Introduction
Sepsis, which has a worldwide mortality rate of 10%,
is induced by infection and leads to life-threatening organ
dysfunction (1). Clinical and
basic research have shown that the cardiovascular system is
affected during sepsis (2), but
the exact mechanism is not fully understood. Dexmedetomidine (Dex),
which has been recommended for patients with sepsis since 1999, has
been proven to be a selective α2-adrenergic receptor (α2-AR)
(3,4). Clinical studies have revealed that
Dex inhibits the activity of sympathetic nerves via analgesic,
sedative and antisialogogue effects (5,6),
while it also exerts protective effects on other organs, such as
the kidney, brain and heart (7).
Inflammation is the primary characteristic of sepsis; the
expression of inflammatory factors, such as tumor necrosis factor α
(TNF-α), interleukin 1 (IL-1), IL-6 and methyl-accepting chemotaxis
protein-1, are significantly increased during sepsis, and Dex is
able to decrease these expression levels (8).
Ferroptosis was first reported by Dolma et al
(9) in cancer cells and was shown
to be different from the known cell death pathways, apoptosis,
pyroptosis and necroptosis. Ferroptosis has since been identified
to be involved in various pathological processes, including
neurotoxicity, acute kidney failure, liver injury and heart disease
(10), as well as myocardial
ischemia reperfusion injury (11,12).
Furthermore, the development of sepsis has been proposed to involve
ferroptosis (13). However, the
role of ferroptosis in septic heart injury remains unknown. The
mechanism of ferroptosis mainly involves increases in lipid
peroxidation and further release of lipid reactive oxygen species
(ROS) (14). It has also been
reported that ferroptosis occurs when the activity of glutathione
peroxidase 4 (GPX4) or glutathione (GSH) decreases (13). In addition, iron chelation has also
been shown to inhibit ferroptosis, thus indicating that ferroptosis
is closely associated with ROS and iron (15,16).
Furthermore, other factors, such as voltage-dependent anion channel
2, heat shock protein β-1, nuclear factor E2-related factor 2
(Nrf2), NADPH oxidase, P53 and heme oxygenase-1 (HO-1), also
participate in ferroptosis (17,18).
The HO system, which includes HO-1 and HO-2, acts as
a defense system against various stimuli, such as oxidants and
hypoxia (19). Moreover, HO-1
degrades heme into carbon monoxide, biliverdin and ferrous iron,
and confers cardioprotection via antiapoptotic, antioxidant and
other effects. HO is one of the intracellular sources of iron
(20), and its overexpression and
activation have been shown to accelerate ferroptotic cell death
(21). Furthermore, HO-1
participates in ferroptosis via its association with iron and its
antioxidant effects (22), but the
exact mechanism remains unknown. As previously reported, Dex
reduces H2O2-induced oxidative stress in
neonatal rat cardiomyocytes by decreasing ROS and GSH (23). According to a previous study
(22), it is hypothesized that
HO-1-mediated regulation of ferroptosis may play a role during
sepsis and that Dex confers cardioprotective effects by influencing
this regulation.
Materials and methods
Cecal ligation and puncture (CLP)
operation
A total of 32 male C57BL/6 mice (25 g, 8 weeks old)
were obtained from the Guangdong Medical Lab Animal Center and
housed in the Laboratory Animal Service Center (Jinan University,
Guangdong, China). Mice received standard care under a 12-h
dark/light cycle (23°C with an atmosphere of 60%) and were given
free access to food and water, in accordance with the Animal Care
guidelines of the Jinan University. The study was approved by the
Institutional Ethics Committee of The Medical Committee of Shenzhen
People's Hospital (approval ID: LL-KY-2019604). Sepsis was induced
by CLP, as previously described (24). Briefly, mice were anesthetized with
isoflurane (RWD Life Science) inhalation at the concentration of
2.5% for anesthetic induction and then at 1% for anesthetic
maintenance until the end of the CLP. During the experiment, the
body temperature was kept at 36–38°C with a heating pad.
Anesthetized mice were subjected to midline laparotomy. The cecum
was carefully separated to avoid blood vessels damage and the cecum
was identified and punctured twice with a 22-gauge needle. Then,
the abdominal cavity was closed with two epithelium layers,
followed by a normal saline injection subcutaneously for
resuscitation before mice were returned to the cage. The duration
of the whole experiment was ~25 h and the CLP model was finished
within 15 min. The cecum was exposed immediately following
laparotomy, which was conducted using a 2-cm lower midline
incision, followed by the use of a 22-gauge needle to impale the
cecum. Before the cecum was returned to the abdominal cavity,
pieces of feces were extruded. The abdomen was closed with 4-0 silk
sutures in two layers. The health and behavior of the animal was
monitored at 1 h intervals over 12 h. During the experiment, the
survival rate (rate of successful CLP model establishment) was 60%
in the CLP group, 86% in CLP + Dex group and 70% in CLP + Dex +
yohimbine hydrochloride (YOH) group. It was found that the primary
reason for mortality was the individual variation in the reaction
to CLP-induced sepsis. To ensure a minimum of six live animals per
group for the experimental period, ten, seven and nine mice were
initially used in the CLP, CLP + Dex and CLP + Dex + YOH groups,
respectively. At the end of the experiments, an overdose of
phenobarbital sodium was used for euthanasia and then blood (500
ml) and heart samples were collected.
Animal experimental protocol
The mice were randomly divided into 4 groups (n=6
per group): Control (Ctrl), CLP, CLP + Dex and CLP + Dex + YOH
groups. Dex was administered 15 min before inducing sepsis at a
dose 50 µg/kg and 1 mg/kg YOH was given 30 min before the
administration of Dex, according to our preliminary experiments and
previous reports (25–27). Survival was 90% in the Dex
administration group. A total of 500 ml of blood was collected to
separate serum and the hearts were harvested after 24 h of sepsis.
Dex was obtained from Jiangsu Hengrui Medicine Co., Ltd., and YOH
from MedChem Express.
Determination of troponin-I (TN-I),
IL-6, methyl-accepting chemotaxis protein 1 (MCP-1), malonaldehyde
(MDA), superoxide dismutase (28),
8-hydroxy-2′-deoxyguanosine (8-OHDG) and GSH expression
After 24 h of CLP, blood samples were collected and
then centrifuged (5,000 g, 10 min, 4°C) to separate the serum for
the detection of TN-I (cat. no. JL31923), MDA (cat. no. JL13329),
SOD (cat. no. JL12237), 8-OHDG (cat. no. JL12294), GSH (cat. no.
JL20360), IL-6 (cat. no. JL20268) and MCP-1 (cat. no. JL20304).
These ELISA kits were obtained from Shanghai Jianglai Biological
Technology Co., Ltd., and used following the manufacturer's
instructions.
Iron concentration detection
To detect iron concentration in the serum during
sepsis, an iron assay kit (cat. no. ab83366; Abcam) was used
according to the manufacturer's protocol.
Hematoxylin and eosin (H&E)
staining
Briefly, the heart was fixed with 4%
paraformaldehyde overnight at room temperature, embedded in
paraffin and sliced into 4 µm-thick slices. The tissues were
stained with hematoxylin for 5 min and eosin for 1 min at room
temperature using the H&E Staining kit (cat. no. C0105;
Beyotime Institute of Biotechnology). The slide was observed under
a fluorescence microscope (DMi8 DFC7000 T; Leica Microsystems,
Inc.).
Western blot analysis
Frozen ventricular tissue samples were homogenized
in RIPA buffer (Cell Signaling Technology, Inc.; cat. no. 9806) and
centrifuged at 4°C, 13,200 × g for 30 min. The supernatant was
collected for total protein analysis and the protein concentration
was determined using a Bradford protein assay. Equal amounts of
protein (30 µg) from mouse heart homogenate were resolved by
7.5–3.5% SDS-PAGE and subsequently transferred to PVDF membranes.
The membranes were then blocked in TBS with 0.1% Tween-20 (TBST;
cat. no. P9416; Sigma-Aldrich; Merck KGaA) containing 5% (w/v)
non-fat milk for 1 h at room temperature, after which the membranes
were incubated with primary antibodies overnight at 4°C. Primary
antibodies against Bax (cat. no. 2772; Cell Signaling Technology,
Inc.), Bcl-2 (cat. no. 210774; Merck KGaA), HO-1 (cat. no. ab13248;
Abcam), GPX4 (cat. no. PAS79321; Thermo Fisher Scientific, Inc.),
gasdermin D (GSDMD; cat. no. ab219800; Abcam), nitric oxide
synthase (iNOS; cat. no. ab3523; Abcam), cleaved caspase 3 (cat.
no. 836; Cell Signaling Technology, Inc.), P53 (cat. no. NB200-103;
Novus Biologicals; Bio-Techne Ltd.), transferrin receptor (TFR;
cat. no. MCA155R; Bio-Rad Laboratories, Inc.) ferritin (cat. no.
ab75973; Abcam), Caspase 9 (cat. no. ab184786; Abcam), Nrf2 (cat.
no. ab31163; Abcam) and GAPDH (cat. no. 5174; Cell Signaling
Technology, Inc.) were used at the dilution 1:1,000. After washing
with TBST three times for 10 min per wash, the membrane strips were
incubated at room temperature with a 1:10,000 dilution of an
anti-rabbit IgG (cat. no. 7074S; Cell Signaling Technology, Inc.)
or anti-mouse IgG (cat. no. 7076S; Cell Signaling Technology, Inc.)
secondary antibody conjugated to horseradish peroxidase for 1 h.
Protein bands were detected by chemiluminescence (cat. no. P90720;
EMD Millipore) and the images were quantified using ImageJ version
1.51 software (National Institutes of Health).
Statistical analysis
Data are presented as the mean ± SEM. All data were
normally distributed, as confirmed by the GraphPad Prism normality
test. Differences among multiple groups and the effects of
treatment for data obtained from in vivo and in vitro
studies were analyzed by one-way ANOVA, followed by the Tukey's
test for multiple comparisons (GraphPad Prism version 7; GraphPad
Prism Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Effects of Dex on myocardial injuries
in sepsis
H&E staining showed changes in the hearts of the
Ctrl, CLP, CLP + Dex and CLP + Dex + YOH mice (Fig. 1A). Moreover, cell nuclei appeared
as blue dominant dots due to inflammation induced by CLP, and
healthy cell nuclei were navy-blue in the control group, Dex
treatment reduced the number of inflammatory cells, while YOH
reversed the protective effects of Dex. TN-I is a classic marker of
myocardial injury (29) and it was
identified that CLP significantly increased TN-I (CLP vs. Ctrl,
P<0.01) (Fig. 1B), together
with elevations in 8-OHDG and MDA (CLP vs. Ctrl, P<0.01)
(Fig. 1C and D), which are markers
of oxidative stress. Moreover, the antioxidant factor SOD (Fig. 1E) was significantly reduced in the
CLP group compared with the Ctrl group (P<0.05). Following
treatment with Dex, the CLP-induced increase in TN-I, MDA and
8-OHDG was significantly attenuated (CLP + Dex vs. CLP, P<0.05).
Furthermore, SOD was significantly elevated following treatment
with Dex (CLP + Dex vs. CLP, P<0.05). After co-treatment with
YOH, 8-OHDG levels were significantly increased and SOD decreased
significantly (Fig. 1C and E);
however, TN-I and MDA levels were not influenced by YOH (CLP + Dex
+ YOH vs. CLP + Dex). Therefore, the present results indicated that
the protective effects of Dex may be partly achieved by reducing
oxidative stress.
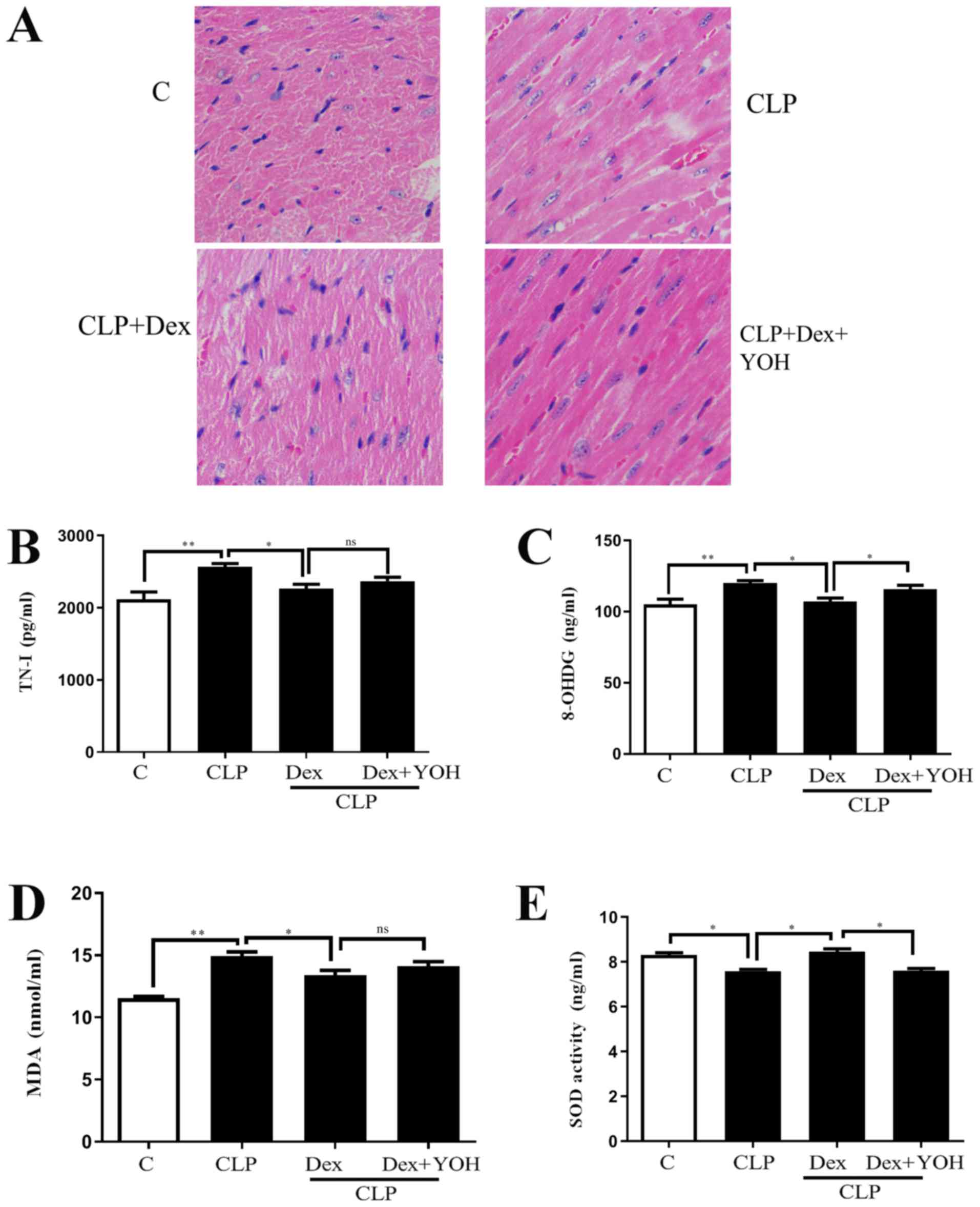 | Figure 1.Effects of Dex on septic myocardial
function. (A) Heart hematoxylin and eosin staining. Cell nuclei
appeared as blue dominant dots due to CLP-induced inflammation,
while normal cell nuclei were navy-blue. Dex treatment reduced
inflammatory cells and YOH reversed the protective effects of Dex.
Changes in (B) TN-I, (C) 8-OHDG, (D) SOD and (E) MDA release.
Sepsis was achieved by CLP. Data are presented as the mean ± SEM.
n=6 per group. *P<0.05 and **P<0.01. ns, not
significant; Dex, dexmedetomidine; CLP, cecal ligation and
puncture; YOH, yohimbine hydrochloride; TN-I, troponin-I; 8-OHDG,
8-hydroxy-2′-deoxyguanosine; SOD, superoxide dismutase; MDA,
malonaldehyde; C, control. |
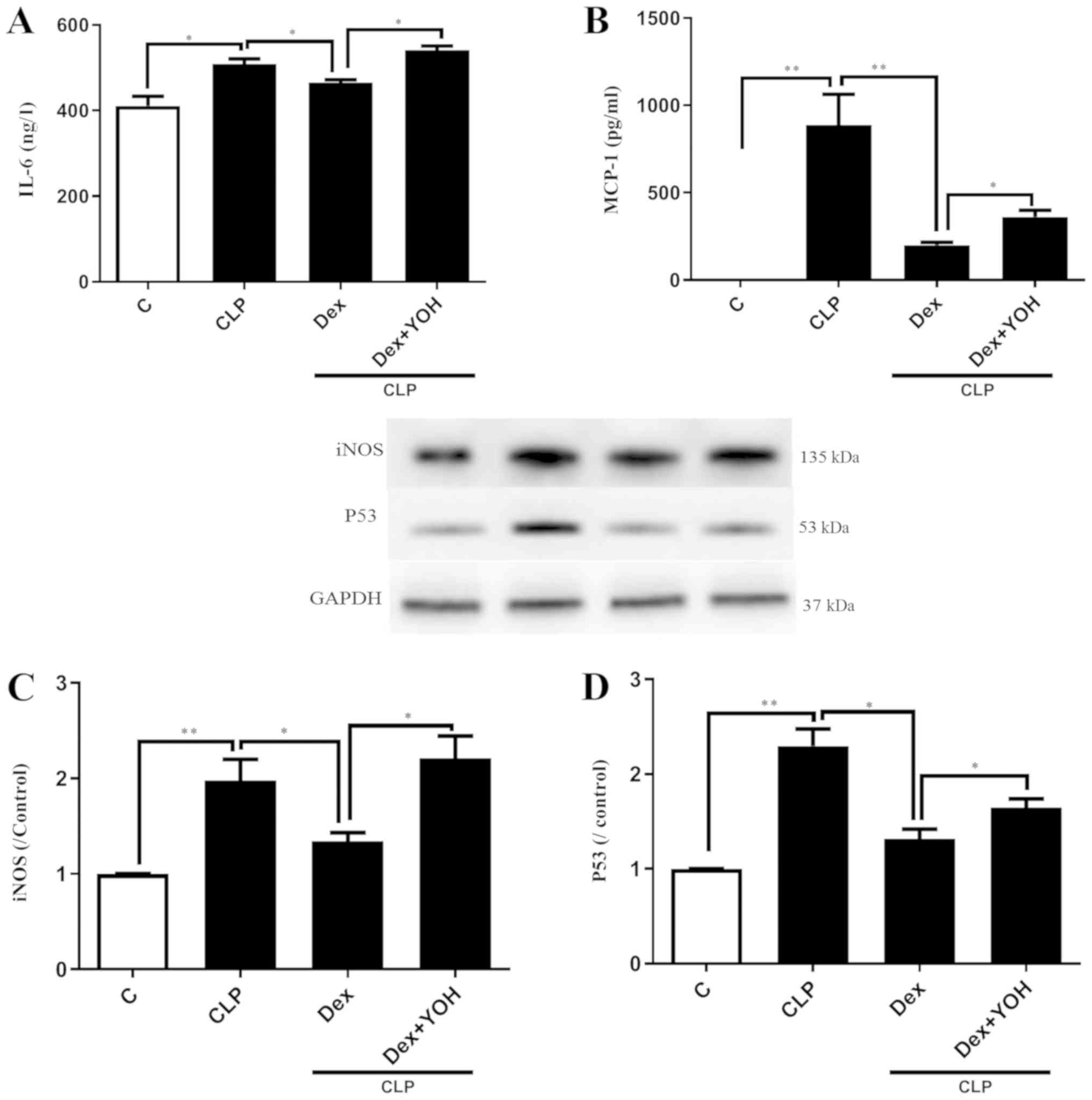
Effects of Dex on reducing the
expression of inflammatory factors
It was demonstrated that the expression levels of
inflammatory factors IL-6 and MCP-1 were significantly increased in
the CLP group compared with the Ctrl group (P<0.05 and
P<0.01, respectively) (Fig. 2A and
B). Furthermore, CLP significantly increased the protein
expression levels of iNOS and P53 (CLP vs. Ctrl, P<0.01)
(Fig. 2C and D). Following
pre-treatment with Dex, the CLP-induced elevations of IL-6 and
MCP-1 were significantly attenuated in the CLP + Dex group compared
with the CLP group (P<0.05) (Fig.
2A and B). In addition, Dex significantly decreased the protein
expression levels of iNOS and P53 compared with CLP alone
(P<0.05) (Fig. 2C and D).
However, these protective effects of Dex were prevented by the
α2-AR inhibitor YOH (CLP + Dex + YOH vs. CLP + Dex, P<0.05).
Changes of ferroptosis with or without
Dex
GPX4 is an indicator of and plays a central role in
ferroptosis (13). The present
results indicated that the protein expression of GPX4 and GSH
release were significantly decreased by CLP (CLP vs. Ctrl,
P<0.05) (Fig. 3A and B), and
that Dex significantly attenuated this decrease (CLP + Dex vs. CLP,
P<0.05). After co-treatment with YOH, Dex did not increase the
protein expression of GPX4 or GSH level (CLP + Dex + YOH vs. CLP +
Dex, P<0.01 and P<0.05, respectively). It was found that
following CLP, iron concentration was elevated from 8.1 to 15.66 µM
(CLP vs. Ctrl, P<0.01) (Fig.
3C). Furthermore, protein expression levels of HO-1 (Fig. 3D), Nrf2 (Fig. 3E) and TFR (Fig. 3F and H) were increased (CLP vs.
Ctrl, P<0.01), while that of ferritin, which accumulates iron
(18), significantly decreased
(CLP vs. Ctrl, P<0.05) (Fig.
3G). Furthermore, Dex treatment significantly decreased iron
concentration (from 15.66 to 9.7 µM, P<0.01) (Fig. 3C) and the protein expression of
HO-1 following CLP compared with CLP alone (P<0.05) (Fig. 3D). Dex treatment also moderately
increased the protein expression of Nrf2, but the difference was
not statistically significant (CLP + Dex vs. CLP). The expression
of ferritin was significantly increased (Fig. 3G), while TFR expression was
significantly decreased (Fig. 3H)
by Dex treatment (CLP + Dex vs. CLP, P<0.05). However, it was
identified that YOH reversed these changes and led to myocardial
injury (CLP + Dex + YOH vs. CLP + Dex, P<0.05) (Fig. 3).
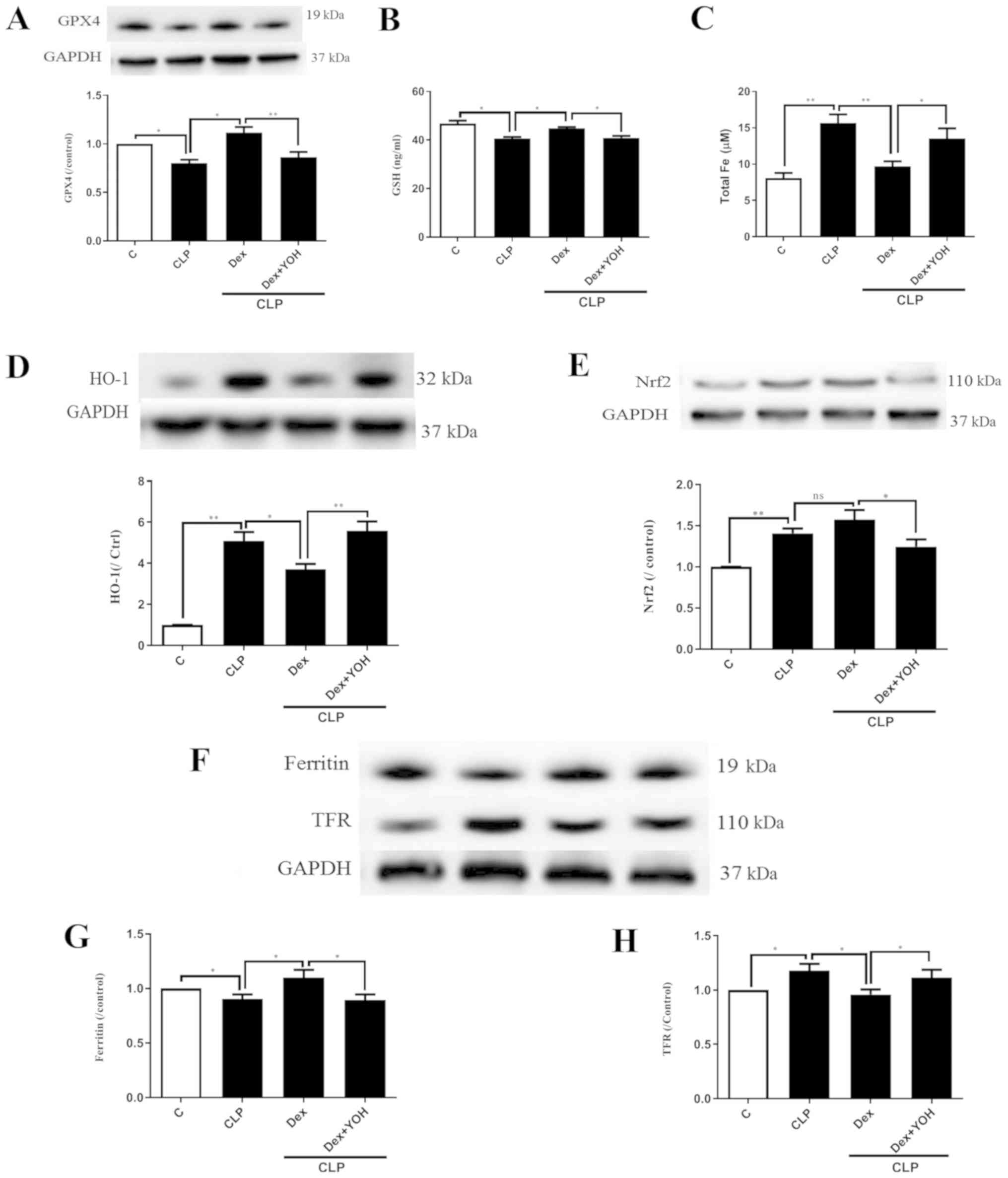 | Figure 3.Changes in ferroptosis with or
without Dex. (A) Changes in the protein expression of GPX4. (B)
Changes in GSH release. (C) Changes in iron concentration. Changes
in the protein expression levels of (D) HO-1 and (E) Nrf2. (F)
Western blot band of (G) ferritin and (H) TFR. Sepsis was achieved
by CLP. Data are presented as the mean ± SEM. n=6 per group.
*P<0.05 and **P<0.01. Dex, dexmedetomidine; CLP, cecal
ligation and puncture; YOH, yohimbine hydrochloride; GPX4,
glutathione peroxidase 4; GSH, glutathione; HO-1, heme oxygenase-1;
Nrf2, nuclear factor E2-related factor 2; TFR, transferrin
receptor; ns, normal control; C, control. |
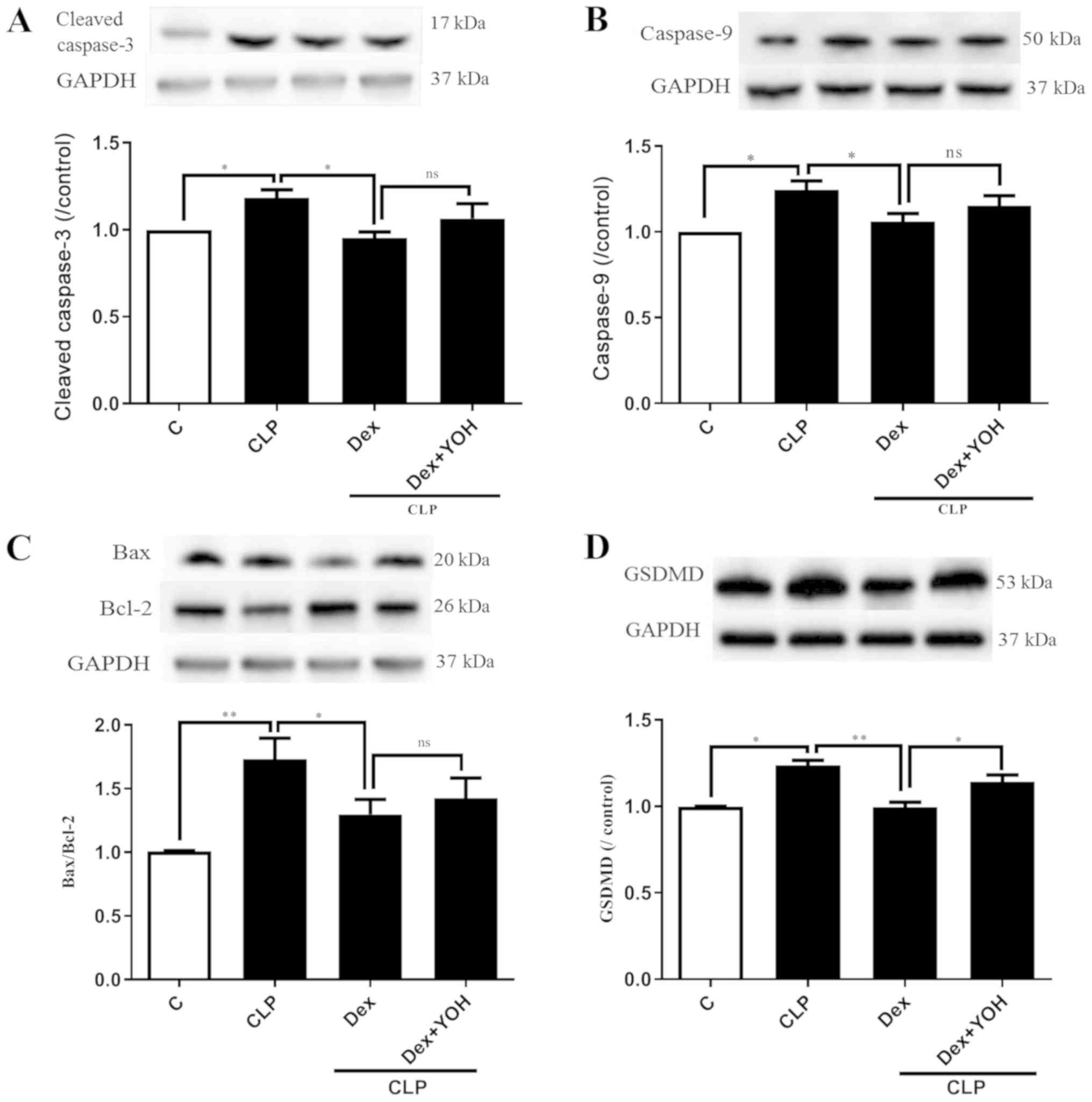
Effects of Dex on apoptosis and
pyroptosis during sepsis
Cleaved caspase 3, caspase 9, Bax and Bcl-2 are
markers of apoptosis. The expression levels of cleaved caspase 3
and caspase 9 were both significantly increased by CLP (CLP vs.
Ctrl, P<0.05) (Fig. 4A and B).
Moreover, it was found that the Bax/Bcl-2 ratio was significantly
elevated (CLP vs. Ctrl, P<0.01) (Fig. 4C). It was identified that Dex
treatment reduced the expression levels of cleaved caspase-3,
caspase-9 and Bax/Bcl-2 to exert cardioprotective effects (CLP +
Dex vs. CLP, P<0.05). However, YOH did not prevent the
Dex-induced reduction in the expression levels of apoptotic
proteins. Following treatment with YOH, cleaved caspase-3 and
caspase-9 expression levels, and the Bax/Bcl-2 ratio were
moderately elevated, but the difference was not significant. GSDMD
plays a key role in pyroptosis (30) and it was identified that CLP
significantly increased the protein expression of GSDMD (CLP vs.
Ctrl, P<0.01) (Fig. 4D).
Furthermore, Dex treatment significantly reduced the CLP-induced
increase in GSDMD (CLP + Dex vs. CLP, P<0.01), but YOH abolished
this Dex-induced reduction of the protein expression of GSDMD (CLP
+Dex + YOH vs. CLP + Dex, P<0.01).
Discussion
Sepsis causes whole body dysfunction due to
infection and is one of the leading causes of mortality worldwide
(31,32). While it has been studied for ~2,000
years, the incidence of sepsis has not decreased over time
(33). Inflammation is the first
characteristic feature of sepsis. Previous studies have revealed
that oxidative stress caused by inherent inflammatory responses can
lead to the initiation of lipid peroxidation, DNA damage and
mitochondrial function deterioration, as well as further the
development of organ dysfunction and failure (14,34).
Therefore, increases in inflammation and oxidative stress both
contribute to the damage caused by sepsis (32). Moreover, oxidative stress has been
reported to participate in numerous pathological conditions, such
as diabetes and myocardial ischemia reperfusion injury (35,36).
To maintain the balance of oxidants and antioxidants, an
antioxidant defense system that involves GPX, SOD, catalase
(37), ascorbic acid, GSH and
α-tocopherol plays crucial roles in decreasing oxidative stress
levels. During sepsis, reactive nitrogen species, including the
free radical NO and the non-radical peroxynitrite, was increased
(38,39). Dex, which exhibits superior
anxiolytic and sedative effects without causing respiratory
depression and with minor adverse effects, has been widely used in
intensive care units and clinical anesthesia (40). Furthermore, Dex has been proven to
exert protective effects against H2O2-induced
myocardial cell injury by reducing oxidative stress; it can also
reduce lung injury induced by lipopolysaccharide (23,41)
and confer other protective effects. In the present study, Dex was
found to reduce CLP-induced heart injury by increasing SOD and GSH
levels, and decreasing inflammatory factor levels, which was
consistent with the findings of previous studies (42,43).
Cell death, which mainly occurs via apoptosis,
necrosis, pyroptosis and the recently discovered ferroptosis, not
only kills cells, but can also play unique roles in various
physiological and pathological conditions to maintain homeostasis
(44). Apoptosis was recognized as
the first type of programmed cell death as early as in 1960; its
main characteristics include chromatin condensation and nuclear
fragmentation, cell shrinkage, plasma membrane blebbing and
apoptotic body formation without plasma membrane breakdown
(45–47). In the beginning of the apoptotic
process, caspases cleave and activate downstream factors, such as
Bcl-2 and ROS, which can affect other types of cell death (47,48).
In addition to apoptosis, other types of regulated cell death, such
as pyroptosis and ferroptosis, have also been shown to participate
in different pathological processes (49). Pyroptosis is recognized as
inflammasome-dependent cell death; during this process, the cell
membrane loses its integrity, leading to the dissolution of the
cell membrane and further induction of an inflammatory response.
Activated NACHT-, LRR- and PYD domains-containing protein 3
(NALP3), a marker of pyroptosis, leads to the aggregation of
cytokines IL-1β and caspase-1, which can be followed by tissue
injury and organ dysfunction (50,51).
Moreover, inflammatory factors released during apoptosis and
necroptosis lead to the induction of pyroptosis via inflammasome
formation and caspase-1 activation, which, in turn, leads to the
activation of GSDMD, another marker of pyroptosis (30). As previously reported, excessive
ROS can activate NLR family pyrin domain containing 3 and lead to
pyroptosis (52).
Ferroptosis is an iron-dependent form of cell death
induced by erastin (15), and was
recently shown to also be ROS-dependent. Furthermore, ferroptosis
is different from apoptosis and pyroptosis. Antioxidants, such as
N-acetylcysteine and iron chelators, prevent ferroptosis, thus
indicating that ferroptosis is a non-apoptotic but iron- and
oxidative stress-dependent form of cell death (18). Ferroptosis mainly relies on
regulators, such as GPX, HO-1, ferritin, transferrin, Nrf2, NADPH
and voltage-dependent anion channel (12). Based on this, excessive ROS can
lead to apoptosis, pyroptosis and ferroptosis, and once cell death
has occurred, the subsequent release of inflammatory cytokines and
overproduction of ROS plays a crucial role in inducing other types
of cell death. Moreover, these types of cell death interact with
each other (53). In the present
study, it was found that during sepsis, GPX4 and GSH protein
expression levels were decreased. In addition, it was demonstrated
that Dex restored the expression levels of GPX4 and GSH, thus
revealing that ferroptosis may be involved in sepsis. Furthermore,
the expression of the antioxidant protein HO-1 was increased in the
septic group compared with the Ctrl group. The present results also
indicated that, following treatment with Dex, the expression of
HO-1 was decreased. The major function of HO-1 is to metabolize
heme to produce carbon monoxide (4), biliverdin and iron, while it also
exerts anticancer, anti-inflammatory, antiapoptotic,
antiproliferative and antioxidant effects. However, it is unclear
whether the sustained overexpression of HO-1 is beneficial, as HO-1
can regulate iron homeostasis (22).
Iron is crucial for oxygen transport, ATP production
and DNA synthesis, which help maintain cellular function. However,
excessive iron is detrimental to the redox balance and can further
enhance the production of inflammatory factors, such as IL-1β, IL-6
and TNF-α, leading to more damage (14). In the present study, iron
concentration was found to be increased during sepsis and the
iron-related proteins TFR and ferritin were increased and
decreased, respectively. It was speculated that these effects may
possibly be due to the overexpression of HO-1, further
demonstrating that ferroptosis participates in sepsis, but the
definite relationship between iron and HO-1 requires further
research. To date, previous studies have focused on the beneficial
effects of HO-1, but have ignored the iron-downstream of HO-1,
which may be toxic. Thus, the effects of a sustained increase in
HO-1 expression under pathological conditions require further
study. Previous studies have demonstrated that HO-1 regulates
ferroptosis via iron, thus leading to septic heart injury (54,55).
The present results suggested that Dex reduced CLP-induced
overexpression of HO-1 and increased the expression levels of GPX4
and Nrf2 to exert a protective effect, which may contradict
previous findings that HO-1 is further enhanced following
antioxidant treatment (56). One
of the reasons for these differences could because different models
were used in each study. Additionally, the primary focus in the
present study was the relationship between HO-1 and ferroptosis,
whereas their study focused on the role of HO-1 in antioxidant
treatment. Therefore, future studies should further explore the
role of HO-1. Hu et al (57) showed that Dex alleviates iron
overload-induced injury in SH-SY5Y cells via antioxidative,
anti-inflammatory and antiapoptotic mechanisms. Therefore, these
findings provided evidence that iron overload is detrimental and
that the effects of Dex are associated with iron. In the present
study, it was speculated that Dex may exert its protective effects
partly by reducing the expression of HO-1, thus also decreasing
iron overload.
Moreover, YOH, an α2-AR antagonist, was used in the
present study and was found to reverse the effects of Dex on HO-1
and GPX4. Therefore, Dex may exert its protective role mainly via
α2-AR to reduce ferroptosis. However, YOH did not prevent Dex from
reducing the expression of the apoptotic protein cleaved caspase 3
or the Bax/Bcl-2 ratio. This result was consistent with the results
of our preliminary study, in which YOH also did not affect
H2O2 induced apoptosis. Therefore, the
present results indicated that Dex exerted its cardioprotective
effects partly by decreasing apoptosis in an α2-AR-independent
manner. The primary aim of the present study was to investigate the
effects of Dex on sepsis-induced ferroptosis, while the
effectiveness of Dex in relation to oxidative stress among others
was used as supporting data for comparison with previous studies.
Thus, the present study only assessed the representative oxidative
related parameters 8-OHDG, SOD and MDA. Therefore, further studies
are required to examine other oxidative stress parameters, such as
4-HNE, catalase and NO estimation for a complete understanding of
the effects of Dex on oxidative stress.
Sepsis causes significant adverse effects on both
the cardiac myocytes and the myocardial vasculature, following its
effects on systemic inflammation. The present study was a pilot
study investigating the effects of Dex on sepsis-induced myocardial
injury. In relation to ferroptosis, future studies should focus on
the effects of Dex on sepsis-induced changes to the vasculature,
including adhesion molecules, such as intercellular adhesion
molecule-1. Dex is a selective α2-AR and was also previously proven
to be an imidazoline receptor (58). Moreover, YOH is a potent α2-AR
antagonist, which was used in the present study. Furthermore,
further studies assessing Dex will use the agonist of α2-AR to
clarify the effects of Dex and to identify the role of imidazoline
receptor, which may facilitate the understanding of the underlying
mechanism of Dex.
In conclusion, it was found that HO-1 regulated
ferroptosis by regulating iron concentrations. Moreover, Dex
exerted cardioprotective effects via ferroptosis reduction, by
reducing iron concentration, the protein expression of HO-1 and
inflammatory factors, as well as increasing the expression of GPX4.
To the best of our knowledge, the present study is the first to
focus on the association between Dex and ferroptosis in septic
heart injury, and these results may provide further insights into
the mechanism of the protective effects of Dex.
Acknowledgements
Not applicable.
Funding
This study was supported by funding from the
National Natural Science Foundation of China (grant no. 81801947),
Shenzhen Science and Technology Innovation Committee (grant no.
JCYJ20180305180809671) and the Cooperative Research and Development
Program of Shenzhen People's Hospital (grant no. SYJY201709).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
CW, YL and ZZ designed the experiments. CW, WY, AH
and JL performed the experiments. CW, WY and AH analyzed the data.
CFY and ZX contributed to data interpretation and manuscript
revision. CW wrote the paper.
Ethics approval and consent to
participate
This study was approved by the Institutional Ethics
Committee of: The Medical Committee of Shenzhen People's Hospital
(approval ID: LL-KY-2019599).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sun X, Dai Y, Tan G, Liu Y and Li N:
Integration analysis of m(6)A-SNPs and eQTLs associated with sepsis
reveals platelet degranulation and staphylococcus aureus infection
are mediated by m(6)A mRNA methylation. Front Genet. 11:72020.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Parrillo JE: The cardiovascular
pathophysiology of sepsis. Annu Rev Med. 40:469–485. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ernsberger P, Giuliano R, Willette RN and
Reis DJ: Role of imidazole receptors in the vasodepressor response
to clonidine analogs in the rostral ventrolateral medulla. J
Pharmacol Exp Ther. 253:408–418. 1990.PubMed/NCBI
|
|
4
|
Barr J, Fraser GL, Puntillo K, Ely EW,
Gélinas C, Dasta JF, Davidson JE, Devlin JW, Kress JP, Joffe AM, et
al: Clinical practice guidelines for the management of pain,
agitation, and delirium in adult patients in the intensive care
unit. Crit Care Med. 41:263–306. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Taniguchi T, Kidani Y, Kanakura H,
Takemoto Y and Yamamoto K: Effects of dexmedetomidine on mortality
rate and inflammatory responses to endotoxin-induced shock in rats.
Crit Care Med. 32:1322–1326. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Venn RM, Bradshaw CJ, Spencer R, Brealey
D, Caudwell E, Naughton C, Vedio A, Singer M, Feneck R, Treacher D,
et al: Preliminary UK experience of dexmedetomidine, a novel agent
for postoperative sedation in the intensive care unit. Anaesthesia.
54:1136–1142. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kong W, Kang K, Gao Y, Liu H, Meng X, Yang
S, Yu K and Zhao M: Dexmedetomidine alleviates LPS-induced septic
cardiomyopathy via the cholinergic anti-inflammatory pathway in
mice. Am J Transl Res. 9:5040–5047. 2017.PubMed/NCBI
|
|
8
|
Ji F, Li Z, Nguyen H, Young N, Shi P,
Fleming N and Liu H: Perioperative dexmedetomidine improves
outcomes of cardiac surgery. Circulation. 127:1576–1584. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dolma S, Lessnick SL, Hahn WC and
Stockwell BR: Identification of genotype-selective antitumor agents
using synthetic lethal chemical screening in engineered human tumor
cells. Cancer Cell. 3:285–296. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gao M, Monian P, Quadri N, Ramasamy R and
Jiang X: Glutaminolysis and transferrin regulate ferroptosis. Mol
Cell. 59:298–308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell Death
Differ. 23:369–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhu H, Santo A, Jia Z and Robert Li Y:
GPx4 in bacterial infection and polymicrobial sepsis: Involvement
of ferroptosis and pyroptosis. React Oxyg Species (Apex).
7:154–160. 2019.PubMed/NCBI
|
|
14
|
Zhou B, Zhang JY, Liu XS, Chen HZ, Ai YL,
Cheng K, Sun RY, Zhou D, Han J and Wu Q: Tom20 senses
iron-activated ROS signaling to promote melanoma cell pyroptosis.
Cell Res. 28:1171–1185. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dixon SJ and Stockwell BR: The role of
iron and reactive oxygen species in cell death. Nat Chem Biol.
10:9–17. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baba Y, Higa JK, Shimada BK, Horiuchi KM,
Suhara T, Kobayashi M, Woo JD, Aoyagi H, Marh KS, Kitaoka H and
Matsui T: Protective effects of the mechanistic target of rapamycin
against excess iron and ferroptosis in cardiomyocytes. Am J Physiol
Heart Circ Physiol. 314:H659–H668. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou B, Liu J, Kang R, Klionsky DJ,
Kroemer G and Tang D: Ferroptosis is a type of autophagy-dependent
cell death. Semin Cancer Biol. Mar 14–2019.doi:
10.1016/j.semcancer.2019.03.002 (Epub ahead of print). View Article : Google Scholar
|
|
19
|
Jin X, Xu Z, Cao J, Yan R, Xu R, Ran R, Ma
Y, Cai W, Fan R, Zhang Y, et al: HO-1/EBP interaction alleviates
cholesterol-induced hypoxia through the activation of the AKT and
Nrf2/mTOR pathways and inhibition of carbohydrate metabolism in
cardiomyocytes. Int J Mol Med. 39:1409–1420. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Andreas M, Oeser C, Kainz FM, Shabanian S,
Aref T, Bilban M, Messner B, Heidtmann J, Laufer G, Kocher A and
Wolzt M: Intravenous heme arginate induces HO-1 (Heme Oxygenase-1)
in the human heart. Arterioscler Thromb Vasc Biol. 38:2755–2762.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kwon MY, Park E, Lee SJ and Chung SW: Heme
oxygenase-1 accelerates erastin-induced ferroptotic cell death.
Oncotarget. 6:24393–24403. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
NaveenKumar SK, Hemshekhar M, Kemparaju K
and Girish KS: Hemin-induced platelet activation and ferroptosis is
mediated through ROS-driven proteasomal activity and inflammasome
activation: Protection by melatonin. Biochim Biophys Acta Mol Basis
Dis. 1865:2303–2316. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu XR, Li T, Cao L, Yu YY, Chen LL, Fan
XH, Yang BB and Tan XQ: Dexmedetomidine attenuates
H2O2-induced neonatal rat cardiomyocytes
apoptosis through mitochondria- and ER-medicated oxidative stress
pathways. Mol Med Rep. 17:7258–7264. 2018.PubMed/NCBI
|
|
24
|
Qiu R, Yao W, Ji H, Yuan D, Gao X, Sha W,
Wang F, Huang P and Hei Z: Dexmedetomidine restores septic renal
function via promoting inflammation resolution in a rat sepsis
model. Life Sci. 204:1–8. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu L, Bao H, Si Y and Wang X: Effects of
dexmedetomidine on early and late cytokines during polymicrobial
sepsis in mice. Inflamm Res. 62:507–514. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen JH, Yu GF, Jin SY, Zhang WH, Lei DX,
Zhou SL and Song XR: Activation of alpha2 adrenoceptor attenuates
lipopolysaccharide-induced hepatic injury. Int J Clin Exp Pathol.
8:10752–10759. 2015.PubMed/NCBI
|
|
27
|
Zhao W, Jia L, Yang HJ, Xue X, Xu WX, Cai
JQ, Guo RJ and Cao CC: Taurine enhances the protective effect of
Dexmedetomidine on sepsis-induced acute lung injury via balancing
the immunological system. Biomed Pharmacother. 103:1362–1368. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rinaldi B, Di Filippo C, Capuano A,
Donniacuo M, Sodano L, Ferraraccio F, Rossi F and D'Amico M:
Adiponectin elevation by telmisartan ameliorates ischaemic
myocardium in Zucker diabetic fatty rats with metabolic syndrome.
Diabetes Obes Metab. 14:320–328. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Szekely L, Vijay P, Sharp TG, Bando K and
Brown JW: Correlation of plasma adrenomedullin to myocardial
preservation during open-heart surgery. Pediatr Cardiol.
21:228–233. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shi J, Gao W and Shao F: Pyroptosis:
Gasdermin-mediated programmed necrotic cell death. Trends Biochem
Sci. 42:245–254. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mantzarlis K, Tsolaki V and Zakynthinos E:
Role of oxidative stress and mitochondrial dysfunction in sepsis
and potential therapies. Oxid Med Cell Longev. 2017:59852092017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee WJ, Chen YL, Chu YW and Chien DS:
Comparison of glutathione peroxidase-3 protein expression and
enzyme bioactivity in normal subjects and patients with sepsis.
Clin Chim Acta. 489:177–182. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Y, Mao X, Chen H, Feng J, Yan M, Wang
Y and Yu Y: Dexmedetomidine alleviates LPS-induced apoptosis and
inflammation in macrophages by eliminating damaged mitochondria via
PINK1 mediated mitophagy. Int Immunopharmacol. 73:471–481. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kaludercic N and Di Lisa F: Mitochondrial
ROS formation in the pathogenesis of diabetic cardiomyopathy. Front
Cardiovasc Med. 7:122020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ighodaro OM: Molecular pathways associated
with oxidative stress in diabetes mellitus. Biomed Pharmacother.
108:656–662. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jin JK, Blackwood EA, Azizi K, Thuerauf
DJ, Fahem AG, Hofmann C, Kaufman RJ, Doroudgar S and Glembotski CC:
ATF6 decreases myocardial ischemia/reperfusion damage and links ER
stress and oxidative stress signaling pathways in the heart. Circ
Res. 120:862–875. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Catalao CHR, Santos-Júnior NN, da Costa
LHA, Souza AO, Alberici LC and Rocha MJA: Brain oxidative stress
during experimental sepsis is attenuated by simvastatin
administration. Mol Neurobiol. 54:7008–7018. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Droge W: Free radicals in the
physiological control of cell function. Physiol Rev. 82:47–95.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Webster NR and Nunn JF: Molecular
structure of free radicals and their importance in biological
reactions. Br J Anaesth. 60:98–108. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Arcangeli A, D'Alò C and Gaspari R:
Dexmedetomidine use in general anaesthesia. Curr Drug Targets.
10:687–695. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fu C, Dai X, Yang Y, Lin M, Cai Y and Cai
S: Dexmedetomidine attenuates lipopolysaccharide-induced acute lung
injury by inhibiting oxidative stress, mitochondrial dysfunction
and apoptosis in rats. Mol Med Rep. 15:131–138. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sha J, Zhang H, Zhao Y, Feng X, Hu X, Wang
C, Song M and Fan H: Dexmedetomidine attenuates
lipopolysaccharide-induced liver oxidative stress and cell
apoptosis in rats by increasing GSK-3β/MKP-1/Nrf2 pathway activity
via the α2 adrenergic receptor. Toxicol Appl Pharmacol.
364:144–152. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yazar E, Er A, Uney K, Bulbul A, Avci GE,
Elmas M and Tras B: Effects of drugs used in endotoxic shock on
oxidative stress and organ damage markers. Free Radic Res.
44:397–402. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fearnhead HO, Vandenabeele P and Vanden
Berghe T: How do we fit ferroptosis in the family of regulated cell
death? Cell Death Differ. 24:1991–1998. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhu H and Sun A: Programmed necrosis in
heart disease: Molecular mechanisms and clinical implications. J
Mol Cell Cardiol. 116:125–134. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mughal W, Dhingra R and Kirshenbaum LA:
Striking a balance: Autophagy, apoptosis, and necrosis in a normal
and failing heart. Curr Hypertens Rep. 14:540–547. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Konstantinidis K, Whelan RS and Kitsis RN:
Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc
Biol. 32:1552–1562. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Galluzzi L, Kepp O, Krautwald S, Kroemer G
and Linkermann A: Molecular mechanisms of regulated necrosis. Semin
Cell Dev Biol. 35:24–32. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li Z, Jia Y, Feng Y, Cui R, Miao R, Zhang
X, Qu K, Liu C and Zhang J: Methane alleviates sepsis-induced
injury by inhibiting pyroptosis and apoptosis: In vivo and in vitro
experiments. Aging (Albany NY). 11:1226–1239. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fu Q, Wu J, Zhou XY, Ji MH, Mao QH, Li Q,
Zong MM, Zhou ZQ and Yang JJ: NLRP3/caspase-1 pathway-induced
pyroptosis mediated cognitive deficits in a mouse model of
sepsis-associated encephalopathy. Inflammation. 42:306–318. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Patel S: Inflammasomes, the cardinal
pathology mediators are activated by pathogens, allergens and
mutagens: A critical review with focus on NLRP3. Biomed
Pharmacother. 92:819–825. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhou W, Chen C, Chen Z, Liu L, Jiang J, Wu
Z, Zhao M and Chen Y: NLRP3: A novel mediator in cardiovascular
disease. J Immunol Res. 2018:57021032018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bruni A, Bornstein S, Linkermann A and
Shapiro AMJ: Regulated cell death seen through the lens of islet
transplantation. Cell Transplant. 27:890–901. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li C, Lönn ME, Xu X, Maghzal GJ, Frazer
DM, Thomas SR, Halliwell B, Richardson DR, Anderson GJ and Stocker
R: Sustained expression of heme oxygenase-1 alters iron homeostasis
in nonerythroid cells. Free Radic Biol Med. 53:366–374. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Soares MP, Seldon MP, Gregoire IP,
Vassilevskaia T, Berberat PO, Yu J, Tsui TY and Bach FH: Heme
oxygenase-1 modulates the expression of adhesion molecules
associated with endothelial cell activation. J Immunol.
172:3553–3563. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mao X, Wang T, Liu Y, Irwin MG, Ou JS,
Liao XL, Gao X, Xu Y, Ng KF, Vanhoutte PM and Xia Z:
N-acetylcysteine and allopurinol confer synergy in attenuating
myocardial ischemia injury via restoring HIF-1α/HO-1 signaling in
diabetic rats. PLoS One. 8:e689492013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hu XB, Xi ZY, Liu LQ, Kang K, Li WH, Shen
YX, Kang F and Li J: Dexmedetomidine promotes SH-SY5Y cell
resistance against impairment of iron overload by inhibiting NF-κB
Pathways. Neurochem Res. 44:959–967. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang F, Ding T, Yu L, Zhong Y, Dai H and
Yan M: Dexmedetomidine protects against oxygen-glucose
deprivation-induced injury through the I2 imidazoline
receptor-PI3K/AKT pathway in rat C6 glioma cells. J Pharm
Pharmacol. 64:120–127. 2012. View Article : Google Scholar : PubMed/NCBI
|