The number of migrated or invaded
cells were counted in ten randomly selected fields (BX51; Olympus
Corporation; magnification, ×200) and the rates were calculated by
comparing with the control cells.
Statistical analysis
The statistical analysis was performed using SPSS
13.0 software (SPSS, Inc.). SHARPIN expression levels in BCC
subtypes and normal skin were analyzed using Kruskal-Wallis tests
followed by Dunn's post hoc test. Age and sex data in BCC were
analyzed using Mann-Whitney U tests. The data on the cell
biological functions assays were analyzed using an independent
sample t-tests. P<0.05 was considered to indicate a
statistically significant differences.
Results
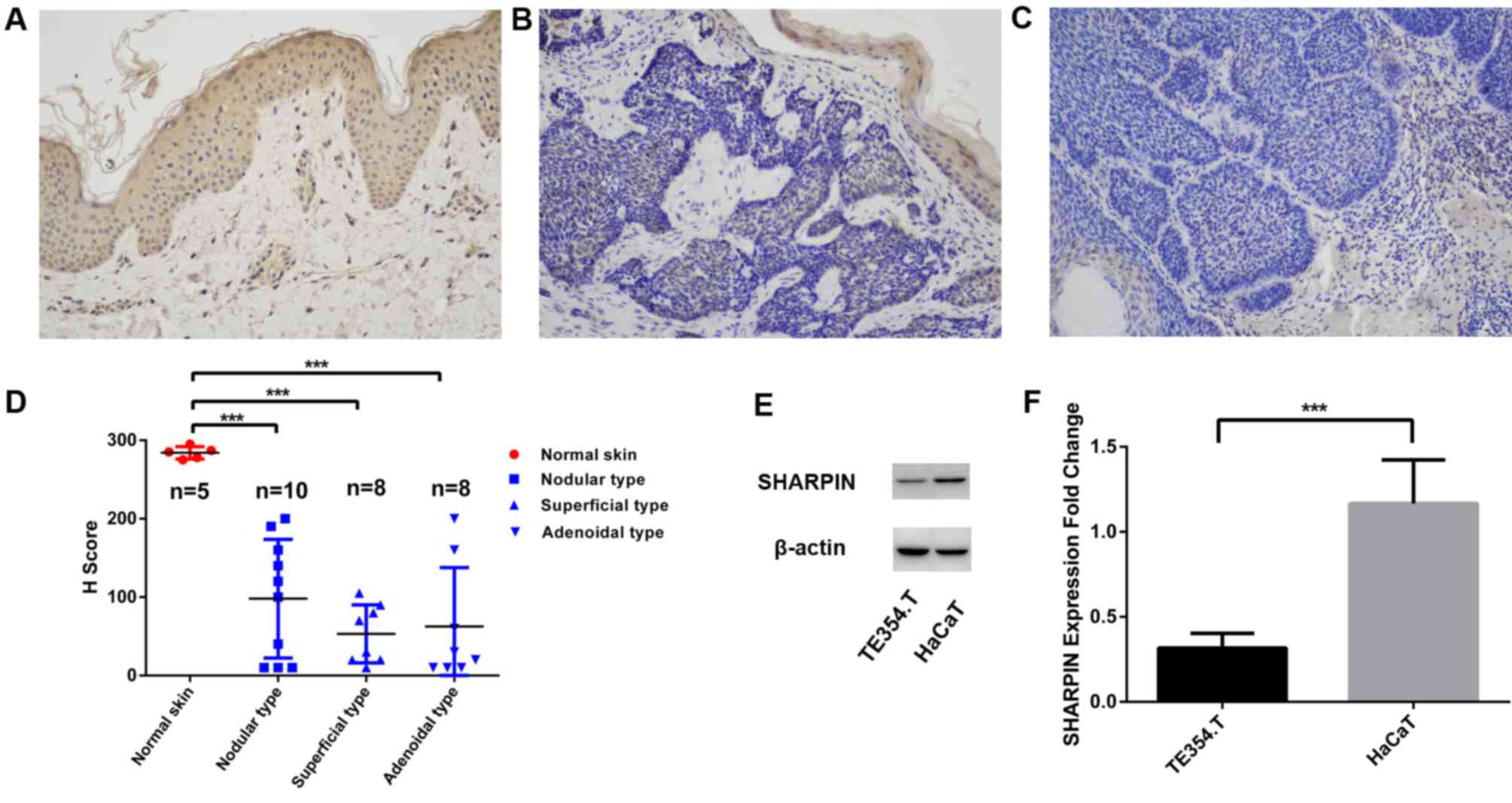
Reduced or absent expression of
SHARPIN in BCC tissues and cell lines
Subcategories of BCC tissues in the donor
demographic profile, risk factors, subtypes and tissue-biopsy
locations are summarized in Table
SI. The present study assessed the expression level of SHARPIN
in 26 BCC tissues compared with five healthy skin tissues, and also
in two cell lines. The present results suggested that SHARPIN
expression was not significantly associated with age, sex or
subtype in BCC tissue samples (Table
I). However, SHARPIN protein expression level was downregulated
or absent in precancerous lesions and cancer nests of BCCs compared
with the expression levels in healthy skin tissues (Fig. 1A-D). In addition, SHARPIN protein
expression level was significantly decreased in TE354.T cells
compared with HaCaT cells (Fig. 1E and
F). To assess these results, the SHARPIN expression level in
breast cancer was used as a control for the assay (Fig. S1). To investigate the expression
levels of cyclin D1, CDK4, c-JUN and GLI2 in BCC tissues, the HPA
website was searched and the following results were showed: i)
Cyclin D1 was low in three cases; ii) CDK4, six cases with one low,
one high, three medium and one unknown; iii) JUN, six cases with
one medium and five high; and iv) GLI2, five cases all high,
compared with healthy skin tissues. The present results suggested
that cyclin D1 was highly expressed in three BCC samples and
moderately expressed in two BCC samples compared with healthy skin.
CDK4 expression in BCC tissues (one low, three medium and one high)
exhibited 80% high expression compared with healthy skin tissues.
In addition, c-JUN expression was one high and four medium cases
compared with healthy skin tissues, and GLI2 expression in BCC
tissues was two high and three medium cases compared with healthy
skin tissues (Fig. S2).
 | Table I.Subcategory-analysis in BCC. |
Table I.
Subcategory-analysis in BCC.
| Characteristic | H Score | P-value |
|---|
| Sex |
| >0.05 |
|
Female | 92.69±68.27 |
|
|
Male | 53.85±61.45 |
|
| Age |
| >0.05 |
|
<60 | 55.00±63.17 |
|
|
≥60 | 85.36±71.32 |
|
| BCC subtype |
| >0.05 |
| Nodular
type | 98.00±75.69 |
|
|
Superficial type | 53.13±37.12 |
|
|
Adenoidal type | 62.50±75.17 |
|
Activation of tumor cell proliferation
after SHARPIN knockdown
SHARPIN expression was decreased in BCC tissue and
TE354.T cells compared with normal tissue and cells; thus, the
present study generated a stable SHARPIN knockdown cell subline and
performed cell viability and proliferation assays. The present
results suggested that the mRNA and protein expression levels of
SHARPIN in sh-SHARPIN group were significantly decreased compared
with the negative control (Fig. 2A and
B). The stable knockdown of SHARPIN significantly increased BCC
cell proliferation and viability (Fig.
2C-E), while the cell cycle distribution assay results
suggested that more sh-SHARPIN-infected cells were in the S phase
compared with negative control cells (Fig. 2F). However, there were no
significant differences in tumor cell apoptosis, migration or
invasion between the SHARPIN knockdown and negative control cells
(Fig. S3).
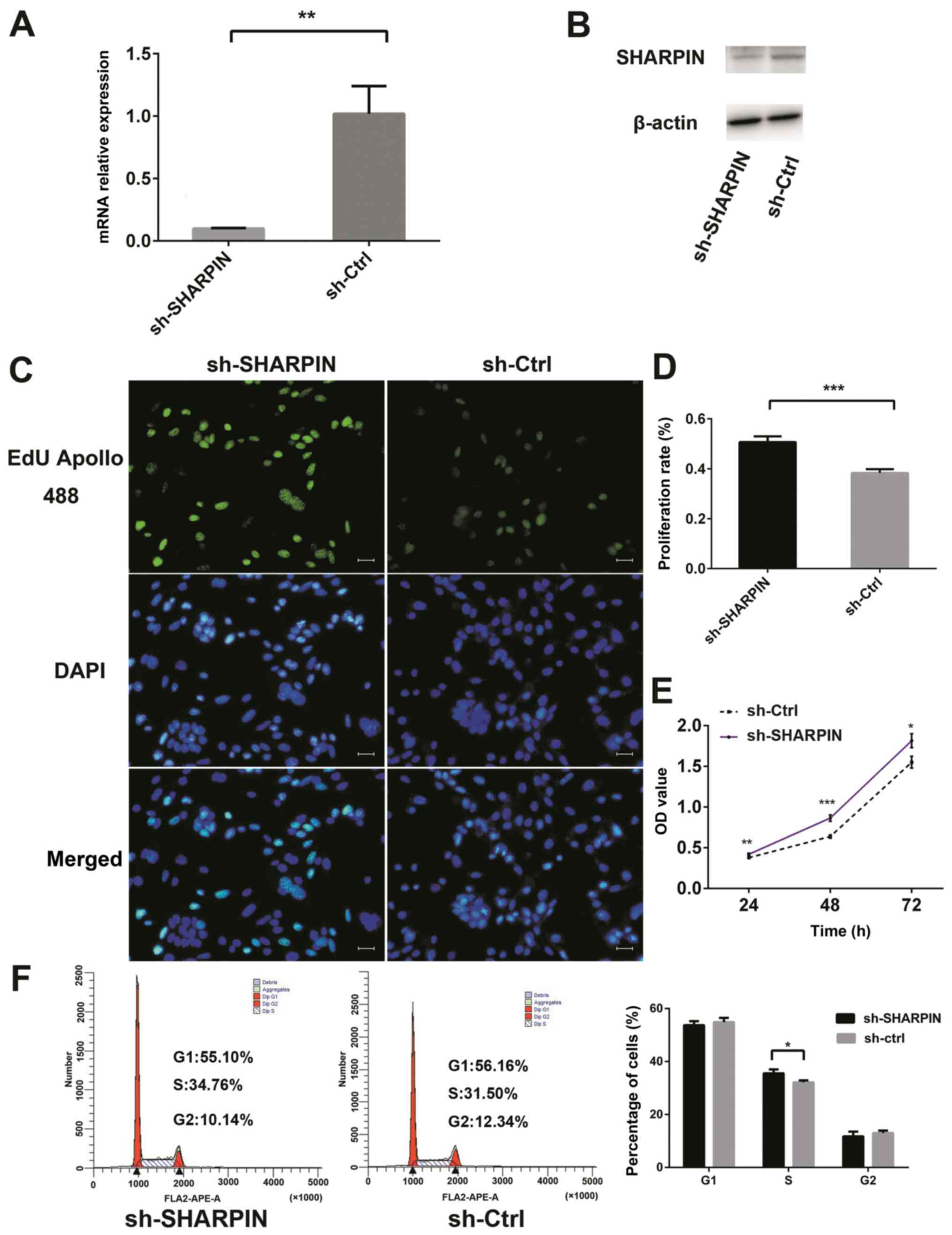 | Figure 2.Effect of SHARPIN on the regulation
of BCC cell proliferation. TE354.T cells were cultured, infected
with SHARPIN shRNA and subjected to various assays. Stable TE354.T
cells were grown, and the expression levels of SHARPIN were
examined via (A) reverse transcription-quantitative PCR and (B)
western blotting. (C) EdU incorporation assay in TE354.T-sh-SHARPIN
transfected cells and negative controls. Magnification, ×200. (D)
Quantified data from EdU assay. (E) Cell viability was assessed
using Cell Counting Kit-8 assay. (F) Flow cytometric cell cycle
assay. *P<0.05, **P<0.01, ***P<0.001. SHARPIN,
SHANK-associated RH domain-interacting protein; BCC, cutaneous
basal cell carcinoma; sh(RNA), short hairpin RNA; EdU,
5-ethynyl-2′-deoxyuridine; sh-Ctrl, shRNA control; OD, optical
density. |
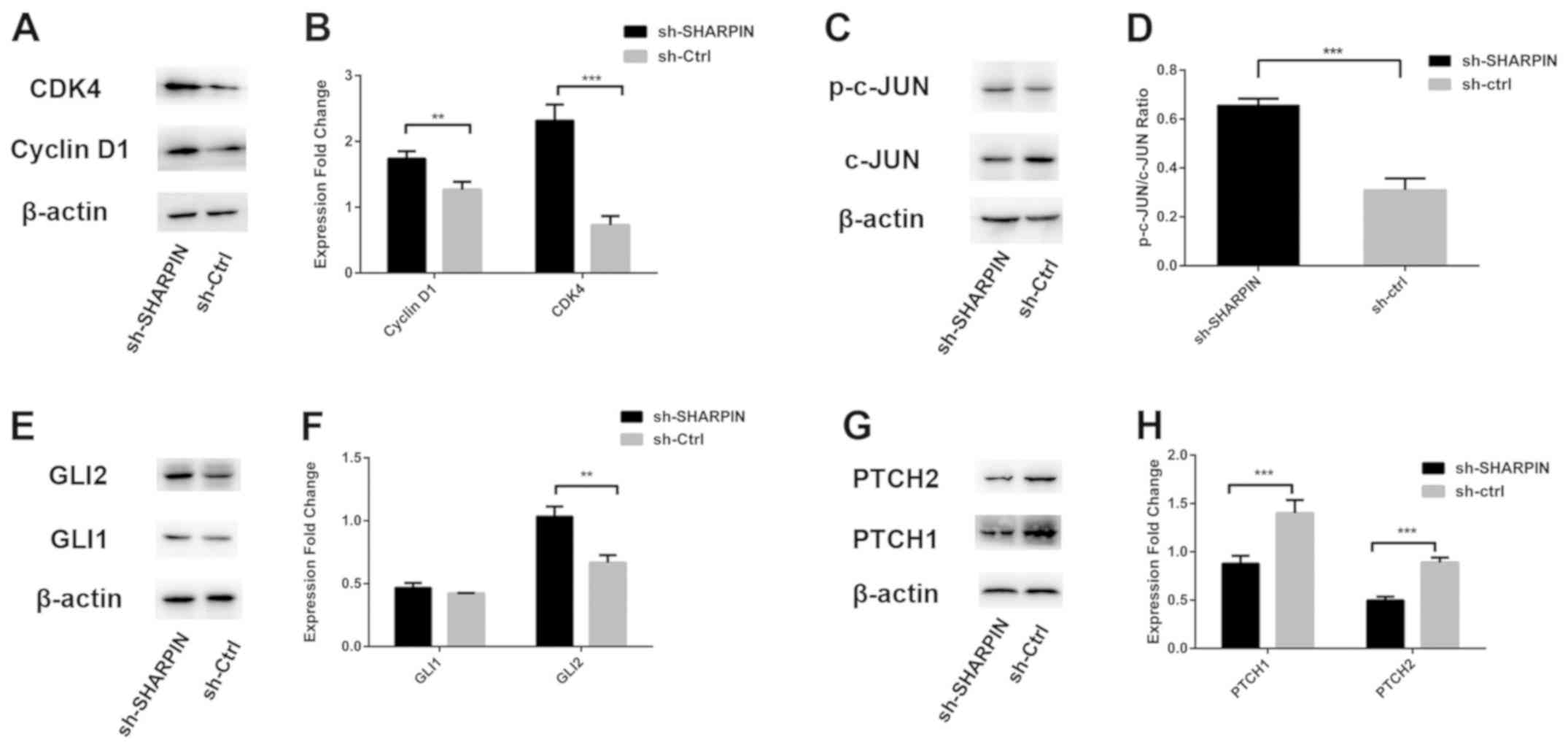
At the molecular level, the expression levels of
cyclin D1 and CDK4 in sh-SHARPIN group were increased compared with
the negative control group (Fig. 3A
and B).
JNK/GLI2 signaling pathway is
activated in SHARPIN-silenced TE354.T cells
The present study investigated the underlying
molecular mechanism of the effects of SHARPIN in BCC cells by
analyzing the protein expression levels of c-JUN, p-c-JUN, GLI1,
GLI2, PTCH1 and PTCH2. It was shown that, compared with the
negative control group, the protein expression levels of p-c-JUN
and GLI2 were significantly increased, while PTCH1 and PTCH2 were
decreased in SHARPIN knockdown cells. In addition, the protein
expression levels of c-JUN were decreased, while GLI1 expression
was not significantly different between the two groups (Fig. 3C-H).
Discussion
The present study identified that SHARPIN expression
level was decreased in BCC tissues and TE354.T cell lines. Using
the stable SHARPIN knockdown cell subline, it was identified that
SHARPIN shRNA enhanced TE354.T tumor cell proliferation and the
number of cells in the S phase of the cell cycle. At the molecular
level, SHARPIN knockdown promoted the protein expression levels of
cyclin D1, CDK4, p-c-JUN and GLI2. Collectively, the present
results suggested that SHARPIN may act as a tumor suppressor gene
during BCC tumorigenesis.
Previous studies have shown that SHARPIN is involved
in the development of various human cancer types. For example,
overexpression of SHARPIN contributed to prostate cancer
development by activating survivin and livin, which are downstream
targets of the NF-κB signaling pathway (45). Additionally, SHARPIN can promote
the activation of the NF-κB/ERK/Akt signaling pathway and induce
prostate cancer tumorigenesis by regulating the expression of the
transforming apoptosis-associated protein (28). A significant elevation of SHARPIN
mRNA expression has been reported in breast cancer (46). SHARPIN expression is associated
with poor prognosis in patients with breast cancer (19,27).
These studies reporting SHARPIN overexpression have provided
evidence of its function as an oncogene in the specific cancer
types described above. The present results suggested that SHARPIN
was downregulated in BCC tissues. BCC is different from other human
cancer types, as it proliferates slow, locally and rarely
metastasizes to other organs; thus, BCC has a more favorable
prognosis (1,47). However, further study is required
to investigate the tissue-specific role of SHARPIN in
tumorigenesis.
The hallmarks of cancer cells encompass six
biological capabilities, including limitless replicative potential,
evading apoptosis, self-sufficiency in growth signals,
insensitivity to antigrowth signals, sustained angiogenesis, and
tissue invasion and metastasis (48,49).
Upon stimulation by internal and external factors, normal cells
show a series of abnormal activities, such as genetic mutations,
altered cell cycle, unlimited cell proliferation and reduced cell
apoptosis; these cells eventually transform into tumor cells
(50). The present study generated
a stable SHARPIN-silencing TE354.T cell line and assessed tumor
cell proliferation, apoptosis, invasion and migration. The present
results suggested that the proliferative ability of
silenced-SHARPIN BCC cells was significantly increased compared
with corresponding control cells. Cell proliferation refers to the
ordered, tightly regulated process involving cell number increases,
nutrients accumulation and cell division (51). Purba et al (52) demonstrated that an increase in the
distribution of cells in the S phase of the cell cycle could be
sensitively and accurately assessed in tumor cells using BrdU or
EdU incorporation assays. The present study identified that
EdU-positive cells were more numerous in sh-SHARPIN-transfected BCC
cells compared with negative control transfected cells. Therefore,
the loss of function of SHARPIN could induce cell proliferation via
an increase in S phase cell cycle stage.
The cell cycle is a complicated dynamic process that
generates daughter cells via DNA replication and cell division
(53). The cell cycle can be
divided into the resting phase (G0), the beginning stage of DNA
synthesis (G1), DNA replication stage (S) and the late stage of DNA
synthesis (G2) (53). The
transition from the G1 phase to S phase is a critical step in
determining the rate of cell proliferation, and is modulated by the
phosphorylation of retinoblastoma (Rb) (54). CDKs are serine/threonine kinases
that are regulated by interactions with cyclins and CDK inhibitors,
and are the key regulators of the G1-to-S checkpoint (55). After CDK4 binds with its catalytic
partner cyclin D to form an active complex, Rb is consecutively
phosphorylated, thus relieving its inhibition on E2F to allow
target genes to promote the G1-to-S phase entry (56). The present results indicated that
the expression levels of cyclin D1 and CDK4 were increased in
SHARPIN knockdown BCC cells, which suggested that silencing SHARPIN
could promote cell proliferation via the overexpression of cyclin
D1 and CDK4.
The HH signaling pathway plays a critical role in
the development of BCC (57).
Decreased expression levels of PTCH1 and PTCH2, which are tumor
suppressor genes, can promote the development of BCC (58). Moreover, the activation of GLI has
been identified to be responsive to certain pathways, such as the
AKT/PI3K, RAS/RAF/mitogen-activated protein kinase (MAPK) kinase
(MEK)/ERK and epidermal growth factor (EGF) signaling pathways
(59–61). Schnidar et al (62) revealed that EGF receptor
(EGFR)/MEK/ERK signaling in combination with the GLI activator
results in GLI1- and GLI2-induced activation of JUN/activator
protein 1 (AP-1), which is essential for oncogenic transformation
in human keratinocytes cells. Moreover, the inhibition of EGFR and
HH/GLI was able to effectively slow the growth of a mouse BCC cell
line (62). Laner-Plamberger et
al (63) showed significantly
increased expression levels of c-JUN in human BCC tissue samples.
In addition, GLI1 and GLI2 can directly upregulate the expression
levels of AP-1 family members c-JUN and GLI2 more effectively than
the expression of GLI1 (63),
which is consistent with the present results.
The JUN family includes c-JUN, JUN B and JUN D as
downstream proteins of the JNK signaling pathway (64). A number of stimulators, such as
growth factors, cytokines, UV light and stress, can activate JNK
via the MAPK cascade to increase the transcription of AP-1 by
phosphorylating c-JUN at the serine 63 and serine 73 sites
(65). The JNK pathway regulates
cell proliferation, differentiation and apoptosis, as well as tumor
cell invasion and metastasis (66,67).
c-JUN is essential in embryonic development (68). Zenz et al (69) showed that cell proliferation was
reduced in JUN−/− keratinocytes, and that c-JUN-promoted
cell proliferation occurred via the activation of cyclin D1 and
other cell cycle-related proteins in keratinocytes. Moreover, the
regulation of cyclin D1 expression was shown to be caused by
binding to the cyclin D1 promoter (64,70).
In addition, GLI2 has been confirmed to accelerate the
transformation of the G1 phase of the cell cycle to the S phase via
the activation of E2F1, cyclin D1, CDC45L and other cell
cycle-related proteins (71). In
the present study, the knockdown of SHARPIN induced cyclin D1 and
c-JUN activation, which is inconsistent with the previous results
showing that SHARPIN induced NF-κB and thus promoted AP-1 activity
in various cells (72). LUBAC can
activate the MAPK cascade signaling pathway, which involves JNK,
p38 and ERK1/2, to increase the expression levels of AP-1 and c-JUN
(73). TNF-α-induced JNK/ERK
activation is enhanced in chronic proliferative dermatitis mice
(cpdm) embryonic fibroblasts (MEFs) compared with wild-type MEFs
(73). Knockdown of cpdm partially
results in an increase in JUN phosphorylation to cause signal
dependence or determine the specificity of the cell type (21,73).
However, the expression levels of cyclin D1, CDK4,
c-JUN and GLI2 in present study were not consistent with previous
studies or those from The Human Protein Atlas. Sivrikoz and
Kandiloğlu (74), Liang et
al (75) and Huang et
al (76) showed that the
expression level of cyclin D1 was higher in BCC tissues compared
with healthy skin. Laner-Plamberger et al (63) reported a higher expression level of
JUN in BCC tissues. Data on the expression of GLI2 in a previous
study (77) and the analysis in
present study were consistent. It was demonstrated that cyclin D1,
c-JUN and GLI2 were highly expressed in BCC samples compared with
healthy skin, which was mostly consistent with previous studies
(74–77). The expression of CDK4 in present
study was similar to The Human Protein Atlas.
In conclusion, SHARPIN expression was reduced in BCC
tissues. SHARPIN knockdown increased the expression levels of
p-c-JUN and GLI2 in BCC cells. The binding of p-c-JUN and GLI2 may
have induced the expression of cyclin D1 and CDK4, promoting cell
proliferation and BCC development. However, the present study did
not investigate the expression of molecules in the Sonic HH signal
pathway, which is as a limitation and requires further
research.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Jiaman Wang from
the Department of Dermatology, Cosmetology and Venereology,
Shenzhen Hospital, Southern Medical University for help in
reviewing the BCC pathology and immunohistochemical data. The
authors would also like to thank Professor Fu Xiong from the
Department of Medical Genetics, Southern Medical University, for
providing the research facilities.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81371724) and
Shenzhen Science and Technology Innovation Commission (grant no.
JCYJ20160429161334124).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YHL designed the experiments, guided the study and
revised the draft. YY performed the experiments, analyzed the data
and prepared the draft. YZ analyzed the data and revised the
article. LJT and STZ performed parts of experiments. JNZ collected
the tissue samples and performed part of the immunohistochemistry.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by The Ethics Committee of
Shenzhen Hospital, Southern Medical University. Written informed
consent was obtained from patients and individuals involved in the
present study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Roewert-Huber J, Lange-Asschenfeldt B,
Stockfleth E and Kerl H: Epidemiology and aetiology of basal cell
carcinoma. Br J Dermatol. 157 (Suppl 2):S47–S51. 2007. View Article : Google Scholar
|
|
2
|
Correia de Sá TR, Silva R and Lopes JM:
Basal cell carcinoma of the skin (part 1): Epidemiology, pathology
and genetic syndromes. Future Oncol. 11:3011–3021. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wong CS, Strange RC and Lear JT: Basal
cell carcinoma. BMJ. 327:794–798. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lomas A, Leonardi-Bee J and Bath-Hextall
F: A systematic review of worldwide incidence of nonmelanoma skin
cancer. Br J Dermatol. 166:1069–1080. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rubin AI, Chen EH and Ratner D: Basal-cell
carcinoma. N Engl J Med. 353:2262–2269. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Martincorena I, Fowler JC, Wabik A, Lawson
ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton
MR, et al: Somatic mutant clones colonize the human esophagus with
age. Science. 362:911–917. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Martincorena I, Roshan A, Gerstung M,
Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB,
Tubio JM, et al: Tumor evolution. High burden and pervasive
positive selection of somatic mutations in normal human skin.
Science. 348:880–886. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xie J, Aszterbaum M, Zhang X, Bonifas JM,
Zachary C, Epstein E and McCormick F: A role of PDGFRalpha in basal
cell carcinoma proliferation. Proc Natl Acad Sci USA. 98:9255–9259.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Svärd J, Heby-Henricson K, Persson-Lek M,
Rozell B, Lauth M, Bergström A, Ericson J, Toftgård R and Teglund
S: Genetic elimination of Suppressor of fused reveals an essential
repressor function in the mammalian hedgehog signaling pathway. Dev
Cell. 10:187–197. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vogt A, Chuang PT, Hebert J, Hwang J, Lu
Y, Kopelovich L, Athar M, Bickers DR and Epstein EJ Jr:
Immunoprevention of basal cell carcinomas with recombinant
hedgehog-interacting protein. J Exp Med. 199:753–761. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Athar M, Tang X, Lee JL, Kopelovich L and
Kim AL: Hedgehog signalling in skin development and cancer. Exp
Dermatol. 15:667–677. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Epstein EH: Basal cell carcinomas: Attack
of the hedgehog. Nat Rev Cancer. 8:743–754. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Athar M, Li C, Tang X, Chi S, Zhang X, Kim
AL, Tyring SK, Kopelovich L, Hebert J, Epstein EJ Jr, et al:
Inhibition of smoothened signaling prevents ultraviolet B-induced
basal cell carcinomas through regulation of Fas expression and
apoptosis. Cancer Res. 64:7545–7552. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xie J, Murone M, Luoh SM, Ryan A, Gu Q,
Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, et al: Activating
smoothened mutations in sporadic basal-cell carcinoma. Nature.
391:90–92. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lum L and Beachy PA: The hedgehog response
network: Sensors, switches, and routers. Science. 304:1755–1759.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim MY, Park HJ, Baek SC, Byun DG and Houh
D: Mutations of the p53 and PTCH gene in basal cell carcinomas: UV
mutation signature and strand bias. J Dermatol Sci. 29:1–9. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gailani MR, Ståhle-Bäckdahl M, Leffell DJ,
Glynn M, Zaphiropoulos PG, Pressman C, Undén AB, Dean M, Brash DE,
Bale AE and Toftgård R: The role of the human homologue of
Drosophila patched in sporadic basal cell carcinomas. Nat Genet.
14:78–81. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim J, Aftab BT, Tang JY, Kim D, Lee AH,
Rezaee M, Kim J, Chen B, King EM, Borodovsky A, et al: Itraconazole
and arsenic trioxide inhibit Hedgehog pathway activation and tumor
growth associated with acquired resistance to smoothened
antagonists. Cancer Cell. 23:23–34. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bii VM, Rae DT and Trobridge GD: A novel
gammaretroviral shuttle vector insertional mutagenesis screen
identifies SHARPIN as a breast cancer metastasis gene and
prognostic biomarker. Oncotarget. 6:39507–39520. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stieglitz B, Haire LF, Dikic I and
Rittinger K: Structural analysis of SHARPIN, a subunit of a large
multi-protein E3 ubiquitin ligase, reveals a novel dimerization
function for the pleckstrin homology superfold. J Biol Chem.
287:20823–20829. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ikeda F, Deribe YL, Skånland SS, Stieglitz
B, Grabbe C, Franz-Wachtel M, van Wijk SJ, Goswami P, Nagy V,
Terzic J, et al: SHARPIN forms a linear ubiquitin ligase complex
regulating NF-κB activity and apoptosis. Nature. 471:637–641. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu J, Wang Y, Gong Y, Fu T, Hu S, Zhou Z
and Pan L: Structural insights into SHARPIN-mediated Activation of
HOIP for the linear ubiquitin Chain assembly. Cell Rep. 21:27–36.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fujita H, Tokunaga A, Shimizu S, Whiting
AL, Aguilar-Alonso F, Takagi K, Walinda E, Sasaki Y, Shimokawa T,
Mizushima T, et al: Cooperative domain formation by homologous
motifs in HOIL-1L and SHARPIN plays a crucial role in LUBAC
stabilization. Cell Rep. 23:1192–1204. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shimizu S, Fujita H, Sasaki Y, Tsuruyama
T, Fukuda K and Iwai K: Differential involvement of the Npl4 zinc
finger domains of SHARPIN and HOIL-1L in linear ubiquitin Chain
assembly complex-mediated cell death protection. Mol Cell Biol.
36:1569–1583. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liang Y, Seymour RE and Sundberg JP:
Inhibition of NF-κB signaling retards eosinophilic dermatitis in
SHARPIN-deficient mice. J Invest Dermatol. 131:141–149. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jung J, Kim JM, Park B, Cheon Y, Lee B,
Choo SH, Koh SS and Lee S: Newly identified tumor-associated role
of human Sharpin. Mol Cell Biochem. 340:161–167. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang H, Yu S, Wang W, Li X, Hou Y, Liu Z,
Shi Y, Mu K, Niu G, Xu J, et al: SHARPIN facilitates p53
degradation in breast cancer cells. Neoplasia. 19:84–92. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li J, Lai Y, Cao Y, Du T, Zeng L, Wang G,
Chen X, Chen J, Yu Y, Zhang S, et al: SHARPIN overexpression
induces tumorigenesis in human prostate cancer LNCaP, DU145 and
PC-3 cells via NF-κB/ERK/Akt signaling pathway. Med Oncol.
32:4442015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Queisser MA, Dada LA, Deiss-Yehiely N,
Angulo M, Zhou G, Kouri FM, Knab LM, Liu J, Stegh AH, DeCamp MM, et
al: HOIL-1L functions as the PKCζ ubiquitin ligase to promote lung
tumor growth. Am J Respir Crit Care Med. 190:688–698. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen B, Zheng Y, Zhu J and Liang Y:
SHARPIN overexpression promotes TAK1 expression and activates JNKs
and NF-κB pathway in Mycosis Fungoides. Exp Dermatol. 28:1279–1288.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou S, Liang Y, Zhang X, Liao L, Yang Y,
Ouyang W and Xu H: SHARPIN promotes melanoma progression via Rap1
signaling pathway. J Invest Dermatol. Aug 8–2019.(Epub ahead of
print).
|
|
32
|
Kasperczyk H, Baumann B, Debatin KM and
Fulda S: Characterization of sonic hedgehog as a novel NF-kappaB
target gene that promotes NF-kappaB-mediated apoptosis resistance
and tumor growth in vivo. Faseb J. 23:21–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schumacher MA, Feng R, Aihara E, Engevik
AC, Montrose MH, Ottemann KM and Zavros Y: Helicobacter
pylori-induced Sonic Hedgehog expression is regulated by NFκB
pathway activation: The use of a novel in vitro model to study
epithelial response to infection. Helicobacter. 20:19–28. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yamasaki A, Kameda C, Xu R, Tanaka H,
Tasaka T, Chikazawa N, Suzuki H, Morisaki T, Kubo M, Onishi H, et
al: Nuclear factor kappaB-activated monocytes contribute to
pancreatic cancer progression through the production of Shh. Cancer
Immunol Immunother. 59:675–686. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vile GF, Tanew-Ilitschew A and Tyrrell RM:
Activation of NF-kappa B in human skin fibroblasts by the oxidative
stress generated by UVA radiation. Photochem Photobiol. 62:463–468.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tang SC, Liao PY, Hung SJ, Ge JS, Chen SM,
Lai JC, Hsiao YP and Yang JH: Topical application of glycolic acid
suppresses the UVB induced IL-6, IL-8, MCP-1 and COX-2 inflammation
by modulating NF-κB signaling pathway in keratinocytes and mice
skin. J Dermatol Sci. 86:238–248. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Simon MM, Aragane Y, Schwarz A, Luger TA
and Schwarz T: UVB light induces nuclear factor kappa B (NF kappa
B) activity independently from chromosomal DNA damage in cell-free
cytosolic extracts. J Invest Dermatol. 102:422–427. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Budwit-Novotny DA, McCarty KS, Cox EB,
Soper JT, Mutch DG, Creasman WT, Flowers JL and McCarty KJ:
Immunohistochemical analyses of estrogen receptor in endometrial
adenocarcinoma using a monoclonal antibody. Cancer Res.
46:5419–5425. 1986.PubMed/NCBI
|
|
39
|
Bollag G, Hirth P, Tsai J, Zhang J,
Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, et al:
Clinical efficacy of a RAF inhibitor needs broad target blockade in
BRAF-mutant melanoma. Nature. 467:596–599. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bai J, Kito Y, Okubo H, Nagayama T and
Takeuchi T: Expression of ZNF396 in basal cell carcinoma. Arch
Dermatol Res. 306:399–404. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Di Gennaro P, Sestini R, Bacci S, Pacini
A, Pinzani P, Domenici L, Toscano A, Massi D, Carli P, Genuardi M
and Romagnoli P: Tacrolimus causes reduced GLI1 expression and
phenotypic changes in the TE 354.T basal cell carcinoma cell line.
J Dermatol Sci. 54:52–54. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Colombo I, Sangiovanni E, Maggio R,
Mattozzi C, Zava S, Corbett Y, Fumagalli M, Carlino C, Corsetto PA,
Scaccabarozzi D, et al: HaCaT cells as a reliable in vitro
differentiation model to dissect the inflammatory/repair response
of human keratinocytes. Mediators Inflamm. 2017:74356212017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang XY, Wu B, Ma S, Yin L, Wu MC and Li
AJ: Decreased expression of ZWINT is associated with poor prognosis
in patients with HCC after surgery. Technol Cancer Res Treat.
17:15330338187941902018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang Y, Huang H, Zhou H, Du T, Zeng L,
Cao Y, Chen J, Lai Y, Li J, Wang G and Guo Z: Activation of nuclear
factor κB pathway and downstream targets survivin and livin by
SHARPIN contributes to the progression and metastasis of prostate
cancer. Cancer. 120:3208–3218. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
De Melo J and Tang D: Elevation of SIPL1
(SHARPIN) increases breast cancer risk. PLoS One. 10:e1275462015.
View Article : Google Scholar
|
|
47
|
Miller SJ: Biology of basal cell carcinoma
(Part I). J Am Acad Dermatol. 24:1–13. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Susnow N, Zeng L and Margineantu D: Bcl-2
family proteins as regulators of oxidative stress. Semin Cancer
Biol. 19:42–49. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ho A and Dowdy SF: Regulation of G(1)
cell-cycle progression by oncogenes and tumor suppressor genes.
Curr Opin Genet Dev. 12:47–52. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Purba TS, Brunken L, Hawkshaw NJ, Peake M,
Hardman J and Paus R: A primer for studying cell cycle dynamics of
the human hair follicle. Exp Dermatol. 25:663–668. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Dante RA, Larkins BA and Sabelli PA: Cell
cycle control and seed development. Front Plant Sci. 5:4932014.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Fernández-Hernández R, Rafel M, Fusté NP,
Aguayo RS, Casanova JM, Egea J, Ferrezuelo F and Gari E: Cyclin D1
localizes in the cytoplasm of keratinocytes during skin
differentiation and regulates cell-matrix adhesion. Cell Cycle.
12:2510–2517. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xu W and McArthur G: Cell cycle regulation
and melanoma. Curr Oncol Rep. 18:342016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Duronio RJ and Xiong Y: Signaling pathways
that control cell proliferation. Cold Spring Harb Perspect Biol.
5:a0089042013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Otsuka A, Levesque MP, Dummer R and
Kabashima K: Hedgehog signaling in basal cell carcinoma. J Dermatol
Sci. 78:95–100. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Daya-Grosjean L and Couvé-Privat S: Sonic
hedgehog signaling in basal cell carcinomas. Cancer Lett.
225:181–192. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mimeault M, Johansson SL, Vankatraman G,
Moore E, Henichart JP, Depreux P, Lin MF and Batra SK: Combined
targeting of epidermal growth factor receptor and hedgehog
signaling by gefitinib and cyclopamine cooperatively improves the
cytotoxic effects of docetaxel on metastatic prostate cancer cells.
Mol Cancer Ther. 6:967–978. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Neill GW, Harrison WJ, Ikram MS, Williams
TDL, Bianchi LS, Nadendla SK, Green JL, Ghali L, Frischauf AM,
O'Toole EA, et al: GLI1 repression of ERK activity correlates with
colony formation and impaired migration in human epidermal
keratinocytes. Carcinogenesis. 29:738–746. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Riobó NA, Lu K, Ai X, Haines GM and
Emerson CP Jr: Phosphoinositide 3-kinase and Akt are essential for
sonic hedgehog signaling. Proc Natl Acad Sci USA. 103:4505–4510.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Schnidar H, Eberl M, Klingler S,
Mangelberger D, Kasper M, Hauser-Kronberger C, Regl G, Kroismayr R,
Moriggl R, Sibilia M and Aberger F: Epidermal growth factor
receptor signaling synergizes with Hedgehog/GLI in oncogenic
transformation via activation of the MEK/ERK/JUN pathway. Cancer
Res. 69:1284–1292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Laner-Plamberger S, Kaser A, Paulischta M,
Hauser- Kronberger C, Eichberger T and Frischauf AM: Cooperation
between GLI and JUN enhances transcription of JUN and selected GLI
target genes. Oncogene. 28:1639–1651. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Albanese C, Johnson J, Watanabe G, Eklund
N, Vu D, Arnold A and Pestell RG: Transforming p21ras mutants and
c-Ets-2 activate the cyclin D1 promoter through distinguishable
regions. J Biol Chem. 270:23589–23597. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Johnson GL and Nakamura K: The c-jun
kinase/stress-activated pathway: Regulation, function and role in
human disease. Biochim Biophys Acta. 1773:1341–1348. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Morrison DK: MAP kinase pathways. Cold
Spring Harb Perspect Biol. 4:2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhao HF, Wang J, Jiang HR, Chen ZP and To
SS: PI3K p110β isoform synergizes with JNK in the regulation of
glioblastoma cell proliferation and migration through Akt and FAK
inhibition. J Exp Clin Cancer Res. 35:782016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Johnson RS, Van Lingen B, Papaioannou VE
and Spiegelman BM: A null mutation at the c-jun locus causes
embryonic lethality and retarded cell growth in culture. Genes Dev.
7:1309–1317. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zenz R, Scheuch H, Martin P, Frank C,
Eferl R, Kenner L, Sibilia M and Wagner EF: c-Jun regulates eyelid
closure and skin tumor development through EGFR signaling. Dev
Cell. 4:879–889. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zenz R and Wagner EF: Jun signalling in
the epidermis: From developmental defects to psoriasis and skin
tumors. Int J Biochem Cell Biol. 38:1043–1049. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Regl G, Kasper M, Schnidar H, Eichberger
T, Neill GW, Ikram MS, Quinn AG, Philpott MP, Frischauf AM and
Aberger F: The zinc-finger transcription factor GLI2 antagonizes
contact inhibition and differentiation of human epidermal cells.
Oncogene. 23:1263–1274. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Gerlach B, Cordier SM, Schmukle AC,
Emmerich CH, Rieser E, Haas TL, Webb AI, Rickard JA, Anderton H,
Wong WW, et al: Linear ubiquitination prevents inflammation and
regulates immune signalling. Nature. 471:591–596. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Tokunaga F, Nakagawa T, Nakahara M, Saeki
Y, Taniguchi M, Sakata S, Tanaka K, Nakano H and Iwai K: SHARPIN is
a component of the NF-κB-activating linear ubiquitin chain assembly
complex. Nature. 471:633–636. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Sivrikoz NO and Kandiloğlu G: The effects
of cyclin D1 and Bcl-2 expression on aggressive behavior in basal
cell and basosquamous carcinoma. Iran J Pathol. 10:185–191.
2015.PubMed/NCBI
|
|
75
|
Liang SB, Furihata M, Takeuchi T, Iwata J,
Chen BK, Sonobe H and Ohtsuki Y: Overexpression of cyclin D1 in
nonmelanocytic skin cancer. Virchows Arch. 436:370–376. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Huang K, Huang C, Shan K, Chen J and Li H:
Significance of PC cell-derived growth factor and cyclin D1
expression in cutaneous squamous cell carcinoma. Clin Exp Dermatol.
37:411–417. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Bladen JC, Moosajee M, Tracey-White D,
Beaconsfield M, O Toole EA and Philpott MP: Analysis of hedgehog
signaling in periocular sebaceous carcinoma. Graefes Arch Clin Exp
Ophthalmol. 256:853–860. 2018. View Article : Google Scholar : PubMed/NCBI
|