Introduction
The inflammatory response that follows tissue injury
mobilizes a large network of molecular mediators. The coordinated
and finely tuned actions of these mediators culminate in the
recruitment of leukocytes to the site of injury; a cascade of
events regulates leukocyte recruitment and trafficking to the site
of injury. Adhesion molecules, such as selectins and integrins,
control the process of initial rolling, firm adhesion, crawling and
transmigration (1,2). Intracellular adhesion molecule-1
(ICAM-1) expressed on the surface of endothelial cells interacts
with activated αMβ2 and αLß2 integrins and allows neutrophils to
crawl and firmly adhere to activated endothelium (3). Furthermore, disulfide bonds are known
to be a key regulator of protein flexibility and function (4), and it has been reported that disulfide
bond reduction (5,6) or cysteine mutation (7,8)
modulates integrin activation and neutrophil adhesion. A previous
study by Hahm et al (9)
demonstrated that extracellular protein disulfide isomerase (PDI)
regulated the ligand-binding activity of αMβ2 integrin and
neutrophil recruitment during vascular inflammation through its
isomerase activity. Another recent study by Rosenberg et al
(10) using three different cell
lines that depend on adhesion for survival indicated that PDI was
involved in integrin-mediated adhesion, through the catalysis of
disulfide bond exchange and enhancement of cell adhesion by both
its oxidoreductase and chaperone activities (10). These data suggest that extracellular
PDI may be a novel target for the modulation of leukocyte
trafficking.
PDI, also known as the β subunit of prolyl
4-hydroxylase, is a 55-kDa soluble protein that constitutes the
archetype of the PDI family of proteins, which contains a
thioredoxin-like βαβαβαββα fold motif and acts as a
dithiol-disulfide oxidoreductase to catalyze the reduction,
oxidization and isomerization of disulfide bonds (11). PDI also acts as a molecular
chaperone both in vitro (12) and in vivo (13). The PDI family comprises >20
members that vary in length and structural arrangement, with most
PDI members sharing catalytic and non-catalytic thioredoxin-like
domains (14). All members are
localized in the ER where they contribute to ER homeostasis by
maintaining an oxidative environment (15). PDI is organized into four
thioredoxin-like domains, a, a′, b and b′, in addition to a linker
domain, x. The catalytic domains a and a′ contain Cys-Gly-His-Cys
motifs that react with thiol groups in substrate proteins, whereas
b and b′ are considered as non-catalytic domains and are involved
in substrate recognition and recruitment (14). The most commonly studied members of
the PDI family after PDI are endoplasmic reticulum resident protein
(ERp)57, ERp72, ERp29, ERp44, and PDIA2 (11). The difference between the PDI family
members is protein length and structural arrangement of active and
inactive domains (14).
Although the chaperone function of PDIs is generally
mapped to the endoplasmic reticulum (ER) (16), one previous study demonstrated
extracellular localization and function of PDI on the surface of
several cell types, suggesting an enzymatic mediation for disulfide
exchange in the cell-surface receptors (17). Indeed, PDIs expressed on the surface
of leukocytes and platelets are involved in hemostasis, vascular
inflammation and thrombosis (18–20).
Moreover, PDI, ERp5 and ERp57 are involved in the initiation of
thrombus formation following laser-induced vascular injury in
vivo (21–23), in which endothelial cells and
platelets are activated and secrete PDI and other thiol isomerases
(24). ERp72 was shown to initiate
coagulation and promote thrombosis formation through a cascade of
reactions in platelets (25), which
promotes tumor progression (26–28).
In addition, ERp72 knockdown impaired platelet function and fibrin
formation in mice (29). PDI is
also associated with thrombus growth through the regulation of β3
integrins (17,18,30,31).
Inhibition of PDI with a blocking antibody completely inhibits both
platelet thrombus formation and fibrin generation (17,22,32).
To the best of the authors' knowledge, the majority
of studies are focused on PDIs in leukocytes and little to almost
no information is available on the role played by endothelial PDIs
during cellular activation and recruitment. The present study was
initiated following a yeast two-hybrid screening of the binding
partner of a biological compound (SI/0220) that is currently under
development (Patent FR2909672A1, pending). SI/0220 is a bispecific
integrin/selectin biological compound that was engineered to target
ICAM1 and P-selectin glycoprotein ligand-1 (1,2). These
two endothelial cell surface receptors are the natural ligands of
the leukocyte adhesion molecules CD11b/CD18 (33) and L-selectin (34), respectively, which are the main
contributors to leukocyte extravasation in inflamed tissues.
Unpublished data have suggested that SI/0220 has anti-inflammatory
activity and modulates polymorphonuclear neutrophil transmigration.
The screening against a library of TNFα-activated human umbilical
vein endothelial cells (HUVECs) identified ERp72 as a major binding
partner of SI/0220. To validate this observation, ERp72 gene
expression was assessed in vascular endothelial cells in a rat
model of inflammatory skeletal muscle injury (35). ERp72 was overexpressed from the
early phase following injury. For validation, the present study
used western blotting, immunohistochemistry and immunofluorescence
experiments in the endothelium of blood vessels post-injury, in
addition to adhesion inhibition assays of neutrophils to
TNFα-activated HUVECs. These findings suggested that endothelial
ERp72 may mediate the inflammatory response through the regulation
of neutrophil adhesion during their recruitment to the inflammation
site and could constitute a novel target to modulate the overflux
of activated leukocytes to the site of injury.
Materials and methods
Yeast two-hybrid analysis
Yeast two-hybrid screening was performed using the
Hybrigenics Services technical platform (http://www.hybrigenics-services.com). The technology
is based on the reconstitution of a functional transcription factor
followed by the expression of a reporter gene in genetically
modified yeast cells. The coding sequence for SI/0220 (data not
shown; patent pending) was amplified by PCR and cloned into pB27
plasmids as a C-terminal fusion to LexA (N-LexA-SI/0220-C). The
construct was checked by sequencing the entire insert and used as a
bait to screen a randomly-primed HUVEC cDNA library constructed
into pP6 and pB27 plasmids derived from the original pBTM116
(36) and pGADGH (37) plasmids, respectively. A total of 113
million clones (10-fold the complexity of the library) were
screened using a mating approach with YHGX13 (Y187
ade2-101:loxP-kanMX-loxP, MATα) and L40ΔGal4 (MATa) yeast strains
as previously described (38).
Cells were incubated in rich medium for 4.5 h at 30°C and
His+ colonies were selected on a plates made from medium
lacking tryptophan, leucine and histidine. The prey fragments of
the positive clones were amplified by PCR and sequenced at their 5′
and 3′ junctions. The resulting sequences were used to identify the
corresponding interacting proteins in the GenBank database
(National Center for Biotechnology Information) using a fully
automated procedure. A confidence score (Predicted Biological
Score; PBS) was attributed to each interaction as previously
described (39). For protein
annotation, conserved domains were predicted using the Pfam v.32.0
(https://pfam.xfam.org) and SMART v.8 (http://smart.embl-heidelberg.de) servers. The
transmembrane domain was predicted using the TMHMM server v.2.0
(http://www.cbs.dtu.dk/services/TMHMM). The signal
peptide was predicted using the SignalP v. 3.0 server (http://www.cbs.dtu.dk/services/SignalP-3.0). The
coiled coil domain was predicted using the COILS v.2.2 server
(https://embnet.vital-it.ch/software/COILS_form.html).
Primer design
Rattus norvegicus mRNA sequences for ERP72
(PDIA4), ICAM1, VCAM1, and SELE were downloaded from the
Uniprot server (release 2019_11, http://www.uniprot.org). Specific primers were
designed using the Primer3 web server v.0.4.0 (http://bioinfo.ut.ee/primer3-0.4.0) with default
parameters, and their specificity was assessed using BLAST v.2.9.0
(https://blast.ncbi.nlm.nih.gov) for
cross-priming. Primers used for reverse-transcription-quantitative
(RT-q) PCR (Table I) were purchased
from Biolegio B.V. in lyophilized form. All primers were
synthesized at a 40 nmol scale and purified by high-performance
liquid chromatography. The primers were resuspended in
nuclease-free water at a concentration of 100 µM.
 | Table I.List of primers used for reverse
transcription-quantitative PCR. |
Table I.
List of primers used for reverse
transcription-quantitative PCR.
| Primer name | Target gene | Primer sequence
(5′→3′) | Melting
temperature, °C | Product size,
bp |
|---|
| PDI4F1 | ERP72 |
TGCAGCCTGAGAAGTTCCAG | 59.96 | 200 |
| PDI4R1 | ERP72 |
GCTGAAGTCCACGCTGTAGT | 60.04 | 200 |
| ICAMF | ICAM1 |
GGTATCCATCCATCCCACAG | 60.01 | 208 |
| ICAMR | ICAM1 |
GCCACAGTTCTCAAAGCACA | 60.03 | 208 |
| VCAMF | VCAM1 |
ACAAAACGCTCGCTCAGATT | 60.02 | 152 |
| VCAMR | VCAM1 |
GTCCATGGTCAGAACGGACT | 59.97 | 152 |
| E-seleF | SELE |
TTTTTGGCACGGTATGTGAA | 59.97 | 168 |
| E-seleR | SELE |
AGGTTGCTGCCACAGAGAGT | 60.06 | 168 |
| GAPDHF | GAPDH |
CTCATGACCACAGTCCATGC | 59.80 | 155 |
| GAPDHR | GAPDH |
TTCAGCTCTGGGATGACCTT | 59.90 | 155 |
Animals
A total of 30 female inbred Wistar rats weighing
200–220 g were used to develop the muscle injury model, following
institutional guidelines and in conformity with the international
standards recommended for animal experimentation. All animal
experimental protocols were approved by the Research and Ethics
Committee of Arabian Gulf University (Manama, Bahrain). All methods
were carried out in accordance with the committee's relevant
guidelines and regulations in March 2019. A total of 25 rats were
used for the monitoring of gene expression and immunohistochemistry
experiments post-injury. The remaining five rats did not undergo
muscle injury and were used as an untreated control group.
Rat skeletal muscle injury model
Rat skeletal muscle injury was performed as
described previously (35). Animals
were anesthetized intraperitoneally using a mixture of 90 mg/kg
ketamine and 10 mg/kg xylazine. Anesthesia induction was confirmed
by lack of pedal reflex. The muscles in both limbs were punctured
using a 20-gauge needle mounted on a manual leather-puncturing
device to create a hematoma as previously described (35). The rats were euthanized using
CO2 at a displacement at a flow rate of 50% at different
timepoints varying from 15 min to 4 h post-injury. Animal death was
verified by ascertaining cardiac and respiratory arrest. A surgical
procedure was then used to extract blood vessels in the injured
area from all rats, vessels were further dissected under a
microscope to remove all irrelevant tissues. Blood vessels were
then stored in TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.) for RNA and protein extraction. For histology,
wounded muscles were resected, fixed in 10% formalin overnight at
4°C and paraffin-embedded, then cut to 4–5 µm sections. The
sections were used for immunohistochemistry and stained at room
temperature for 3 min and 45 sec with hematoxylin and eosin,
respectively, for light microscopy examination. Histological
observations were performed on a Zeiss Axioskop light microscope
(Carl Zeiss AG) using an eyepiece graticule grid.
Gene expression analysis
Total mRNA was extracted from homogenized tissues
using the TRIzol® extraction protocol (Invitrogen;
Thermo Fisher Scientific, Inc.) and reverse transcribed using the
ProtoScript® First Strand cDNA Synthesis kit (New
England BioLabs, Inc.) according to the manufacturer's
instructions. Primer sets for RT-qPCR (Table I) were used to amplify target
regions from cDNA as templates using the GoTaq DNA polymerase
(Promega Corporation). For qPCR, the PowerUp SYBR Green Master Mix
(Applied Biosystems; Thermo Fisher Scientific, Inc.) was used to
measure ERP72, ICAM1, VCAM1, and SELE gene expression
levels 15, 30, 90 and 120 min post-injury. Fluorescence was
monitored for 40 cycles (95°C for 3 sec, 60°C for 30 sec) on a 7500
fast real-time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Experiments were run in triplicates for all
groups. The data were quantified using the 2−ΔΔCq method
(40), and the results are
presented as the fold change relative to GAPDH, using the untreated
control group as reference.
Western blotting
Total proteins were extracted from homogenized
vessels using TRIzol as recommended by the manufacturer
(Invitrogen; Thermo Fisher Scientific, Inc.). Protein extracts from
two rats of each group were resuspended in 1% SDS supplemented with
phosphatase and a protease inhibitor cocktail (New England Biolabs,
Inc.). The soluble protein concentration was determined using a BCA
Protein Assay kit (Thermo Fisher Scientific, Inc.). For western
blotting analysis, 20 µg of total protein extract were resolved by
SDS-PAGE on 12% gels (41). The
proteins were transferred to nitrocellulose membranes, blocked for
1 h at room temperature in PBS containing 5% non-fat dry milk and
0.1% Tween-20, then incubated overnight at 4°C with the anti-ERp72
rabbit polyclonal antibody (1:1,000; Abcam; cat. no. ab82587) or
anti-β-actin mouse antibody (1:1,000; BD Biosciences; cat. no.
612656) (41). The respective
HRP-conjugated secondary antibodies (anti-rabbit IgG HRP-linked
antibody cat. no. 7074 and anti-mouse IgG HRP-linked antibody cat.
no. 7076; Cell Signaling Technology, Inc.) were used at a dilution
of 1:1,000 for 2 h at room temperature for the detection step. The
bands were detected using an Enhanced Chemiluminescence kit
(Cytiva) and images were acquired using an LAS-1000 plus image
analyzer with Image Reader LAS-1000 Lite software ver. 2.2
(Fujifilm Wako Pure Chemical Corporation).
Multiplex immunohistochemistry
Immunohistochemistry staining was performed on the
Ventana Discovery Ultra Chromogenic AmpHQ automated immunostainer
(Ventana Medical Systems, Inc.) to determine ERp72 protein
expression 2 h post-injury in the aforementioned sections. The
multiplex technology uses the sequential application of unmodified
primary antibodies with specific heat deactivation steps in between
that does not affect the epitope in the tissue (42). In a sequential staining procedure,
deactivation of the primary antibody and HRP/AP-conjugated
secondary antibody bound to the first biomarker, before the
application of subsequent biomarker(s), is critical to reducing
cross-reactivity and facilitating downstream image analysis
(43). Deparaffinization and
on-board antigen retrieval were performed for 64 min at 95°C using
the CC1 reagent (Ventana Medical Systems, Inc.; cat. no. 950-500).
Blocking buffer (Invitrogen; Thermo Fisher Scientific, Inc.; cat.
no. 00-4952-54) was used for section blocking at 37°C for 20 min.
Slides were processed using Ventana Medical Systems reagents except
as noted, according to the manufacturer's instructions. The Cell
Conditioning (CC) 2 buffer (Ventana Medical Systems, Inc.; cat. no.
950-123) was used for deactivation of the bound primary antibody
and secondary antibody-HRP while maintaining the integrity of the
tissue morphology and the subsequent epitopes (42).
The pre-diluted anti-ERp72 primary antibody
(1:10,000; Abcam; cat. no. ab109869), was applied first at 37°C for
60 min, followed by 16 min incubation at 37°C with ready-to-use
OmniMap anti-Rabbit HRP solution (Ventana Medical Systems, Inc.;
cat. no. 760-4311) followed by 8 min with ready-to-use ChromoMap
DAB kit (Ventana Medical Systems, Inc.; cat. no. 760-159) for
single IHC or DISCOVERY Teal HRP Kit (RUO; Ventana Medical Systems,
Inc.; cat. no. 760-247) for duplex IHC (42). For duplex IHC, a supplementary
treatment with mouse anti-CD34 (1:400; Abcam; cat. no. ab8536) for
40 min at 37°C was performed and followed by 16 min incubation with
a secondary ready-to-use OmniMap anti-Ms HRP antibody (Ventana
Medical Systems, Inc.; cat. no. 760-4310) and 8 min with
ready-to-use ChromoMap DAB kit (Ventana Medical Systems, Inc.; cat.
no. 760-159). Finally, the slides were counterstained with
hematoxylin and bluing reagent according to the manufacturer's
recommendations. Images of IHC specimens were captured on an
Olympus BX-51 microscope (Olympus Corporation) fitted with a
CoolSNAP ES2 CCD camera with 1,392×1,040 pixels and 12-bit
resolution (Teledyne Photometrics) and Olympus UPlanSApo 20× (NA
0.75) and 10× (NA 0.40) air objectives (Olympus Corporation). Image
acquisition and quantitative analysis were performed using HALO™
Image Analysis software Cytonuclear module v. 1.6 (Indica Labs).
Blood vessels were manually delimited on the acquired images and
endothelial cells were identified by their morphology and counted,
then classified according to the level of expression of ERp72 into
low (1+), medium (2+) and high expression cells (3+) by quantifying
color intensity of each counted cell.
Multiplex immunofluorescence
For immunofluorescence, deparaffinization and
on-board antigen retrieval were carried out for 64 min at 95°C
using the CC1 reagent (Ventana Medical Systems, Inc.; cat. no.
950-500). Blocking buffer (Invitrogen; Thermo Fisher Scientific,
Inc.; cat. no. 00-4952-54) was used for section blocking at 37°C
for 20 min. Slides were processed using Ventana Medical Systems
reagents according to the manufacturer's instructions. A duplex
protocol was used; two pre-diluted primary antibodies were
sequentially applied for 40 min at 37°C each, in the following
order: Rabbit anti-ERp72 (1:10,000, Abcam; cat. no. ab109869)
followed by mouse anti-CD34 (1:400; Abcam; cat. no. ab8536) for 40
min at 37°C. Secondary antibodies were then added in the following
order using the indicated chromogenic detection: OmniMap anti-Rb
HRP (Ventana Discovery; cat. no. 760-4310) and Discovery Rhodamine
kit (Ventana Medical Systems, Inc.; cat. no. 760-233), then
ready-to-use OmniMap anti-Ms HRP (Ventana Medical Systems, Inc.;
cat. no. 760-4310) and Discovery Cy5 kit (Ventana Medical Systems,
Inc.; cat. no. 60-238). Sections were then counterstained with DAPI
and mounted using Vectashield mounting medium (Vector Laboratories,
Inc.). Images were acquired on a Zeiss Axio Observer Z1 Inverted
Fluorescence Microscope (Carl Zeiss AG). Image acquisition was
performed using HALO™ Image Analysis software v. 2.1 (PerkinElmer,
Inc.).
Plating and maintenance of HUVECs
HUVECs were grown in EGM-2 medium (Lonza Group
Ltd.). When cells reached 80–90% confluence, they were trypsinized
and resuspended at 120,000 cells/ml. Then, 36,000 cells/well (0.3
ml) were plated into a 48-well polystyrene tissue culture plate.
For the treatment, HUVEC monolayers were washed once with Hank's
balanced salt solution (HBSS) and supplemented with 0.3 ml DMEM
media containing 100 ng/ml TNFα (R&D Systems, Inc.; cat. no.
210-TA-100) for 3 h before neutrophils addition. Negative controls
were supplemented with DMEM alone.
Neutrophil isolation and labeling
Experimental protocols were approved by the Research
and Ethics committee at Arabian Gulf University. All methods were
carried out in accordance with the committee's relevant guidelines
and regulations. Participants provided written informed consent
prior to enrolment in the study and for the publication of the
data. Isolation of neutrophils was performed using Polymorphprep™
density gradient solution (Progen Biotechnik GmbH) to isolate
polymorphonuclear granulocytes from whole blood (44). Briefly, 30 ml whole blood from a
healthy human volunteer were collected in EDTA and 5 ml of whole
blood were layered over 5 ml Polymorphprep™ solution and
centrifuged at 450 × g for 30 min at 18–22°C. The plasma and upper
leukocyte phase containing peripheral blood mononuclear cells were
removed and the lower leukocyte layer containing neutrophils was
recovered. Cells were washed in PBS (without Ca2+ and
Mg2+) and centrifuged at 450 × g for 10 min at 18–22°C.
After red blood cell lysis, cells were washed in 1X PBS (without
Ca2+ and Mg2+), then centrifuged at 250 × g
for 5 min at 18–22°C. After a final wash in PBS (without
Ca2+ and Mg2+), cells were resuspended at
2×106 cells/ml in RPMI-1640 (Sigma-Aldrich; Merck
KGaA).
2′,7′-Bis-(2-carboxyethyl)-5(and-6)-carboxyfluorescein,
acetoxymethyl (BCECF-AM; Invitrogen; Thermo Fisher Scientific,
Inc.) was used for neutrophil labeling. Briefly, BCECF-AM was added
to the cells at a final concentration of 1 µM and incubated for 30
min at 37°C. Cells were washed twice in HBSS/BSA and resuspended in
serum-free RPMI-1640 medium (Sigma-Aldrich; Merck KGaA) to achieve
a final concentration of 1×106 cells/ml.
Cell adhesion assay
Adherence of BCECF-AM-labeled neutrophils to HUVEC
monolayers was evaluated as described previously (45). Confluent HUVECs were prepared as
aforementioned and incubated at 37°C 24 h before use. Rabbit
polyclonal antibodies specific for ERp72 (Abcam; cat. no. ab109869)
were added to HUVECs in serum-free RPMI-1640 medium at different
concentrations (5–100 µg/ml) and incubated for 20 min at 37°C.
Rabbit anti-ERp5 polyclonal antibody (Abcam; cat. no. ab11432) was
used as an isotype control at 100 µg/ml. HUVEC monolayers (≥80%
confluence) were then washed twice with warm serum-free RPMI-1640,
then added to BCECF-AM-labeled neutrophils (1.0×105
neutrophils/well in 100-µl total volume). After a 15-min
incubation, non-adherent neutrophils were removed using a gentle
wash with PBS. After washing, 100 µl PBS was added to each well and
fluorescence was determined using a fluorescence plate-reader
(excitation filter was a 20-nm bandwidth filter centered at 485 nm,
and the emission filter was a 25-nm bandwidth filter centered at
530 nm). All experiments were performed in triplicate. Wells with
HUVECs that were not incubated with neutrophils were used as
controls in order to obtain a background fluorescence reading.
Neutrophil adhesion percentages were calculated relatively to
HUVECs stimulated with TNFα (as aforementioned) as a positive
control of adhesion.
Statistical analysis
Gene expression analyses were set up in triplicates
for each rat. Cell adhesion assay were run in three independent
experiments. One-way ANOVA followed by Tukey's post hoc test was
used to compare experimental groups to the control for all
experiments. All analyses were performed using SPSS statistics
software version 27 (IBM Corp.). P<0.05 was considered to
indicate a statistically significant difference.
Results
Yeast two-hybrid screening
Yeast two-hybrid screening was performed using the
coding sequence of SI/0220 as a bait to screen a randomly primed
HUVEC cDNA library. The screen identified ERp72 (GenBank ID
157427676) as a prey with a Predicted Biological Score (PBS) of B
(high confidence in the interaction). The interacting domain
mapping identified the ERp72 domain ranging from amino acids (aa)
172 to 352 as the selected interacting domain (SID) shared by all
fragments matching the same reference protein (Fig. 1). This SID covers the ‘a’ catalytic
domain and part of the ‘b’ substrate-binding domain.
Gene expression analysis
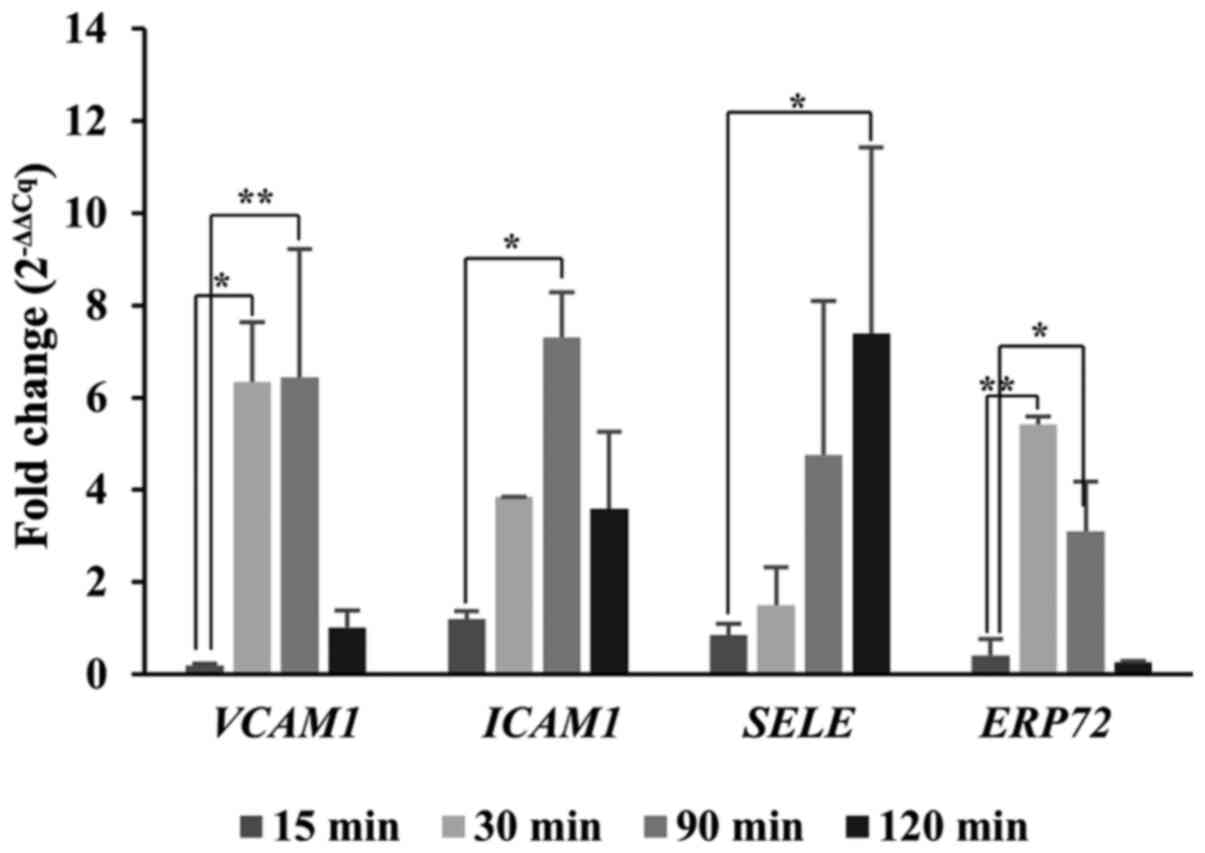
RT-qPCR was performed to determine the gene
expression levels of ERP72, ICAM1, VCAM1 and SELE in
rat blood vessels at the site of injury at different timepoints
post-injury: 15, 30, 90 and 120 min (Fig. 2). VCAM1, ICAM1 and
SELE were selected to monitor the inflammation process
(46). VCAM1 and
ICAM1 expression was upregulated starting from 30 min until
90 min post-injury and reaching up to an 8-fold increase compared
to the non-injured group. The SELE expression profile showed
a small delay in expression, compared with VCAM1 and
ICAM1. Indeed, SELE expression reached a 7-fold
increase at 120 min. For ERP72, a significant upregulation
was observed at 30 min (6-fold) and at 90 min post-injury
(4-fold).
Multiplex immunohistochemistry and
immunofluorescence
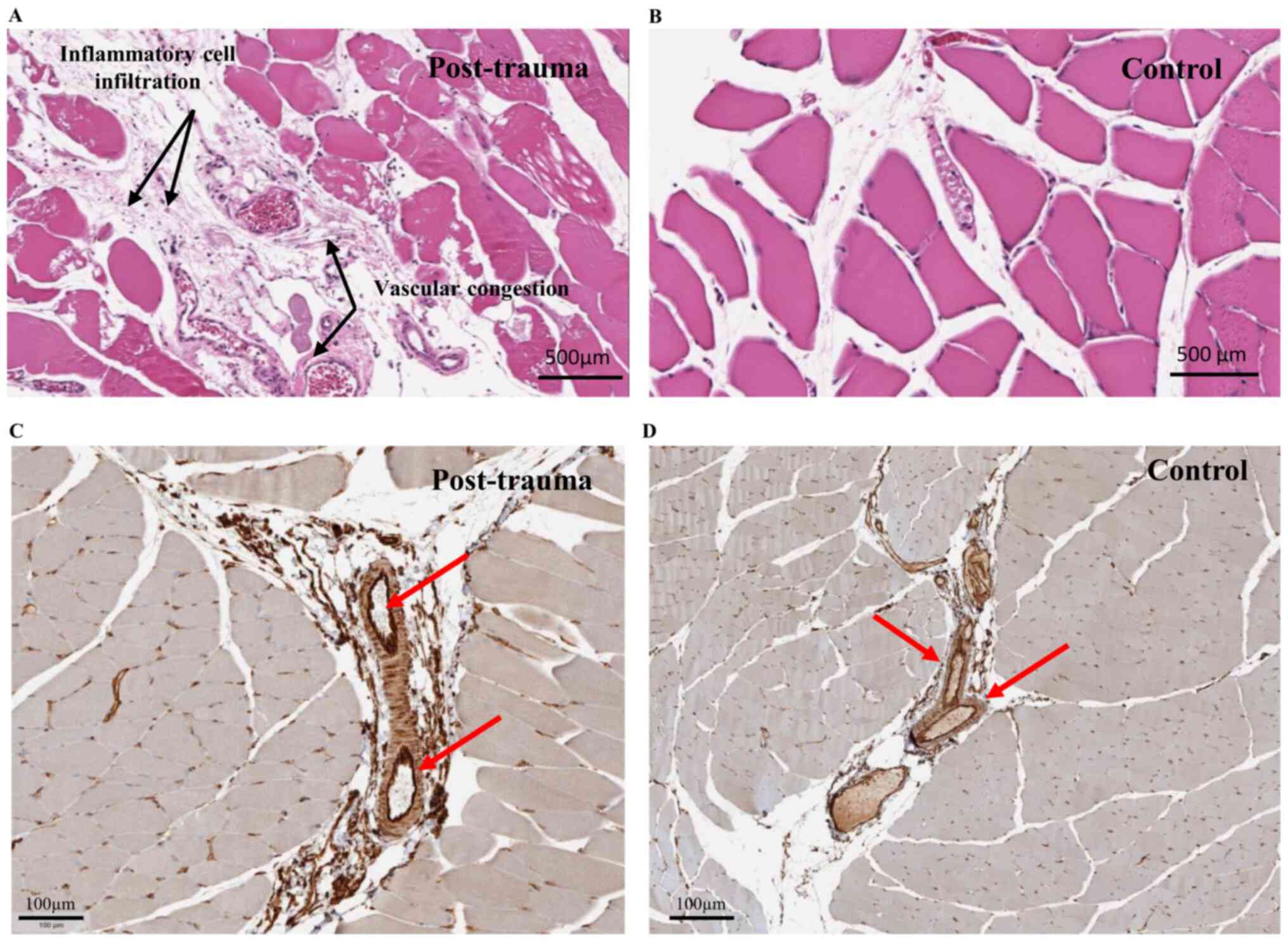
For in situ investigation, H&E staining
of muscle sections from model rat injury sites and controls was
performed (Fig. 3A and B,
respectively). Congestion in blood vessels was observed at the site
of injury, together with infiltration of inflammatory cells in the
site 2 h post-injury, as reported previously (35). Single immunohistochemical labeling
of ERp72 indicated detectable expression of the protein mainly in
the endothelial cells of the blood vessels in control sample
tissues (Fig. 3D) and post-trauma
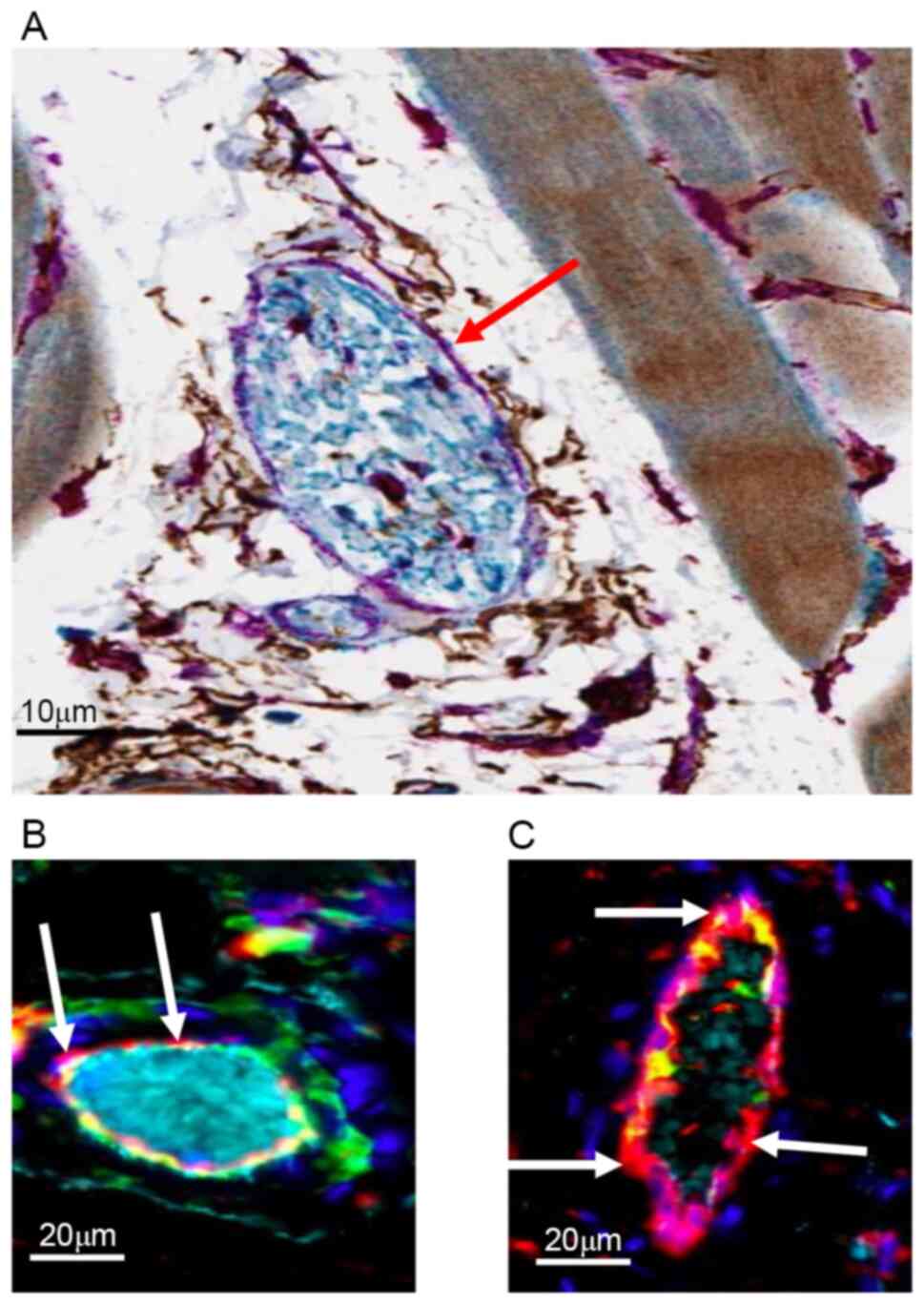
tissues (Fig. 3C). CD34 (a specific
marker of microvascular endothelial cells) was co-expressed with
ERp72 in the endothelial cells of the blood vessels in the control
group (Fig. 4A). Indeed, multiplex
immunofluorescence confirmed this colocalization at the level of
endothelial cells, as well as the increase of specific ERp72 signal
2 h post-injury (Fig. 4C) compared
with the control group (Fig.
4B).
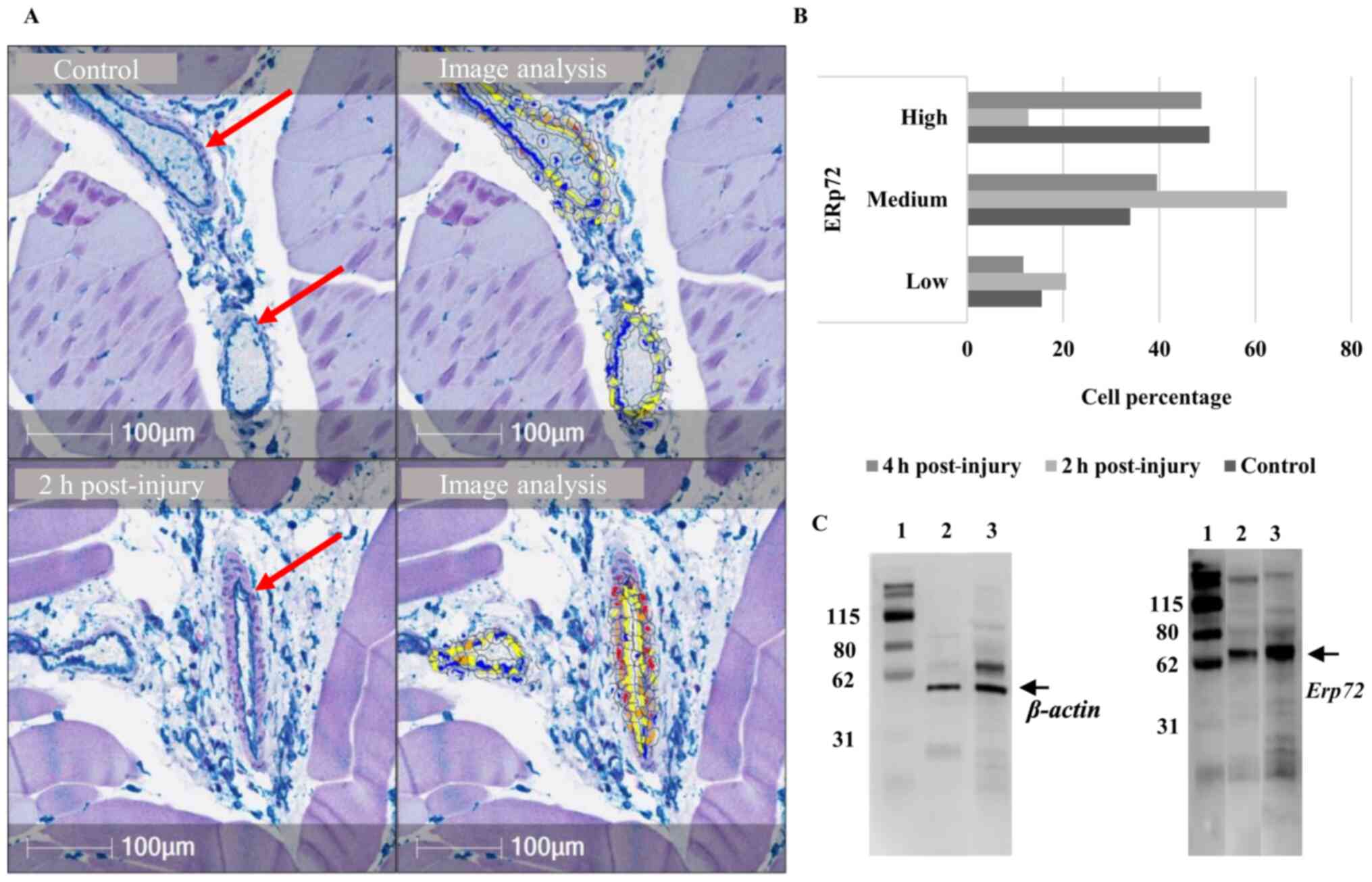
To measure differential expression of these target
proteins, quantification of their cellular expression in vessels at
the site of injury was performed (Fig.
5A and B). A notable increase in the percentage of cells with
medium ERp72 expression (33-66%) was observed 2 h post-injury.
Western blot analysis also indicated ERp72 upregulation in total
vessel extracts at 2 h post-injury compared to untreated rats
(Figs. 5C, S1 and S2).
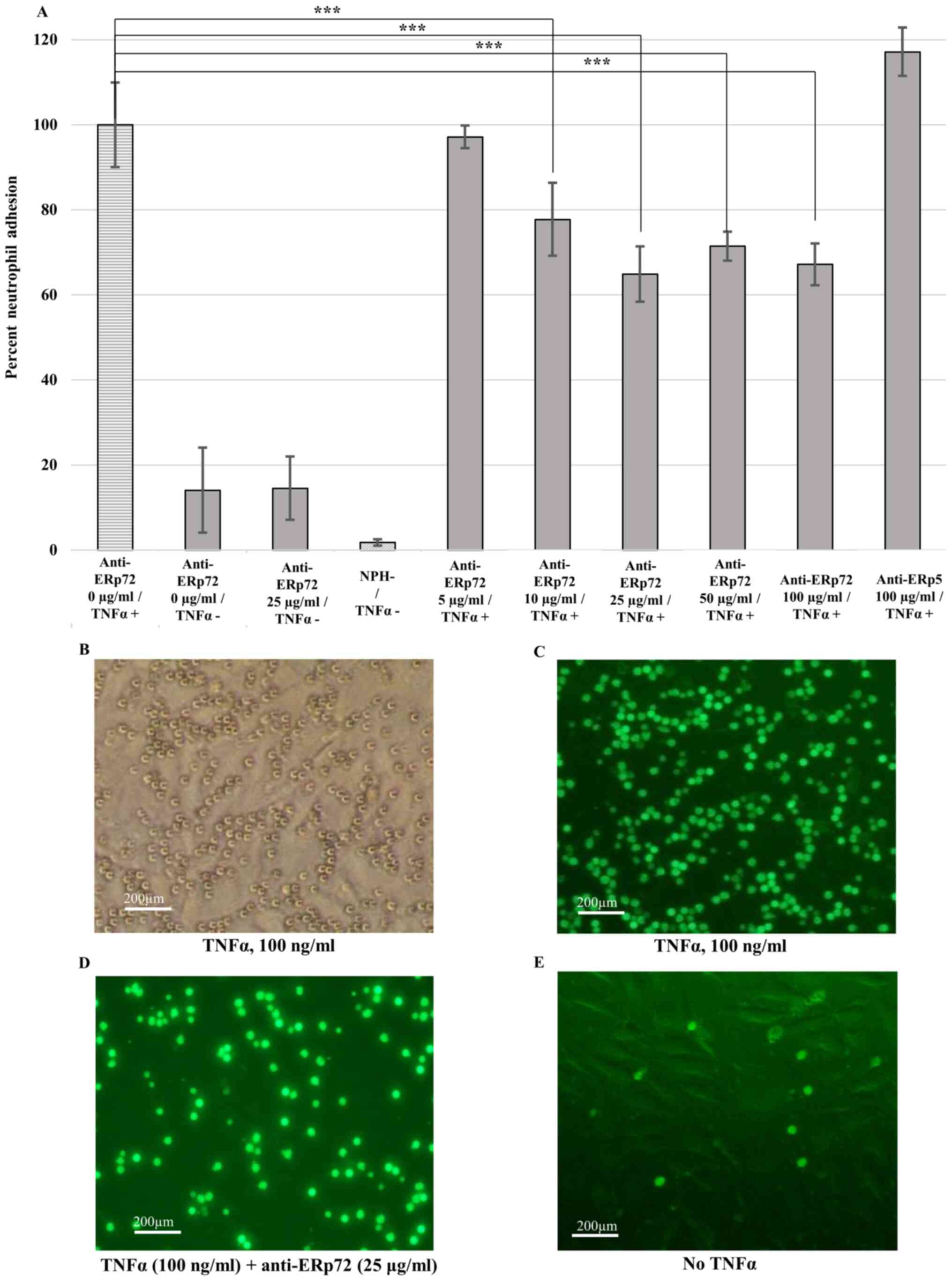
Adhesion assay
Adhesion of human neutrophils to TNFα-activated
HUVECs was used to examine the role of ERp72 in cellular adhesion
(Fig. 6A). Anti-ERp72
antibody-treated HUVECs that did not receive TNFα treatment
displayed relatively low neutrophil adhesion levels (6-14%). There
was little background signal in control cells without addition of
neutrophils (media only, NPH-). Interestingly, anti-ERp72 antibody
significantly inhibited neutrophil adhesion starting from 10 µg/ml
(P<0.001). The highest adhesion inhibition was observed for
antibody concentration of 25 µg/ml (35% inhibition of neutrophil
adhesion). However, no inhibition was observed for the anti-ERp5
isotype control (anti-ERp5 100 µg/ml/TNF +) for the tested
concentration. Light (Fig. 6B) and
fluorescence microscopy (Fig. 6C-E)
of neutrophils stained with BCECF-AM-labeled dye confirm the
results obtained with fluorescence measurement.
Discussion
The development and progression of
leukocyte-mediated tissue injury in inflammatory diseases is a
multi-step processes that involves several adhesion molecules and
neutrophil extravasation through the endothelial lining (47,48).
The present study was based upon an observation made during the
development of a biological anti-inflammatory compound, SI/0220.
This compound has the capacity to modulate PMN transmigration and
to downregulate the expression of proinflammatory soluble mediators
in ex vivo (data not shown; patent pending). SI/0220 was
used as a prey in a yeast two-hybrid system to screen a library of
HUVECs for a binding partner. This screening unexpectedly revealed
ERp72 as a major binding partner of SI/0220, with a high-confidence
interaction. Erp72 comprises 645 aa and is one of the largest PDI
family members. The protein structural organization contains three
classical Cys-Gly-His-Cys active sites, where the a- and a′-type
domains are separated by the non-catalytic b-type domain (14). Unlike other PDI family members,
ERp72 possesses a C-terminal Lys-Glu-Glu-Leu ER retention sequence.
The protein associates with ER heat shock protein 70 (also known as
binding immunoglobulin protein), Grp94, PDI and ERp29 to form a
multiprotein chaperone complex that can bind to unfolded protein
substrates (49). This finding
suggested endothelial ERp72 might be involved in the neutrophil
adhesion process acting from the endothelial side, since PDI
expression in leukocytes has been demonstrated to play a role in
this process (9).
To investigate the potential role of vascular ERp72
in neutrophil recruitment, ERp72 gene expression was examined in a
previously developed rat model of skeletal muscle injury (35). In this mechanical trauma model, the
acute inflammatory response was characterized by an early wave of
PMN influx (within 30 min post-injury) into the injured site and
the endomysium 5–10 mm from the immediate site of hematoma
formation, followed by a second phase 3 h post-trauma that lasts up
to 24 h (35). Histological
examination of inflamed muscle of rats that received the
anti-CD11b-blocking monoclonal antibody OX4238 also indicated a
significant decrease in the number of infiltrating PMN in this
model (35). Neutrophils are
amongst the first cells to arrive at the site of muscle injury
(50). In the present study, a
significant upregulation in ERp72 expression in capillary
endothelial tissues was observed following injury, compared with
control rats. ERp72 protein expression was observed in the blood
vessels within the site of injury, particularly on the vascular
endothelial cells. ERp72 upregulation was also confirmed using
western blotting 2 h post-injury. Moreover, ERp72 blockade using
specific antibodies inhibited neutrophil adhesion to TNFα-activated
HUVECs in a plate-based assay, similar to what has been shown with
PDI (9). Thus, these data support
the role of ERp72, a thiol isomerase on the surface of endothelial
cells, in neutrophil recruitment under inflammatory conditions.
The role of PDIs in inflammation and cellular
adhesion has been investigated in several previous studies. Passam
et al (51) demonstrated
that platelet- and endothelial cell-derived ERp5 support thrombus
formation in a laser-induced mouse model of thrombosis. Moreover,
PDI on the surface of leukocytes regulates neutrophil recruitment
during vascular inflammation through binding to αMβ2 (9). Moreover, the deletion of the
PDI protein in neutrophils inhibited cell adhesion to
inflamed endothelium without affecting the synthesis of αMβ2 and
other integrins, which demonstrates that PDI is essential to
adhesion of human neutrophils under shear and static conditions and
for binding of soluble fibrinogen to activated αMβ2 integrin
(9). In addition, Bennett et
al (52) reported that
neutrophil PDIs may influence L-selectin shedding by regulating the
activity of TNFα-converting enzyme, suggesting a role of PDI in
neutrophil rolling in inflammation. However, very few studies have
highlighted the role of PDIs in endothelial cells during vascular
inflammation.
Gene expression profiling demonstrated that the
majority of upregulated genes in activated human coronary artery
endothelial cells exposed to proinflammatory stimuli encode
surface, adhesion, and receptor proteins (53). Cell adhesion molecules (CAMs) and
selectins are among the most important molecules required for
leukocyte adhesion, rolling and arrest (54). In the present study, upregulation of
VCAM1 (30 min post-injury, ICAM1 (90 min
post-injury), and SELE (120 min post-injury) were observed,
which validates the inflammatory state of the isolated tissues in
this study. In addition, a previous study (35) reported that the adhesion process
occurs in the studied model and led to the infiltration of
neutrophils to the site of injury. For ERp72, upregulation of
expression was observed at the same timepoints (30 and 90 min
post-injury) in the same tissues and demonstrated that ERp72
protein was exclusively expressed on the endothelial cell lining of
the vessels, as demonstrated by co-staining with CD34, a specific
marker for endothelial cells in blood vessels (55). A significant increase in the
percentage of cells with medium expression of ERp72 in blood
vessels at 2 h post-injury. Although western blotting was conducted
only 2 h post-injury, which constitutes a limitation to this work,
it confirmed ERp72 significant expression upregulation at this time
point. These results are in line with the inflammatory pattern of
leukocyte infiltration in the rat model; the latter occurs in two
phases: At 1 and 3 h post-injury (35). In addition, an antibody against
ERp72 inhibited neutrophil adhesion to TNFα-activated HUVECs by
35%.
It has been reported that thiol exchange on
integrins regulates their adhesive function (10,56,57).
For example, cleavage of two disulfide bonds in the cysteine-rich
domain of αIIbβ3, induces conformational changes in the subunits
and exposure of ligand-binding sites (58). PDI was demonstrated to interact with
αMβ2 integrin in a charge-dependent manner and to regulate thiol
exchange on αMβ2 integrin and its adhesive activity (9). Indeed, Holbrook et al and
Mor-Cohen et al (25,57)
suggested that anti-ERp72 antibodies inhibited platelet
aggregation, granule secretion, calcium mobilization, and integrin
activation, revealing an important role for extracellular ERp72 in
the regulation of platelet activation. Nevertheless, a similar
mechanism may be proposed, whereby ERp72 similarly interacts with
neutrophil integrins. ERp72 knockdown and mouse knockout
experiments will help elucidate this mechanism. In addition,
further characterization of ERp72 binding partners on the surface
of leukocytes and identification of specific modulators for each of
the PDI family members could provide further insight into the
mechanisms through which this class of proteins regulates
neutrophil recruitment and adhesion on the endothelial cells during
vascular inflammation, and may lead to the identification of novel
site-specific targets for modulation of cell-mediated chronic
inflammatory diseases.
Supplementary Material
Supporting Data
Acknowledgements
The authors thank Mr. Ammar Marweni from Arabian
Gulf University (Manama, Bahrain) for his support in the work
involving animals and Mrs. Luma Fayez Al Salah from Arabian Gulf
University (Manama, Bahrain) for language editing.
Funding
This work was funded by an internal research grant
from Arabian Gulf University (grant no. LS_NB_18).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
NBK conceived the study and designed the
experimental work and data analysis. DAM carried out experimental
work and data analysis. DMF conceived the study and analyzed the
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Experimental protocols were approved by the Research
and Ethics committee at Arabian Gulf University (Manama, Bahrain).
All methods were carried out in accordance with the committee's
relevant guidelines and regulations. Participants provided written
informed consent prior to enrolment in the study and for the
publication of the data.
Patient consent for publication
Participants provided written informed consent for
the publication of the data.
Competing interests
The current work used information related to a
biological compound cited in patent FR2909672A1 submitted by
Professor Dahmani Fathallah (Department of Life Sciences, Arabian
Gulf University, Bahrain), Dr M. Ali Jarboui (Institut Pasteur,
Tunis, Tunisia) and Professor Koussay Dellagi (Institut Pasteur,
Tunis, Tunisia) on 11/12/2006 and filed by Institut Pasteur of
Tunis, which is currently undergoing renewal.
Glossary
Abbreviations
Abbreviations:
|
BCECF-AM
|
2′,7′-bis-(2-carboxyethyl)-5(and-6)-carboxyfluorescein
acetoxymethyl
|
|
ER
|
endoplasmic reticulum
|
|
HBSS
|
Hank's balanced salt solution
|
|
HUVEC
|
human umbilical vein endothelial
cell
|
|
NPH
|
neutrophil
|
|
PDI
|
protein disulfide isomerase
|
|
PMN
|
polymorphonuclear neutrophil
|
References
|
1
|
Mitroulis I, Alexaki VI, Kourtzelis I,
Ziogas A, Hajishengallis G and Chavakis T: Leukocyte integrins:
Role in leukocyte recruitment and as therapeutic targets in
inflammatory disease. Pharmacol Ther. 147:123–135. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kansas GS: Selectins and their ligands:
Current concepts and controversies. Blood. 88:3259–3287. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zarbock A and Ley K: Neutrophil adhesion
and activation under flow. Microcirculation. 16:31–42. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chiu J and Hogg PJ: Allosteric disulfides:
Sophisticated molecular structures enabling flexible protein
regulation. J Biol Chem. 294:2949–2960. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Edwards BS, Southon EA, Curry MS, Salazar
F, Gale JM, Robinson MK, Graf LH Jr and Born JL: Oxidant inhibition
of alphaLbeta2 integrin adhesion: Evidence for coordinate effects
on conformation and cytoskeleton linkage. J Leukoc Biol.
63:190–202. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schwartz BR and Harlan JM: Sulfhydryl
reducing agents promote neutrophil adherence without increasing
surface expression of CD11b/CD18 (Mac-1, Mo1). Biochem Biophys Res
Commun. 165:51–57. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shimaoka M, Lu C, Palframan RT, von
Andrian UH, McCormack A, Takagi J and Springer TA: Reversibly
locking a protein fold in an active conformation with a disulfide
bond: Integrin alphaL I domains with high affinity and antagonist
activity in vivo. Proc Natl Acad Sci USA. 98:6009–6014. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shimaoka M, Lu C, Salas A, Xiao T, Takagi
J and Springer TA: Stabilizing the integrin alpha M inserted domain
in alternative conformations with a range of engineered disulfide
bonds. Proc Natl Acad Sci USA. 99:16737–16741. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hahm E, Li J, Kim K, Huh S, Rogelj S and
Cho J: Extracellular protein disulfide isomerase regulates
ligand-binding activity of αMβ2 integrin and neutrophil recruitment
during vascular inflammation. Blood. 121:3789–3800, S1-S15. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rosenberg N, Mor-Cohen R, Sheptovitsky VH,
Romanenco O, Hess O and Lahav J: Integrin-mediated cell adhesion
requires extracellular disulfide exchange regulated by protein
disulfide isomerase. Exp Cell Res. 381:77–85. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Parakh S and Atkin JD: Novel roles for
protein disulphide isomerase in disease states: A double edged
sword? Front Cell Dev Biol. 3:302015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cai H, Wang CC and Tsou CL: Chaperone-like
activity of protein disulfide isomerase in the refolding of a
protein with no disulfide bonds. J Biol Chem. 269:24550–24552.
1994.PubMed/NCBI
|
|
13
|
McLaughlin SH and Bulleid NJ:
Thiol-independent interaction of protein disulphide isomerase with
type X collagen during intra-cellular folding and assembly. Biochem
J. 331:793–800. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kozlov G, Määttänen P, Thomas DY and
Gehring K: A structural overview of the PDI family of proteins.
FEBS J. 277:3924–3936. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Anelli T, Alessio M, Mezghrani A, Simmen
T, Talamo F, Bachi A and Sitia R: ERp44, a novel endoplasmic
reticulum folding assistant of the thioredoxin family. EMBO J.
21:835–844. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vaux D, Tooze J and Fuller S:
Identification by anti-idiotype antibodies of an intracellular
membrane protein that recognizes a mammalian endoplasmic reticulum
retention signal. Nature. 345:495–502. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cho J, Furie BC, Coughlin SR and Furie B:
A critical role for extracellular protein disulfide isomerase
during thrombus formation in mice. J Clin Invest. 118:1123–1131.
2008.PubMed/NCBI
|
|
18
|
Cho J: Protein disulfide isomerase in
thrombosis and vascular inflammation. J Thromb Haemost.
11:2084–2091. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xiong B, Jha V, Min JK and Cho J: Protein
disulfide isomerase in cardiovascular disease. Exp Mol Med.
52:390–399. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou J, Wu Y, Wang L, Rauova L, Hayes VM,
Poncz M and Essex DW: The C-terminal CGHC motif of protein
disulfide isomerase supports thrombosis. J Clin Invest.
125:4391–4406. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Furie B and Flaumenhaft R: Thiol
isomerases in thrombus formation. Circ Res. 114:1162–1173. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li J, Kim K, Jeong SY, Chiu J, Xiong B,
Petukhov PA, Dai X, Li X, Andrews RK, Du X, et al: Platelet protein
disulfide isomerase promotes glycoprotein Ibalpha-mediated
platelet-neutrophil interactions under thromboinflammatory
conditions. Circulation. 139:1300–1319. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim K, Hahm E, Li J, Holbrook LM,
Sasikumar P, Stanley RG, Ushio-Fukai M, Gibbins JM and Cho J:
Platelet protein disulfide isomerase is required for thrombus
formation but not for hemostasis in mice. Blood. 122:1052–1061.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Flaumenhaft R and Furie B: Vascular thiol
isomerases. Blood. 128:893–901. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Holbrook LM, Watkins NA, Simmonds AD,
Jones CI, Ouwehand WH and Gibbins JM: Platelets release novel thiol
isomerase enzymes which are recruited to the cell surface following
activation. Br J Haematol. 148:627–637. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stopa JD and Zwicker JI: The intersection
of protein disulfide isomerase and cancer associated thrombosis.
Thromb Res. 164 (Suppl 1):S130–S135. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Flaumenhaft R, Furie B and Zwicker JI:
Therapeutic implications of protein disulfide isomerase inhibition
in thrombotic disease. Arterioscler Thromb Vasc Biol. 35:16–23.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Z, Zhang H and Cheng Q: PDIA4: The
basic characteristics, functions and its potential connection with
cancer. Biomed Pharmacother. 122:1096882020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou J, Wu Y, Chen F, Wang L, Rauova L,
Hayes VM, Poncz M, Li H, Liu T, Liu J and Essex DW: The disulfide
isomerase ERp72 supports arterial thrombosis in mice. Blood.
130:817–828. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bowley SR, Fang C, Merrill-Skoloff G,
Furie BC and Furie B: Protein disulfide isomerase secretion
following vascular injury initiates a regulatory pathway for
thrombus formation. Nat Commun. 8:141512017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cho J, Kennedy DR, Lin L, Huang M,
Merrill-Skoloff G, Furie BC and Furie B: Protein disulfide
isomerase capture during thrombus formation in vivo depends on the
presence of β3 integrins. Blood. 120:647–655. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jasuja R, Furie B and Furie BC:
Endothelium-derived but not platelet-derived protein disulfide
isomerase is required for thrombus formation in vivo. Blood.
116:4665–4674. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Diamond MS, Staunton DE, de Fougerolles
AR, Stacker SA, Garcia-Aguilar J, Hibbs ML and Springer TA: ICAM-1
(CD54): A counter-receptor for Mac-1 (CD11b/CD18). J Cell Biol.
111:3129–3139. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ivetic A, Hoskins Green HL and Hart SJ:
L-selectin: A major regulator of leukocyte adhesion, migration and
signaling. Front Immunol. 10:10682019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zerria K, Jerbi E, Hammami S, Maaroufi A,
Boubaker S, Xiong JP, Arnaout MA and Fathallah DM: Recombinant
integrin CD11b A-domain blocks polymorphonuclear cells recruitment
and protects against skeletal muscle inflammatory injury in the
rat. Immunology. 119:431–440. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vojtek AB and Hollenberg SM: Ras-Raf
interaction: Two-hybrid analysis. Methods Enzymol. 255:331–342.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bartel PL and Fields S: Analyzing
protein-protein interactions using two-hybrid system. Methods
Enzymol. 254:241–263. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fromont-Racine M, Rain JC and Legrain P:
Toward a functional analysis of the yeast genome through exhaustive
two-hybrid screens. Nat Genet. 16:277–282. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Formstecher E, Aresta S, Collura V,
Hamburger A, Meil A, Trehin A, Reverdy C, Betin V, Maire S, Brun C,
et al: Protein interaction mapping: A Drosophila case study. Genome
Res. 15:376–384. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu ZQ, Mahmood T and Yang PC: Western
blot: Technique, theory and trouble shooting. N Am J Med Sci.
6:1602014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ilié M, Beaulande M, Ben Hadj S, Chamorey
E, Schiappa R, Long-Mira E, Lassalle S, Butori C, Cohen C, Leroy S,
et al: Chromogenic multiplex immunohistochemistry reveals
modulation of the immune microenvironment associated with survival
in elderly patients with lung adenocarcinoma. Cancers. 10:3262018.
View Article : Google Scholar
|
|
43
|
Dixon AR, Bathany C, Tsuei M, White J,
Barald KF and Takayama S: Recent developments in multiplexing
techniques for immunohistochemistry. Expert Rev Mol Diagn.
15:1171–1186. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nuzzi PA, Lokuta MA and Huttenlocher A:
Analysis of neutrophil chemotaxis. Methods Mol Biol. 370:23–36.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kilgore KS, Ward PA and Warren JS:
Neutrophil adhesion to human endothelial cells is induced by the
membrane attack complex: The roles of P-selectin and platelet
activating factor. Inflammation. 22:583–598. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Granger DN and Senchenkova E: Inflammation
and The Microcirculation. Morgan & Claypool Life Sciences; San
Rafael, CA: 2010, View Article : Google Scholar
|
|
47
|
Issekutz AC and Issekutz TB: The
contribution of LFA-1 (CD11a/CD18) and MAC-1 (CD11b/CD18) to the in
vivo migration of polymorphonuclear leucocytes to inflammatory
reactions in the rat. Immunology. 76:655–661. 1992.PubMed/NCBI
|
|
48
|
Issekutz TB: Leukocyte adhesion and the
anti-inflammatory effects of leukocyte integrin blockade. Agents
Actions Suppl. 46:85–96. 1995.PubMed/NCBI
|
|
49
|
Meunier L, Usherwood YK, Chung KT and
Hendershot LM: A subset of chaperones and folding enzymes form
multiprotein complexes in endoplasmic reticulum to bind nascent
proteins. Mol Biol Cell. 13:4456–4469. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Londhe P and Guttridge DC: Inflammation
induced loss of skeletal muscle. Bone. 80:131–142. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Passam FH, Lin L, Gopal S, Stopa JD,
Bellido-Martin L, Huang M, Furie BC and Furie B: Both platelet- and
endothelial cell-derived ERp5 support thrombus formation in a
laser-induced mouse model of thrombosis. Blood. 125:2276–2285.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bennett TA, Edwards BS, Sklar LA and
Rogelj S: Sulfhydryl regulation of L-selectin shedding:
Phenylarsine oxide promotes activation-independent L-selectin
shedding from leukocytes. J Immunol. 164:4120–4129. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Franscini N, Bachli EB, Blau N, Leikauf
MS, Schaffner A and Schoedon G: Gene expression profiling of
inflamed human endothelial cells and influence of activated protein
C. Circulation. 110:2903–2909. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rao RM, Yang L, Garcia-Cardena G and
Luscinskas FW: Endothelial-dependent mechanisms of leukocyte
recruitment to the vascular wall. Circ Res. 101:234–247. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sidney LE, Branch MJ, Dunphy SE, Dua HS
and Hopkinson A: Concise review: Evidence for CD34 as a common
marker for diverse progenitors. Stem Cells. 32:1380–1389. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mor-Cohen R, Rosenberg N, Einav Y, Zelzion
E, Landau M, Mansour W, Averbukh Y and Seligsohn U: Unique
disulfide bonds in epidermal growth factor (EGF) domains of β3
affect structure and function of αIIbβ3 and αvβ3 integrins in
different manner. J Biol Chem. 287:8879–8891. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mor-Cohen R, Rosenberg N, Landau M, Lahav
J and Seligsohn U: Specific cysteines in beta3 are involved in
disulfide bond exchange-dependent and -independent activation of
alphaIIbbeta3. J Biol Chem. 283:19235–19244. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yan B and Smith JW: Mechanism of integrin
activation by disulfide bond reduction. Biochemistry. 40:8861–8867.
2001. View Article : Google Scholar : PubMed/NCBI
|