Introduction
Alzheimer's disease (AD) is the most common type of
neurodegenerative disorder that affects the older population; it is
pathologically characterized by the formation of extracellular
senile plaques (SP) and intraneuronal neurofibrillary tangles,
which result from the deposition of β-amyloid peptide (Aβ) and the
aggregation of hyperphosphorylated tau protein, respectively
(1). Aβ, a peptide of 38–43 amino
acids, serves an important role in the pathogenesis of AD as it
aggregates to form soluble oligomers, fibrils and SPs (2). Previous studies have reported that
the soluble Aβ oligomer, amyloid peptide oligomers (AβOs), is more
toxic than insoluble fibrils (3,4) and
is more closely associated with the progression of AD (4,5). The
deposition of Aβ plaques is also associated with cross-sectional
synaptic network dysfunction, progressive brain atrophy, neuronal
calcium homeostasis (6) and long
term cognitive decline (7).
Studies conducted on both postmortem brains of
individuals with AD and AD model animals have reported that that
the synapses are affected during the early stages of
neurodegenerative processes (8,9). The
primary function of the synapse is to mediate intracellular
communication by releasing neurotransmitters from the presynaptic
terminal, as well as to modulate neural plasticity (10,11).
Synaptic proteins, including vesicle-associated and pre- and
post-synaptic membrane proteins, are closely related to cognitive
function, and previous studies have shown that the loss of synapses
in the brain tissues of patients with AD was associated with
cognitive impairment (12–14).
Synaptophysin (SYP), a specific marker of vesicles
that reflects the density and distribution of synapses, serves a
crucial role in neural plasticity by influencing the synaptic
structure and mediating neurotransmitter release via
phosphorylation (15).
Synaptosomal-associated protein of 25 kDa (SNAP-25), a component of
the complex of soluble N-ethylmaleimides sensitive factor
attachment protein receptor proteins, is central to synaptic
vesicle exocytosis, a process that regulates intracellular calcium
dynamics by negatively modulating neuronal voltage-gated calcium
channels (16). Postsynaptic
density of 95 kDa (PSD-95), a neuronal PSD-95/Dlg/ZO-1 protein,
associates with receptors and cytoskeletal elements at synapses,
orchestrates synaptic development and regulates synapse
stabilization and plasticity (17). Notably, numerous studies have
demonstrated that certain chemicals, such as AβOs, can alter the
levels of SYP in SH-SY5Y cells, which may be related to the
pathogenesis of AD (18–20).
Previous studies have revealed that cholinergic
deficits, such as the modification of acetylcholine (ACh) and
cholinesterase (ChE), as well as the downregulated expression of
nicotinic ACh receptors (nAChRs), are all significant contributors
to cognitive impairment in AD (21,22).
ACh is stored in the synaptic vesicles and is exocytosed and
released into the synaptic cleft to activate nAChRs; activated
nAChRs are highly permeable to Ca2+, which is an
important signaling molecule involved in the long-lasting
modulatory effects of the receptors (21). There are two forms of ChE,
acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE). It
was previously demonstrated that AChE C-terminal peptides modify
the regulation of nAChRs (23),
which is consistent with another study that reported treatment with
an AChE inhibitor increased nAChR expression levels (24). Neuronal nAChRs, members of the
super-family of ligand-gated ion channels, are involved in a number
of important physiological functions, such as memory and cognition
(25). The loss of these receptors
from several regions of brains affected by AD is observed in
receptor-ligand binding studies, and this loss is positively
correlated with the presence of SPs in the temporal lobe (26). The α (α2-α10) and β (β2-β4)
subunits of these receptors combine to form both hetero- and
homo-pentameric subtypes, of which α4β2 and α7 nAChRs are the
predominant subtypes in human brains (27). Moreover, neuronal nAChRs are
expressed pre-, post- and extra-synaptically in the brains and
mediate postsynaptic responses (28). Notably, electrophysiological
studies have shown that the formation of glutamatergic synapses in
the hippocampus is promoted by α7 nAChR (29). It has been suggested that α3 and α7
nAChR may change the neuroinflammatory status by inhibiting NF-κB
activation and reducing the production of inflammatory cytokines
(30–32).
An early sign of AD is the loss of synapses, which
is most strongly correlated with cognitive decline (33). Another characteristic of AD, as
demonstrated by positron emission tomography, is a deficit of
nAChRs in the brain, which is also closely related to cognitive
dysfunction (34). In a previous
study conducted in α7 nAChR knockout mice, the number of
glutamatergic synapses in the hippocampus was severely reduced,
whereas the activation of endogenous α7 nAChR was found to increase
this number (29); however, the
effects of altered α7 nAChR expression on the synaptic proteins are
relatively unknown.
The present study aimed to determine whether the
altered expression levels of α7 nAChR in SH-SY5Y cells may regulate
the expression levels of SYP, PSD-95 and SNAP-25, in addition to
their influence on the neurotoxic effect of Aβ on these synaptic
proteins, cell apoptosis, and the levels and activity of ACh and
AChE.
Materials and methods
Chemicals and reagents
The anti-α7 nAChR rabbit polyclonal antibody (cat.
no. GTX10096), anti-α7 nAChR rabbit polyclonal antibody (cat. no.
GTX64511), fluorescent FITC-conjugated anti-rabbit IgG antibody
(cat. no. BA1105), fluorescence Cy3-conjugated anti-mouse IgG
antibody (cat. no. BA1031), anti-SNAP-25 rabbit monoclonal antibody
(cat. no. GTX62566) and the anti-SD-95 rabbit monoclonal antibody
(cat. no. GTX61948) were all purchased from GeneTex, Inc. The
anti-Aβ mouse polyclonal antibody (cat. no. 803001) was purchased
from BioLegend, Inc., and the anti-SYP rabbit monoclonal antibody
(cat. no. ab32127) was obtained from Abcam. The horseradish
peroxidase (HRP)-conjugated anti-rabbit IgG antibody (cat. no.
7074) was purchased from Cell Signaling Technology, Inc., while the
HRP-conjugated anti-mouse IgG antibody (cat. no. SC-2005) and
anti-β-actin mouse monoclonal antibody (cat. no. SC-81178) were
obtained from Santa Cruz Biotechnology, Inc.
Control short hairpin RNA (shRNA) plasmid-B (cat.
no. SC-108065), α7 nAChR shRNA plasmid (h; cat. no. SC-42532-sh)
and GFP shRNA plasmid (A.vic; cat. no. SC-45924-sh) were purchased
from Santa Cruz Biotechnology, Inc. E.coli DH5α competent
cells purchased from Beijing Solarbio Science and Technology Co.,
Ltd. The Alexa 488-conjugated goat anti-rabbit IgG antibody, DMEM
and FBS were obtained from Thermo Fisher Scientific, Inc. The
pcDNA3.1 plasmid, G418 solution, X-treme GENE HP DNA Transfection
reagent, First Start Universal SYBR Green Master mix (Rox) and
First Strand cDNA Synthesis kit were obtained from Roche
Diagnostics GmbH. The Annexin V-APC/7-AAD apoptosis kit was
purchased from Multi Sciences, while the Hyper Performance
Chemiluminescence film and ECL Plus reagent were obtained from
Cytiva. AChE assay kits (cat. no. A024-1-1) and ACh assay kits
(cat. no. A105-3) were obtained from Nanjing Jiancheng
Bioengineering Institute and the quantitative (q)PCR primer pairs
for α7 nAChR, SYP, PSD-95, SNAP-25 and β-actin were designed based
on the corresponding gene sequences and synthesized by Shanghai
Genecore Bio Technologies Co., Ltd. DMEM and FBS purchased from
Thermo Fisher Scientific, Inc. The pcDNA3.1-GFP plasmid was kindly
donated by Professor Jian-Jiang Zhou of Guizhou Medical University
(Guizhou, China), while Aβ1-42, puromycin, 1,1,1,3,3,3-
hexafluoro-2-propanol (HFIP), goat serum, DMSO and the other
general chemicals used were purchased from Sigma-Aldrich; Merck
KGaA.
Patient samples
The study was approved by the Ethics Committee of
the Affiliated Hospital of Guizhou Medical University. All the
brain tissue blocks had been donated voluntarily by the patient or
with the consent of the family. Post-mortem brain samples from the
Netherlands Brain Bank were obtained and characterized according to
the specific clinical and neuropathological criteria (35). According to their medical history,
clinical manifestations and laboratory tests, the donor was
diagnosed as ‘probable AD’ by excluding other possible causes of
dementia, such as stroke. The clinical diagnosis was performed
according to the National Institute of Neurological and
Communication Disorders and Stroke and the Association of
Alzheimer's Disease and Related Diseases criteria (36), and the severity of dementia was
assessed according to the Global Deterioration Scale (37). The control brain samples had no
known history or symptoms of any neurological or psychiatric
disorder.
The temporal cortex, frontal cortex and hippocampus
of nine patients with AD (men, 3; women, 6; age range, 70–100
years) and nine controls (men, 4; women, 5; age range, 65–100
years) were studied; the mean age at mortality for the patients
with AD was 81.5±7.1 years, while the mean age at mortality of the
controls was 79.4±9.2 years. The interval between mortality and
autopsy in the patients with AD and the controls was 5.1±1.0 and
8.0±3.4 h, respectively.
AβO preparation
Synthetic human Aβ1-42 was suspended in
pre-cooled HFIP at a final concentration of 1 mM; the solution was
incubated at room temperature for 60 min and subsequently on ice
for 10 min (38). Aliquots were
transferred into non-siliconized microcentrifuge tubes and the HFIP
was allowed to evaporate overnight in a hood at room temperature,
which was then stored at −80°C (38). Prior to the treatment of cells,
Aβ1-42 was dissolved in DMSO to obtain a final
concentration of 5 mmol/l. For the preparation of oligomers, the
solution was subsequently diluted with modified DMEM and incubated
for 24 h at 4°C.
Double-labelling
immunofluorescence
The co-localization of α7 nAChR and Aβ was analyzed
using double staining. Human brain tissue wax blocks were provided
by the Netherlands Brain Bank, and were cut into 6-µm sections. The
sections were heated at 58°C for 1 h, placed in xylene I, xylene
II, xylene III (15 min each) and 100% ethanol (10 min), 90% ethanol
(10 min), 70% ethanol (10 min), deparaffinized and hydrated. After
three washes in PBS and microwaving in 0.01 M citric acid buffer
(pH 6.0) at 70°C for 20 min for the antigen repair, sections were
subsequently blocked with goat serum for 1 h at 37°C prior to
incubation with primary antibodies against α7 nAChR (1:100) and Aβ
(1:100) overnight at 4°C. Following the primary antibody
incubation, the sections were washed three times in PBS and
incubated with either a FITC-conjugated goat anti-mouse IgG
antibody (1:100) or a Alexa 488-conjugated goat anti-rabbit IgG
antibody (1:100) for 1 h at room temperature. The sections were
subsequently rinsed three times in PBS and mounted with VECTASHIELD
(Vector Laboratories, Inc.).
The immunofluorescence procedure was performed by an
independent neuropathologist and the analysis of the results were
performed by another researcher. The brain regions analyzed
included the hippocampus, temporal lobe and frontal lobe. The
integrated optical density (IOD) of immunofluorescence staining for
α7 nAChR and Aβ was determined by counting randomly selected nine
fields of vision using a fluorescent microscope (magnification,
×200 or ×400).
Cell culture and treatments
SH-SY5Y cells, derived from human neuroblastoma
cells, were purchased from the German Collection of Microorganisms
and Cell Cultures GmbH. Cells were cultured in DMEM, supplemented
with 10% FBS, 100 mg/ml streptomycin and 100 U/ml penicillin, and
maintained in a humidified atmosphere containing 5% CO2
at 37°C. To maintain their neuronal characteristics, the cells were
treated following ≤3 passages with 1 mM AβO at 37°C for 24 h, in
combination with the knockdown or overexpression of α7 nAChR.
Construction of the pcDNA3.1-α7 nAChR
expression plasmid
Total RNA was extracted from the SH-SY5Y cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. A total of 1 µg
RNA was reverse transcribed into cDNA using a First Strand cDNA
synthesis kit. The following primers pairs were used for reverse
transcription (RT-)qPCR, which were designed according to the
GenBank sequence no. BC-037571: α7 nAChR forward,
5′-CCCAAGCTTATGCAGGAGGCAGATATCAGTGGC-3′ and reverse,
5′-CGCGGATCCTTACGCAAAGTCTTTGGACACG-3′. The final PCR product
obtained was 966-bp long. The final PCR products were subsequently
separated by 1% agarose gel electrophoresis and the 966-bp fragment
was purified using an agarose gel extraction kit. This fragment was
then ligated into the pcDNA3.1 plasmid to create the recombinant
pcDNA3.1-α7 nAChR plasmid, which was amplified in competent E.
coli DH5a cells. The pcDNA3.1-α7 nAChR plasmid was subsequently
purified, digested with HindIII (1 µl) and BamHI (1
µl) at 37°C for 1 h in a water bath, visualized with ethidium
bromide and detected using agarose gel electrophoresis. The plasmid
was sequenced by Sangon Biotech Co., Ltd. to validate its
identity.
Transfection of the pcDNA3.1-α7 nAChR
or α7 nAChR shRNA plasmid (h) into SH-SY5Y cells and selection of
stable clones
SH-SY5Y cells, maintained and cultured
aforementioned, were stably transfected with the pcDNA3.1-α7 nAChR,
empty pcDNA 3.1, pcDNA 3.1-GFP, control shRNA, α7 nAChR shRNA
plasmid (h) or GFP shRNA plasmid using X-treme GENE HP DNA
Transfection reagent, according to the manufacturer's protocol. The
pcDNA 3.1 and control shRNA plasmid-B, which do not target any
endogenous transcript, were used as negative controls, and pcDNA
3.1-GFP and GFP shRNA plasmids served as the controls for the
efficiency of transfection.
After 24 h of transfection incubation at 37°C,
1×103 cells/ml transfected with pcDNA 3.1-α7 nAChR (1.5
mg/ml) and pcDNA 3.1 (1.5 mg/ml) were selected for 800 µg/ml G418
administration for 14 days at 37°C, while cells transfected with
control shRNA plasmid-B or α7 nAChR shRNA plasmid (h) were selected
for 4 µg/ml puromycin administration 14 days at 37°C. When
resistant cell clones appeared, individual clones were collected
and re-cultured to 60–80% confluence in successive passages until
cells demonstrated the overexpression or genetic silencing of α7
nAChR. Subsequent experimentations were performed after 24 h.
Analysis of mRNA encoding the α7 nAChR
subunit, SYP, PSD-95 and SNAP-25 using RT-qPCR
The mRNA expression levels of the α7 nAChR subunit,
SYP, PSD-95, SNAP-25 and β-actin were determined using RT-qPCR,
according to the manufacturer's instructions of each kit or
reagent. Cells were collected for RNA extraction when the cell
density reached 80%. In brief, 1 µg total RNA was extracted from
the SH-SY5Y cells using TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) and the reverse transcription of
RNA to cDNA was performed using a First Strand cDNA synthesis kit
at 42°C for 60 min, 70°C for 5 min and 4°C for 10 min. qPCR was
subsequently performed using an ABI Step One Plus system and First
Start Universal SYBR Green Master (ROX), with the following
thermocycling conditions: Initial denaturation at 95°C 10 min,
followed by 40 cycles at 95°C for 15 sec, annealing temperature for
60 sec and 72°C for 60 sec. The primer pairs for α7 nAChR, SYP,
PSD-95, SNAP-25 or β-actin are presented in Table I. Expression levels were determined
using the 2−ΔΔCq method (39). The experiment was repeated nine
times independently and were quantified using SDS v1.4 software
(Applied Biosystems; Thermo Fisher Scientific, Inc.).
 | Table I.Primers used for determining the
level of mRNA encoding α7nAChR, SYP, PSD-95, SNAP-25 or β-actin by
reverse transcription-quantitative PCR. |
Table I.
Primers used for determining the
level of mRNA encoding α7nAChR, SYP, PSD-95, SNAP-25 or β-actin by
reverse transcription-quantitative PCR.
| Gene | Primer
sequence | Amplicon size
(bp) | Annealing
temperature (°C) |
|---|
| α7nAChR |
5′ACCACTCACCGTCTACTTCTCC 3′ | 167 | 58 |
|
|
5′CATCTGGGAAACGAACAGTCTT 3′ |
|
|
| SYP |
5′GCTGTCAGATGTGAAGATGGC 3′ | 192 | 52 |
|
|
5′CCTGTCTCCTTAAACACGAACC 3′ |
|
|
| PSD-95 | 5′
CTCAGGGTCAACGACAGCAT 3′ | 114 | 58 |
|
| 5′
GACATAGAGGCGAACGATGG 3′ |
|
|
| SNAP-25 | 5′
GGATATGGGCAATGAGATCG 3′ | 115 | 58 |
|
| 5′
CAGCATCTTTGTTGCACGTT 3′ |
|
|
| β-actin |
5′TGGCACCACACCTTCTACAATG3′ | 103 | 58 |
|
|
5′TCATCTTCTCGCGGTTGGC 3′ |
|
|
Analysis of the α7 nAChR subunit, SYP,
PSD-95 and SNAP-25 expression levels using western blotting
The protein expression levels of α7 nAChR, SYP,
PSD-95 and SNAP-25 in cell lysates were quantified using western
blotting. Cells were washed three times with pre-chilled PBS in a
6-well plate. Then, cells were incubated on ice for 3 h with 60
µl/well RIPA buffer containing protease and phosphatase inhibitors
(cat. no. P0013B; Beyotime Institute of Biotechnology). The
supernatant was collected after centrifugation at 4°C for 15 min at
4,024.8 × g, and the concentration of the supernatant protein was
assessed with a Bradford Protein Assay kit (cat. no. P0006;
Beyotime Institute of Biotechnology), according to the
manufacturer's instructions. Proteins were loaded (12 µg/lane)and
proteins were separated by 10% SDS-PAGE. The separated proteins
were transferred to PVDF membranes and blocked with 5% fat-free dry
milk at room temperature for 2 h. The membranes were incubated with
primary antibodies against α7 nAChR (1:200), SYP(1:500), PSD-95
(1:500) and SNAP-25 (1:200) at 4°C overnight. Following the primary
antibody incubation, Membranes were washed three times with TBS
buffer with 0.1% Tween-20 and subsequently incubated with a
HRP-conjugated secondary antibody [1:5,000 (anti-mouse) or 1:2,000
(anti-rabbit)] at room temperature for 1 h. Protein bands were
detected with an ECL kit, according to the manufacturer's
instructions. After stripping these antibodies with buffer
containing detergent (cat. no. P0025; Beyotime Institute of
Biotechnology), the membranes were probed again with antibodies
against β-actin (1:6,000) at 37°C for 2 h and then incubated with a
HRP-conjugated anti-mouse IgG secondary antibody 37°C for 60 min.
Expression levels of the target proteins were normalized to β-actin
and the resulting ratio was presented as a percentage of the
corresponding control value. Semi-quantification was performed
using ImageJ software (v1.46; National Institutes of Health).
Flow cytometric analysis of
apoptosis
Cell apoptosis was determined using an Annexin
V-APC/7-AAD apoptosis kit, according to the manufacturer's
protocol. Cells were harvested and washed three times with
pre-cooled PBS. Following centrifugation at 400.8 × g at room
temperature for 5 min, cells were resuspended in 500 µl binding
buffer containing 5 µl Annexin V-APC and 10 µl 7-ADD and incubated
in the dark at room temperature for 5 min. Apoptotic cells were
analyzed using a BD FACSVerse flow cytometer (BD Biosciences) and
FlowJo software (FlowJo LLC; v.10.6.2). Results are presented as
the percentage of early + late apoptotic cells.
Analysis of the concentration of ACh
and the activity of AChE in cell supernatant using
spectrophotometry
The concentration of ACh and activity of AChE were
measured according to manufacturer's protocol of the AChE assay
kits and ACh assay kits. The substrate solution was reacted with
ACh at room temperature for 15 min to produce a brown compound. ACh
was synthesized by choline and acetyl-CoA under the catalytic
action of choline acetyl translocase (choline acetylase). The
absorbance OD value was measured at 550 nm, with the depth of color
being proportional to the concentration of ACh.
AChE is mainly located in the synaptic space of
cholinergic nerve terminals and is released under biological
stimulation (40). AChE hydrolyzed
ACh to cholic acid and acetic acid, and subsequently choline was
reacted with mercapto color reagent to produce a
5-thio-2-nitro-benzoic acid anion, which produced a yellow color.
The catalytic activity of AChE was measured at a wavelength of 412
nm.
Statistical analysis
Statistical analysis was performed using SPSS v22.0
software (IBM Corp.) and data are presented as the mean ± standard
deviation. Pearson correlation analysis was used to determine the
correlation, statistical differences between two groups were
determined using a two-tail Student's t-test and statistical
differences between multiple groups were determined using a one-way
ANOVA followed by a Student-Newman-Keuls or Tukey's post hoc test
(n=9). P<0.05 was considered to indicate a statistically
significant difference.
Results
Expression levels of α7 nAChR and Aβ
in the brains of patients with AD and healthy brains
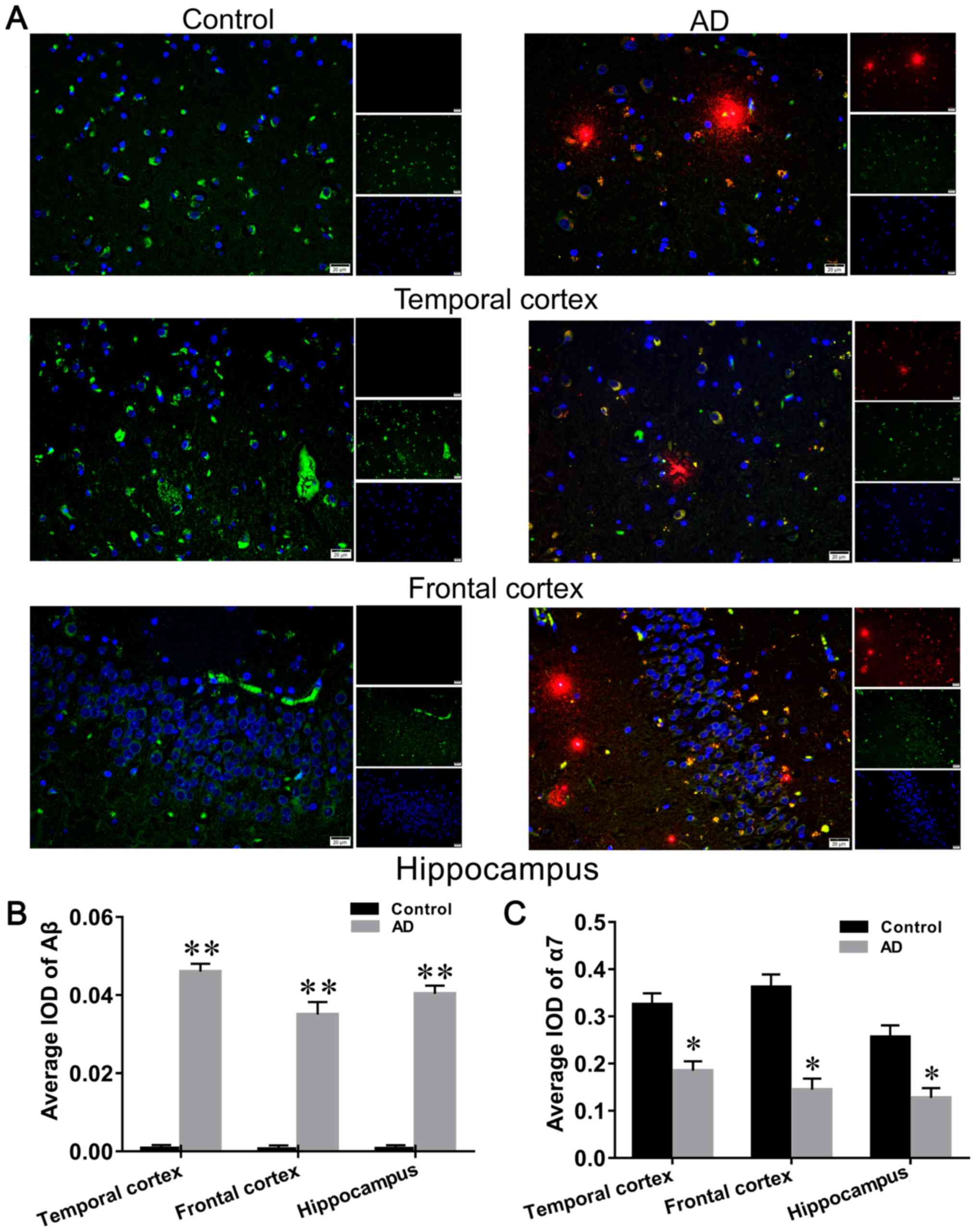
Aβ was observed to be expressed mainly in the
cytoplasmic and extracellular forms of both hippocampal and
cortical neurons (Fig. 1A), and
was found to be expressed at high levels in the brain of patients
with AD compared with the controls (Fig. 1B). α7nAChR was observed to be
mainly expressed in the cytoplasm of hippocampal and cortical
nerves (Fig. 1A), and its
distribution in the brain of patients with AD was significantly
decreased compared with the healthy control group (Fig. 1C).
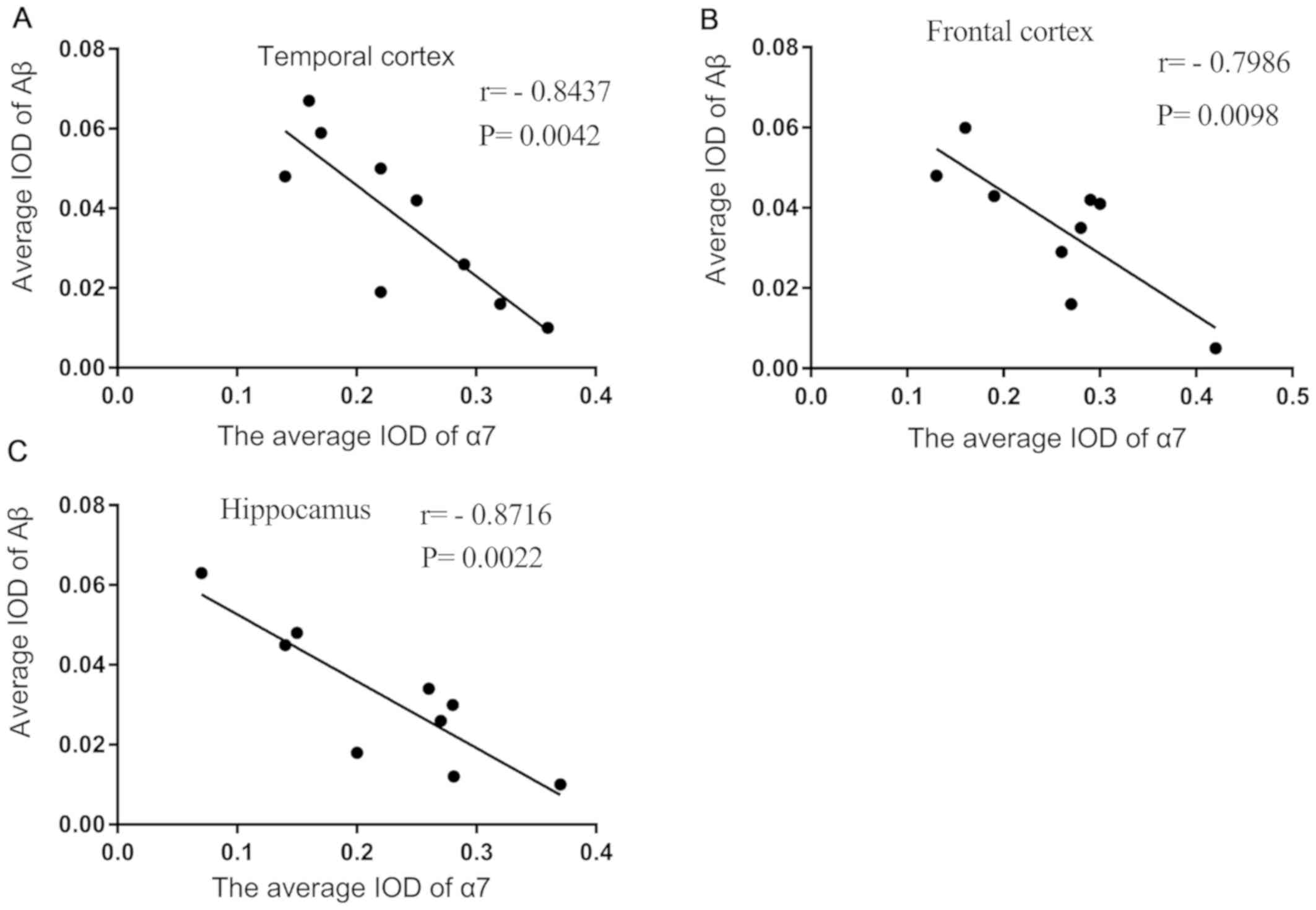
Mean IOD of α7nAChR is negatively
correlated with the mean IOD of Aβ in the brains of patients with
AD
The mean IOD of α7 nAChR and Aβ observed by
immunofluorescence double staining were demonstrated to be strongly
negatively correlated with each other in the temporal cortex
(Fig. 2A), frontal cortex
(Fig. 2B) and hippocampus
(Fig. 2C) of brain sections of
patients with AD.
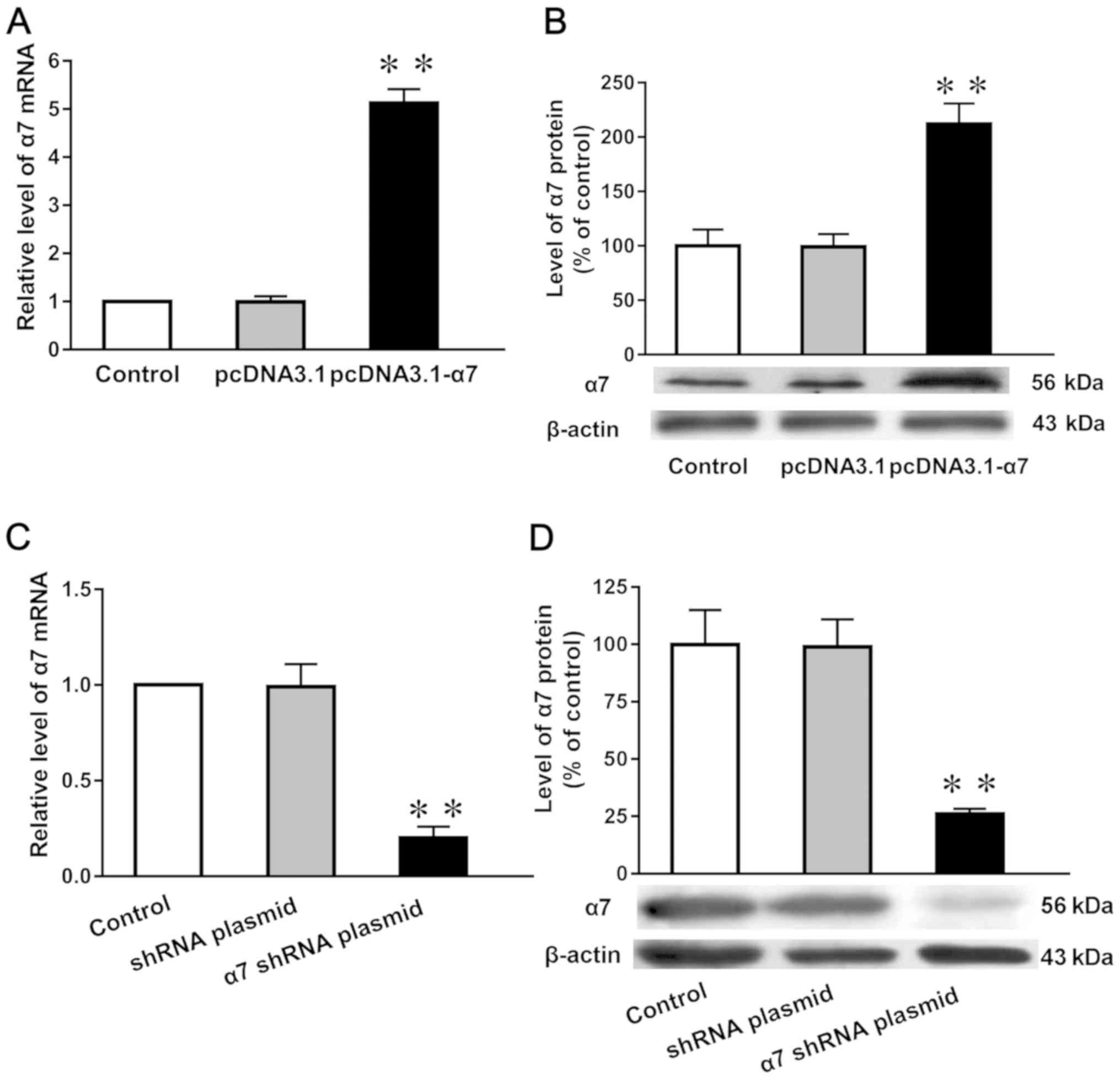
Expression levels of α7 nAChR mRNA and
protein in SH-SY5Y cells in which α7 nAChR is overexpressed or
silenced
Compared with both untransfected cells (normal
control) and those transfected with the empty plasmid (negative
control), the mRNA and protein expression levels of the α7 nAChR
subunit (Fig. 3A and B) in cells
overexpressing the receptor were increased by 433 and 110%,
respectively, in SH-SY5Y cells. Moreover the mRNA and protein
expression levels were reduced by 95% (Fig. 3C) and 80% (Fig. 3D), respectively, following the
genetic silencing of α7 nAChR.
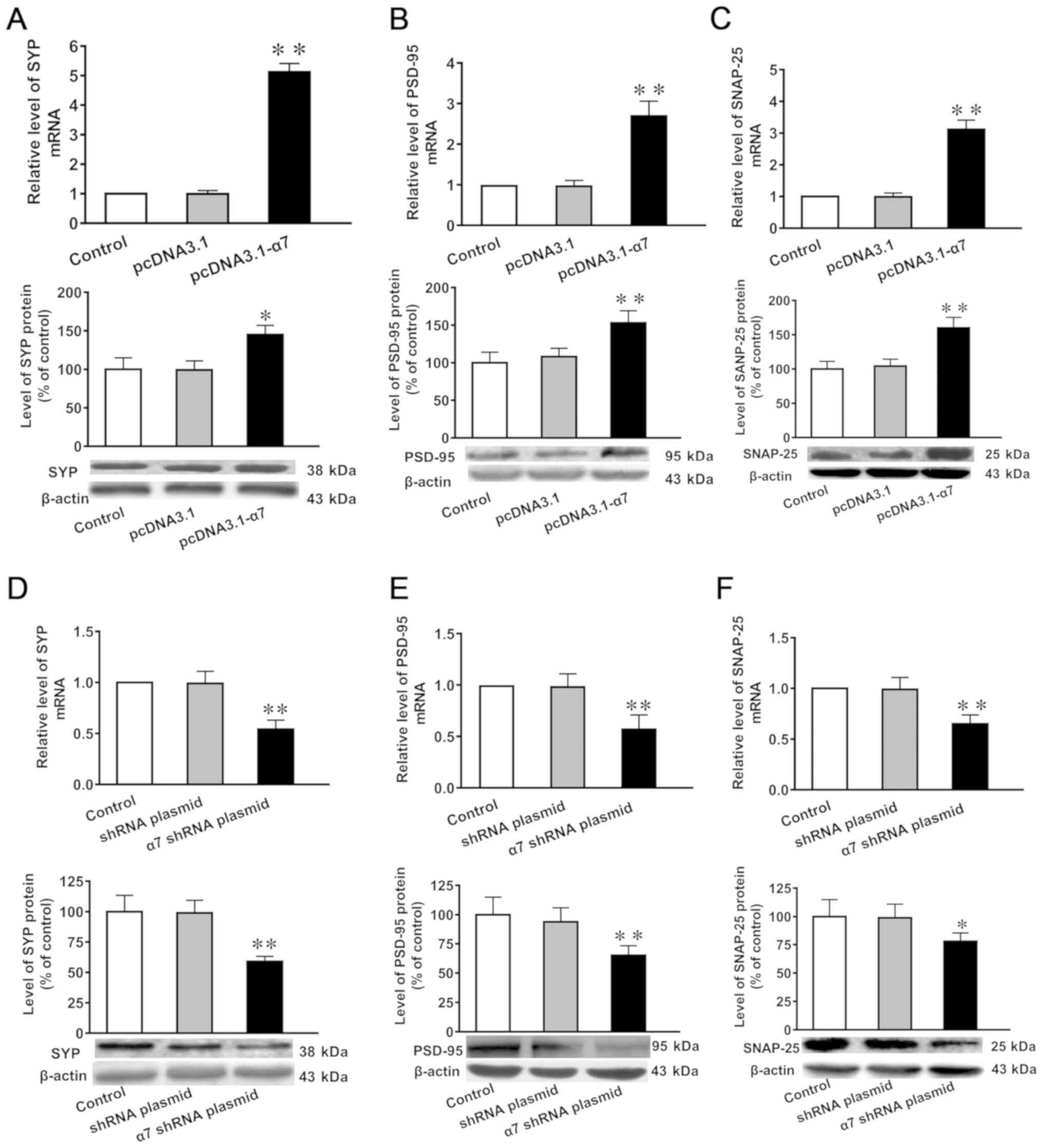
mRNA and protein expression levels of
SYP, PSD-95 and SNAP-25 in SH-SY5Y cells in which α7 nAChR is
overexpressed or silenced
Compared with untransfected controls and cells
transfected with the empty plasmid, the mRNA and protein expression
levels of SYP, PSD-95 and SNAP-25 were significantly increased in
SH-SY5Y cells overexpressing α7 nAChR (Fig. 4A-C). Furthermore, the mRNA and
protein expression levels of SYP, PSD-95 and SNAP-25 were
significantly decreased in SH-SY5Y cells in which α7 nAChR was
silenced (Fig. 4D-F).
Concentration of ACh and the activity
of AChE in SH-SY5Y cells in which the expression of α7 nAChR is
overexpressed or silenced
No significant differences were observed in the
concentration of ACh or the activity of AChE in SH-SY5Y cells with
overexpressed or silenced α7 nAChR expression compared with the
control cells (Table II).
| Table II.Concentration of ACh and the activity
of AChE in SH-SY5Y cells in which expression of α7 nAChR was
overexpressed or silenced. |
Table II.
Concentration of ACh and the activity
of AChE in SH-SY5Y cells in which expression of α7 nAChR was
overexpressed or silenced.
| Group | ACh concentration
(µg/ml) | AChE activity
(U/ml) |
|---|
| Control | 25.32±1.82 | 0.79±0.18 |
| pcDNA 3.1 | 25.75±2.07 | 0.85±0.23 |
| α7 nAChR-pcDNA
3.1 | 27.72±4.02 | 0.71±0.20 |
| Control shRNA
plasmid B | 26.59±3.82 | 0.74±0.16 |
| α7 nAChR shRNA
plasmid B | 29.97±2.78 | 0.82±0.22 |
mRNA and protein expression levels of
SYP, PSD-95 and SNAP-25 in SH-SY5Y cells in which α7 nAChR is
overexpressed or silenced and exposed to AβO
Compared with the untransfected control or negative
control, the mRNA and protein expression levels of SYP, PSD-95 and
SNAP-25 were significantly decreased following the exposure to AβO
compared with the control group (Fig.
5A-F). In addition, the overexpression of α7 nAChR attenuated
the toxicity of AβO (Fig. 5A-C),
while α7 nAChR silencing potentiated the toxic effects induced by
AβO (Fig. 5D-F).
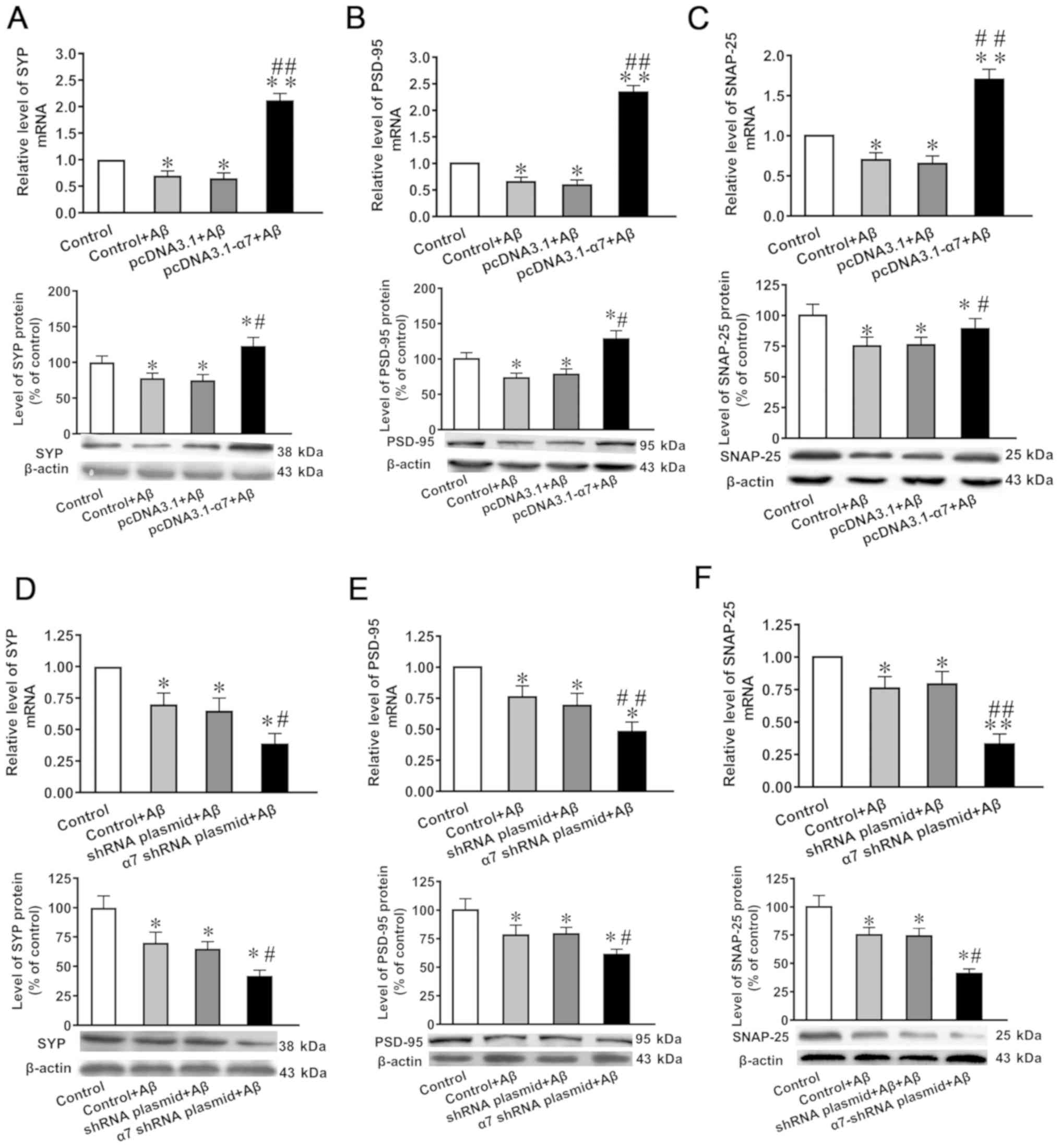 | Figure 5.Expression levels of SYP, PSD-95 and
SNAP-25 in SH-SY5Y cells following the overexpression of α7 nAChR
or silencing of α7 nAChR, and exposure to AβO. mRNA and protein
expression levels of (A) SYP, (B) PSD-95 and (C) SNAP-25 after α7
nAChR overexpression. mRNA and protein expression levels of (D)
SYP, (E) PSD-95 and (F) SNAP-25 after α7 nAChR silencing. The
expression levels of mRNA and protein were determined using reverse
transcription-quantitative PCR and western blotting, respectively.
Data are presented as the mean ± standard deviation (n=9).
*P<0.05 and **P<0.01 vs. control; ##P<0.01,
#P<0.05 vs. empty plasmid. SYP, synaptophysin;
PSD-95, postsynaptic density of 95 kDa; SNAP-25,
synaptosomal-associated protein of 25 kDa; α7 nAChR, nicotinic
acetylcholine receptor α7 subunit; AβO, Aβ1-42 oligomer; shRNA,
short hairpin RNA. |
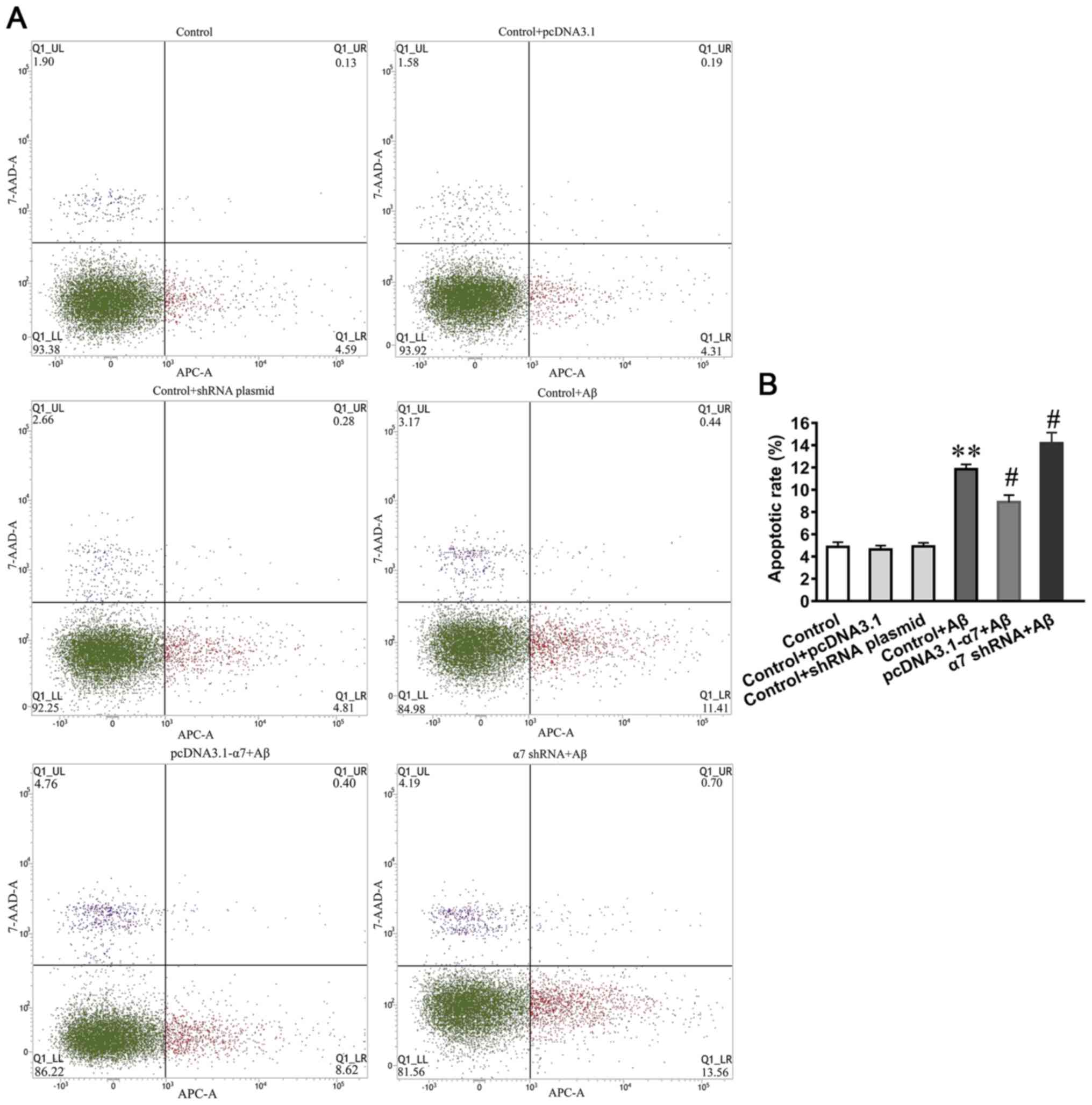
Apoptotic rate of SH-SY5Y cells in
which the expression of α7 nAChR is overexpressed or silenced and
simultaneously exposed to AβO
To investigate the neurotoxic effects of AβO on cell
apoptosis and the neuroprotective function of α7 nAChR, the
apoptotic rate was determined using an Annexin V-APC/7AAD apoptosis
kit and flow cytometry (Fig. 6A).
The results demonstrated that the apoptotic rates were
significantly increased in SH-SY5Y cells treated with AβO, and that
the overexpression of α7 nAChR attenuated the AβO-induced apoptotic
rate, whereas α7 nAChR silencing increased the AβO-induced
apoptotic rate (Fig. 6B).
Discussion
The present study successfully established cell
lines stably transfected with either the pcDNA3.1-α7 nAChR or α7
nAChR shRNA plasmid, and subsequently demonstrated that the mRNA
and protein expression levels of α7 nAChR were significantly
increased and decreased, respectively. In the present study, the
levels of upregulation and downregulation of α7 nAChR were not
consistent. Since not all post-transcriptional products are
translated into proteins, the increase of mRNA is not necessarily
synchronized with the expression of proteins. Under different
conditions of up- and downregulation, the efficiency of
transcription and translation differs.
Previous studies have shown that alterations in
nicotinic cholinergic mechanisms contribute to brain dysfunction
(41). ACh is degraded by AChE to
choline at cholinergic synapses to modulate synaptic transmission,
and choline is suggested to be a full agonist of α7 nAChR (42). A previous study revealed that an α7
nAChR agonist increased the release of ACh transiently (43). In the present study, the effects of
the altered expression levels of α7 nAChR on the concentration of
ACh and the activity of AChE was investigated in SH-SY5Y cells. The
findings indicated that there were no significant differences in
either ACh concentration or AChE activity in the treated cells. The
possible reason for the emergence of this result is that α-7nAChR
can increase the release of ACh and affect the activity of AChE
under short-term conditions; however, under the conditions of the
present study, this change was not observed. The present results
suggested that changes in the expression levels of α7 nAChR in
SH-SY5Y cells may not affect ACh concentrations and AChE activity,
which requires further investigation.
Numerous studies have reported that α7 nAChR served
an important role in cognitive processes in brains of patients with
AD, which may be related to the inhibition of Aβ toxicity and
modulation of synaptic transmission and plasticity (44–46).
The findings of the present study suggested that the
expression levels of Aβ in the brains of patients with AD were
increased compared with the healthy control group, and the
expression levels of α7 nAChR in brain sections of patients with AD
were decreased. Immunofluorescence double staining identified that
the expression levels of Aβ were decreased in the brain regions
exhibiting higher expression levels of α7 nAChR; a negative
correlation was found between α7 nAChR and Aβ in the temporal lobe,
frontal lobe and hippocampus of patients with AD, which suggested
that α7 nAChR may have a neuroprotective effect in the brains of
patients with AD. Subsequently, the neuroprotective effect of α7
nAChR in SH-SY5Y was investigated. It was speculated that the
correlation may be due to the neuronal loss induced by Aβ by Aβ
itself. However, due to the lack of human brain tissue specimens,
the present study did not perform hematoxylin-eosin staining.
In the present study, mRNA and protein expression
levels of SYP, SNAP-25 and PSD-95 were significantly increased in
the neuronal cells overexpressing α7 nAChR, while decreased
expression levels were observed following α7 nAChR RNA silencing.
These results suggested that α7 nAChR may be involved in regulating
the expression of these proteins, in addition to other synaptic
proteins, which would establish a link to synaptic stabilization
and plasticity. It has been revealed that SYP levels in the
superior temporal and inferior parietal cortex are ~5% lower in
patients with severe AD compared with individuals with no cognitive
impairment (47). Our previous
study reported significantly decreased expression levels of SYP
protein and mRNA in the brains of patients with AD, mice carrying
the amyloid protein precursor (APP)SWE mutation and rats
injected with Aβ1-42 (48). Furthermore, the expression levels
of PSD-95 and SNAP-25 are decreased in the cerebral cortex of
3×Tg-AD mice (49). The findings
of the present study also indicated that α7 nAChR may provide a
link to synaptic stabilization, plasticity and transmission in the
nervous system, which may be related to the protective function of
this receptor against cognitive decline during the pathogenesis of
AD. However, α7 nAChR may regulate synaptic plasticity by
regulating the level of calcium ions, which will be the subject of
future studies.
Aβ exerts its synaptotoxicity by suppressing
long-term potentiation, impairing synaptic plasticity and inducing
neuronal dysfunction (50). The
synaptotoxic effect of Aβ42 peptides was suggested to be
one of the most important aspects of the pathogenic process
resulting in AD (51), and
significantly reduced expression levels of SYP, PSD-95 and SNAP-25
have been revealed in the brains of mice influenced by Aβ (52–54).
Moreover, the present study detected significant reductions in the
expression levels of SYP, PSD-95 and SNAP-25 in SH-SY5Y cells
exposed to AβO, which may reflect the synaptotoxic effects induced
by Aβ.
In the present study, the inhibitory effects of Aβ
on the expression levels of SYP, PSD-95 and SNAP-25 were
significantly attenuated following the increased expression levels
of α7 nAChR and potentiated following α7 nAChR RNA silencing. These
observations indicated that α7 nAChR may also prevent the loss of
synaptic proteins caused by Aβ in vivo. It has been
previously reported that the inhibition of the expression levels of
α7 nAChR in SH-SY5Y cells by RNA interference increases the
toxicity and expression levels of Aβ, which subsequently elevates
the expression levels of β-site APP-cleaving enzyme 1 (BACE1) and
decreases the expression levels of BACE2 (38,55).
In the present study, the expression levels of synaptic proteins
were reduced to a greater extent by Aβ in the presence of α7 nAChR
silencing, which may be related to the reduced production of Aβ
caused by α7 nAChR. In addition, it was found that the treatment of
SH-SY5Y cells with AβO significantly increased the apoptotic rate,
while the proliferation of the cells was poor (data not shown),
which was attenuated by α7 nAChR overexpression and potentiated by
α7 nAChR silencing. These findings suggested that α7 nAChR may
effectively decrease AβO-induced neuronal apoptosis, which may be
connected to the increased expression of synaptic proteins.
In conclusion, the expression levels of Aβ in the
brain of patients with AD were significantly increased compared
with the healthy control group, whereas the expression levels of α7
nAChR in the brain of patients with AD were significantly decreased
compared with the healthy control group. Furthermore, it was
demonstrated that the expression levels of α7 nAChR and Aβ were
negatively correlated with each other in the brain of patients with
AD. The mRNA and protein expression levels of SYP, SNAP-25 and
PSD-95 were all significantly increased following the
overexpression of α7 nAChR, but were reduced following RNA
silencing of this receptor. In un-transfected or negative control
cells, the expression levels of these factors and the apoptotic
rate were significantly reduced following the exposure to AβO,
which was found to be attenuated by α7 nAChR overexpression, but
potentiated by α7 nAChR RNA silencing. Overall, these findings
suggested that α7 nAChR may serve an important neuroprotective role
related to the pathogenesis of AD and it may be useful for future
drug developments in AD.
Acknowledgements
The authors would like to thank the Netherlands
Brain Bank for providing the brain tissue samples.
Funding
This work was supported by grants from the Chinese
National Natural Science Foundation (grant no. 81860207), the
Program for Changjiang Scholars and Innovative Research Team in
University (grant no. IRT13058), the Guizhou Foundation [grant.
nos. (2014)06 and 2014(6008)], The Guizhou Science and Technology
Foundation [grant no. (2019)1437] and the Guizhou Province Graduate
Education Innovation Program Foundation [grant nos.
YJSCXJH(2018)040 and YJSCXJH(2019)070].
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request
Authors' contributions
XLQ and JMR designed the study, JMR wrote and
revised the manuscript. JMR and SLZ analyzed the data and revised
the manuscript. SLZ, XLW and ZZG performed the experiments. ZZG
helped perform the analysis of the data and provided constructive
discussions. All authors read and approved the final manuscript
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
the Affiliated Hospital of Guizhou Medical University. All the
brain tissue blocks had been donated voluntarily by the patient or
with the consent of the family.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bloom GS: Amyloid-β and tau: The trigger
and bullet in Alzheimer disease pathogenesis. JAMA Neurol.
71:505–508. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Amemori T, Jendelova P, Ruzicka J,
Urdzikova LM and Sykova E: Alzheimer's disease: Mechanism and
approach to cell therapy. Int J Mol Sci. 16:26417–26451. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferreira ST, Lourenco MV, Oliveira MM and
De Felice FG: Soluble amyloid-β oligomers as synaptotoxins leading
to cognitive impairment in Alzheimer's disease. Front Cell
Neurosci. 9:1912015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Viola KL and Klein WL: Amyloid β oligomers
in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta
Neuropathol. 129:183–206. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lesné SE, Sherman MA, Grant M, Kuskowski
M, Schneider JA, Bennett DA and Ashe KH: Brain amyloid-β oligomers
in ageing and Alzheimer's disease. Brain. 136:1383–1398. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Magi S, Castaldo P, Macrì ML, Maiolino M,
Matteucci A, Bastioli G, Gratteri S, Amoroso S and Lariccia V:
Intracellular calcium dysregulation: Implications for Alzheimer's
disease. BioMed Res Int. 2016:67013242016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jagust W: Is amyloid-β harmful to the
brain? Insights from human imaging studies. Brain. 139:23–30. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Overk CR and Masliah E: Pathogenesis of
synaptic degeneration in Alzheimer's disease and Lewy body disease.
Biochem Pharmacol. 88:508–516. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sheng M, Sabatini BL and Südhof TC:
Synapses and Alzheimer's disease. Csh Perspect Biol.
4:doi.org/10.1101/cshperspect.a005777.
|
|
10
|
Harrill JA, Chen H, Streifel KM, Yang D,
Mundy WR and Lein PJ: Ontogeny of biochemical, morphological and
functional parameters of synaptogenesis in primary cultures of rat
hippocampal and cortical neurons. Mol Brain. 8:102015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
García-Morales V, Montero F,
González-Forero D, Rodríguez-Bey G, Gómez-Pérez L,
Medialdea-Wandossell MJ, Domínguez-Vías G, García-Verdugo JM and
Moreno-López B: Membrane-derived phospholipids control synaptic
neurotransmission and plasticity. PLoS Biol. 13:e10021532015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang DB, Kinoshita Y, Kinoshita C, Uo T,
Sopher BL, Cudaback E, Keene CD, Bilousova T, Gylys K, Case A, et
al: Loss of endophilin-B1 exacerbates Alzheimer's disease
pathology. Brain. 138:2005–2019. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marcello E, Epis R, Saraceno C and Di Luca
M: Synaptic dysfunction in Alzheimer's disease. Adv Exp Med Biol.
970:573–601. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang DB, Kinoshita Y, Kinoshita C, Uo T,
Sopher BL, Cudaback E, Keene CD, Bilousova T, Gylys K, Case A, et
al: Loss of endophilin-B1 exacerbates Alzheimer's disease
pathology. Brain. 138:2005–2019. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sivanesan S, Tan A and Rajadas J:
Pathogenesis of Abeta oligomers in synaptic failure. Curr Alzheimer
Res. 10:316–323. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Antonucci F, Corradini I, Fossati G,
Tomasoni R, Menna E and Matteoli M: SNAP-25, a known presynaptic
protein with emerging postsynaptic functions. Front Synaptic
Neurosci. 8:72016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang J, Yuan J, Pang J, Ma J, Han B, Geng
Y, Shen L, Wang H, Ma Q, Wang Y and Wang M: Effects of chronic
stress on cognition in male SAMP8 mice. Cell Physiol Biochem.
39:1078–1086. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xi YD, Zhang DD, Ding J, Yu HL, Yuan LH,
Ma WW, Han J and Xiao R: Genistein inhibits Aβ25-35-induced
synaptic toxicity and regulates CaMKII/CREB pathway in SH-SY5Y
cells. Cell Mol Neurobiol. 36:1151–1159. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ito S, Ménard M, Atkinson T, Brown L,
Whitfield J and Chakravarthy B: Relative expression of the p75
neurotrophin receptor, tyrosine receptor kinase A, and insulin
receptor in SH-SY5Y neuroblastoma cells and hippocampi from
Alzheimer's disease patients. Neurochem Int. 101:22–29. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gray NE, Zweig JA, Kawamoto C, Quinn JF
and Copenhaver PF: STX, a novel membrane estrogen receptor ligand,
protects against amyloid-β toxicity. J Alzheimers Dis. 51:391–403.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ferreira-Vieira TH, Guimaraes IM, Silva FR
and Ribeiro FM: Alzheimer's disease: Targeting the cholinergic
system. Curr Neuropharmacol. 14:101–115. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Park D, Choi EK, Cho TH, Joo SS and Kim
YB: Human neural stem cells encoding ChAT gene restore cognitive
function via acetylcholine synthesis, Aβ elimination, and
neuroregeneration in APPswe/PS1dE9 mice. Int J Mol Sci. 21:212020.
View Article : Google Scholar
|
|
23
|
Badin AS, Eraifej J and Greenfield S:
High-resolution spatio-temporal bioactivity of a novel peptide
revealed by optical imaging in rat orbitofrontal cortex in vitro:
Possible implications for neurodegenerative diseases.
Neuropharmacology. 73:10–18. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Galimberti D and Scarpini E: Old and new
acetylcholinesterase inhibitors for Alzheimer's disease. Expert
Opin Investig Drugs. 25:1181–1187. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fukunaga K and Yabuki Y: SAK3-induced
neuroprotection is mediated by nicotinic acetylcholine receptors.
In: Nicotinic Acetylcholine Receptor Signaling in Neuroprotection.
Akaike A, Shimohama S and Yoshimi Misu Y: Springer; Berlin: pp.
159–171. 2018, PubMed/NCBI
|
|
26
|
Hernandez CM, Kayed R, Zheng H, Sweatt JD
and Dineley KT: Loss of alpha7 nicotinic receptors enhances
beta-amyloid oligomer accumulation, exacerbating early-stage
cognitive decline and septohippocampal pathology in a mouse model
of Alzheimer's disease. J Neurosci. 30:2442–2453. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gil SM and Metherate R: Enhanced
sensory-cognitive processing by activation of nicotinic
acetylcholine receptors. Nicotine Tob Res. 21:377–382. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gu Z and Yakel JL: Timing-dependent septal
cholinergic induction of dynamic hippocampal synaptic plasticity.
Neuron. 71:155–165. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lozada AF, Wang X, Gounko NV, Massey KA,
Duan J, Liu Z and Berg DK: Glutamatergic synapse formation is
promoted by α7-containing nicotinic acetylcholine receptors. J
Neurosci. 32:7651–7661. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yue Y, Liu R, Cheng W, Hu Y, Li J, Pan X,
Peng J and Zhang P: GTS-21 attenuates lipopolysaccharide-induced
inflammatory cytokine production in vitro by modulating the Akt and
NF-κB signaling pathway through the α7 nicotinic acetylcholine
receptor. Int Immunopharmacol. 29:504–512. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tyagi E, Agrawal R, Nath C and Shukla R:
Inhibitory role of cholinergic system mediated via alpha7 nicotinic
acetylcholine receptor in LPS-induced neuro-inflammation. Innate
Immun. 16:3–13. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liao Y, Qi XL, Cao Y, Yu WF, Ravid R,
Winblad B, Pei JJ and Guan ZZ: Elevations in the levels of NF-κB
and inflammatory chemotactic factors in the brains with Alzheimer's
disease - One mechanism may involve α3 nicotinic acetylcholine
receptor. Curr Alzheimer Res. 13:1290–1301. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Domínguez-Álvaro M, Montero-Crespo M,
Blazquez-Llorca L, Insausti R, DeFelipe J and Alonso-Nanclares L:
Three-dimensional analysis of synapses in the transentorhinal
cortex of Alzheimer's disease patients. Acta Neuropathol Commun.
6:202018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bauwens M, Mottaghy FM and Bucerius J: PET
imaging of the human nicotinic cholinergic pathway in
atherosclerosis. Curr Cardiol Rep. 17:672015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dubois B, Feldman HH, Jacova C, Dekosky
ST, Barberger-Gateau P, Cummings J, Delacourte A, Galasko D,
Gauthier S, Jicha G, et al: Research criteria for the diagnosis of
Alzheimer's disease: Revising the NINCDS-ADRDA criteria. Lancet
Neurol. 6:734–746. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McKhann G, Drachman D, Folstein M, Katzman
R, Price D and Stadlan EM: Clinical diagnosis of Alzheimer's
disease: Report of the NINCDS-ADRDA work group under the auspices
of Department of Health and Human Services Task Force on
Alzheimer's disease. Neurology. 34:939–944. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Reisberg B, Ferris SH, de Leon MJ and
Crook T: The Global Deterioration Scale for assessment of primary
degenerative dementia. Am J Psychiatry. 139:1136–1139. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang XL, Deng YX, Gao YM, Dong YT, Wang F,
Guan ZZ, Wei H and Qi XL: Activation of α7 nAChR by PNU-282987
improves synaptic and cognitive functions through restoring the
expression of synaptic-associated proteins and the CaM-CaMKII-CREB
signaling pathway. Aging (Albany NY). 12:543–570. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dunant Y and Gisiger V: Ultrafast and slow
cholinergic transmission. Different involvement of
acetylcholinesterase molecular forms. Molecules. 22:13002017.
View Article : Google Scholar
|
|
41
|
Dani JA and Bertrand D: Nicotinic
acetylcholine receptors and nicotinic cholinergic mechanisms of the
central nervous system. Annu Rev Pharmacol Toxicol. 47:699–729.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Albiñana E, Luengo JG, Baraibar AM, Muñoz
MD, Gandía L, Solís JM and Hernández-Guijo JM: Choline induces
opposite changes in pyramidal neuron excitability and synaptic
transmission through a nicotinic receptor-independent process in
hippocampal slices. Pflugers Arch. 469:779–795. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Huang M, Felix AR, Kwon S, Lowe D, Wallace
T, Santarelli L and Meltzer HY: The alpha-7 nicotinic receptor
partial agonist/5-HT3 antagonist RG3487 enhances cortical and
hippocampal dopamine and acetylcholine release. Psychopharmacology
(Berl). 231:2199–2210. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Stoiljkovic M, Kelley C, Nagy D, Hurst R
and Hajós M: Activation of α7 nicotinic acetylcholine receptors
facilitates long-term potentiation at the hippocampal-prefrontal
cortex synapses in vivo. Eur Neuropsychopharmacol. 26:2018–2023.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lagostena L, Trocme-Thibierge C, Morain P
and Cherubini E: The partial alpha7 nicotine acetylcholine receptor
agonist S 24795 enhances long-term potentiation at CA3-CA1 synapses
in the adult mouse hippocampus. Neuropharmacology. 54:676–685.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Inestrosa NC, Godoy JA, Vargas JY,
Arrazola MS, Rios JA, Carvajal FJ, Serrano FG and Farias GG:
Nicotine prevents synaptic impairment induced by amyloid-β
oligomers through α7-nicotinic acetylcholine receptor activation.
Neuromolecular Med. 15:549–569. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Counts SE, Nadeem M, Lad SP, Wuu J and
Mufson EJ: Differential expression of synaptic proteins in the
frontal and temporal cortex of elderly subjects with mild cognitive
impairment. J Neuropathol Exp Neurol. 65:592–601. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cao Y, Xiao Y, Ravid R and Guan ZZ:
Changed clathrin regulatory proteins in the brains of Alzheimer's
disease patients and animal models. J Alzheimers Dis. 22:329–342.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Carvalho C, Santos MS, Oliveira CR and
Moreira PI: Alzheimer's disease and type 2 diabetes-related
alterations in brain mitochondria, autophagy and synaptic markers.
Biochim Biophys Acta. 1852:1665–1675. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ferreira ST and Klein WL: The Aβ oligomer
hypothesis for synapse failure and memory loss in Alzheimer's
disease. Neurobiol Learn Mem. 96:529–543. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tu S, Okamoto S, Lipton SA and Xu H:
Oligomeric Aβ-induced synaptic dysfunction in Alzheimer's disease.
Mol Neurodegener. 9:482014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang S, Yu L, Yang H, Li C, Hui Z, Xu Y
and Zhu X: Oridonin attenuates synaptic loss and cognitive deficits
in an Aβ1-42-induced mouse model of Alzheimer's disease. PLoS One.
11:e01513972016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liu SJ, Yang C, Zhang Y, Su RY, Chen JL,
Jiao MM, Chen HF, Zheng N, Luo S, Chen YB, et al: Neuroprotective
effect of β-asarone against Alzheimer's disease: Regulation of
synaptic plasticity by increased expression of SYP and GluR1. Drug
Des Devel Ther. 10:1461–1469. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chauhan NB, Lichtor T and Siegel GJ: Aging
potentiates Abeta-induced depletion of SNAP-25 in mouse
hippocampus. Brain Res. 982:219–227. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Qi XL, Nordberg A, Xiu J and Guan ZZ: The
consequences of reducing expression of the alpha7 nicotinic
receptor by RNA interference and of stimulating its activity with
an alpha7 agonist in SH-SY5Y cells indicate that this receptor
plays a neuroprotective role in connection with the pathogenesis of
Alzheimer's disease. Neurochem Int. 51:377–383. 2007. View Article : Google Scholar : PubMed/NCBI
|