Introduction
Pneumonia, a common infectious disease, is
characterized by infection of pathogens such as viruses, bacteria
and fungi in the lower respiratory tract, and is increasing in
incidence and mortality worldwide (1). The typical symptoms of pneumonia are
chills, fever, pleuritic chest pain and cough productive of
purulent sputum (1). Pneumonia is
a major cause of mortality in children <5 years worldwide,
accompanied with approximately 1.3 million mortalities in children
each year (2,3). A better understanding of the
mechanism underlying pneumonia in children may aid the development
of effective and novel therapeutic approach to overcome this
burden.
High-temperature requirement A2 (HtrA2), also known
as Omi, is a ubiquitously expressed protein that encodes a serine
protein kinase. HtrA2, produced in the endoplasmic reticulum, is
located in the inner membrane of the mitochondrial intermembrane
space (4). Under apoptotic
stimuli, HtrA2 translocates to the cytosol and then promotes cell
death by binding to an inhibitor of apoptosis proteins (IAPs),
causing caspase activity or promoting caspase-independent cell
death (5,6). UCF-101 (5-[5-(2-nitrophenyl) furfuryl
iodine]-1, 3-diphenyl-2-thiobarbituric acid) is a specific
inhibitor of HtrA2. Previous studies have demonstrated that UCF-101
has a clear protective capacity in organ injury in
streptozotocin-induced mouse cardiomyocyte contractile dysfunction
(7), dextran sulfate
sodium-induced colitis (8) and
cecal ligation and puncture-induced septic shock in rats (9). Studies of HtrA2 on pulmonary injury
are rare and whether UCF-101 protects lung against apoptotic
stimuli remains to be elucidated. Thus, the present study explored
the effect of UCF-101 in pneumonia.
The present study identified significant changes of
HtrA2 in lung tissues of lipopolysaccharide (LPS)-treated rats. The
pathological symptoms and pulmonary injuries were ameliorated by
treatment with UCF-101. Investigation of the mechanism suggested
that UCF-101 may exert its function by regulating mitochondrial
activity and associated gene expression.
Materials and methods
Animals and experimental design
To induce the pneumonia model, 32 newborn
Sprague-Dawley rats (male, 3–8 days old, 8–14 g) were obtained from
Zhejiang Center of Laboratory Animals and kept in an animal room
with prespecified conditions (temperature: 22°C; relative humidity,
40–50%; 12 h light/dark cycle) with free access to water and food.
The rats were randomly divided into four groups: The control group,
without any treatment; the LPS group, which received
saline-dissolved LPS (2 mg/kg; Sigma-Aldrich; Merck KGaA)
intratracheally using a miniature nebulizer; the LPS+UCF-101 group,
which was injected intraperitoneally with UCF-101 (2 µmol/kg rats,
dissolved in distilled water containing 10% DMSO) 30 min after LPS
administration or the same amount of 10% DMSO as control for
intraperitoneal injection 30 min after LPS administration for the
LPS+DMSO group. Then, 24 h after LPS administration, rats were
anesthetized with 30 mg/kg pentobarbital sodium and abdominal aorta
blood was collected. Finally, rats were euthanized using 30 mg/kg
pentobarbital sodium and cervical dislocation. When the rats were
verified to have succumbed by lack of heartbeat and cold body, the
lung tissue samples were collected for further experiments. All
animal experiments were performed in accordance with the animal
experimental guidelines set by the National Institutes of Health
Guide for the Care and Use of Laboratory Animals (10). The present study was approved by
Ethics Committee of Shengjing Hospital of China Medical University
(Shenyang, Liaoning, China).
Western blot analysis
The total protein of lung tissue was extracted using
RIPA lysis buffer (Invitrogen; Thermo Fisher Scientific, Inc.). The
mitochondrial and cytosolic fraction were prepared according to a
previous study (11). The
concentration of protein was determined using a BCA protein assay
kit (Beyotime Institute of Biotechnology). Total protein (25
µg/lane) was separated with 12% SDS-PAGE and transferred onto a
PVDF membrane. The membrane was blocked with 5% skimmed milk for 1
h at room temperature, and subsequently incubated with the primary
antibodies with a dilution of 1:1,000 overnight at 4°C. The
antibodies against HtrA2 (cat. no. ab32092), Bcl-2 (cat. no.
ab59348), Bax (cat. no. ab32503), cleaved caspase-3 (cat. no.
ab49822), caspase-3 (cat. no. ab13847), cleaved caspase-9 (cat. no.
ab2324), caspase-9 (cat. no. ab52298), survivin (cat. no. ab469),
X-linked inhibitor of apoptosis protein (XIAP; cat. no. ab2541),
cytochrome c (cat. no. ab90529), β-actin (cat. no. ab8227)
and cyclooxygenase (Cox) IV (cat. no. ab33985) were obtained from
Abcam. Then, the membranes were incubated with horseradish
peroxidase bound to secondary antibody (1:5,000; cat. no. 7074;
Cell Signaling Technology, Inc.) at room temperature for 2 h. The
protein bands were visualized by an enhanced chemiluminescence
(ECL) kit (Bio-Rad Laboratories, Inc.) and quantified using
Quantity one 4.6.2 software (Bio-Rad Laboratories, Inc.). β-actin
and Cox-IV were used as the internal controls.
Lung edema
After the rats were sacrificed, the right lung was
removed and weighed on an electronic balance to obtain wet weight.
After being held at 60°C for 48 h, the lung was weighed to obtain
the dry weight. The ratio of wet weight/dry weight (W/D) was
considered as the degree of lung edema.
Histopathology
The lung tissues were collected and fixed with 4%
paraformaldehyde at room temperature overnight. After dehydrating
in graded (70–100%) ethanol and embedding in paraffin, samples were
sectioned at 5 µm. The sections were then stained using hematoxylin
and eosin for 5 min at room temperature. The histopathological
changes were assayed under a light microscope (magnification, ×400)
from at least three random fields.
Inflammatory cytokine assay
To determine the levels of tumor necrosis factor
(TNF)-α, interleukin (IL)-6, IL-1β and monocyte chemoattractant
protein-1 (MCP-1), ELISA kits (R&D Systems, Inc.) for IL-6
(cat. no. R6000B), IL-1β (cat. no. RLB00), TNF-α (cat. no. RTA00)
and MCP-1 (cat. no. DY3144-05) were obtained and conducted
according to the manufacturers instructions.
Oxidative stress factor assay
To determine the levels of malondialdehyde (MDA),
reactive oxide species (ROS), lactate dehydrogenase (LDH) and
superoxide dismutase (SOD) the corresponding test kits (Nanjing
Jiancheng Bioengineering Institute) were obtained for analysis
according to the manufacturer's instructions.
TUNEL assay
Paraffin sections of lung tissue were dewaxed,
hydrated, washed with xylene for 5 min and rehydrated with graded
(100-50%) ethanol. Then, 1% Triton-100 and 3%
H2O2-methanol solution was added to the lung
tissue sections. After 15 min, the sections were incubated with
proteinase K solution at 37°C for 30 min. Subsequently, the lung
tissue sections were incubated with TUNEL solution at 37°C in the
dark for 1 h, then DAPI was added to sections for 10 min incubation
at room temperature. The sections were analyzed with a confocal
microscope (magnification, ×400) from at least three random
fields.
ATP level assay
To determine the mitochondrial
adenosine-5′-triphosphate (ATP) level, an ATP-Luciferase Based
Bioluminescence Assay kit (Sigma-Aldrich; Merck KGaA) was applied
to determine the ATP level in accordance with the instructions of
the manufacturer.
Statistical analysis
Data are given as mean ± standard deviation from ≥3
independent experiments. Comparisons among groups were evaluated
with one-way analysis of variance followed by the Tukey-Kramer post
hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
HtrA2 inhibition ameliorates
histopathological changes in lung tissues in the LPS-treated
rats
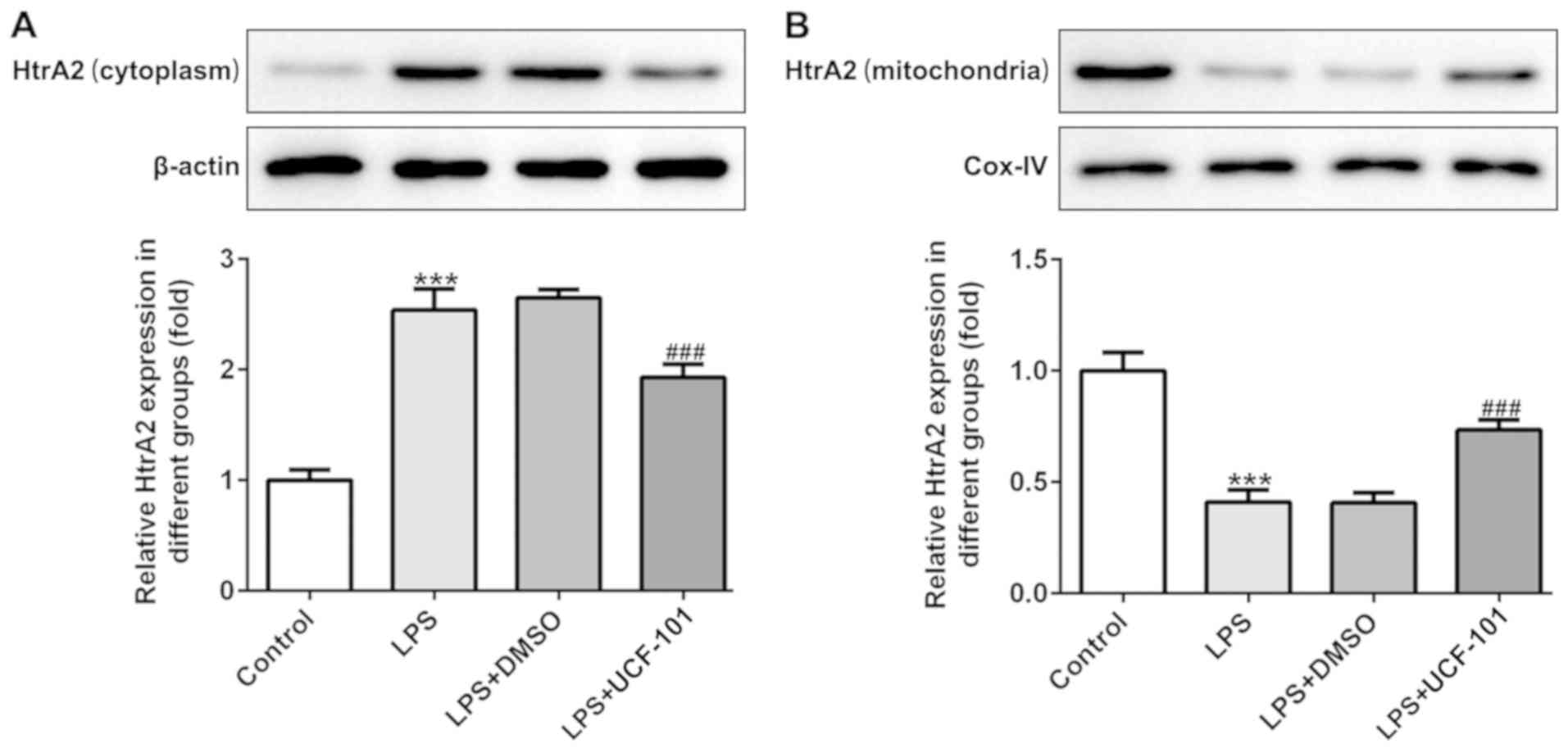
To identify whether HtrA2 was dysregulated in acute
pneumonia, the present study utilized a pneumonia rat model induced
by LPS to detect the expression level of HtrA2. As shown in
Fig. 1A and B, there was an
increased expression of HtrA2 in the cytoplasm but a decreased
expression of HtrA2 in mitochondria, indicating that HtrA2 is
released from the mitochondria to the cytoplasm in pneumonia. The
administration of UCF-101 effectively reversed the dysregulated
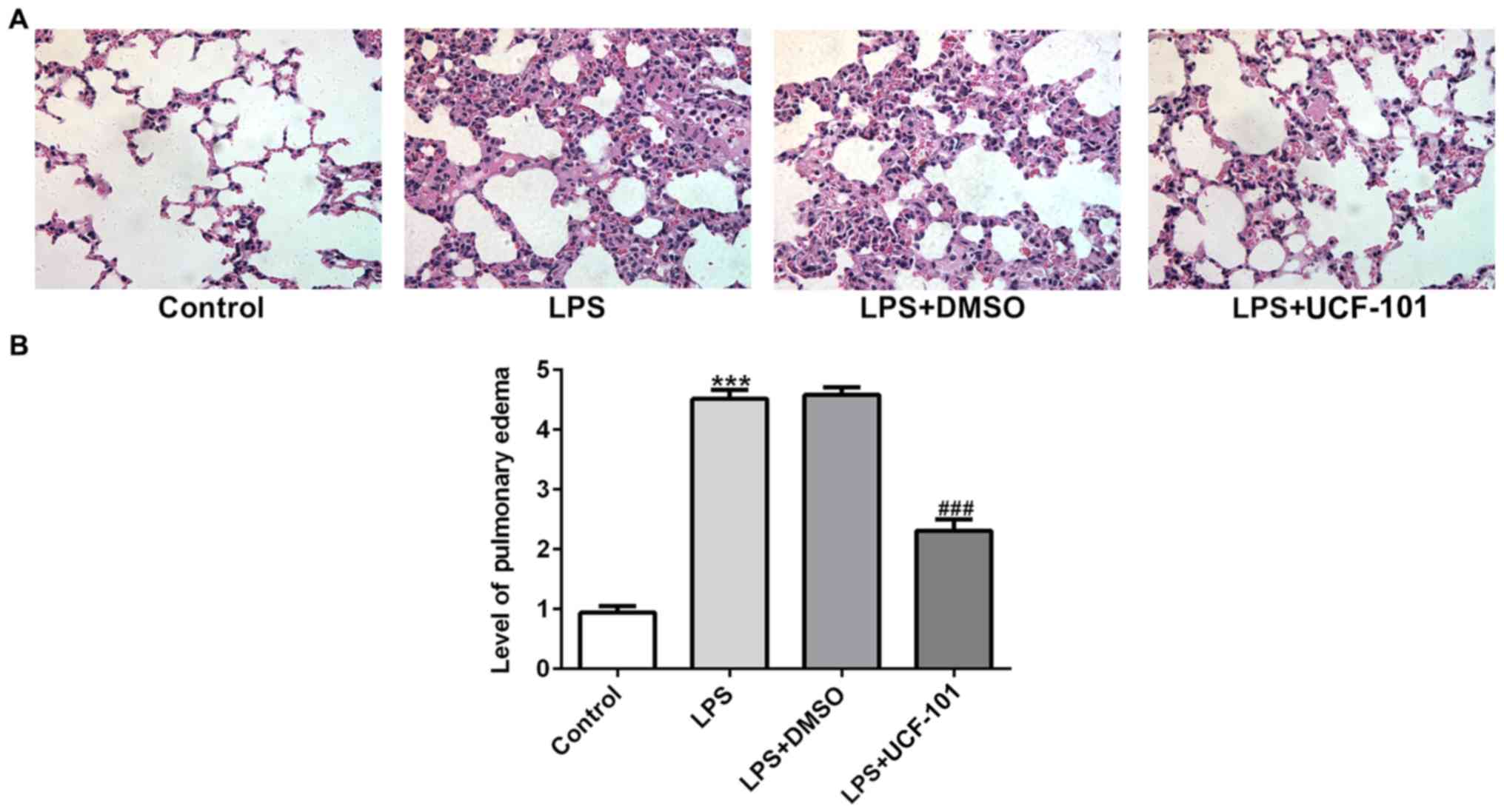
HtrA2. The histopathological analysis from Fig. 2A showed that the control group had
a normal lung tissue structure, and intensive inflammation was
observed in LPS-induced acute pneumonia rats. The administration of
UCF-101 significantly ameliorated these histopathological changes.
The level of pulmonary edema calculated by the W/D ratio was
significantly increased after LPS stimulation, which was
significantly inhibited by treatment of UCF-101 (Fig. 2B).
UCF-101 decreases inflammatory
cytokines in the LPS-treated rats
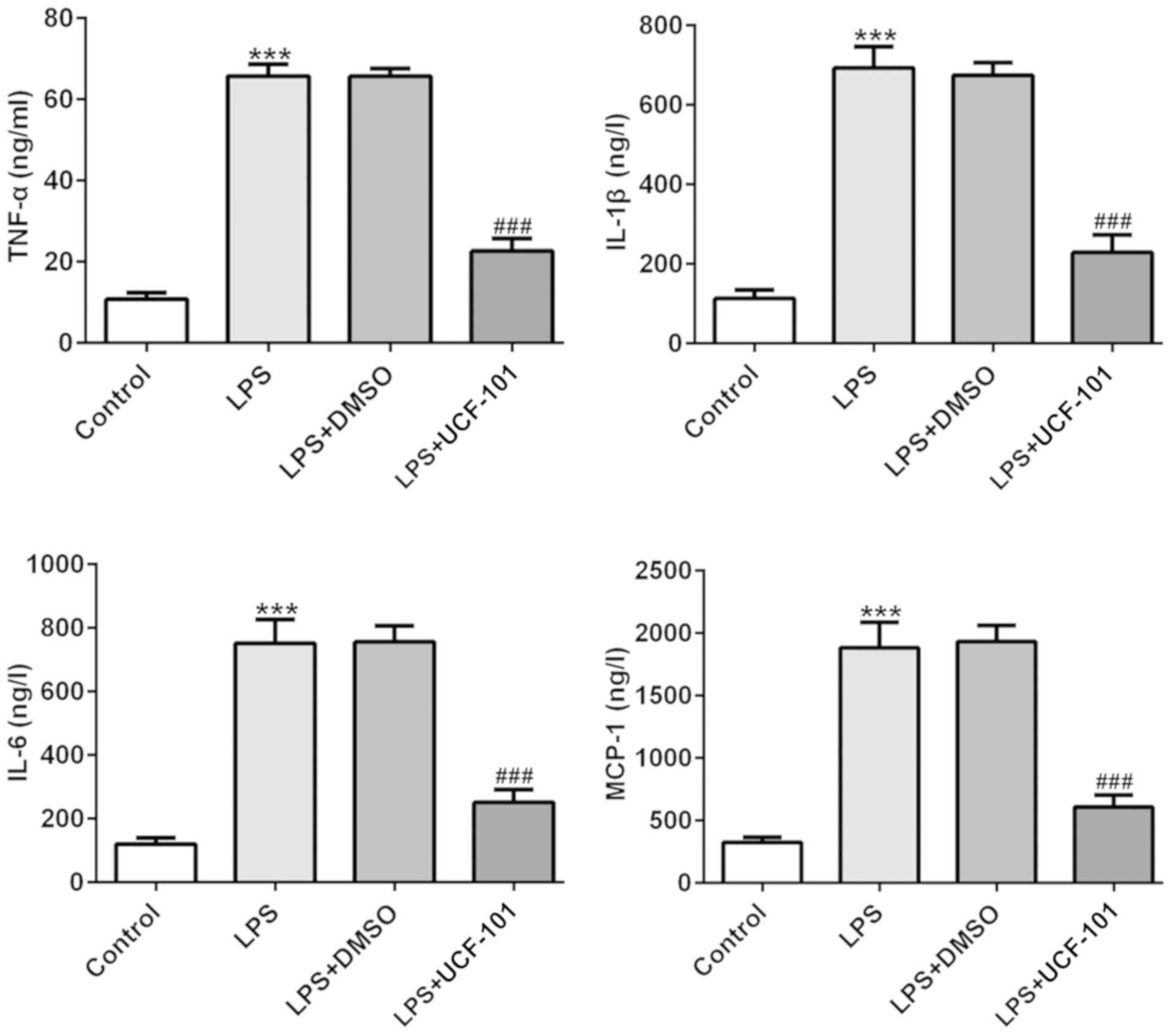
The production of TNF-α, IL-6, IL-1β and MCP-1 were
examined to discover the effect of UCF-101 on LPS-induced
inflammatory response in pneumonia. In Fig. 3 the levels of TNF-α, IL-6, IL-1β
and MCP-1 were significantly increased in lung tissues of the
pneumonia rat after LPS induction. Treatment with UCF-101 reduced
the levels of these LPS-induced cytokines, indicating that UCF-101
alleviated inflammatory response by decreasing inflammatory
cytokine expression in LPS-induced pneumonia.
UCF-101 alleviates oxidative stress in
LPS-treated rats
The effect of UCF-101 on oxidative stress in lung
tissues in LPS-induced pneumonia was also observed. As shown in
Fig. 4, the activities of ROS, MDA
and LDH were significantly increased, while the activity of SOD was
declined, upon induction of LPS. However, these changes were
significantly rescued by the administration of UCF-101. These
results indicated that LPS triggered a severe oxidative stress in
lung tissue, while UCF-101 was able to alleviate the oxidative
stress in the LPS-induced pneumonia rats.
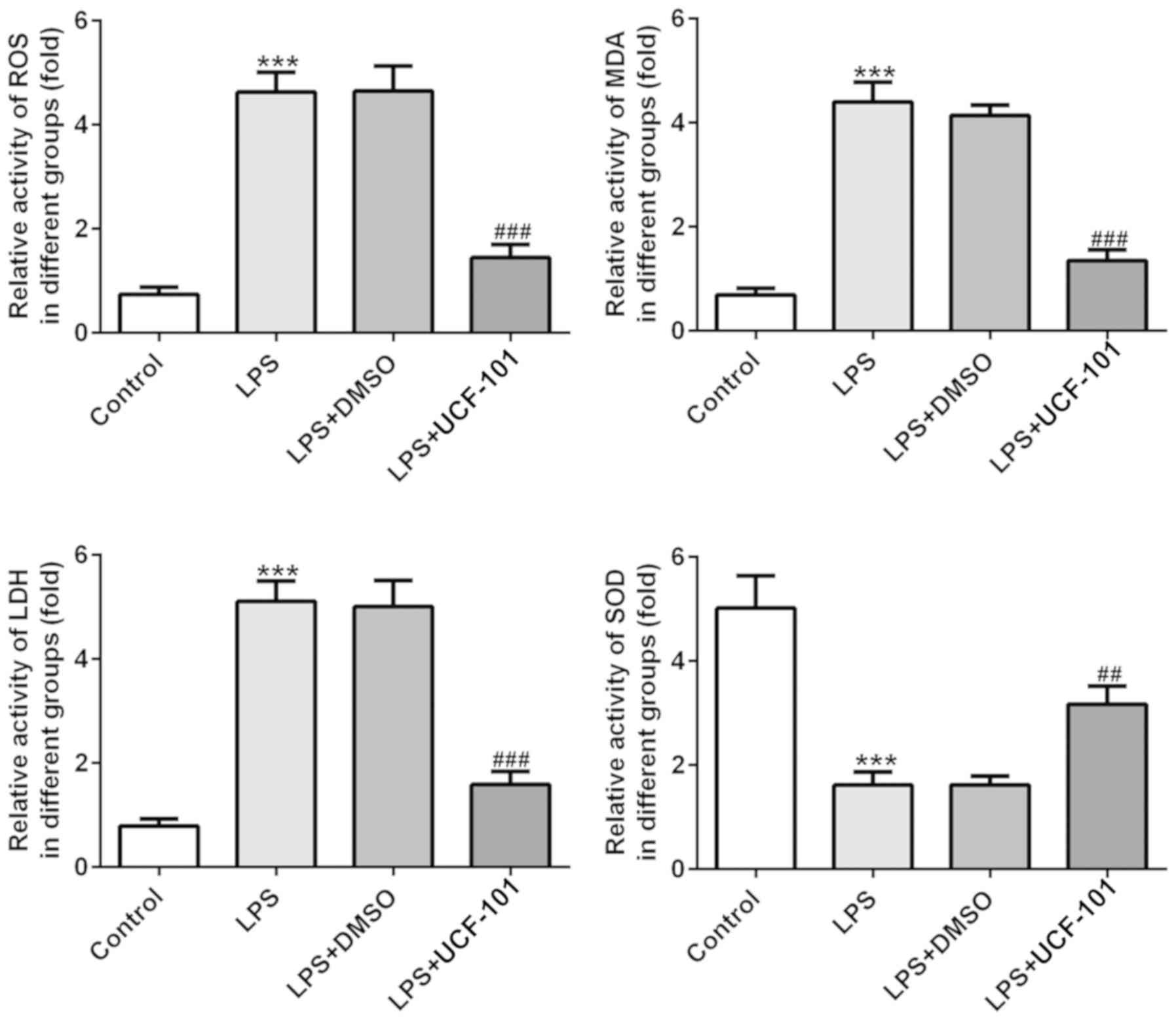 | Figure 4.Anti-oxidant effect of UCF-101 in
pneumonia. The levels of ROS, MDA, LDH and SOD were measured by
corresponding test kits. Data are expressed as mean ± standard
deviation (n=8). ***P<0.001 vs. control, ##P<0.01
and ###P<0.001 vs. the LPS+DMSO group. UCF-101,
5-[5-(2-nitrophenyl)furfuryl iodine]-1,3-diphenyl-2-thiobarbituric
acid; ROS, reactive oxide species; MDA, malondialdehyde; LDH,
lactate dehydrogenase; SOD, superoxide dismutase; LPS,
lipopolysaccharide. |
UCF-101 reduces cell apoptosis in
LPS-treated rats
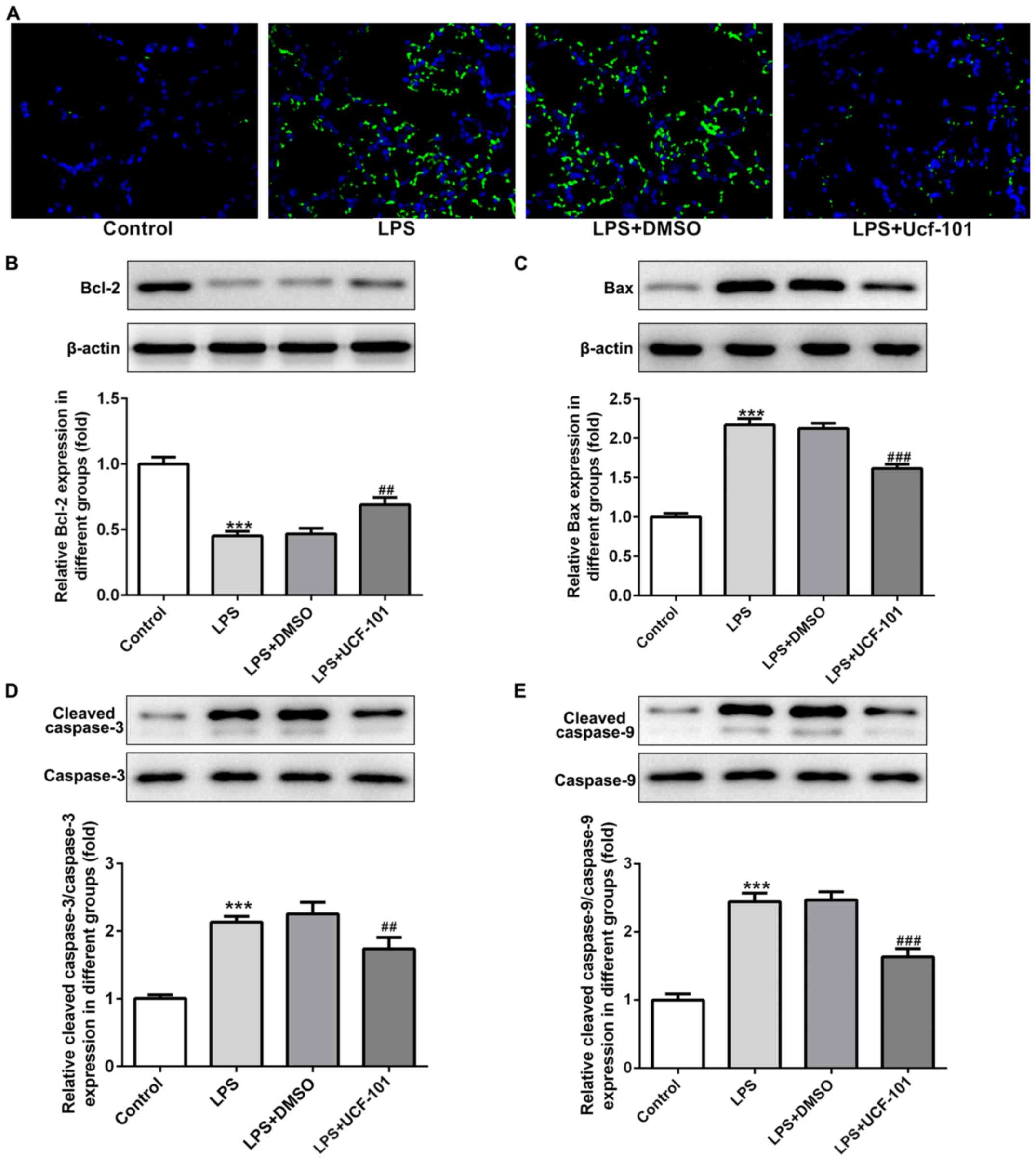
As apoptosis represents an important phenomenon in
pneumonia (12), the apoptosis
condition of lung tissue was explored to examine the anti-apoptotic
capacity of UCF-101 in pneumonia. A large number of apoptotic cells
was found upon the induction of LPS, while the amount of apoptotic
cells was decreased by the administration of UCF-101 (Fig. 5A). The expression of
apoptosis-related proteins showed that Bcl-2 was decreased, and
Bax, cleaved caspase-3 and cleaved caspase-9 were increased upon
the induction of LPS, which were rescued by the treatment of
UCF-101 (Fig. 5B-E), suggesting
that marked apoptosis occurred in LPS-induced acute pneumonia, and
UCF-101 had a potent anti-apoptotic capacity to alleviate the
apoptosis condition.
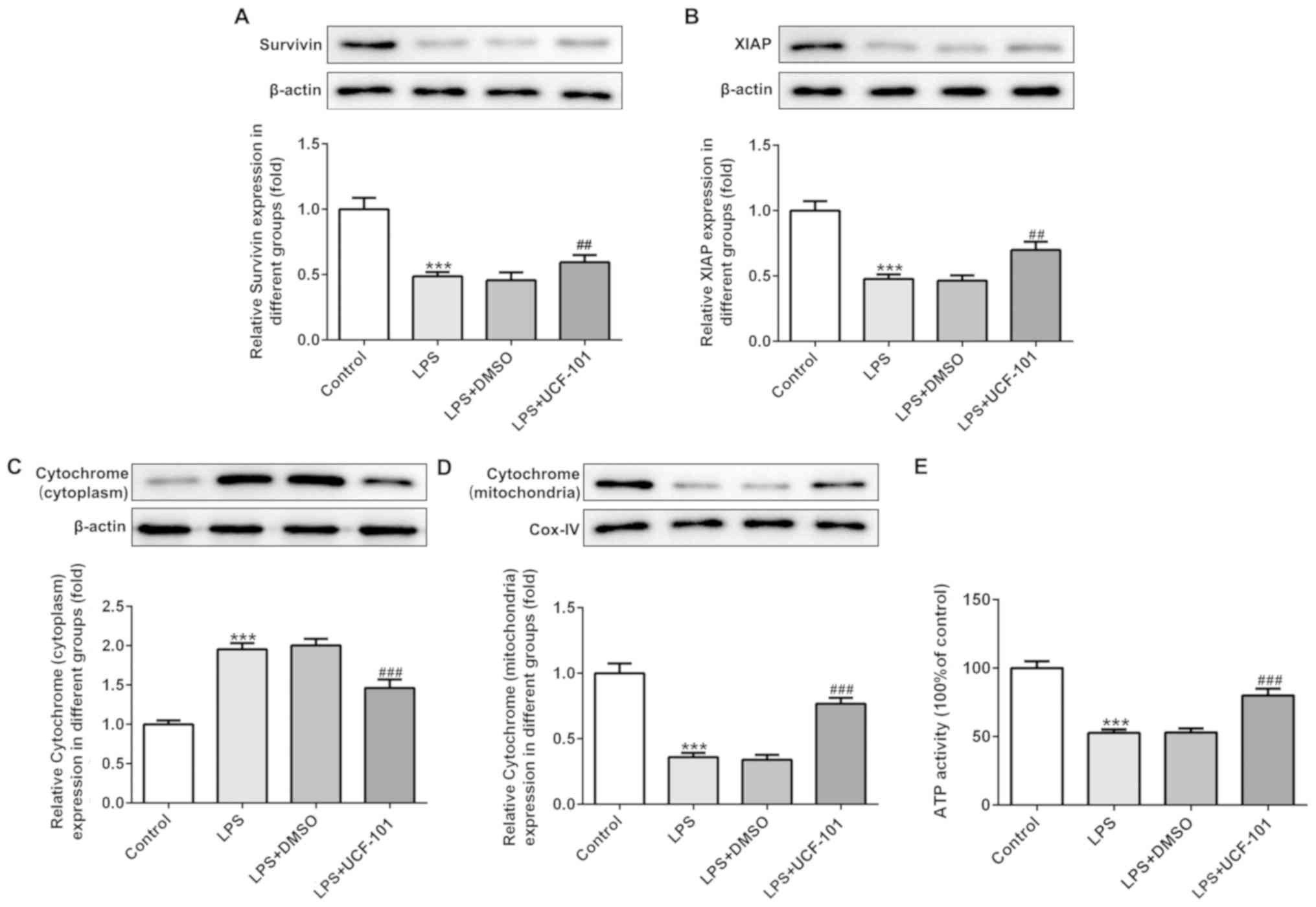
UCF-101 regulates the mitochondrial
apoptosis in pneumonia
The present study next examined the regulatory
effect of UCF-101 on mitochondrial apoptosis in LPS-induced
pneumonia rats. IAPs can regulate caspase activity, thus affecting
apoptosis. Therefore, the expression levels of survivin and XIAP,
two IAP family proteins, were detected. As shown in Fig. 6A and B, both survivin and XIAP were
downregulated upon the induction of LPS, which was reversed by the
administration of UCF-101. The expression of cytochrome c in
the cytoplasm and mitochondria was also detected with western blot
assay. The results showed that LPS significantly increased the
protein expression of cytochrome c in the cytoplasm and
decreased cytochrome c in the mitochondria (Fig. 6C and D), suggesting that LPS
triggered the release of cytochrome c from mitochondria into
cytoplasm. Nevertheless, UCF-101 effectively inhibited the
translocation of cytochrome c from the mitochondria into the
cytoplasm. Additionally, LPS significantly reduced ATP generation,
and the effect caused by LPS was inhibited by the administration of
UCF-101 to a certain extent (Fig.
6E).
Discussion
Pneumonia, a common infectious diseases along with
lower respiratory tract infection, is showing increased incidence
and mortality all over the world, especially in children (13). Thus, the search for novel and
effective therapeutic targets or drugs is urgently required. The
aim of the present study was to understand whether UCF-101, a
specific inhibitor of high-temperature requirement A2 (HtrA2), has
a protective property in lipopolysaccharide (LPS)-induced pneumonia
rats. The present study demonstrated that UCF-101 could effectively
protect lung tissues against LPS-induced inflammatory response,
oxidative stress and cell apoptosis, providing a potential
therapeutic drug for acute pneumonia.
Pulmonary inflammation has always been a
characteristic of pneumonia (14).
Although UCF-101 has been indicated to be anti-inflammatory in the
pathogenesis of sepsis complications and colitis (8,9,15),
the special anti-inflammatory role of UCF-101 in pneumonia remains
to be elucidated. Consistent with the reported studies that UCF-101
suppressed the release of pro-inflammatory cytokines such as TNF-α,
IL-6 and IL-1β in colon tissue in colitis, UCF-101 also decreased
the levels of TNF-α, IL-6, IL-1β and MCP-1 in LPS-induced pneumonia
rats in the present study. Therefore, UCF-101 appeared to alleviate
pulmonary inflammation by inhibiting the production of inflammatory
cytokines.
Reactive oxygen species (ROS) play a pivotal role in
inflammation (16). Under normal
condition, ROS are neutralized by antioxidants so as to maintain
the balance of the oxidant and antioxidant system; under pathologic
conditions, the level of ROS is expected to be increased and the
activity of the antioxidant system decreased, directly leading to
an imbalance trending to oxidation, termed oxidative stress
(17,18). As oxidative stress is an important
process during the pathophysiology of inflammatory diseases,
including pulmonary inflammation (19), the present study examined the
levels of ROS and antioxidant-oxidant enzymes including MDA, LDH
and SOD to verify the effect of UCF-101 on oxidative stress. In
agreement with the literature, the induction of LPS significantly
induced an elevated level of ROS, MDA, LDH and a decrease of SOD in
pneumonia. The administration of UCF-101 effectively alleviated the
production of ROS, MDA, LDH and a decrease of SOD.
Cell function and apoptosis are commonly regulated
by mitochondria in response to stress stimuli such as inflammatory
response and oxidative stress (20). The potential mechanism of how
mitochondria function during the process of apoptosis may be
important for explaining the protective property of UCF-101 on
oxidative stress and inflammatory response in LPS-induced
pneumonia. Previous studies have demonstrated that LPS can lead to
mitochondrial damage in microglial, blood-brain barrier and even
lung tissue, accompanied with injuries, inflammation and cell
apoptosis (21–23). In the present study, evident
apoptosis occurred in lung tissue upon the induction of LPS,
promoting the release of HtrA2 and cytochrome c from
mitochondria to the cytoplasm and increasing the expression of
pro-apoptotic proteins, which may have been responsible for the
subsequent inflammatory response and oxidative stress. The evidence
revealed that HtrA2 was able to trigger apoptosis upon its release
from mitochondria into the cytoplasm, as well as to trigger the
concomitant degradation of XIAP and active caspase-9 and then
downstream caspase-3. Under physiological conditions, caspase-9 and
caspase-3 are retarded by XIAP to block apoptosis at the
post-mitochondrial level (9).
UCF-101 was found to suppress the protease activity of HtrA2 to
reduce caspase-independent apoptosis (24). Wang et al (15) reported that mitochondrial HtrA2
expression is reduced in murine sepsis, together with a
translocation of HtrA2 from mitochondria to the cytoplasm, while
UCF-101 blocks the mobilization of HtrA2 from mitochondria to the
cytoplasm, and reduces XIAP, cleaved caspase-3 and caspase-9 to
alleviate sepsis-associated encephalopathy. Hu et al
(9) demonstrated the
neuroprotective effect of UCF-101 by inhibiting caspase activity
and cell apoptosis to attenuate sepsis-induced cognitive
dysfunction. As expected, the present study found that cleaved
caspase-9 and cleaved caspase-3 were increased in LPS-induced
pneumonia, and this change was reversed by UCF-101. Cytochrome
c is considered a caspase activator and is involved in the
mitochondrial apoptotic pathway, which is modulated by Bcl-2 family
members (25–27). In addition, mitochondria produce
ATP for cellular metabolism under a normal condition (28). The reduced ATP level directly
modulated mitochondrial dysfunction upon LPS stimulation, which was
also reversed by UCF-101. Therefore, the inhibition of HtrA2 was
effective in recovering mitochondrial function and restoring
mitochondrial-related apoptosis. The administration of UCF-101 had
a protective role in LPS-pneumonia by regulating mitochondrial
apoptosis.
Overall, the present study indicated that UCF-101
acted as a positive regulator of acute pneumonia by inhibiting the
inflammatory response, oxidative stress and mitochondrial
apoptosis. The findings suggest UCF-101 as a potential candidate
for pneumonia therapy.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors contributions
XW designed and performed the experiments, and wrote
the manuscript. The author read and approved the final
manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Shengjing Hospital of China Medical University.
Patient consent for publication
Not applicable.
Competing interests
The author declares that he has no competing
interests.
References
|
1
|
Lutfiyya MN, Henley E, Chang LF and
Reyburn SW: Diagnosis and treatment of community-acquired
pneumonia. Am Fam Physician. 73:3127–450. 2006.
|
|
2
|
Agweyu A, Kibore M, Digolo L, Kosgei C,
Maina V, Mugane S, Muma S, Wachira J, Waiyego M and Maleche-Obimbo
E: Prevalence and correlates of treatment failure among Kenyan
children hospitalised with severe community-acquired pneumonia: A
prospective study of the clinical effectiveness of WHO pneumonia
case management guidelines. Trop Med Int Health. 19:1310–1320.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bhutta ZA, Das JK, Walker N, Rizvi A,
Campbell H, Rudan I and Black RE; Lancet Diarrhoea and Pneumonia
Interventions Study Group, : Interventions to address deaths from
childhood pneumonia and diarrhoea equitably: What works and at what
cost? Lancet. 381:1417–1429. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clausen T, Southan C and Ehrmann M: The
HtrA family of proteases: Implications for protein composition and
cell fate. Mol Cell. 10:443–455. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bhuiyan MS and Fukunaga K: Mitochondrial
serine protease HtrA2/Omi as a potential therapeutic target. Curr
Drug Targets. 10:372–383. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ding X, Patel M, Shen D, Herzlich AA, Cao
X, Villasmil R, Klupsch K, Tuo J, Downward J and Chan CC: Enhanced
HtrA2/Omi expression in oxidative injury to retinal pigment
epithelial cells and murine models of neurodegeneration. Invest
Ophthalmol Vis Sci. 50:4957–4966. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li Q, Hueckstaedt LK and Ren J: The
protease inhibitor UCF-101 ameliorates streptozotocin-induced mouse
cardiomyocyte contractile dysfunction in vitro: Role of
AMP-activated protein kinase. Exp Physiol. 94:984–994. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang C, He A, Liu S, He Q, Luo Y, He Z,
Chen Y, Tao A and Yan J: Inhibition of HtrA2 alleviated dextran
sulfate sodium (DSS)-induced colitis by preventing necroptosis of
intestinal epithelial cells. Cell Death Dis. 10:3442019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hu Y, Huang M, Wang P, Xu Q and Zhang B:
Ucf-101 protects against cerebral oxidative injury and cognitive
impairment in septic rat. Int Immunopharmacol. 16:108–113. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the care and use of laboratory animals (8th
edition). National Academies Press (US); Washington, DC: 2011
|
|
11
|
Kowluru RA and Abbas SN: Diabetes-induced
mitochondrial dysfunction in the retina. Invest Ophthalmol Vis Sci.
44:5327–5334. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Qin Z, Yang Y, Wang H, Luo J, Huang X, You
J, Wang B and Li M: Role of autophagy and apoptosis in the
postinfluenza bacterial pneumonia. Biomed Res Int.
2016:38010262016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Walker CLF, Rudan I, Liu L, Nair H,
Theodoratou E, Bhutta ZA, OBrien KL, Campbell H and Black RE:
Global burden of childhood pneumonia and diarrhea. Lancet.
381:1405–1416. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wolf L, Sapich S, Honecker A, Jungnickel
C, Seiler F, Bischoff M, Wonnenberg B, Herr C, Schneider-Daum N,
Lehr CM, et al: IL-17A-mediated expression of epithelial IL-17C
promotes inflammation during acute pseudomonas aeruginosa
pneumonia. Am J Physiol Lung Cell Mol Physiol. 311:L1015–L1022.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang P, Hu Y, Yao D and Li Y: Omi/HtrA2
regulates a mitochondria- dependent apoptotic pathway in a murine
model of septic encephalopathy. Cell Physiol Biochem. 49:2163–2173.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hussain T, Tan B, Yin Y, Blachier F,
Tossou MC and Rahu N: Oxidative stress and inflammation: What
polyphenols can do for us? Oxid Med Cell Longev. 2016:74327972016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Floyd RA: Antioxidants, oxidative stress,
and degenerative neurological disorders. Proc Soc Exp Biol Med.
222:236–245. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Boskabadi J, Mokhtari-Zaer A, Abareshi A,
Khazdair MR, Emami B, Roshan NM, Hosseini M and Boskabady MH: The
effect of captopril on lipopolysaccharide-induced lung
inflammation. Exp Lung Res. 44:191–200. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ahmad A, Shameem M and Husain Q: Relation
of oxidant-antioxidant imbalance with disease progression in
patients with asthma. Ann Thorac Med. 7:226–232. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tao-Cheng JH: Stimulation-induced
structural changes at the nucleus, endoplasmic reticulum and
mitochondria of hippocampal neurons. Mol Brain. 11:442018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou D and Jiang Y: Sirtuin 3 attenuates
neuroinflammation- induced apoptosis in BV-2 microglia. Aging
(Albany NY). 11:9075–9089. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
You M, Miao Z, Pan Y and Hu F:
Trans-10-hydroxy-2-decenoic acid alleviates LPS-induced blood-brain
barrier dysfunction by activating the AMPK/PI3K/AKT pathway. Eur J
Pharmacol. 865:1727362019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Luo X, Liu R, Zhang Z, Chen Z, He J and
Liu Y: Mitochondrial division inhibitor 1 attenuates mitophagy in a
rat model of acute lung injury. Biomed Res Int. 2019:21937062019.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cilenti L, Lee Y, Hess S, Srinivasula S,
Park KM, Junqueira D, Davis H, Bonventre JV, Alnemri ES and Zervos
AS: Characterization of a novel and specific inhibitor for the
pro-apoptotic protease Omi/HtrA2. J Biol Chem. 278:11489–11494.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Saelens X, Festjens N, Walle LV, van Gurp
M, van Loo G and Vandenabeele P: Toxic proteins released from
mitochondria in cell death. Oncogene. 23:2861–2874. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brunelle JK and Letai A: Control of
mitochondrial apoptosis by the Bcl-2 family. J Cell Sci.
122:437–441. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin PY, Tsai CT, Chuang WL, Chao YH, Pan
IH, Chen YK, Lin CC and Wang BY: Chlorella sorokiniana induces
mitochondrial-mediated apoptosis in human non-small cell lung
cancer cells and inhibits xenograft tumor growth in vivo. BMC
Complement Altern Med. 17:882017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Krause J, Löser A, Lemoine MD, Christ T,
Scherschel K, Meyer C, Blankenberg S, Zeller T, Eschenhagen T and
Stenzig J: Rat atrial engineered heart tissue: A new in vitro model
to study atrial biology. Basic Res Cardiol. 113:412018. View Article : Google Scholar : PubMed/NCBI
|