Introduction
Traumatic brain injury (TBI) is one of the primary
causes of death and disability among individuals aged 1–45 years
(1). It is estimated that 69
million people suffer from TBI each year, among these, 81% exhibit
mild and 11% exhibit moderate symptoms (2). Furthermore, the incidence rate among
males is twice that among females (3). Notably, patients with TBI frequently
experience persistent personality changes and severe neurological
dysfunction, including cognitive loss, as well as physical and
psychological deficiency (4).
Research has confirmed that TBI can trigger a series of
pathological and physiological reactions, ultimately leading to
neurological outcomes (5,6). Moreover, TBI is one of the primary
risk factors for a number of neurological diseases, including
stroke, epilepsy and neurodegenerative disease (7). Therefore, it is important to
understand the molecular mechanisms of post-traumatic neurological
dysfunction and to identify novel therapeutic approaches for the
treatment of TBI.
Autophagy, a conserved catabolic process, maintains
cellular homoeostasis by degeneration of unfolded/misfolded protein
and damaged organelles in a lysosome-dependent pathway (8). The presence of autophagosomes and the
increase in microtubule-associated protein 1 light chain 3 (LC3)-II
expression levels in damaged brain tissue have been observed in
specimens from patients with TBI (9) and TBI models (10). These results suggest that the
autophagy pathway participates in the pathophysiological process of
TBI. However, the precise role of autophagy in TBI-induced
histological and neurological outcomes is complicated and remains
unknown. It has been reported that persistent activation of
autophagy is a protective mechanism for neurological recovery
following TBI (11), and the
inhibition of TBI-induced autophagy may enhance neuronal apoptosis
and microglia activation (12).
Conversely, certain studies have reported that autophagy induction
post-TBI aggravates brain injury, neuroinflammation, neuronal death
and long-term neurological outcomes (13–15).
These findings indicate that autophagy induction serves a
detrimental role in the pathogenesis of TBI. Previous studies have
revealed that the induction of neuronal autophagy in the
hippocampal region can lead to behavioral and cognitive impairments
following TBI (16,17). However, the upstream regulatory
mechanism and the function of autophagy induction remain
unclear.
Evidence has revealed that there is a potential
interaction between the activation of autophagy and endoplasmic
reticulum (ER) stress (18). ER
dysfunction, also known as ER stress, is caused by the accumulation
of misfolded and unfolded proteins (19). In order to restore ER function and
cellular homeostasis, the unfolded protein response (UPR) is
induced via activation of three ER transmembrane proteins,
including ER to nucleus signaling 1 (ERN1), eukaryotic translation
initiation factor 2 α kinase 3 (EIF2AK3) and activating
transcription factor 6 (ATF6) (20). ER stress and autophagy dysfunction
serve significant roles in exacerbating lipid metabolic disorder
and steatohepatitis (21). Recent
evidence has demonstrated that ER stress contributes to the loss of
newborn hippocampal neurons and alteration of dendritic arbors
following TBI (22). It has been
revealed that impaired ER stress can give rise to brain injury
expansion and behavioral and cognitive deficits following juvenile
TBI (23). Nevertheless, activation
of ER stress-associated ATF6 in post-ischemic neurons has been
revealed to significantly decrease infarct volume and neurologic
dysfunction within 24 h of a stroke (24). It has been reported that ER stress
is closely associated with the occurrence of autophagy in numerous
types of disease, including cerebral ischemia, neurodegeneration
and brain injury (25,26). The present study aimed to
investigate the roles and the molecular mechanisms of ER
stress-mediated autophagy in a rat model of TBI and to identify
novel therapeutic targets for the treatment of patients with
TBI.
Materials and methods
Animals and experimental groups
A total of 160 adult (age, 2–3 months) male
Sprague-Dawley rats, (weight, 280–300 g; Beijing Vital River
Laboratory Animal Technology Co., Ltd.) were maintained under
standard laboratory conditions (12-h light/dark cycle; temperature,
21±1°C; humidity, 60%) with free access to water and food. Rats
were randomly assigned to the following groups: Sham (n=20), TBI
(n=30), 3-MA (n=30), 4-phenylbutyric acid (4-PBA; n=30), saline
(n=30), lentivirus (LV)-ATF6 short hairpin (sh)RNA (n=10) and
LV-scrambled shRNA (n=10). Each experiment was performed ≥3
times.
Controlled cortical impact (CCI) model
for TBI
A CCI rat model of TBI was used in the present study
according to a previous study (27). Animals were initially anesthetized
with isoflurane (5%) in oxygen for 2 min and maintained with
isoflurane (3%) in oxygen (0.8 l/min) for the duration of the
procedure. Following exposure of the skull, a 6-mm craniotomy was
performed lateral to the sagittal suture midway between lambda and
bregma. Cortical injury was delivered using an electronic
controlled pneumatic impact device (TBI0310; Precision Systems and
Instrumentation, LLC) using the following parameters: Strike
velocity, 5.0 m/sec; strike depth, 3.0 mm; dwell time, 500 msec.
Body temperature was maintained at 37°C with a thermoregulating
heating pad. In the Sham group, rats were subjected to craniotomy,
but did not receive CCI treatment.
Drug administration and
intracerebroventricular injection of lentiviral vector
Rats were anesthetized with isoflurane (5%) in
oxygen for 2 min and maintained with isoflurane (3%) in oxygen (0.8
l/min). In the 3-MA group, a single intracerebroventricular
injection of autophagy inhibitor 3-MA (600 nmol, diluted in 0.9%
sterile saline to a final volume of 5 µl; Sigma-Aldrich; Merck
KGaA) was administered at the onset of brain surgery. Subsequently,
4-PBA (ChemeGen (Shanghai) Biotechnology Co.,Ltd.), a common ER
stress inhibitor, was diluted with 0.9% sterile saline to a
concentration of 10 mg/ml, and then intraperitoneally injected into
rats at a dose of 100 mg/kg immediately post-TBI. A total of 6 µl
LV-ATF6 or LV-scramble shRNA (5×109 TU/ml; Beijing
Syngentech Co., Ltd.) was injected into lateral ventricle at 48 h
prior to TBI surgery. Transfection efficiencies were analyzed by
reverse transcription-quantitative (RT-q)PCR and western blot
assay.
Modified neurological severity score
(mNSS)
Neurological measurement was performed using the
mNSS test at 1, 3, 7, 14 and 28 days post-TBI. The mNSS comprises
motor, sensory, reflex and beam walking tests (28). Neurological function was graded on a
scale from 0 to 18 (normal, 0; maximal deficit, 18).
Foot fault test
In order to evaluate sensorimotor function, the foot
fault test was performed before TBI and at 1, 3, 7, 14 and 28 days
post-injury. Rats were allowed to walk on a grid. With each
weight-bearing step, a paw may fall or slip between the wires; if
this occurred, it was recorded as a foot fault (29). A total of 50 steps were recorded for
each right forelimb.
Morris water maze (MWM)
The ability of spatial learning and memory was
assessed using a MWM task. Spatial learning began on days 24–28
post-TBI and consisted of four daily trials. Animals were placed in
the pool facing the wall at each of the four potential start
locations in a randomized manner and had 90 sec to locate a hidden
platform. Rats that failed to find the platform within 90 sec were
recorded as having a maximum latency score of 90 sec. Finally, the
mean time spent in the target quadrant searching for the missing
platform and the percentage of time spent in the correct quadrant
were analyzed.
Hematoxylin and eosin (H&E)
staining
Animals were perfused intracardially with PBS
followed by 4% paraformaldehyde (PFA) in PBS in room temperature
for 30 min. Brain tissue was fixed in 4% PFA at 4°C for 72 h,
embedded in paraffin and cut into sections (thickness, 5 µm). The
slices underwent xylene dewaxing and alcohol gradient rehydration
and were stained with hematoxylin for 10 min followed by eosin for
2 min at room temperature. The sections were observed under a light
microscope (Leica Microsystems GmbH; magnification, ×400).
RT-qPCR assay
Total RNA was extracted from hippocampus tissue
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), and then reverse transcribed to generate cDNA
with the Revert Aid™ First Strand cDNA Synthesis kit (Takara
Biotechnology Co., Ltd.) according to the manufacturer's
instructions. The primers were as follows: ATF6 forward,
5′-TCGCCTTTTAGTCCGGTTCTT-3′ and reverse,
5′-GGCTCCATAGGTCTGACTCC-3′; autophagy-related (ATG)3 forward,
5′-GAGGCTACCCTAGACACAAGG-3′ and reverse,
5′-GGCTGCCGTTGCTCATCATA-3′; ATG9 forward,
5′-AACTTTACGTGGCACGAGGT-3′ and reverse, 5′-TGACGACGGACATTCCAAGG-3′;
ATG12 forward, 5′-TCCCCGGAACGAGGAACTC-3′ and reverse,
5′-TTCGCTCCACAGCCCATTTC-3′; β-actin forward,
5′-CCCATCTATGAGGGTTACGC-3′ and reverse,
5′-TTTAATGTCACGCACGATTTC-3′. RT-qPCR was performed using
SYBR-GreenER™ qPCR SuperMix for the iCycler (Invitrogen; Thermo
Fisher Scientific, Inc.) under the following cycling parameters:
95°C for 5 min, followed by 40 cycles of 15 sec at 94°C, 60°C for
15 sec and 72°C for 30 sec. Relative expression levels were
calculated using the 2−ΔΔCq method (30). β-actin was used as the endogenous
control.
Western blotting
Hippocampal specimens were extracted and homogenized
in RIPA buffer containing protease/phosphatase inhibitors (Beyotime
Institute of Biotechnology). The lysates were centrifuged at 12,000
× g for 15 min at 4°C. The concentrations of proteins were examined
using the BCA assay kit (Beyotime Institute of Biotechnology).
Equal amounts of protein (30 µg) were loaded into the wells of 10%
SDS-PAGE and then transferred to nitrocellulose membranes. Then, 5%
non-fat dry milk in TBST containing 0.1% Tween-20 was used for
blocking non-specific protein binding for 2 h at room temperature.
The following primary antibodies were used: Rabbit polyclonal
anti-LC3 (1:500; cat. no. AF1720; Beyotime Institute of
Biotechnology), Beclin-1 (1:1,000; cat. no. AF5123; Beyotime
Institute of Biotechnology), SQSTM1/p62 (1:400; cat. no. AF5312;
Beyotime Institute of Biotechnology), GRP78 (1:500; cat. no.
AF0171; Beyotime Institute of Biotechnology) and ATF6 (1:500; cat.
no. AF6243; Beyotime Institute of Biotechnology) and monoclonal
anti-β-actin (1:5,000; cat. no. 3700T; Cell Signaling Technology,
Inc.) at 4°C overnight. Then, the membranes were incubated with
horseradish peroxidase-conjugated goat anti-rabbit secondary
antibody (1:10,000; cat. no. sc-2004; Santa Cruz Biotechnology,
Inc.) for 1 h at room temperature. The bands were visualized by
enhanced chemiluminescence detection kit (Thermo Fisher Scientific,
Inc.). The expression levels of proteins were normalized to those
of β-actin and quantified using ImageJ software (version 1.46;
National Institutes of Health).
Immunofluorescence staining
The brains were removed, fixed in 4% PFA for 24 h at
room temperature, immersed in 30% sucrose for 72 h at room
temperature, embedded at optimal cutting temperature (Leica
Microsystems GmbH) and sectioned (thickness, 10 µm) using a Leica
cryostat (Leica Microsystems, Inc.). Sections were treated with 5%
donkey serum and 0.1% Triton X-100 for 1 h at room temperature,
then incubated with the primary antibodies: NeuN (1:100; cat. no.
94403; Cell Signaling Technology, Inc.) and LC3 (1:100; cat. no.
sc-271625; Santa Cruz Biotechnology, Inc.)at 4°C overnight.
Following washing in PBS, sections were incubated with Alexa Fluor
488 donkey anti-mouse IgG (1:200; R37114; Invitrogen; Thermo Fisher
Scientific, Inc.) and Alexa Fluor 594 donkey anti-rabbit IgG
(1:200; cat. no. R37119; Invitrogen; Thermo Fisher Scientific,
Inc.) for 1 h at room temperature. Subsequently, nuclear staining
was performed using DAPI (Beyotime Institute of Biotechnology) for
15 min at room temperature. Images were obtained using a laser
scanning confocal microscope (DM 5000B; Leica Microsystems GmbH;
magnification, ×400).
Statistical analysis
Data are presented as the mean ± SD, and each
experiment was repeated three times. Sigma Plot software (version
18.0; IBM Corp.) was used to analyze all data. The unpaired
Student's t-test was used for comparisons of two groups.
Differences among multiple groups were analyzed by one-way ANOVA
with Bonferroni's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
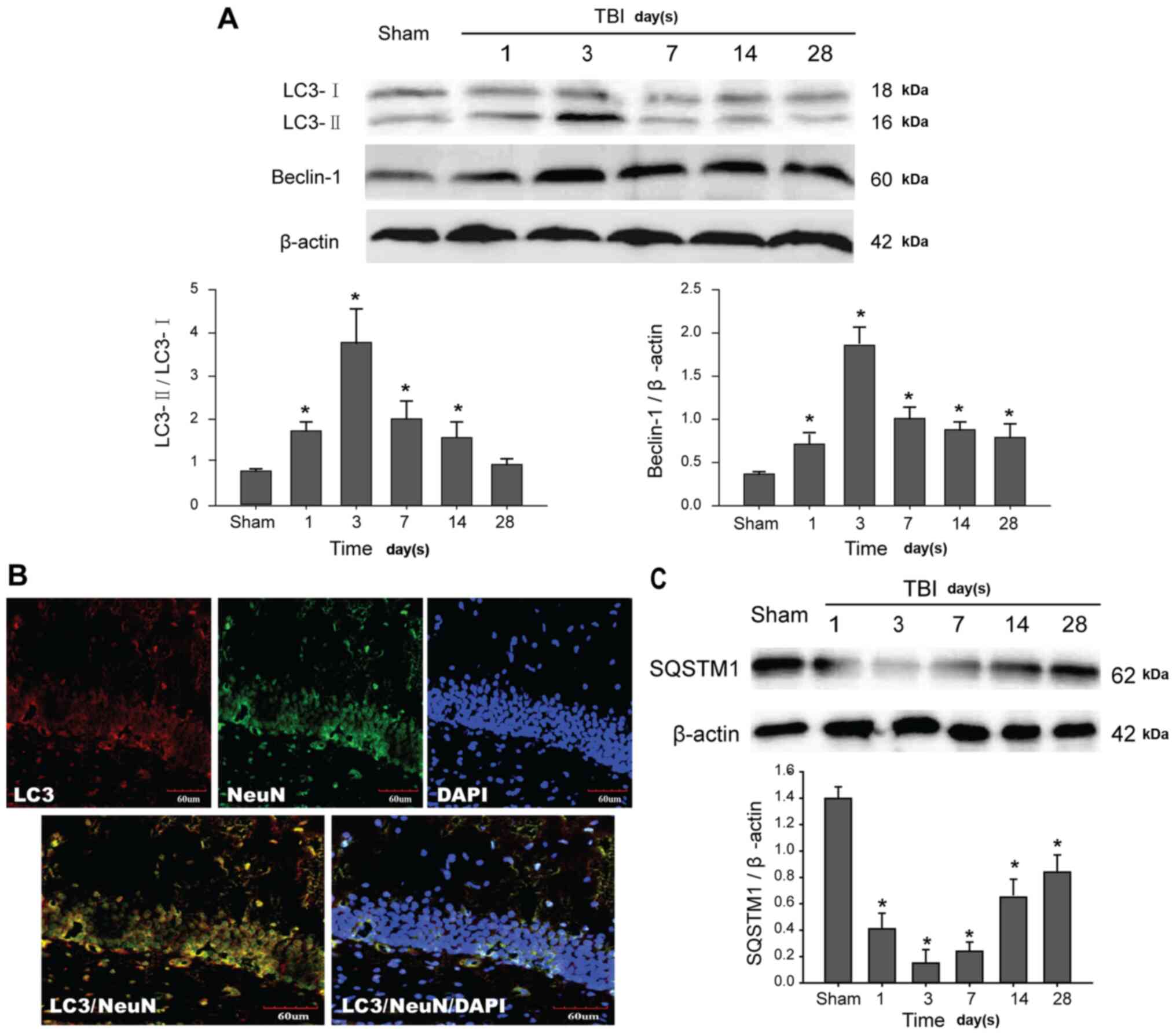
Autophagy is initiated and autophagic
flux is impaired in the hippocampus post-TBI
In order to study the induction of the autophagic
process in the rat hippocampus, western blot analysis was used to
detect the levels of endogenous LC3 conversion and Beclin-1.
Cytosolic LC3-I, a soluble LC3, is formed by ATG4-mediated cleavage
and forms a conjugate with phosphatidylethanolamine to produce the
lipid conjugated LC3-II that is anchored in autophagosome membranes
(10). Therefore, the ratio of
LC3-II to LC3-I is widely recognized and used as a marker for the
formation of autophagosomes. Moreover, Beclin-1 is also essential
for autophagy initiation (8). TBI
increased conversion of LC3-II to LC3-I and Beclin-1 expression
levels in the hippocampal tissue of rats in a time-dependent manner
(Fig. 1A). These results indicated
that the autophagic process was successfully initiated in the
damaged region of the hippocampus. Next, the expression levels and
localization of LC3 in the hippocampus were observed. LC3 protein
was primarily located in the neurons of the hippocampus (Fig. 1B). Next, the changes of autophagic
flux were evaluated by analyzing the degradation of SQSTM1/p62
protein, which is a key indicator of autophagic flux (31). Compared with the Sham group, a
significant decrease of SQSTM1 was observed in post-traumatic
hippocampus tissue, suggesting that autophagic flux was impaired
and selective degradation of SQSTM1 by autophagy was inhibited
(Fig. 1C). Overall, these data
indicated that TBI triggered autophagy initiation, but did not
enhance the degradation of autophagic protein.
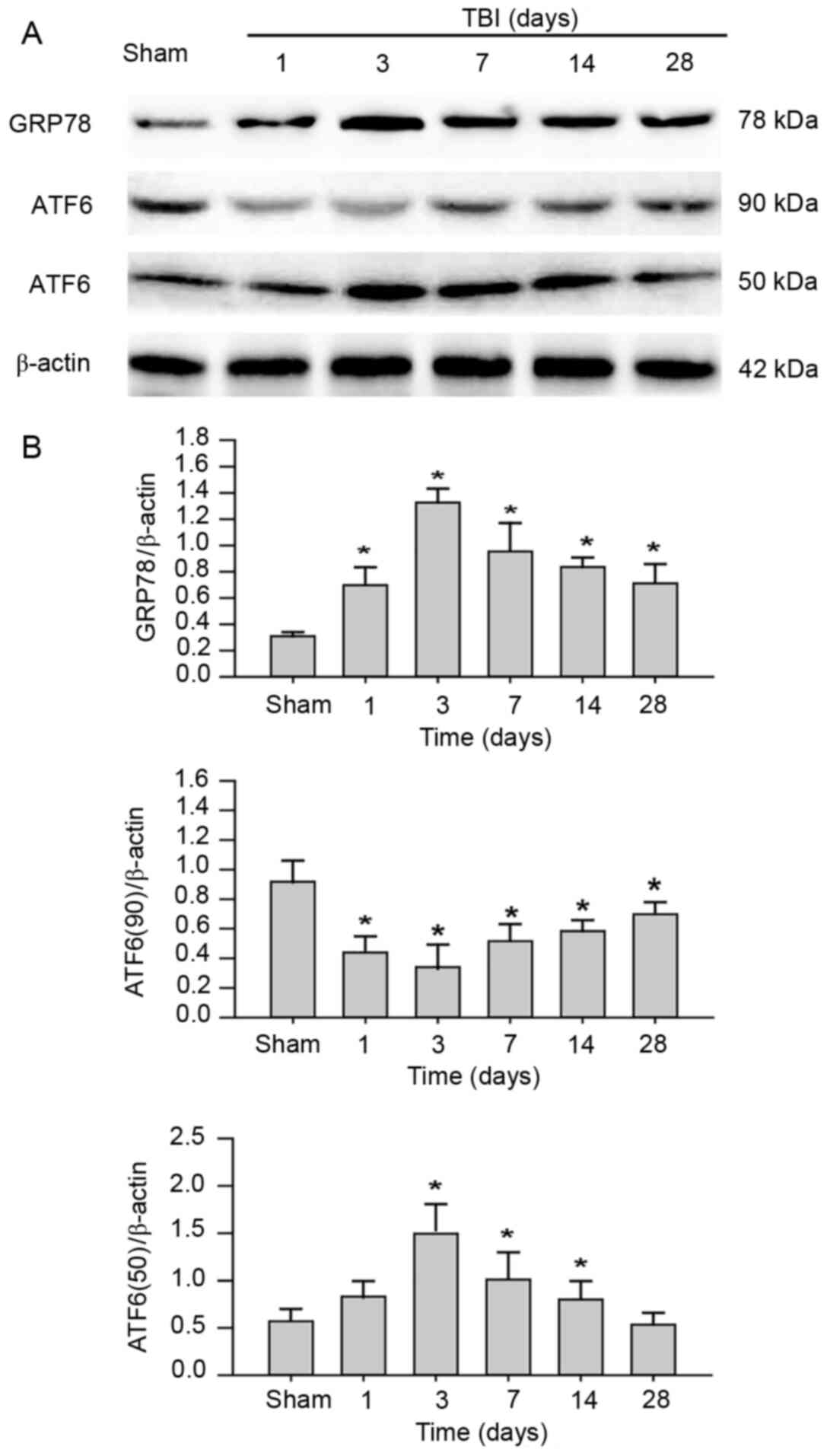
Activation of ER stress is mediated by
ATF6 in the hippocampus post-TBI
In order to investigate whether ER stress was
induced in the hippocampus following TBI, the expression levels of
ER stress-associated proteins GRP78 and ATF6 were examined. GRP78,
an ER chaperone, is important for ER stress signaling, as well as
protein quality control and folding (32). Upon ER stress, GRP78 dissociates on
unfolded proteins to activate EIF2AK3, ERN1 and ATF6 (33). As anticipated, compared with the
Sham group, the protein expression levels of GRP78 were
significantly increased in the hippocampus post-TBI in a
time-dependent manner (Fig. 2).
These results indicated that TBI triggered the activation of ER
stress in the rat hippocampus. In addition, following ER stress,
precursor ATF6 (90 kDa), which is anchored in the ER membrane,
translocates to the Golgi apparatus and is cleaved to produce
transcriptionally active ATF6 (50 kDa) (34). Western blot results revealed that
transcriptionally active ATF6 was upregulated in the hippocampus
tissue, consistent with the expression pattern of GRP78.
Conversely, 90 kDa precursor ATF6 was significantly downregulated
in a time-dependent manner. Collectively, the data indicated that
TBI may induce ER stress via the activation of the ATF6 UFP
pathway.
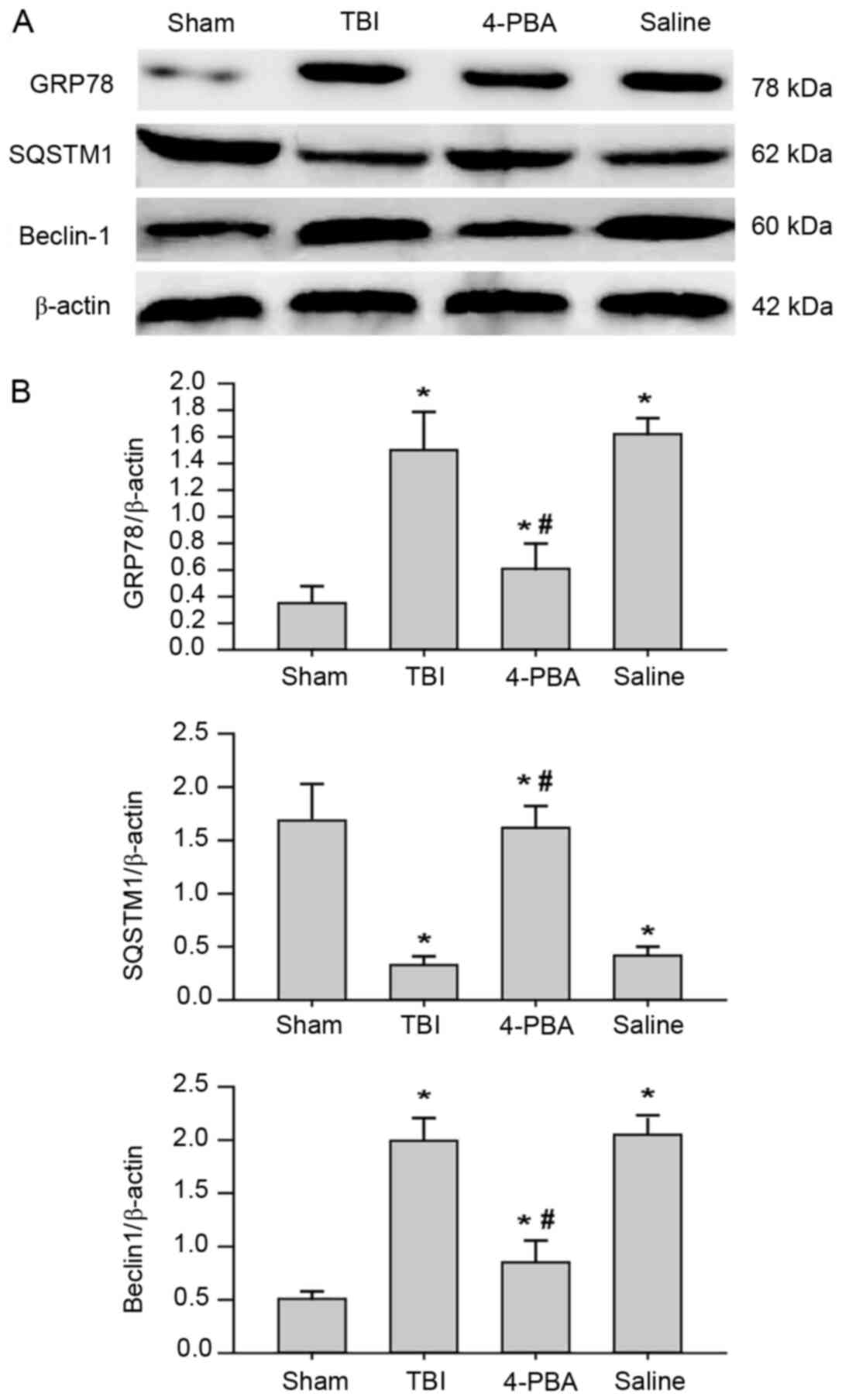
Autophagy is partially induced by
activation of ER stress following TBI
In order to elucidate the association between ER
stress pathways and autophagy activation, ER stress inhibitor 4-PBA
was used to investigate whether the occurrence of autophagy was
caused by ER stress activation. Administration of 4-PBA
significantly decreased the protein expression levels of ER stress
markers in the rat hippocampus at 3 days post-injury (Fig. 3). Notably, TBI-induced autophagy was
significantly prevented in rats pretreated with 4-PBA, as evidenced
by the downregulation of Beclin-1 and upregulation of SQSTM1. These
results indicated that ER stress was a positive regulator of
autophagy induction in the rat model of TBI.
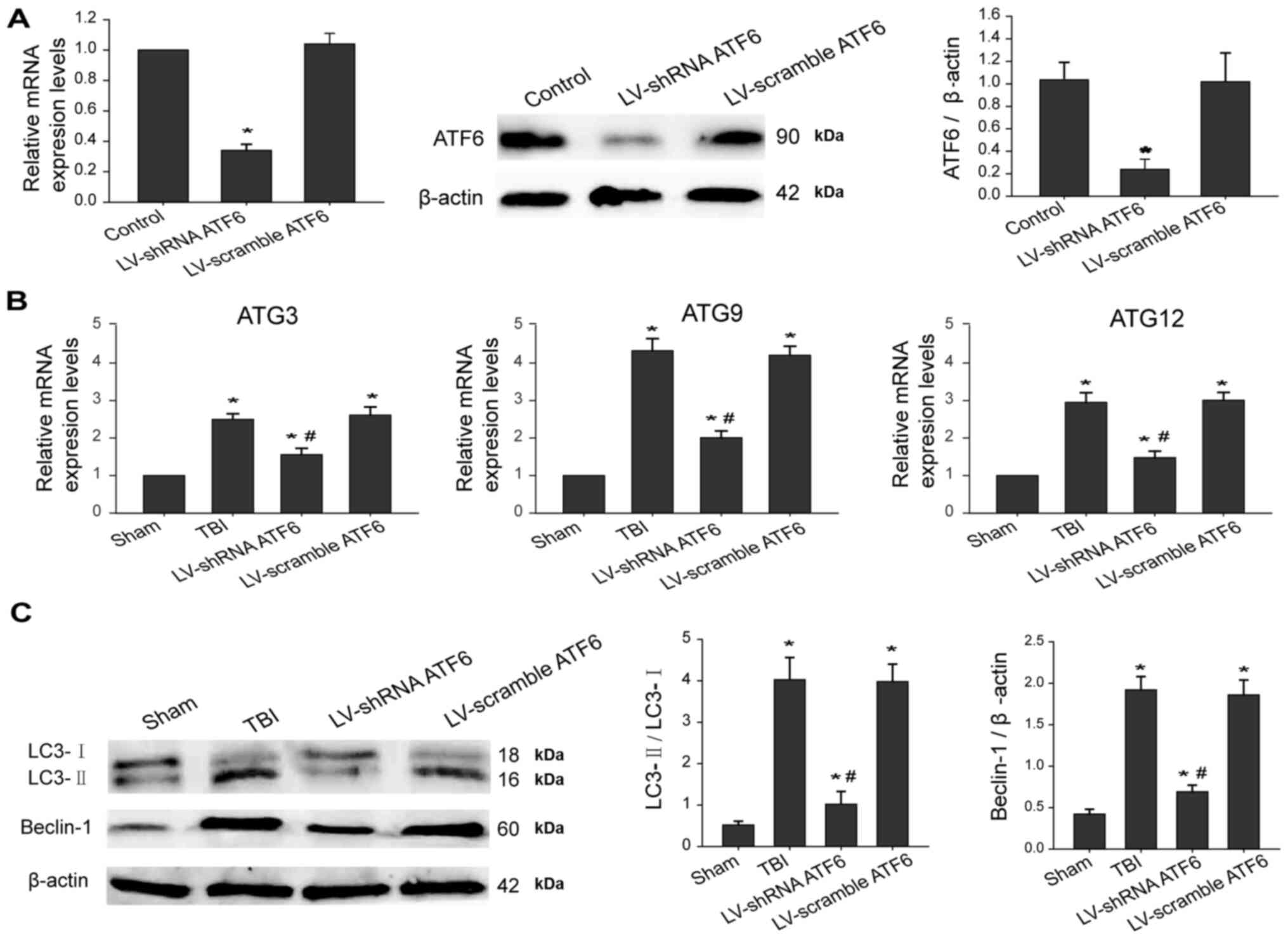
ER stress induces autophagy initiation
via activation of the ATF6 UPR pathway
Next, the effects of ATF6 knockdown on the
expression levels of autophagic makers at 3 days post-TBI were
investigated. Firstly, ATF6 expression in the brain tissues was
silenced using lentiviral-based shRNA. As expected, the stable
decrease in ATF6 mRNA and protein in the hippocampus were mediated
by ATF6 shRNA treatment (Fig. 4A).
Furthermore, RT-qPCR results demonstrated that ATF6 knockdown
significantly decreased mRNA levels of autophagy-associated genes,
including ATG3, ATG9 and ATG12, leading to inactivation of
autophagy (Fig. 4B). Additionally,
western blot results revealed that silencing ATF6 alleviated ER
stress-induced autophagy, as indicated by a significant decrease in
conversion of LC3-I to LC3-II as well as Beclin-1 expression levels
(Fig. 4C). These findings indicated
that the ATF6 pathway was involved in ER stress-induced autophagy
via regulating downstream autophagic genes.
Pharmacological inhibition of ER
stress or autophagy improves TBI and neurological deficiency
The involvement ER stress-mediated autophagy in
post-traumatic histological and neurological impairment was
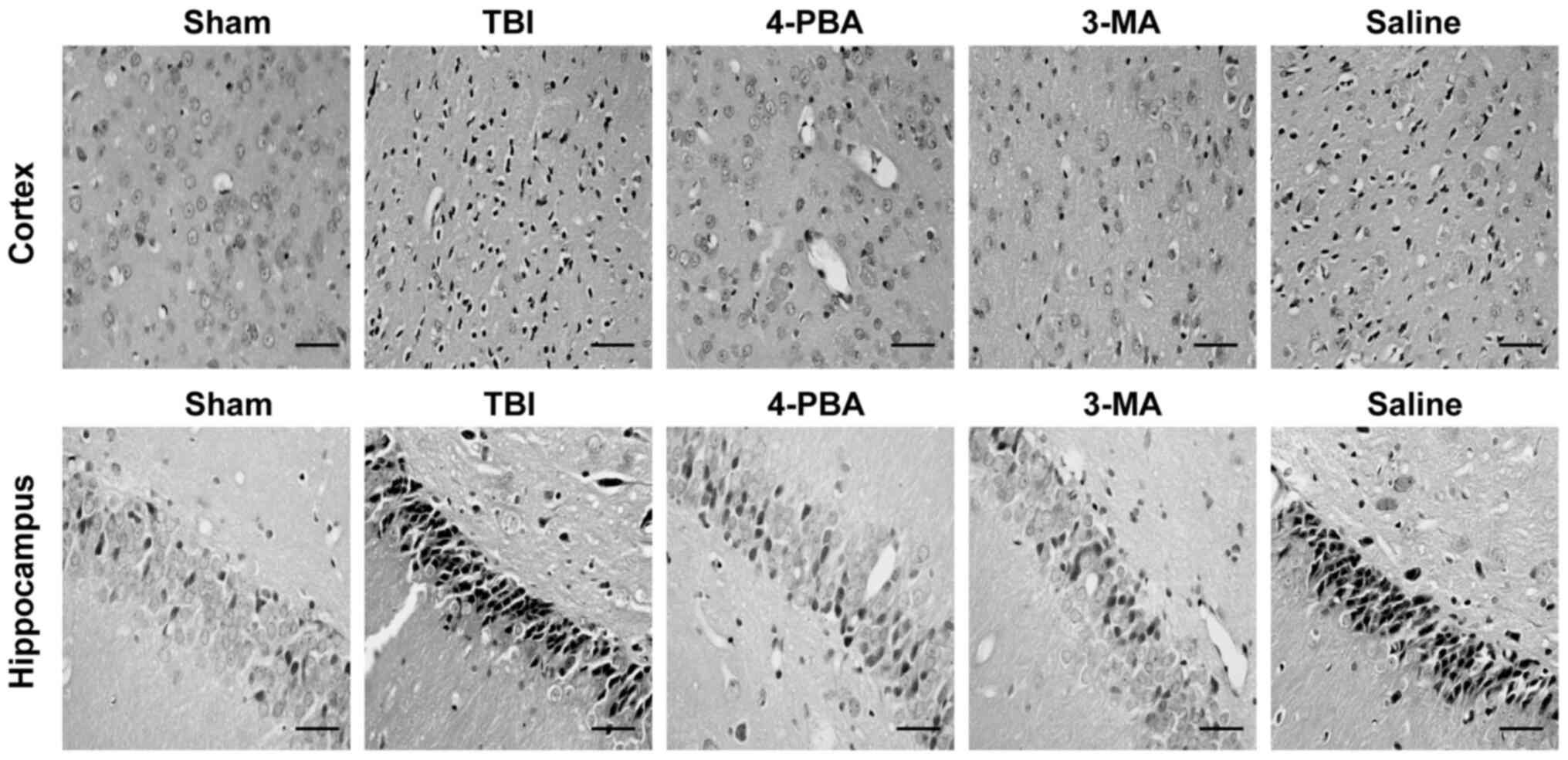
investigated. Compared with the Sham group, a greater number of
damaged neurons were observed in the cortex and hippocampus CA1
region of rats with TBI (Fig. 5).
However, compared with the TBI and saline groups, 4-PBA or 3-MA
treatment was revealed to decrease neuronal death and loss
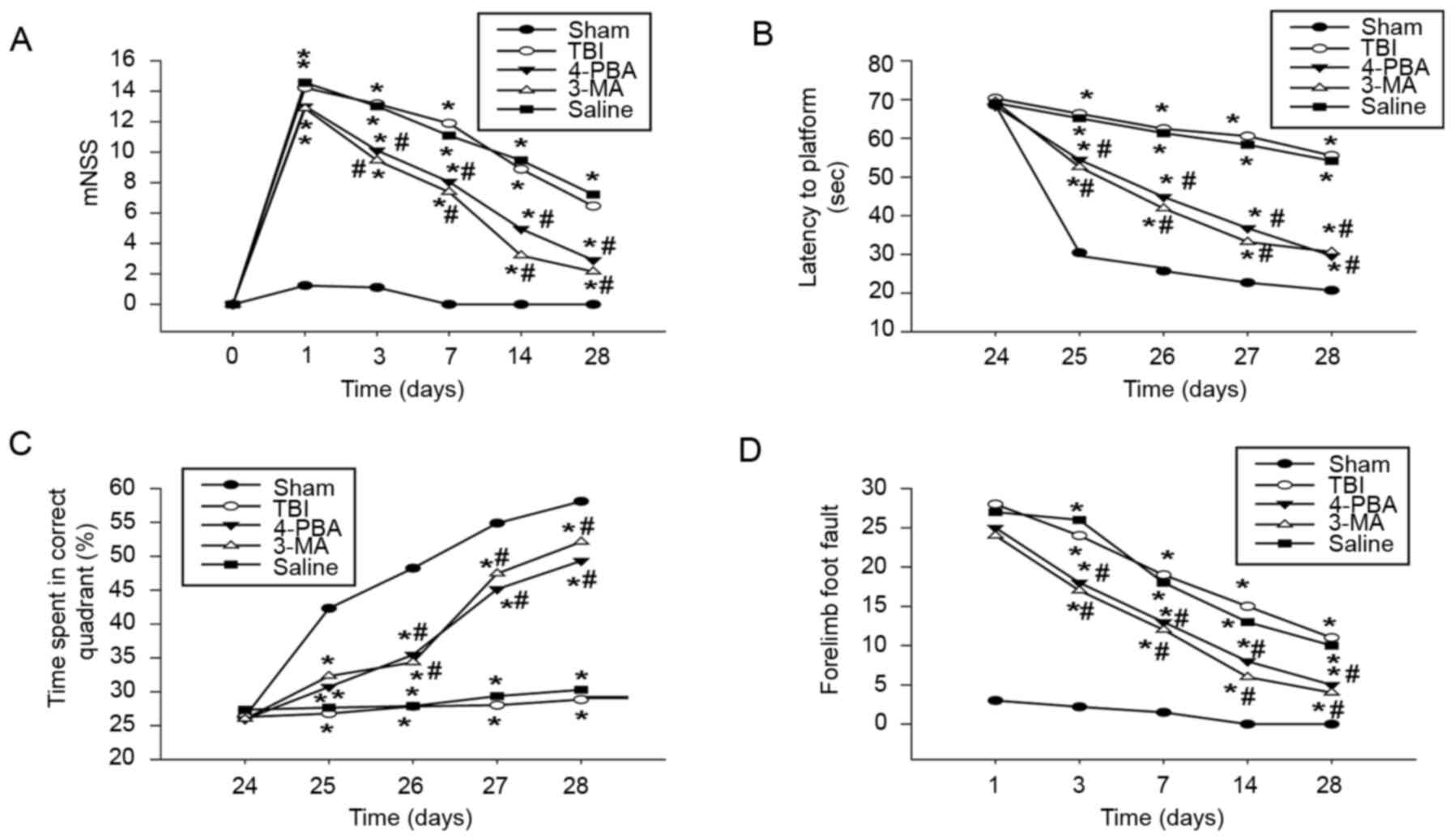
following TBI. Results revealed that treatment with 4-PBA or 3-MA
significantly improved sensorimotor functional recovery, as
indicated by decreased mNSS (Fig.
6A) and frequency of forelimb foot fault occurrence (Fig. 6D). Moreover, 4-PBA or 3-MA
significantly improved cognitive deficits post-TBI, as evidenced by
significantly shortened latency to find the hidden platform
(Fig. 6B) and increased percentage
of time in the correct quadrant (Fig.
6C). Collectively, these results indicated that suppression of
ER stress or autophagy promoted the recovery of neurological
dysfunction following TBI.
Discussion
A number of animal models have been developed to
mimic human TBI, including CCI, lateral fluid percussion injury, as
well as Marmarou's and Feeney's weight-drop model, among them, the
CCI model is widely used to study TBI due to the precise control of
cortical depth penetration, dwell time and speed of impact
(35). A previous study
demonstrated that the rat CCI model replicates clinical TBI
pathophysiology and neurobehavioral impairment (27). In the present study, relevant
phenotypes were demonstrated by brain injury and neuronal loss in
the cortex and hippocampus region of TBI rats, accompanied by
behavioral and cognitive deficits. Autophagy serves a crucial role
in neurobehavioral and cognitive deficiency induced by TBI
(36). Although autophagy is
usually considered to be an essential process for the clearance of
aggregated toxic proteins and damaged organelles, abnormal
autophagy has been implicated in the development of TBI (37). In the present study, the activation
of autophagy was confirmed in the hippocampus, which was reflected
by the increased ratio of LC3-II to LC3-I and Beclin-1 expression
levels, in line with previous studies (38,39).
However, an increased ratio of LC3-II to LC3-I may result from
autophagy activation or autophagic flux defects; therefore, it is
essential to determine whether the accumulation of LC3-II anchored
in autophagosome is due to the increased upstream activation of
autophagy or a blockade of autophagosome-lysosomal fusion.
Therefore, autophagic flux was measured via the preferential
degradation of SQSTM1 following TBI. SQSTM1, a ubiquitin-binding
protein, directly binds to LC3 protein and contributes to its
selective degradation by autophagy (40). The present data revealed that the
protein expression levels of SQSTM1 were decreased in the
post-traumatic hippocampus, suggesting defective autophagic flux
following TBI. Notably, these results revealed that TBI triggered
autophagy initiation and suppressed autophagosome clearance,
leading to autophagic flux impairment. However, the beneficial or
detrimental role of TBI-induced autophagy is controversial. The
results of the present study suggested that pharmacological
inhibition of autophagy significantly ameliorated brain damage,
neurological function score and behavioral and cognitive
impairment. Therefore, it was speculated that TBI-induced autophagy
served a detrimental role in neurological dysfunction and that
targeting autophagy may represent a promising approach for the
treatment of TBI.
Furthermore, accumulating evidence has demonstrated
that ER stress can cause apoptosis, neuronal injury,
neuroinflammation, microglia or macrophage activation and
neurological outcomes following TBI (41,42).
However, the mechanisms by which ER stress mediates autophagy have
not been fully characterized in the rat model of TBI to date. In
the present study, activation of ER stress was verified in the
post-traumatic hippocampus by confirming upregulation of protein
levels of ER stress-associated makers. In order to elucidate the
molecular mechanism underlying upstream activation of autophagy,
rats were pretreated with ER stress inhibitor before TBI induction.
The present results revealed that the suppression of ER stress
significantly decreased the conversion of LC3-I to LC3-II and
Beclin-1 expression levels, as well as neurological dysfunction,
indicating that TBI-induced autophagy activation was mediated by ER
stress.
It is recognized that the ER stress sensors of ATF6
are crucial for autophagy induction in numerous types of disease
including neurodegeneration, stroke, metabolic diseases and cancer
(24,43,44).
ATF6, an ER stress-induced transcription factor, regulates the
expression levels of ER-associated proteins, such as GRP78, DNA
damage inducible transcript 3 and X-box binding protein 1 (45). In the present study, it was revealed
that increased levels ER stress-associated markers were accompanied
by an increase in 50 kDa transcriptionally active ATF6. Therefore,
it was concluded that TBI induced the degradation of 90 kDa ATF6
precursor and generated a 50-kDa cleavage product, known as ATF.
Concurrently, ATF6 knockdown significantly decreased the conversion
of LC3-I to LC3-II and Beclin-1 expression levels, suggesting that
ER stress-induced autophagy was regulated by the ATF6 UPR pathway.
Next, the molecular mechanisms by which ATF6 pathway modulated
autophagy activation were further investigated. A previous study
revealed that depletion of ATF6 decreased transcription of ATG3 and
Beclin-1 in Japanese encephalitis virus-infected cells (46). Another study indicated that hepatic
conditional knockout of ATF6 inhibited autophagy induction by
targeting the mTOR pathway (47).
It has been reported that ER stress triggers autophagy via
ATF6/death-associated protein kinase 1-mediated ATG9 trafficking
and Beclin-1 phosphorylation (48).
Additionally, it has been revealed that LC3 and ATG12 are
transcriptionally upregulated by the ATF6 UPR pathway (49). Accordingly, it was hypothesized that
knockdown of ATF6 suppressed the transcriptional levels of key
autophagic genes, including ATG3, ATG9 and ATG12 post-TBI. To sum
up, these findings indicated that the ATF6 pathway was involved in
ER stress-induced autophagy via transcriptionally activating
crucial autophagic genes, suggesting inhibition of ATF6 could be a
potential therapeutic target for TBI.
In conclusion, ER stress and autophagy were
activated in the hippocampus following TBI. Activation of autophagy
induced by ER stress may contribute to neurological dysfunction
following TBI. Furthermore, it was confirmed that ER stress caused
autophagy activation via ATF6 UPR-mediated transcriptional
activation of autophagic genes.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of Hebei Province (grant no. 16397747D) and the
Health and Family Planning Commission Project of Hebei Province
(grant no. 20171358).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XX designed the research and revised the manuscript.
DYW performed the experiments and prepared the manuscript. MYH, JP,
YHG and YZ analyzed the data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Animal Care and Use Committee of Tangshan Gongren Hospital
(approval no. 2018550). All procedures were performed in accordance
with the Institutional Animal Care and Use Committee of Tangshan
Gongren Hospital and complied with the Declaration of the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ng SY and Lee AYW: Traumatic Brain
Injuries: Pathophysiology and Potential Therapeutic Targets. Front
Cell Neurosci. 13:5282019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dewan MC, Rattani A, Gupta S, Baticulon
RE, Hung YC, Punchak M, Agrawal A, Adeleye AO, Shrime MG, Rubiano
AM, et al: Estimating the global incidence of traumatic brain
injury. J Neurosurg. 1:1–18. 2018.
|
|
3
|
Iaccarino C, Carretta A, Nicolosi F and
Morselli C: Epidemiology of severe traumatic brain injury. J
Neurosurg Sci. 62:535–541. 2018.PubMed/NCBI
|
|
4
|
Pavlovic D, Pekic S, Stojanovic M and
Popovic V: Traumatic brain injury: Neuropathological,
neurocognitive and neurobehavioral sequelae. Pituitary. 22:270–282.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ling H, Hardy J and Zetterberg H:
Neurological consequences of traumatic brain injuries in sports.
Mol Cell Neurosci. 66:114–122. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li D, Ni H, Rui Q, Gao R and Chen G:
Deletion of Mst1 attenuates neuronal loss and improves neurological
impairment in a rat model of traumatic brain injury. Brain Res.
1688:15–21. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wilson L, Stewart W, Dams-O'Connor K,
Diaz-Arrastia R, Horton L, Menon DK and Polinder S: The chronic and
evolving neurological consequences of traumatic brain injury.
Lancet Neurol. 16:813–825. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Z, Miah M, Culbreth M and Aschner M:
Autophagy in neurodegenerative diseases and metal neurotoxicity.
Neurochem Res. 41:409–422. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Clark RS, Bayir H, Chu CT, Alber SM,
Kochanek PM and Watkins SC: Autophagy is increased in mice after
traumatic brain injury and is detectable in human brain after
trauma and critical illness. Autophagy. 4:88–90. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wolf MS, Bayır H, Kochanek PM and Clark
RSB: The role of autophagy in acute brain injury: A state of flux?
Neurobiol Dis. 122:9–15. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu CL, Chen S, Dietrich D and Hu BR:
Changes in autophagy after traumatic brain injury. J Cereb Blood
Flow Metab. 28:674–683. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jin Y, Wang R, Yang S, Zhang X and Dai J:
Role of microglia autophagy in microglia activation after traumatic
brain injury. World Neurosurg. 100:351–360. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zeng Z, Zhang Y, Jiang W, He L and Qu H:
Modulation of autophagy in traumatic brain injury. J Cell Physiol.
235:1973–1985. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li D, Huang S, Zhu J, Hu T, Han Z, Zhang
S, Zhao J, Chen F and Lei P: Exosomes from mir-21-5p-increased
neurons play a role in neuroprotection by suppressing
rab11a-mediated neuronal autophagy in vitro after traumatic brain
injury. Med Sci Monit. 25:1871–1885. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Feldmann LK, Le Prieult F, Felzen V, Thal
SC, Engelhard K, Behl C and Mittmann T: Proteasome and
autophagy-mediated impairment of late long-term potentiation
(l-LTP) after traumatic brain injury in the somatosensory cortex of
mice. Int J Mol Sci. 20:30482019. View Article : Google Scholar
|
|
16
|
Feng Y, Cui C, Liu X, Wu Q, Hu F, Zhang H,
Ma Z and Wang L: Protective role of apocynin via suppression of
neuronal autophagy and TLR4/NF-κB signaling pathway in a rat model
of traumatic brain injury. Neurochem Res. 42:3296–3309. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu K, Wu F, Xu K, Li Z, Wei X, Lu Q, Jiang
T, Wu F, Xu X, Xiao J, et al: NaHS restores mitochondrial function
and inhibits autophagy by activating the PI3K/Akt/mTOR signalling
pathway to improve functional recovery after traumatic brain
injury. Chem Biol Interact. 286:96–105. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cai Y, Arikkath J, Yang L, Guo ML,
Periyasamy P and Buch S: Interplay of endoplasmic reticulum stress
and autophagy in neurodegenerative disorders. Autophagy.
12:225–244. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Su Y and Li F: Endoplasmic reticulum
stress in brain ischemia. Int J Neurosci. 126:681–691. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rashid HO, Yadav RK, Kim HR and Chae HJ:
ER stress: Autophagy induction, inhibition and selection.
Autophagy. 11:1956–1977. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang Q, Li Y, Liang T, Lu X, Zhang C, Liu
X, Jiang X, Martin RC, Cheng M and Cai L: ER stress and autophagy
dysfunction contribute to fatty liver in diabetic mice. Int J Biol
Sci. 11:559–568. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hood KN, Zhao J, Redell JB, Hylin MJ,
Harris B, Perez A, Moore AN and Dash PK: Endoplasmic reticulum
stress contributes to the loss of newborn hippocampal neurons after
traumatic brain injury. J Neurosci. 38:2372–2384. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hylin MJ, Holden RC, Smith AC, Logsdon AF,
Qaiser R and Lucke-Wold BP: Juvenile traumatic brain injury results
in cognitive deficits associated with impaired endoplasmic
reticulum stress and early tauopathy. Dev Neurosci. 40:175–188.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu Z, Sheng H, Liu S, Zhao S, Glembotski
CC, Warner DS, Paschen W and Yang W: Activation of the ATF6 branch
of the unfolded protein response in neurons improves stroke
outcome. J Cereb Blood Flow Metab. 37:1069–1079. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Adhami F, Schloemer A and Kuan CY: The
roles of autophagy in cerebral ischemia. Autophagy. 3:42–44. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yin Y, Sun G, Li E, Kiselyov K and Sun D:
ER stress and impaired autophagy flux in neuronal degeneration and
brain injury. Ageing Res Rev. 34:3–14. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dixon CE, Clifton GL, Lighthall JW,
Yaghmai AA and Hayes RL: A controlled cortical impact model of
traumatic brain injury in the rat. J Neurosci Methods. 39:253–262.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang L, Wang X, Su H, Han Z, Yu H, Wang D,
Jiang R, Liu Z and Zhang J: Recombinant human erythropoietin
improves the neurofunctional recovery of rats following traumatic
brain injury via an increase in circulating endothelial progenitor
cells. Transl Stroke Res. 6:50–59. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Baskin YK, Dietrich WD and Green EJ: Two
effective behavioral tasks for evaluating sensorimotor dysfunction
following traumatic brain injury in mice. J Neurosci Methods.
129:87–93. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Δ Δ C(T)) Method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Evans TD, Sergin I, Zhang X and Razani B:
Target acquired: Selective autophagy in cardiometabolic disease.
Sci Signal. 10:eaag22982017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Z and Li Z: Glucose regulated protein
78: A critical link between tumor microenvironment and cancer
hallmarks. Biochim Biophys Acta. 1826:13–22. 2012.PubMed/NCBI
|
|
33
|
Sprenkle NT, Sims SG, Sánchez CL and
Meares GP: Endoplasmic reticulum stress and inflammation in the
central nervous system. Mol Neurodegener. 12:422017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shen J, Chen X, Hendershot L and Prywes R:
ER stress regulation of ATF6 localization by dissociation of
BiP/GRP78 binding and unmasking of Golgi localization signals. Dev
Cell. 3:99–111. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Whalen MJ, Carlos TM, Kochanek PM, Clark
RS, Heineman S, Schiding JK, Franicola D, Memarzadeh F, Lo W,
Marion DW, et al: Neutrophils do not mediate blood-brain barrier
permeability early after controlled cortical impact in rats. J
Neurotrauma. 16:583–594. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sun L, Zhao M, Liu M, Su P, Zhang J, Li Y,
Yang X and Wu Z: Suppression of FoxO3a attenuates neurobehavioral
deficits after traumatic brain injury through inhibiting neuronal
autophagy. Behav Brain Res. 337:271–279. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zeng XJ, Li P, Ning YL, Zhao Y, Peng Y,
Yang N, Zhao ZA, Chen JF and Zhou YG: Impaired autophagic flux is
associated with the severity of trauma and the role of
A2AR in brain cells after traumatic brain injury. Cell
Death Dis. 9:2522018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun LQ, Gao JL, Cui CM, Cui Y, Jing XB,
Zhao MM, Wang YC, Tian YX, Wang KJ and Cui JZ: Astrocytic
p-connexin 43 regulates neuronal autophagy in the hippocampus
following traumatic brain injury in rats. Mol Med Rep. 9:77–82.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhao M, Liang F, Xu H, Yan W and Zhang J:
Methylene blue exerts a neuroprotective effect against traumatic
brain injury by promoting autophagy and inhibiting microglial
activation. Mol Med Rep. 13:13–20. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Komatsu M, Kageyama S and Ichimura Y:
p62/SQSTM1/A170: Physiology and pathology. Pharmacol Res.
66:457–462. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gao Y, Zhang MY, Wang T, Fan YY, Yu LS, Ye
GH, Wang ZF, Gao C, Wang HC, Luo CL, et al: IL-33/ST2L Signaling
Provides Neuroprotection Through Inhibiting Autophagy, Endoplasmic
Reticulum Stress, and Apoptosis in a Mouse Model of Traumatic Brain
Injury. Front Cell Neurosci. 12:952018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang CF, Zhao CC, He Y, Li ZY, Liu WL,
Huang XJ, Deng YF and Li WP: Mild hypothermia reduces endoplasmic
reticulum stress-induced apoptosis and improves neuronal functions
after severe traumatic brain injury. Brain Behav. 9:e012482019.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sun X, Zhang X, Zhai H, Zhang D and Ma S:
Chicoric acid (CA) induces autophagy in gastric cancer through
promoting endoplasmic reticulum (ER) stress regulated by AMPK.
Biomed Pharmacother. 118:1091442019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Iurlaro R and Muñoz-Pinedo C: Cell death
induced by endoplasmic reticulum stress. FEBS J. 283:2640–2652.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Namba T, Ishihara T, Tanaka K, Hoshino T
and Mizushima T: Transcriptional activation of ATF6 by endoplasmic
reticulum stressors. Biochem Biophys Res Commun. 355:543–548. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sharma M, Bhattacharyya S, Sharma KB,
Chauhan S, Asthana S, Abdin MZ, Vrati S and Kalia M: Japanese
encephalitis virus activates autophagy through XBP1 and ATF6 ER
stress sensors in neuronal cells. J Gen Virol. 98:1027–1039. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sun X, Li W, Deng Y, Dong B, Sun Y, Xue Y
and Wang Y: Hepatic conditional knockout of ATF6 exacerbates liver
metabolic damage by repressing autophage through MTOR pathway.
Biochem Biophys Res Commun. 505:45–50. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhou Y, Zhang S, Dai C, Tang S, Yang X, Li
D, Zhao K and Xiao X: Quinocetone triggered ER stress-induced
autophagy via ATF6/DAPK1-modulated mAtg9a trafficking. Cell Biol
Toxicol. 32:141–152. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wang J, Kang R, Huang H, Xi X, Wang B,
Wang J and Zhao Z: Hepatitis C virus core protein activates
autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B
and ATG12 expression. Autophagy. 10:766–784. 2014. View Article : Google Scholar : PubMed/NCBI
|