Introduction
Studies have reported some key breakthroughs in
tumor immunological therapies for lung cancer (1,2). For
example, a blockade of immune checkpoint therapy regulated T cell
activity to enhance the antibody-tumor immune responses.
Significant advances in the molecular characterizations of lung
cancer have led to the creation of effective immunotherapies that
assist in the recognition of cancer as foreign by the host immune
system, stimulate the immune system and relieve the inhibition that
allows tumor growth and spread (3). However, immunotherapies have a number
of limitations in clinical applications due to the existence of
tumor heterogeneity and immunological suppression (4,5). In
a previous study that focused on tumor immunological genomics, the
assessment of tumor immune infiltrating cells served a key role in
investigating the way in which the interactions between the tumor
and the immune system affect patient outcome (6).
In an investigation into lung cancer and its immune
regulations, a previous study demonstrated the significance of CpG
islands in the lung immune responses of granulomatous lung diseases
(7). Simultaneously, the study
indicated that long non-coding RNAs (lncRNAs) serve an important
role in directing the development of various immune cells and
controlling dynamic transcriptional programs and were hallmarks of
immune cell activation (7). In
addition to the direct regulation of lncRNAs by DNA methylation via
interactions with their promoter regions, previous studies have
revealed several intricate regulatory associations between lncRNAs
and DNA methylation (8,9). In the cis-methylated lncRNAs
group, DNA methylation adjacent to a lncRNA locus, such as the
promoter or the imprinting control region, directly modulate the
expression levels of target lncRNAs as a cis-regulator
(10). In trans-methylation
due to lncRNAs, lncRNAs regulate the DNA methylation of a
trans-genomic locus as an intermedium by recruiting DNA
(cytosine-5-)-methyltransferase (11,12).
In the trans-methylation-regulated lncRNAs group, an
alteration in the DNA methylation state at a specific genomic locus
regulates the transcription of its antisense-oriented lncRNAs
(13,14). The lncRNA extracoding CEBPA
transcribed from the CEBPA gene locus is reported to be
critical for the regulation of DNA methylation at this site through
interactions with DNA methyltransferase 1 (15).
Based on the interactions between DNA methylation
and lncRNAs, it is easy to investigate the role of methylation on
the tumor immune responses from the perspective of lncRNAs.
Currently, the competitive endogenous (ce)RNA hypothesis provides
an approach to study the regulatory mechanisms of RNA molecules
(16). ceRNA is an RNA with a
binding site that competitively binds to a micro (mi)RNA to inhibit
its target gene regulation (16).
Pseudogenes, lncRNAs, miRNAs and other types of RNAs can be
regulated by ceRNA mechanisms (7).
Therefore, the present study investigated the roles of DNA
methylation in tumor immune cell infiltration abundance and its
association with patient prognosis from the novel perspective of
RNA molecules, DNA methylation and their regulatory mechanisms.
The present study investigated the cause behind the
differences in tumor immune responses from a new perspective. The
present study investigated how DNA methylation affected the
interactions between tumors and immune cells and between RNA
molecules. Starting with DNA methylation, the present study
revealed DNA methylation abnormalities, differentially expressed
RNA (DERNA) molecules and mutual regulatory associations between
DNA methylation and RNA molecules and assessed potential
associations between DNA methylation and tumor immune infiltration
in LUAD.
Materials and methods
Acquisition and preprocessing of the
data
LUAD transcriptome, miRNA, DNA methylation and
associated clinical data were downloaded from The Cancer Genome
Atlas (TCGA) website (TCGA-LUAD; portal.gdc.cancer.gov/). The transcriptome data
contained 594 samples (59 normal samples and 535 tumor samples),
the miRNA data contained 577 samples (46 normal samples and 531
tumor samples) and the DNA methylation data included 507 samples
(30 normal samples and 477 tumor samples). These data were
downloaded on April 1, 2018. For the original data, most of the
data with RNA expression level=0 were deleted. For the
transcriptome data, the RSEM-preprocessed per million transcript
method was used to isolate and obtain lncRNA and mRNA expression
data (17).
Extraction of the differential
methylation regions and associated lncRNAs
For the preprocessed DNA methylation data,
differential DNA methylation regions and associated lncRNAs were
identified using the lncDM algorithm in a package in R 3.5.3 in
March 11, 2019. LncDM is a novel computational method for
identifying differentially methylated regions in specific diseases.
LncDM uses the Illumina HumanMethylation450 BeadChip GENCODE of all
annotation information based on the reannotation method and obtains
the CpG values of the differential transcript (18). The gene functional regions were
used to calculate the patterns of differential methylation. The
screening criteria for the differentially methylated regions were
adjusted to P<0.05 and β diffcut >0.3. The β diffcut value
was used to reflect the differential situations between
samples.
Extraction of the abnormally expressed
RNA molecules
For those RNA molecules in original data that were
subjected to deletion processing, DERNA were screened using the
DEseq2 algorithm in R software. The DEseq2 method uses shrinkage to
estimate fold change, thus improving the stability and
interpretability of the DEseq-based estimation (19). The screening criteria for the DERNA
were |log FC|>2 and P<0.05.
Selection of the immune
cell-associated genes
To investigate the cause of tumor heterogeneity and
its association with prognosis, the immune infiltration abundance
of relevant LUAD tumor samples needed to be calculated. In this
process, the TIMER 1.0 algorithm was used (20). First, the CHAT package was used to
estimate the tumor purity of the samples (21), then the COMBAT package was used to
combine all relevant immune cell gene expression data to eliminate
the effect of batch processing of different data (22). Genes negatively correlated with
tumor purity were selected and matched with some known immune cell
(B cell, CD4+ and CD8+ T cell, neutrophil, macrophage and dendritic
cell) tag genes to obtain 6 immune cell-associated genes.
Construction of the DNA methylation
and immune-associated ceRNA network
To construct the ceRNA network, multiple databases
were used to investigate the inter-relationships between DERNA
molecules and DNA methylation. First, the differential methylation
region (DMR)-DElncRNA relationship pairs were identified by
employing the Lnc2Meth 1.0 (bio-bigdata.hrbmu.edu.cn/Lnc2Meth/) database (18). Next, the DElncRNA-DEmiRNA
relationship pairs were identified by searching the miRcode 1.0
(mircode.org/) database (23). miRNAs were then annotated with 3p
and 5p, which were used to distinguish between the 3′ and 5′ arms
of miRNA precursors that target different sites, using the Starbase
v2.0 (starbase.sysu.edu.cn/) database
(24). Finally, the DEmiRNA-DEmRNA
relationship pairs (25–27) were identified using miRDB v3.0
(mirdb.org/), miRTarBase 1.0 (mirtarbase.mbc.nctu.edu.tw/) and TargetScan v7.2
(targetscan.org/vert_72/). The
visualization of this ceRNA network was achieved using Cytoscape
version 3.6.0 software (28). In
order to study the overall survival for the associated RNA
molecules in the network, the present study used the Renyi test
(29), a weighted test method, to
generate survival curves.
Clinical analysis of the immune cell
genes associated with DNA methylation
In order to investigate the effects of DNA
methylation-associated tumor immune infiltration on clinical data,
a multivariate regression model with associated factors, such as
infiltration abundance, tumor purity, stage, infection status, age
and sex was constructed. In this process, a single-factor Cox
analysis was performed on the differential DNA
methylation-associated immune genes. Those associated mRNAs with
P-value <0.0001 were screened as clinically relevant genes. A
multivariate risk regression analysis was then performed based on
these clinically relevant genes. The theoretical basis of this
model was the semiparametric regression model proposed by the
British statistician D.R. Cox (30). The dependent variables of this
model were the overall survival rates and result of survival. This
model had the advantage of analyzing the influences of multiple
factors on overall survival rates and the truncation analysis of
overall survival rates simultaneously. Finally, Kaplan-Meier curves
were used to verify the overall survival rates within 5 years.
Kaplan-Meier curves were generated using the log-rank test. These
aforementioned processes were completed with the survival package
3.2–3 of R software (CRAN.R-project.org/package=survival).
Functional enrichment analysis
For the immune genes associated with DNA
methylation, the biological functions of genes were analyzed using
the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene
Ontology (GO) functional enrichment (geneontology.org/). The KEGG (kegg.jp/) and GO
functional enrichment analyses were performed by using the Database
for Annotation, Visualization and Integrated Discovery (DAVID;
v6.8) website (31).
Results
Extraction of DERNAs and differential
DNA methylation regions
To understand the RNAs and methylation regions that
have undergone significant changes in tumor and control tissues,
differential data were first extracted. The extraction process of
differential data involved extracting DERNAs, DNA methylation
regions and the regulatory correlations between DNA methylation
regions and lncRNAs. In the process of extraction, DERNA molecules
in LUAD, such as DElncRNAs, DEmiRNAs and DEmRNAs were screened
using the DEseq2 method in R software. The screening criteria for
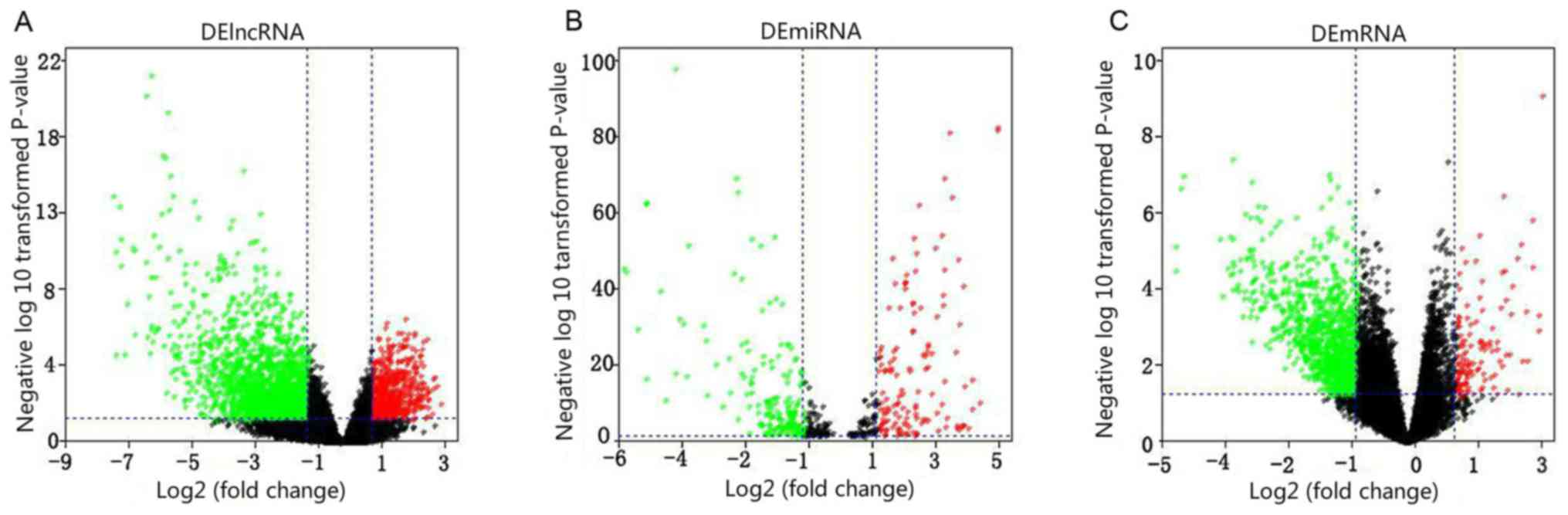
DERNA molecules were |log FC|>2 and P<0.05 (Fig. 1). A total of 1,187 DElncRNAs, 133
DEmiRNAs and 1,474 DEmRNAs were obtained. The extracted RNA
molecules were increased or decreased expressed in the patients
with LUAD. It can be inferred that the abnormal expression levels
of RNA molecules were associated with LUAD.
The processed DNA methylation data were used to
screen for differential DNA methylation regions and the
corresponding lncRNAs using the lncDM method in R software. Those
methylation regions were removed when methylation differences were
<0.05. The detailed information regarding the DMRs included the
methylation regions, the associated lncRNAs and the methylation
difference are presented in Table
SI. During the process, 1,692 DMRs associated with the lncRNAs
were extracted. It was hypothesized that the increased or decreased
expression levels of lncRNAs were associated with abnormal
methylation.
Construction of the immune-associated
ceRNA network
In order to investigate the regulatory mechanisms of
gene expression associated with immune cells from the perspective
of DNA methylation, a DNA methylation and immune-associated ceRNA
network was constructed based on the internal competitive RNA
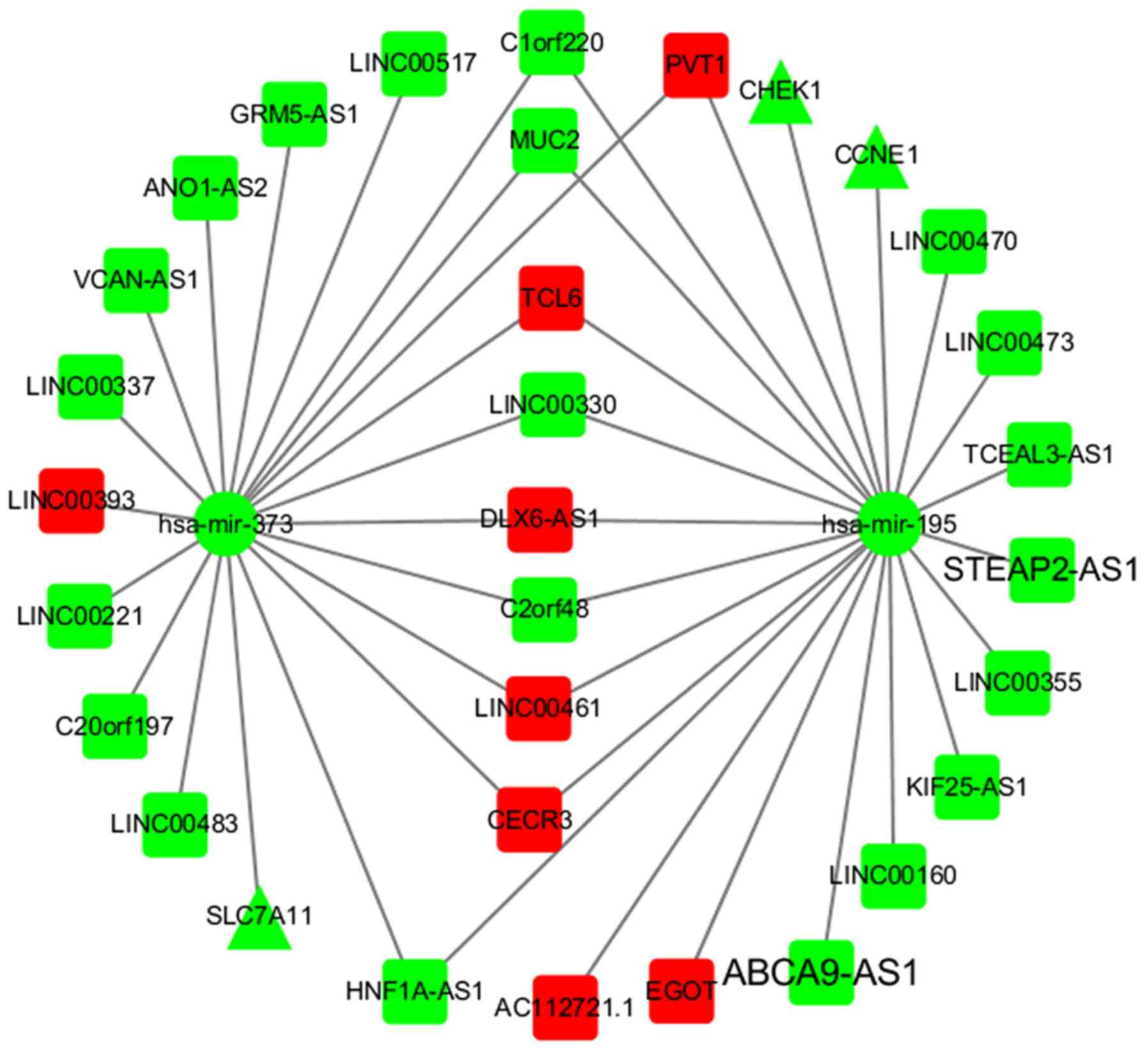
hypothesis (Fig. 2). By matching
the Lnc2Meth 1.0 database, 1,692 DMR-DElncRNA relationship pairs
were identified, of which there were 64 lncRNAs. In addition, 285
DElncRNA-DEmiRNA relationship pairs were identified by matching the
Micode 1.0 database, of which there were 21 miRNAs and 64 lncRNAs
in DElncRNA-DEmiRNA relationship pairs. In addition, 1,177
DEmiRNA-DEmRNA relationship pairs were obtained by searching the
databases of interactions between miRNAs and mRNAs, of which there
were 21 miRNAs and 92 mRNAs.
Then, the RNA molecules in the aforementioned
relationship pairs were matched with the increased or decreased
expressed RNA molecules yielding abnormally expressed relationship
pairs. The abnormal expression levels of RNA molecules in
relationship pairs are presented in Tables SII, SIII and SIV. The expression levels of CHEK1,
SLC7A11 and CCNE1 were much lower in LUAD samples
compared with normal samples (Table
SII). The expression levels of hsa-miR-373 and
hsa-miR-195 were much lower in LUAD samples compared with
normal samples (Table SIII). The
expression levels of LINC00377, LINC00473 and
LINC00355 were much lower in LUAD samples compared with
normal samples (Table SIV). The
changes in DNA methylation regions and corresponding RNAs of the
ceRNA network are presented in Table
SV.
According to the abnormally expressed relationship
pairs, the ceRNA network was constructed. In order to improve the
current understanding of the ceRNA network, the visualization of
this ceRNA network was achieved using Cytoscape version 3.6.0
software (27). This network
included 3 mRNAs, 2 miRNAs and 29 lncRNAs. It can be inferred that
low expression levels of the lncRNA LINC00337 were associated with
hypomethylation in the corresponding DNA regions (Fig. 2). The miRNAs (hsa-miR-372,
hsa-miR-373) can bind to mRNAs (SLC7A11, CHEK1
and CCNE1). The lncRNAs (LINC00337 and
LINC00355) may bind to miRNAs (Fig. 2).
The associated RNA molecules in this network were
critical for investigating immunity and DNA methylation. The
present study used the Renyi test (29), a weighted test method, to generate
survival curves for the associated RNA molecules in the network
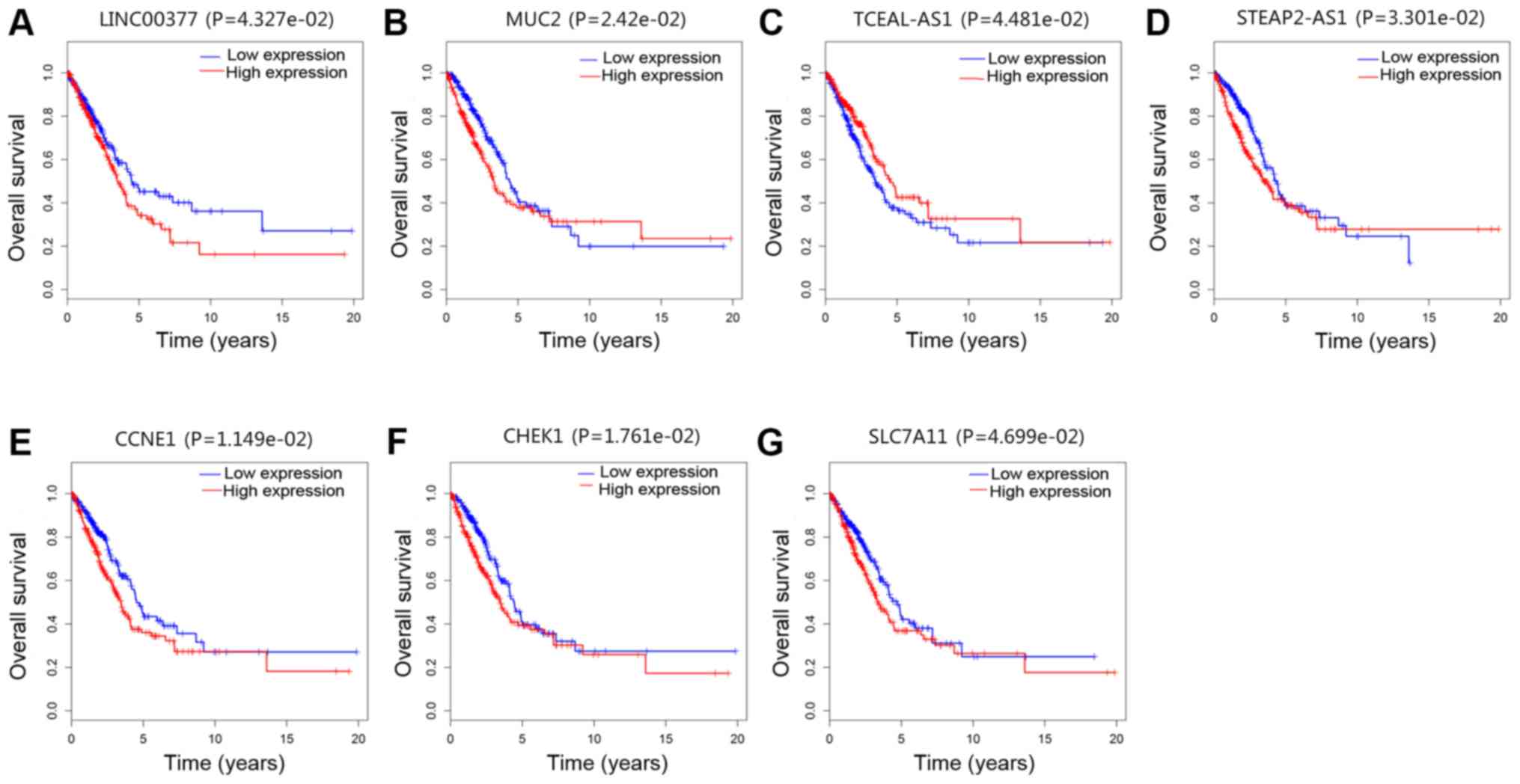
(Fig. 3). In the present study,
the Renyi test weight is 1.
The criterion for expression levels of RNA molecules
was derived from the criteria in the premier increased or decreased
expression level extraction of RNA molecules. The screening
criteria for DERNA molecules were |log FC|>2 and P<0.05,
which indicated that the expression levels of the RNAs were
evidently increased or decreased in LUAD samples. Survival curves
of all the RNA in the network were generated and survival curves
with P<0.05 were selected as this indicated that abnormal RNA
expression levels were significantly associated with survival rates
in the LUAD samples.
The patients with LUAD that exhibited low expression
levels of mRNA CCNE1 (Fig.
3E), CHEK1 (Fig. 3F)
and SLC7A11 (Fig. 3G) had
improved survival rate. The patients with LUAD that exhibited high
expression levels of TCEAL3-AS1 (Fig. 3C) had improved survival time. The
patients with LUAD that exhibited low expression levels of lncRNA
LINC00337 had improved survival rate (Fig. 3A). These results indicated that the
increased or decreased expression levels of immune cells genes
affected the survival rate of patients with LUAD and suggested that
the interactions between DNA methylation and RNA molecules may
affect the responses to immunotherapies. The associated mRNAs can
be considered potential markers of immunotherapies in LUAD. It was
also verified that the RNAs in the network influenced the survival
rate of patients with LUAD.
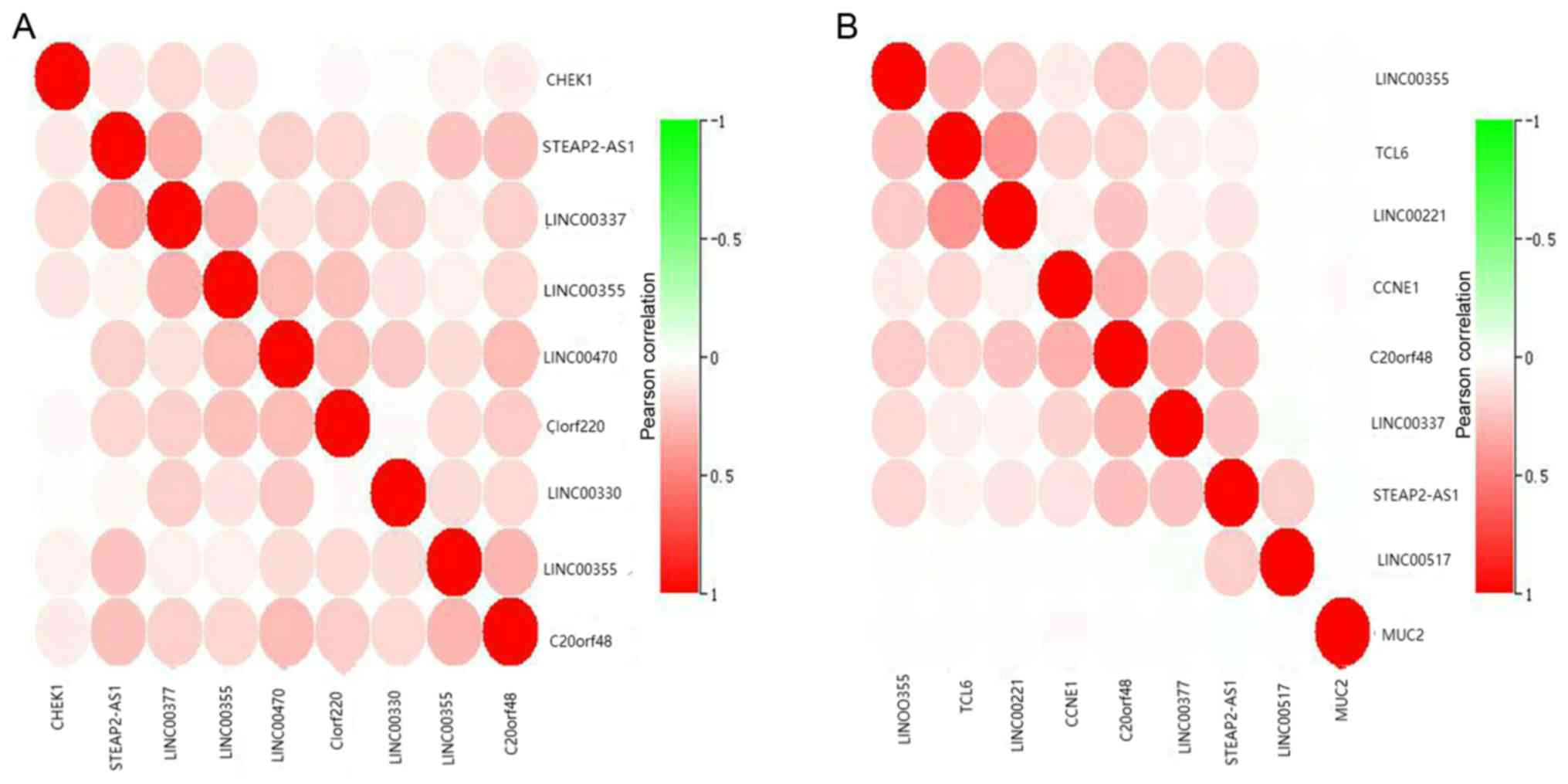
In order to investigate the expressional
relationships between the RNA molecules, correlational analyses of
the potential mRNAs and lncRNAs in the network were performed.
There may be several competitive relationships in these RNA
molecules. The expression levels of CHEK1 were positively
correlated with all the associated lncRNA molecules including
LINC00473, LINC00337 and LINC00355. Among them,
CHEK1 was strongly correlated with LINC00337
(Fig. 4A). There were also
positive correlations between the expression levels of CCNE1
and the majority of its associated lncRNAs, including C20orf48,
LINC00377 and TCL6 (Fig.
4B). These results verified the hypothesis of the present study
based on the ceRNA hypothesis. It can be inferred that
LINC00337 affects the expression levels of CHEK1 by
competing with the binding of hsa-miR-373 and
hsa-miR-195 from the ceRNA network and competitive
relationships. Concerning the changes in DNA methylation regions
and the corresponding lncRNAs, it can be deduced that the
hypomethylation of the 1101320–1104290 regions of chromosome 1
resulted in the low expression levels of LINC00337, and that
LINC00337 may have affected the expression levels of
CHEK1 by competitively binding with hsa-miR-373 and
hsa-miR-195, thereby affecting tumor immune cells and
overall survival rates in patients with LUAD.
DNA methylation-associated immune
genes analysis
In the process of constructing the ceRNA network,
the correlations between RNA molecules were obtained by searching a
large number of databases. In addition, 181 mRNAs associated with
tumor immunity and clinical outcomes were identified. In order to
investigate how these associated mRNAs influence survival rates in
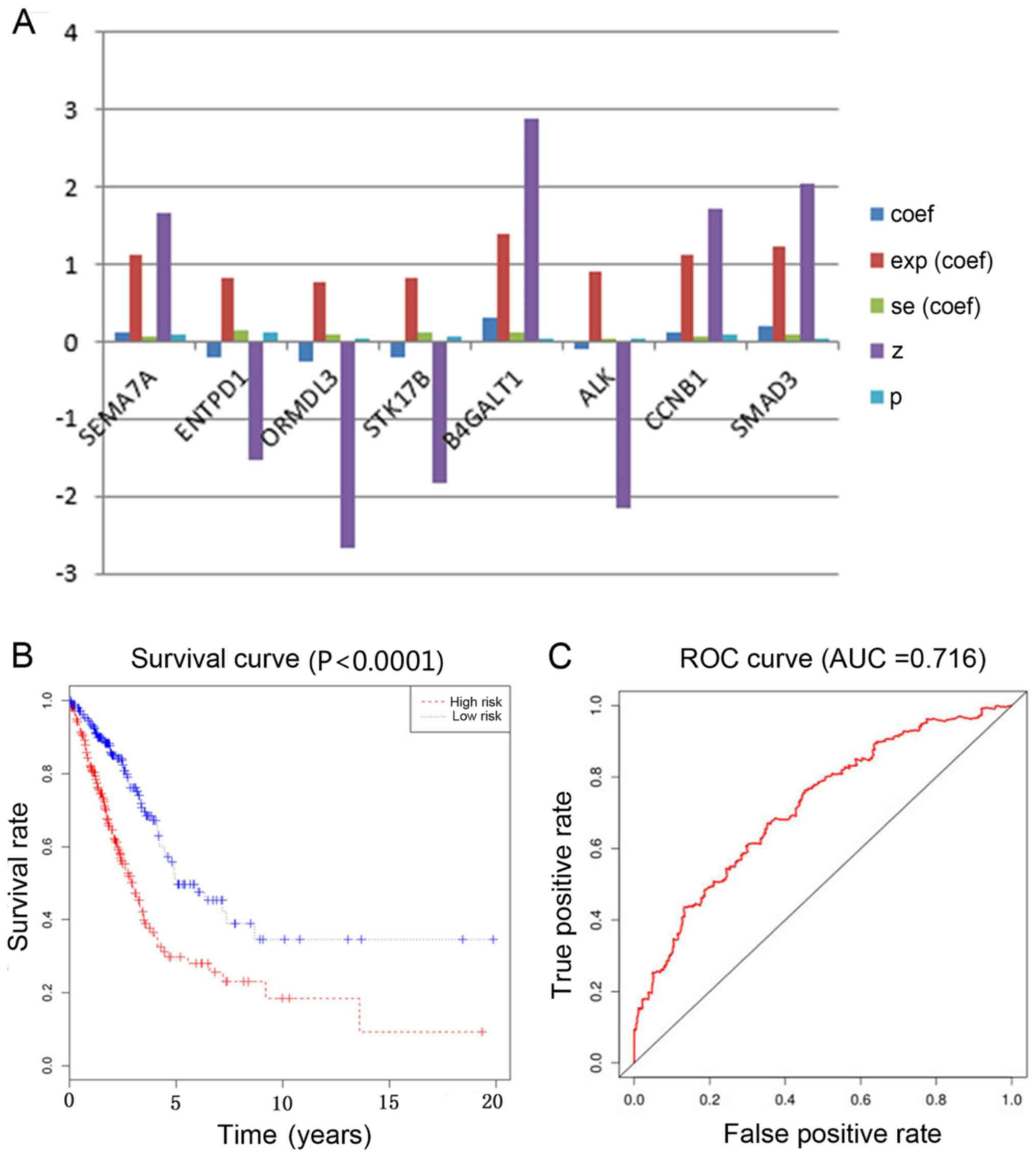
patients with LUAD, a multivariate Cox regression model was
performed using 8 mRNA expression values (SEMA7A, ENTPD1,
ORMDL3, STK17B, B4GALT1, ALK, CCNB1, SMAD3) to reflect the
associations between gene expression levels and the survival rates
in patients with LUAD (Fig. 5A). A
survival rates analysis between the high-risk group and the
low-risk group was conducted (Fig.
5B). The samples were divided based on the median value of the
risk values. The risk value was derived from the multivariate Cox
regression analysis. The cut-off value for high-risk samples was a
risk value >0.956. Based on the survival curves between
high-risk and low-risk patients, it can be seen that low-risk
patients have improved overall survival rates. In the present
study, the area of the ROC curves was 0.716 (Fig. 5C). The area of the ROC curve
indicated the accuracy of diagnostic methods. The criterion of
trustworthy accuracy was when the area of the ROC curves >0.7.
The prediction of survival outcomes had reliable accuracy in
patients with LUAD. The results demonstrated that the expression
levels of the DNA methylation-associated immune cells genes
affected the prognosis of patients with LUAD.
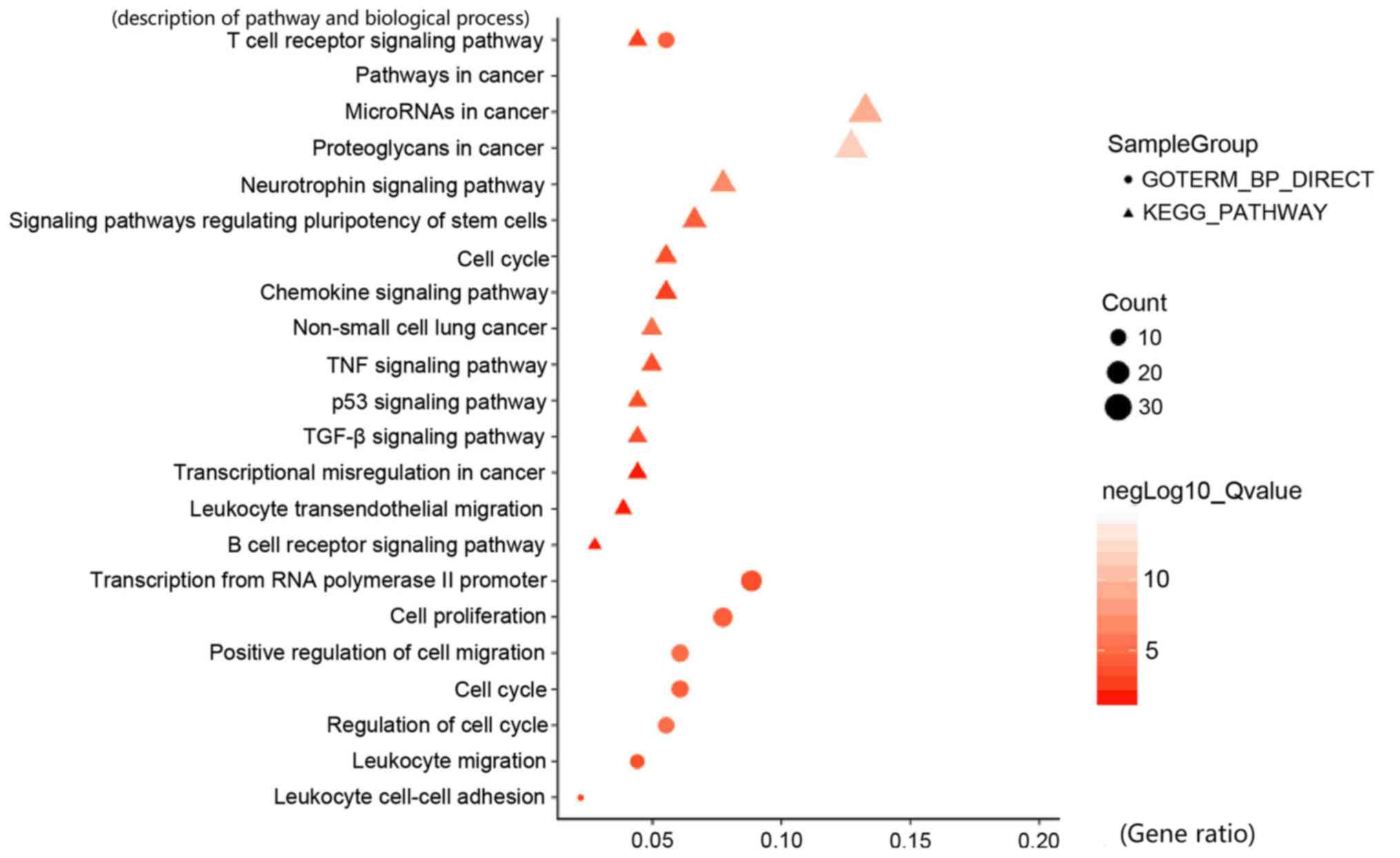
Finally, the functional enrichment analysis of DNA
methylation-associated immune cells genes was conducted using the
DAVID website. Some pathways were found to be associated with
immune processes, including the pathway terms ‘B cell receptor
signaling pathway’ and the ‘T cell receptor signaling pathway’.
Several biological processes involving immune processes were also
obtained, including the GO terms ‘Positive regulation of cell
migration’ and the ‘T cell receptor signaling pathway’ (Fig. 6). The results of the enrichment
analysis also indicated that these associated genes served an
important role in LUAD tumor immunity. From these results, it can
be inferred that DNA methylation may have regulatory effects on the
expression levels of the immune cells genes.
Discussion
Understanding tumor immune cells infiltration is
crucial for investigating the differences in responses to cancer
immunotherapies and for developing effective immunotherapies. DNA
methylation may explain the variabilities in immune cells of lung
cancer in response to antigens (7). However, it is not completely clear
how DNA methylation affected tumor immune cell infiltration. The
present study investigated how DNA methylation affected the
interactions between tumors and immune cells from the perspective
of DNA methylation affecting the interactions between RNA
molecules. A ceRNA network with associated DNA methylation and
immune cell genes was constructed to study the associations between
DNA methylation and the expression levels of immune cell genes.
In the present study, differential DNA methylation
regions and DERNA molecules in lung adenocarcinoma (LUAD) samples
were extracted. lncDM software (lncdm.r-forge.r-project.org/) was used to extract
differential DNA methylation regions and their corresponding
lncRNAs (18). The DEseq2 method
was used to extract DElncRNAs, DEmiRNAs and DEmRNAs in LUAD samples
(19). Then, to study the effects
of differential DNA methylation on tumor immune infiltrating cells,
tumor immune cells infiltration abundance was calculated. When
calculating the infiltration abundance of tumor immune cells,
CIBERSORTx (cibersortx.stanford.edu/) outperformed other
algorithms in eliminating noise, estimating unknown mixture
contents and accuracy. However, the algorithm was prone to bias due
to the statistical collinear effect generated by the regression
analysis, whereas the TIMER 1.0 algorithm (cistrome.dfci.harvard.edu/TIMER/) blocked
immunological marker genes and highly expressed genes to eliminate
bias effects and collinearity between immune cells, ensuring the
accuracy of reasoning (20). Thus,
the TIMER 1.0 algorithm was applied in the present study. Databases
of molecular associations between RNAs (Lnc2Meth 1.0, miRcode 1.0,
Starbase v2.0, miRDB v3.0, miRTarBase 1.0 and TargetScan v7.2) were
used to investigate the associations between RNA molecules and
differential DNA methylation regions (13,23–27).
Based on the ceRNA hypothesis, a ceRNA regulatory network of immune
genes was established to determine the regulatory associations
between DNA methylation and RNA molecules in LUAD. By using mRNAs
expressional data and clinical data associated with differential
DNA methylation regions, a regression model was constructed to
investigate the associations between methylation-associated tumor
immune cells infiltration and overall survival rates (30).
Compared with previous studies (6–8) that
focus on the tumor immune cells infiltration, the present study
further analyzed the regulatory roles in it. It was hypothesized
that the hypomethylation of the 1101320–1104290 regions of
chromosome 1 may affect the expression levels of CHEK1,
thereby affecting tumor immune cells and survival outcomes in
patients with LUAD. The ceRNA network was further enriched from the
perspective of DNA methylation. From the present study, it can be
deduced that the hypomethylation of the 1101320–1104290 regions of
chromosome 1 resulted in the low expression levels of
LINC00337, and that LINC00337 may affect the
expression levels of CHEK1 by competitively binding with
hsa-miR-373 and hsa-miR-195.
The present study was performed based on theoretical
research described in previous studies (7–9,16).
There is evidence that lncRNAs play important roles in directing
the development of various immune cells and controlling dynamic
transcriptional programs, which are hallmarks of immune cell
activation (7). Additionally, a
ceRNA is an RNA with an miRNA binding site that competitively binds
to an miRNA to inhibit its target gene regulation (16). Pseudogenes, lncRNAs, miRNAs and
other types of RNA, can be regulated by ceRNA mechanisms (7). Therefore, the ceRNA hypothesis
provided an approach to investigate the regulatory mechanisms of
RNA molecules. In addition, it was possible to construct a ceRNA
network using existing databases of the interactions between DNA
methylation and RNA molecules. In addition, previous studies have
shown that DNA methylation directly regulates lncRNAs through
interactions with their promoter regions (8,9). For
example, in the cis-methylated lncRNAs group, DNA
methylation adjacent to an lncRNA locus, such as the promoter or
the imprinting control region, directly modulates the expression
levels of target lncRNAs as a cis-regulator (9).
DERNA molecules and DEDNA methylation regions
associated with the lncRNAs were extracted in the present study to
understand the RNAs and methylation regions that have undergone
significant changes in tumor and control tissues. In order to
investigate the regulatory mechanisms of genes expression levels
associated with immune cells from the perspective of DNA
methylation, a DNA methylation and immune-associated ceRNA network
was constructed based on the internal competitive RNA hypothesis.
Next, the associations between the expression levels of RNA
molecules in the network and the survival rates of patients were
investigated. Then, correlational analyses of potential lncRNA-mRNA
pairs in LUAD was conducted to further investigate the competitive
relationships. Subsequently, how these associated genes affected
survival rates was investigated in the present study, and analyses
of the DNA methylation-associated immune cells genes were
performed. Finally, enrichment analyses of DNA
methylation-associated immune cells genes were performed.
According to the analysis of the results, it can be
inferred that hypomethylation of the 1101320–1104290 regions of
chromosome 1 resulted in the low expression levels of
LINC00337, and that LINC00337 may affect the
expression levels of CHEK1 by competitively binding with
hsa-miR-373 and hsa-miR-195, thereby affecting tumor
immune cells and survival outcomes in patients with LUAD.
To verify the hypothesis, several studies (32–42)
on the RNA molecules in the ceRNA network were searched. Previous
studies (32–39) have also indicated that these RNAs
showed important correlations in cancer, tumor immunity and DNA
methylation. For example, CCNE1 is significantly associated
with TRIM58 methylation and the treatment of lung squamous cell
carcinoma and may be used as a potential prognostic biomarker
(32). Previous studies (33–35)
have also shown that CCNE1 is the most common amplified gene
in lung cancer, and that its gene amplification was a therapeutic
target for survival and lung cancer. Therefore,
CCNE1-targeted therapy may be beneficial for CCNE1
amplification in patients with lung cancer (33). HOXC13, which is directly
targeted by miR-141, is highly expressed in LUAD and promotes the
proliferation of LUAD by modulating the expression of CCNE1
(34). Cyclin E1, encoded by the
CCNE1 gene, promotes G1/S phase transition and
chromosome instability (35).
Cyclin E1 is downregulated by both miR-497 and
miR-34a, which synergistically retard the growth of human
lung cancer cells (35).
In addition, smoking can induce the high expression
levels of SLC7A11 in oral cancer cells, which suggested that
the expression levels of SLC7A11 may encourage lung tumor
progression (36–38). In addition, a previous study
(36) demonstrated that
SLC7A11 regulates metabolic requirements during lung cancer
progression and is a potential therapeutic target in non-small cell
lung cancer. The transporter gene SLC7A11 exhibits
significant changes in enhancer DNA methylation and gene expression
levels during pregnancy (37). In
the present study, marker SLC7A11 expression levels were
determined in lung cancer cell lines using microarray data and
Kaplan-Meier survival analyses were performed for each marker using
patient clinical data (38). High
expression levels of the marker SLC7A11 are significantly
associated with worse survival rates (38).
In a previous study (39), among the five associated pathways
in LUAD, the p53 signaling pathway was the most significant, with
CHEK1 being identified as a hub gene. Previous studies
(39–43) have also shown that miRNAs
(hsa-miR-195 and hsa-miR-372) in the network play
important roles in tumor immunity and DNA methylation. For example,
hsa-miR-195 suppresses tumor growth and is associated with
improved survival outcomes in several types of malignancy,
including non-small cell lung cancer. The expression of hsa-miR-195
is lower in tumor tissues and is associated with poor survival
outcomes (39). The overexpression
of miR-195 suppresses tumor cell growth, migration and
invasion. CHEK1 is a direct target of miR-195, which
decreases the expression levels of CHEK1 in lung cancer
cells. Therefore, the high expression levels of CHEK1 in
lung tumors are associated with poor overall survival rates
(39).
In addition, hsa-miR-195 suppresses non-small
lung cell cancer and is a predictive factor for lung cancer
prognosis (39). A previous study
(40) demonstrated that the
expression levels of hsa-miR-195 and O-6-methylguanine-DNA
methyltransferase methylation status are associated with the
clinical outcomes of the primary malignancy (40). In addition, miR-372-3p
enhances the proliferation and invasion of lung squamous cell
carcinoma cells by inhibiting FGF9 (41). A previous study (42) conducted a comparative proteomic
analysis of non-small-cell lung carcinoma (NSCLC) CL1-0 cells
expressing miRNA-372 and/or a vector only using
two-dimensional gel electrophoresis, two-dimensional difference gel
electrophoresis and liquid chromatography tandem mass spectrometry.
Proteins identified as up- or downregulated were further classified
according to their biological functions (42). A number of the proteins identified
in the study (42) may be
potential diagnostic biomarkers of NSCLC, particularly
phosphorylated eIF4A-I. In the corresponding study (43), to clarify the molecular mechanisms
underlying the tumor suppressive roles of propofol in the human
lung cancer cell line A549, the study detected the expression
levels of miR-372 and analyzed the mediating effects of
miR-372 on the proliferation and metastasis of A549 cells
(43). A previous study
demonstrated that the expression levels of LINC00473 can
serve as a robust biomarker for the tumor LKB1 functional status
that can be integrated into clinical trials for patient selection
and treatment evaluation, and implicated LINC00473 as a
therapeutic target for LKB1-inactivated NSCLC (44).
The present study was primarily focused on the
biological information analyses section, and so all the results
were from the biological information analyses without biological
verifications; for example, the direct validation experiments of
the associations between DNA methylation and the expression levels
of immune genes, the direct validation experiments of the
associations between DNA methylation and tumor immune cells and
survival outcomes in patients with LUAD. However, in order to
verify the hypothesis of the present study, relevant studies were
searched and some analyses were added in this discussion
section.
The present study investigated the way in which DNA
methylation affects the reactions between tumors and immune cells
by constructing a ceRNA network. To further determine how the DNA
methylation regions influenced the expression levels of immune
genes by regulating the ceRNA network, the associations between the
expression levels of RNAs, the overall survival rates of patients
with LUAD and the competitive relationships between RNAs were
investigated. Finally, a multivariate Cox regression model was
constructed using 8 mRNAs expression values to investigate how
these associated genes affected survival in patients with LUAD. The
results revealed that hypomethylation of the 1101320–1104290
regions of chromosome 1 resulted in the low expression levels of
LINC00337 and that LINC00337 may affect the
expression levels of CHEK1 by competitively binding with
hsa-miR-373 and hsa-miR-195, thereby affecting tumor
immune cells and survival outcomes in patients with LUAD. The RNA
molecules in the ceRNA network served important roles in DNA
methylation and immunotherapies and can be used as biomarkers of
immunotherapies in patients with LUAD. The results of the present
study may provide a new perspective for the exploration of
cancer-immune interactions.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Dr Deng Jin and Mr
Wei Boyang (both Shanghai Maritime University, Shanghai, China) for
their technical assistance and writing assistance during the
research process.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 61803257) and
Natural Science Foundation of Shanghai (grant no. 18ZR1417200).
Availability of data and materials
LUAD transcriptome, miRNA, DNA methylation data and
associated clinical data can be downloaded from The Cancer Genome
Atlas (TCGA) website (TCGA-LUAD; portal.gdc.cancer.gov/).
Authors' contributions
CC, WK, XM and SW conceived and designed the present
study. CC and WK conducted analyses. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sharma P, Wagner K, Wolchok JD and Allison
JP: Novel cancer immunotherapy agents with survival benefit: Recent
successes and next steps. Nat Rev Cancer. 11:3173–812. 2011.
View Article : Google Scholar
|
|
2
|
Pardoll DM: The blockade of immune
checkpoints in cancer immunotherapy. Nat Rev Cancer. 12:252–264.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carter BW, Halpenny DF, Ginsberg MS,
Papadimitrakopoulou VA and de Groot PM: Immunotherapy in Non-small
cell lung cancer treatment: Current status and the role of imaging.
J Thorac Imaging. 32:300–312. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Di Giacomo AM, Calabrò L, Danielli R,
Fonsatti E, Bertocci E, Pesce I, Fazio C, Cutaia O, Giannarelli D,
Miracco C, et al: Long-term survival and immunological parameters
in metastatic melanoma; patients who responded to ipilimumab 10
mg/kg within an expanded access; programme. Cancer Immunol
Immunother. 62:1021–1028. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Prieto PA, Yang JC, Sherry RM, Hughes MS,
Kammula US, White DE, Levy CL, Rosenberg SA and Phan GQ: CTLA-4
blockade with ipilimumab: Long-term follow-up of 177 patients with
metastatic melanoma. Clin Cancer Res. 18:2039–2047. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu XS and Mardis ER: Applications of
immunogenomics to cancer. Cell. 168:600–612. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang IV, Konigsberg I, Macphail K, Li L,
Davidson EJ, Mroz PM, Hamzeh N, Gillespie M, Silveira LJ, Fingerlin
TE and Maier LA: DNA methylation changes in lung immune cells are
associated with granulomatous lung disease. Am J Respir Cell Mol
Biol. 60:96–105. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gudrun B, Rowley MJ, Kuciński J, Zhu Y,
Amies I and Wierzbicki AT: RNA-directed DNA methylation requires
stepwise binding of silencing factors to long non-coding RNA. Plant
J Cell Mol Biol. 79:181–191. 2014. View Article : Google Scholar
|
|
9
|
Hui Z, Shangwei N, Xiang L, Yuyun L, Wei W
and Xia L: A novel reannotation strategy for dissecting DNA
methylation patterns of human long intergenic non-coding RNAs in
cancers. Nucleic Acids Res. 42:82582014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bohne F, Langer D, Martiné U, Eider CS,
Cencic R, Begemann M, Elbracht M, Bülow L, Eggermann T, Zechner U,
et al: Kaiso mediates human ICR1 methylation maintenance and H19
transcriptional fine regulation. Clin Epigenetics. 8:472016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vennin C, Spruyt N, Robin YM, Chassat T,
Le Bourhis X and Adriaenssens E: The long non-coding RNA 91H
increases aggressive phenotype of breast cancer cells and
up-regulates H19/IGF2 expression through epigenetic modifications.
Cancer Lett. 385:198–206. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu F, Chen B, Dong P and Zheng J: HOTAIR
epigenetically modulates PTEN expression via MicroRNA-29b: A novel
mechanism in regulation of liver fibrosis. Mol Ther. 25:205–217.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gregory MC and Loeb DM: Hypoxia-sensitive
epigenetic regulation of an antisense-oriented lncRNA controls WT1
expression in myeloid leukemia cells. PLoS One. 10:e01198372015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tianyi G, Bangshun H, Yuqin P, Yeqiong X,
Rui L, Qiwen D, Huilin S and Shukui W: Long non-coding RNA 91H
contributes to the occurrence and progression of esophageal
squamous cell carcinoma by inhibiting IGF2 expression. Chinese J
Clin Lab Sci. 54:359–367. 2015.
|
|
15
|
Ruscio AD, Ebralidze AK, Benoukraf T,
Amabile G, Goff LA, Terragni J, Figueroa ME, Pontes LLDF,
Alberich-Jorda M, Zhang P, et al: DNMT1-interacting RNAs block
gene-specific DNA methylation. Nature. 503:371–376. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: ceRNA hypothesis: The Rosetta stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li B and Dewey CN: RSEM: Accurate
transcript quantification from RNA-Seq data with or without a
reference genome. BMC Bioinformatics. 12:323. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhi H, Li X, Wang P, Gao Y, Gao B, Zhou D,
Zhang Y, Guo M, Yue M, Shen W, et al: Lnc2Meth: A manually curated
database of regulatory relationships between long non-coding RNAs
and DNA methylation associated with human disease. Nucleic Acids
Res. 46:D133–D138. 2017. View Article : Google Scholar
|
|
19
|
Love MI, Wolfgang H and Simon A: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li B, Severson E, Pignon JC, Zhao H, Li T,
Novak J, Peng J, Hui S, Aster JC, Rodig S, et al: Comprehensive
analyses of tumor immunity: Implications for cancer immunotherapy.
Genome Biol. 17:1742016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li B and Li JZ: A general framework for
analyzing tumor subclonality using SNP array and DNA sequencing
data. Genome Biol. 15:4732014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chao C, Kay G, Judith B, Dandan Z, Elliot
G, Li J and Chunyu L: Removing batch effects in analysis of
expression microarray data: An evaluation of six batch adjustment
methods. PLoS One. 6:e172382011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ashwini J, Marks DS and Erik L: miRcode: A
map of putative microRNA target sites in the long non-coding
transcriptome. Bioinformatics. 28:2062–2063. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li JH, Liu S, Zhou H, Qu LH and Yang JH:
starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA
interaction networks from large-scale CLIP-Seq data. Nucleic Acids
Res. 42((Database Issue)): D92–D97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. 4:e050052015. View Article : Google Scholar
|
|
26
|
Chou CH, Shrestha S, Yang CD, Chang NW,
Lin YL, Liao KW, Huang WC, Sun TH, Tu SJ, Lee WH, et al: miRTarBase
update 2018: A resource for experimentally validated
microRNA-target interactions. Nucleic Acids Res. 46:D296–D302.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wong N and Wang X: miRDB: An online
resource for microRNA target prediction and functional annotations.
Nucleic Acids Res. 43((Database Issue)): D146–D152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gill RD: Censoring and stochastic
integrals. Statistica Neerlandica. 34:124. 1980. View Article : Google Scholar
|
|
30
|
Fox J: Cox proportional-hazards regression
for survival data. See Also. 371–372. 2012.
|
|
31
|
Huang DW, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang W, Cui Q, Qu W, Ding X, Jiang D and
Liu H: TRIM58/cg26157385 methylation is associated with eight
prognostic genes in lung squamous cell carcinoma. Oncol Rep.
40:206–216. 2018.PubMed/NCBI
|
|
33
|
Nakayama K, Rahman MT, Rahman M, Nakamura
K, Ishikawa M, Katagiri H, Sato E, Ishibashi T, Iida K, Ishikawa N
and Kyo S: CCNE1amplification is associated with aggressive
potential in endometrioid endometrial carcinomas. Int J Oncol.
48:506–516. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yao Y, Luo J, Sun Q, Xu T, Sun S, Chen M,
Lin X, Qian Q, Zhang Y, Cao L, et al: HOXC13 promotes proliferation
of lung adenocarcinoma via modulation of CCND1 and CCNE1. Am J
Cancer Res. 7:1820–1834. 2017.PubMed/NCBI
|
|
35
|
Han Z, Zhang Y, Yang Q, Liu B, Wu J, Zhang
Y, Yang C and Jiang Y: miR-497 and miR-34a retard lung cancer
growth by co-inhibiting cyclin E1 (CCNE1). Oncotarget.
6:13149–13163. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ji X, Qian J, Rahman SMJ, Siska PJ, Zou Y,
Harris BK, Hoeksema MD, Trenary IA, Chen H, Eisenberg R, et al: xCT
(SLC7A11)-mediated metabolic reprogramming promotes non-small cell
lung cancer progression. Oncogene. 37:5007–5019. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Simner C, Novakovic B, Lillycrop KA, Bell
CG, Harvey NC, Cooper C, Saffery R, Lewis RM and Cleal JK: DNA
methylation of amino acid transporter genes in the human placenta.
Placenta. 60:64–73. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cohen AS, Khalil FK, Welsh EA, Schabath
MB, Enkemann SA, Davis A, Zhou JM, Boulware DC, Kim J, Haura EB and
Morse DL: Cell-surface marker discovery for lung cancer.
Oncotarget. 8:113373–113402. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ben L, Jinli Q, Fangxiu X, Yan G, Yu W,
Herbert Y and Biyun Q: MiR-195 suppresses non-small cell lung
cancer by targeting CHEK1. Oncotarget. 6:9445–9456. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lakomy R, Sana J, Hankeova S, Fadrus P,
Kren L, Lzicarova E, Svoboda M, Dolezelova H, Smrcka M, Vyzula R,
et al: MiR-195, miR-196b, miR-181c, miR-21 expression levels and
O-6-methylguanine-DNA methyltransferase methylation status are
associated with clinical outcome in glioblastoma patients. Cancer
Sci. 102:2186–2190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang Y, Hu Y, Wu G, Yang Y, Tang Y, Zhang
W, Wang K, Liu Y, Wang X and Li T: Long noncoding RNA PCAT-14
induces proliferation and invasion by hepatocellular carcinoma
cells by inducing methylation of miR-372. Oncotarget.
8:34429–34441. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lai JH, She TF, Juang YM, Tsay YG, Huang
AH, Yu SL, Chen JJW and Lai CC: Comparative proteomic profiling of
human lung adenocarcinoma cells (CL 1-0) expressing miR-372.
Electrophoresis. 33:675–688. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sun H and Gao D: Propofol suppresses
growth, migration and invasion of A549 cells by down-regulation of
miR-372. BMC Cancer. 18:12522018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen Z, Li JL, Lin S, Cao C, Gimbrone NT,
Yang R, Fu DA, Carper MB, Haura EB, Schabath MB, et al:
cAMP/CREB-regulated LINC00473 marks LKB1-inactivated lung cancer
and mediates tumor growth. J Clin Invest. 126:2267–2279. 2016.
View Article : Google Scholar : PubMed/NCBI
|