Introduction
Being a common outcome of numerous cardiovascular
diseases, heart failure (HF) is characterized by high incidence and
poor prognosis, and it is a serious health problem worldwide
(1,2). Myocardial infarction is the main cause
of HF. A series of pathological lesions following myocardial
infarction, including cardiomyocyte apoptosis, inflammatory
response and overactivation of the neurohumoral system, result in
ventricular remodeling and thus HF (3). Although progress has been made in the
fundamental research of HF following myocardial infarction, a large
number of myocardial infarction patients are not diagnosed and
treated in a timely manner owing to atypical symptoms. They
eventually deteriorate to HF or arrhythmia (4). Therefore, improvement of the
diagnostic rate in the early phase and investigation of relevant
regulatory targets are necessary to enhance the therapeutic
efficacy and prognosis in patients with HF or myocardial
infarction.
Sirtuin 1 (SIRT1) is a highly conserved
NAD-dependent histone deacetylase, which is abundantly expressed in
mammalian hearts. SIRT1 serves a vital role in regulating the
energy metabolism of cardiac muscle cells, production of reactive
oxygen species (ROS), induction of angiogenesis and inhibition of
autophagy (5,6). It is reported that SIRT1 exerts a
protective effect in HF by alleviating oxidative stress, fibrosis
and inflammation through SIRT1/PCG-1α signaling (7). Previous studies have demonstrated that
microRNA-138-5p (miR-138-5p) is able to downregulate SIRT1, its
downstream target. The inhibitory role of miR-138-5p in tumor
progression and metastasis has been identified (8,9).
Recently, experimental studies have demonstrated that miR-138-5p is
involved in the process of heart diseases (10,11).
Wang et al (10) proposed
that overexpression of miR-138-5p leads to aggravation of cardiac
injury through accelerating cardiac hypoxia and reperfusion via
inactivating the SIRT1-PGC-1α axis. However, the potential function
of miR-138-5p in the progression of HF remains largely unclear.
During the progression of HF, cardiomyocyte
apoptosis is of great significance. Due to the non-renewability of
the myocardium, apoptotic or necrotic cardiomyocytes are recognized
by the body and replaced by scar tissue. Subsequently, insufficient
blood supply and ventricular remodeling ultimately lead to HF
(12). A
H2O2-induced HF model was widely used in
in vitro or in vivo HF research (13,14).
In the present study, in vitro HF models were generated by
H2O2 induction in AC-16 and human
cardiomyocyte (HCM) cells. The role of miR-138-5p in influencing
the process of HF and the involvement of SIRT1 were mainly
investigated.
Materials and methods
Cell culture
Human cardiomyocyte cell lines, including AC16 (cat.
no. BNCC337712), HCM (cat. no. BNCC337719), HCFB (cat. no.
BNCC339420) and CCC-HEH-2 (cat. no. BNCC100615) were obtained from
the BeNa culture collection and cardiomyocyte HCM-a cells (cat. no.
XK-1262) were purchased from Shanghai Xuanke Biotechnology Co.,
Ltd. Cells were cultured in Dulbecco's modified Eagle's medium
(DMEM; Gibco; Thermo Fisher Scientific, Inc.) containing 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), and 1%
penicillin and streptomycin in a humidified atmosphere of 5%
CO2 at 37°C. Cell passage was conducted using trypsin
until adherent cells were grown to >80% confluence.
Dual-luciferase reporter assay
Binding sites were predicted between the seed
sequences of miR-138-5p and SIRT1 using TargetScan 7.2 (http://www.targetscan.org/vert_72). Sequences
containing the SIRT1 3′-untranslated region (3′-UTR), mutant SIRT1
3′-UTR and miR-138-5p sequences were constructed by CoBioer
Biosciences Co., Ltd. DNAs were directly synthesized by annealing,
and luciferase vectors were constructed using pmirGLO (Promega
Corporation) following splicing by the restriction endonucleases
NheI and SalI. Plasmids were extracted following
culturing with E. coli, and they were respectively named as
pmirGLO-SIRT1-3′UTR and pmirGLO-SIRT1-mut3′UTR. A plasmid
overexpressing miR-138-5p was generated using pcDNA3.1(+), which
was named as pcDNA3.1(+)-miR-138-5p.
AC-16 cells were transfected with plasmids
classified as follows: i) pmirGLO-SIRT1-3′UTR + pcDNA3.1(+); ii)
pmirGLO-SIRT1-3′UTR + pcDNA3.1(+)-miR-138-5p; iii)
pmirGLO-SIRT1-mut3′UTR + pcDNA3.1(+;) and iv)
pmirGLO-SIRT1-mut3′UTR + pcDNA3.1(+)-miR-138-5p. In brief, AC-16
cells were seeded onto a 96-well plate and cultured to ~80%
confluence. Cell transfection was performed using Lipofectamine
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) for 6 h, then the
medium was replaced with fresh culture medium. At 48 h
post-transfection, 100 µl cell lysate was added to each well, and
the mixture was collected and centrifuged at room temperature at
13,500 × g for 5 min. A total of 50 µl supernatant was incubated in
100 µl Firefly luciferase detection reagent (Promega Corporation).
They were rapidly mixed together and subjected to detection of
relative light units (RLU). Three replicates were set in each
group. Relative firefly luciferase activity was normalized to
Renilla luciferase activity.
Cell transfection
Oligonucleotide sequences of the miR-138-5p mimic
(5′-AGCUGGUGUUGUGAAUCAGGCCG-3′) and miR-138-5p inhibitor
(5′-CGGCCUGAUUCACAACACCAGCU-3′), as well as their negative controls
(NC; miR-138-5p mimic-NC, 5′-UUCUCCGAACGUGUCACGUTT-3′; and
miR-138-5p inhibitor-NC, 5′-UUCUCCGAACGUGUCACGUTT-3′) were
purchased from Guangzhou RiboBio Co., Ltd. All oligonucleotides
were transfected at a final concentration of 50 nM. Cell
transfection was performed using Lipofectamine 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.). Briefly, AC-16 and HCM cells were
seeded onto a 24-well plate and cultured for 24 h. Until cell
confluence reached >70%, cells were cultured in serum-free DMEM
for 4 h. miR-138-5p mimic, miR-138-5p inhibitor or NC was
respectively mixed with polyethylenimine at room temperature for 40
min, followed by application of serum-free Opti-MEM (Gibco; Thermo
Fisher Scientific, Inc.). Cells were incubated in the transfection
mixture at a final concentration of 100 nM at 37°C in a humidified
atmosphere containing 5% CO2 for 6 h, and then the
transfection mixture was replaced with DMEM containing 10% FBS, and
1% penicillin and streptomycin. Transfection efficacy was observed
under a fluorescence microscope (magnification, ×100) using
FAM-labeled miRNAs as internal references. Cells were used
following 24–48 h of transfection for subsequent experiments.
Generation of the in vitro HF
models
AC-16 and HCM cells transfected for 24 h in the
logarithmic growth phase were prepared in suspension at
1×104 cells/ml. Cell suspension was applied to a 6-well
plate with 2 ml/well. Following culturing for 24 h, DMEM containing
2.5% FBS was replaced for 12-h synchronization treatment.
Subsequently, cells were induced in a medium containing or not
containing 200 µM hydrogen peroxide (H2O2)
for 6 h. Successful generation of the in vitro HF models was
defined through detecting the relative level of natriuretic peptide
precursor B (NPPB) (15,16).
Detection of ROS level
AC-16 and HCM cells were collected and inoculated
into a 24-well plate. When the cell density was ~80%, 10 µmol/l
2,7-Dichlorodi-hydrofluorescein diacetate was added into each well,
and the cells were cultured at 37°C for 30 min. Subsequently, five
visual fields were randomly selected under the fluorescence
microscope (magnification, ×100) to analyze the green fluorescence
intensity, and the mean fluorescence intensity (MFI) was used to
demonstrate the ROS level.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Cellular RNA was isolated by TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.). Then, cellular RNA
was processed by gDNA eraser and reverse transcribed at 37°C for 15
min and 85°C for 5 sec (maintained at 4°C) into cDNA using a
PrimeScript™ RT reagent kit with gDNA Eraser (Takara Bio, Inc.).
qPCR was subsequently performed using a SYBR Premix Ex Taq™ II (Tli
RNase H Plus) kit (Takara Bio, Inc.). qPCR was conducted at 95°C
for 15 min, followed by 40 cycles at 95°C for 5 sec, 60°C for 30
sec and 72°C for 40 sec, followed by a final extension at 72°C for
10 min. Relative levels of NPPB, SIRT1, p53 and miR-138-5p were
determined using the internal references of GAPDH and U6. Each
experiment was conducted in triplicate. Primer sequences are listed
in Table I. The data were
quantified using the 2−ΔΔCq method (17).
 | Table I.Sequences of primers. |
Table I.
Sequences of primers.
| Genes | Sequences |
|---|
| miR-138-5p |
5′-AGCTGGTGTTGTGAATCAGG-3′ |
| SIRT1 forward |
5′-GTATTTATGCTCGCCTTGCTG-3′ |
| SIRT1 reverse |
5′-TGACAGAGAGATGGCTGGAA-3′ |
| NPPB forward |
5′-AAGGAGGCACTGGGAGAGGGGAAT-3′ |
| NPPB reverse |
5′-CCCCACCAAGCCAACACAGGATGGA-3′ |
| p53 forward |
5′-GAGCGAATCACGAGGGAC-3′ |
| p53 reverse |
5′-GCACAAACACGGACAGGA-3′ |
| GAPDH forward |
5′-AGCCACATCGCTCAGACAC-3′ |
| GAPDH reverse |
5′-GCCCAATACGACCAAATCC-3′ |
| U6 forward |
5′-CTCGCTTCGGCAGCACA-3′ |
| U6 reverse |
5′-AACGCTTCACGAATTTGCGT-3′ |
Western blotting
Cardiomyocytes were transfected or induced with
H2O2 as described earlier. Subsequently,
protein levels of SIRT1, p53 and acetylated p53 were determined by
western blotting. In brief, cells were lysed to isolate total
proteins using RIPA lysis buffer (Beyotime Institute of
Biotechnology), and total concentrations were determined using the
bicinchoninic acid method. Protein samples (50 µg per lane) were
subjected to 10% SDS-PAGE and transferred onto polyvinylidene
fluoride (PVDF) membranes. Non-specific antigens in the PVDF
membranes were blocked using 5% skimmed milk at room temperature
for 1 h. Following washing in TBS-0.05% Tween-20, the membranes
were incubated with the following primary antibodies (dilution,
1:1,000; all ABclonal Biotech Co., Ltd.) at 4°C overnight:
Anti-SIRT1 (cat. no. A19667), anti-p53 (cat. no. A19585),
anti-acetyl-p53 (cat. no. A16324) and anti-GAPDH (cat. no. AC001).
Following the primary antibody incubation, the membranes were
incubated with the secondary antibody (dilution, 1:1,000; cat. no.
AS014; ABclonal Biotech Co., Ltd.) at room temperature for 1 h.
Band exposure was achieved using enhanced chemiluminescence reagent
(Sangon Biotech Co., Ltd.). Protein expression levels were
quantified using Quantity One® 1-D Analysis software
(version 4.6.8; Bio-Rad Laboratories, Inc.).
Flow cytometric analysis of cell
apoptosis
Following H2O2 induction for 6
h, cells were subjected to apoptosis detection using the Annexin
V-FITC and propidium iodide (PI) Apoptosis kit (US Everbright
Inc.). To begin with, cells were prepared in suspension at
5×105 cells/ml. Cell suspension was incubated with 5 µl
Annexin V-FITC and 5 µl PI in the dark at room temperature for 15
min, and subjected to flow cytometry (FACSCalibur; BD Biosciences).
Apoptotic cells were analyzed using CellQuest Pro software (version
5.1; BD Biosciences). Annexin V-FITC-positive cells were considered
to be apoptotic cells.
Determination of the enzymatic
activity of SIRT1
SIRT1 Direct Fluorescent Screening assay kit (Abcam;
cat. no. ab156915) was used to assess the enzyme activity of SIRT1.
Total protein (30 µg per well in a 96-well plate) was incubated
with the reaction mixture containing buffer, SIRT1 substrate with
fluorophore and quencher, and protease. Absorbances at 360 nm
excitation wavelength and 465 nm emission wavelength were recorded
using the microplate reader (BioTek Instruments, Inc.).
Lentivirus transfection
A lentivirus overexpressing SIRT1 (GV287-SIRT1) was
constructed by cleavage of GV287 plasmid (Shanghai GeneChem Co.,
Ltd.) with AgeI and thus splicing SIRT1. A 2nd generation
system was used to package the lentivirus. The lentiviral plasmid,
packaging vector and envelope vector were mixed at a 4:3:2 ratio
for a total DNA mass of 20 µg. In brief, 293T cells were cultured
to 80% confluence and then DMEM was replaced by Opti-MEM. Following
cell culture for 4 h at 37°C, cells were incubated with Lenti-Easy
Packaging mix and GV287-SIRT1 (or GV287 as negative control) for 5
min at room temperature, then Lipofectamine for a further 20 min,
prior to the addition of 293T fresh culture medium (cat. no.
BNCC352005; BeNa Culture Collection; Beijing Beina Chunglian
Biotechnology Research Institute) for 6 h at 37°C. Following the
incubation, the medium was replaced with DMEM containing 10% FBS,
1% penicillin and streptomycin and cultured to day 3. The
supernatant of transfected 293T cells was collected at day 3, which
was filtered for detecting viral titers. Lentiviral transfection
(at a multiplicity of infection of 5) in AC-16 and HCM cells was
conducted for 12 h at 37°C when cell confluence reached ~80%.
Fluorescence expression was observed 3 days later.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 8 (GraphPad Software, Inc.). Data are expressed as the mean ±
standard deviation. All data conformed to a normal distribution.
Comparisons among multiple groups were analyzed using one-way
analysis of variance followed by Tukey's or Bonferroni's post hoc
test. The correlation analysis was performed by Pearson's
correlation analysis. P<0.05 was considered to indicate a
statistically significant difference. All experiments were
performed in triplicate and repeated three times.
Results
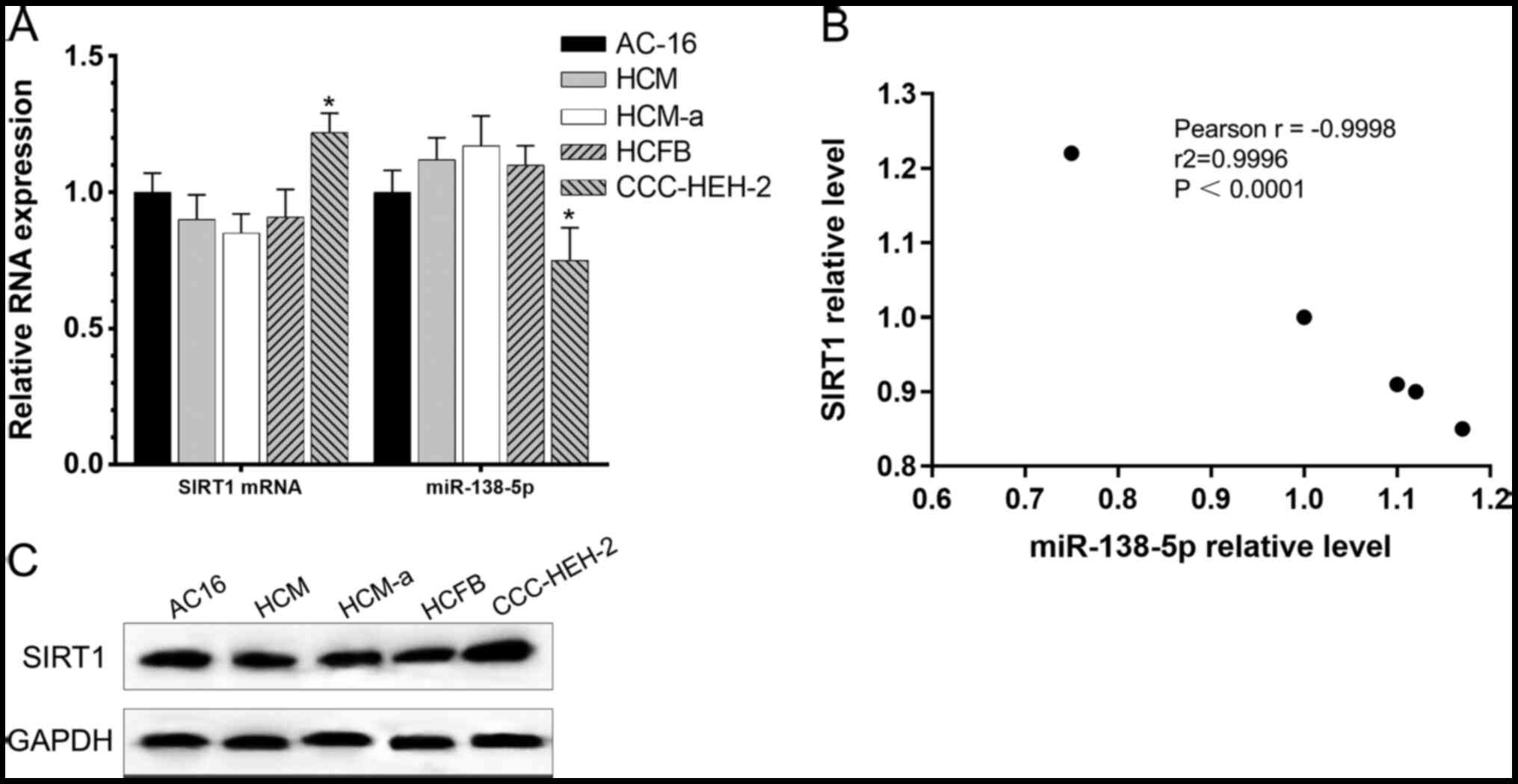
A negative correlation between SIRT1
and miR-138-5p in cardiomyocytes
Following cell culture in DMEM containing 10% FBS
for 48 h, the relative levels of SIRT1 and miR-138-5p were detected
in cardiomyocyte cell lines (AC16, HCM, HCM-a, HCFB and CCC-HEH-2)
by RT-qPCR and Western blotting. Relative RNA expression is
presented in Fig. 1A and B, and
protein expression is presented in Fig.
1C. According to the Pearson's correlation analysis, a negative
correlation was identified between SIRT1 and miR-138-5p at mRNA
levels.
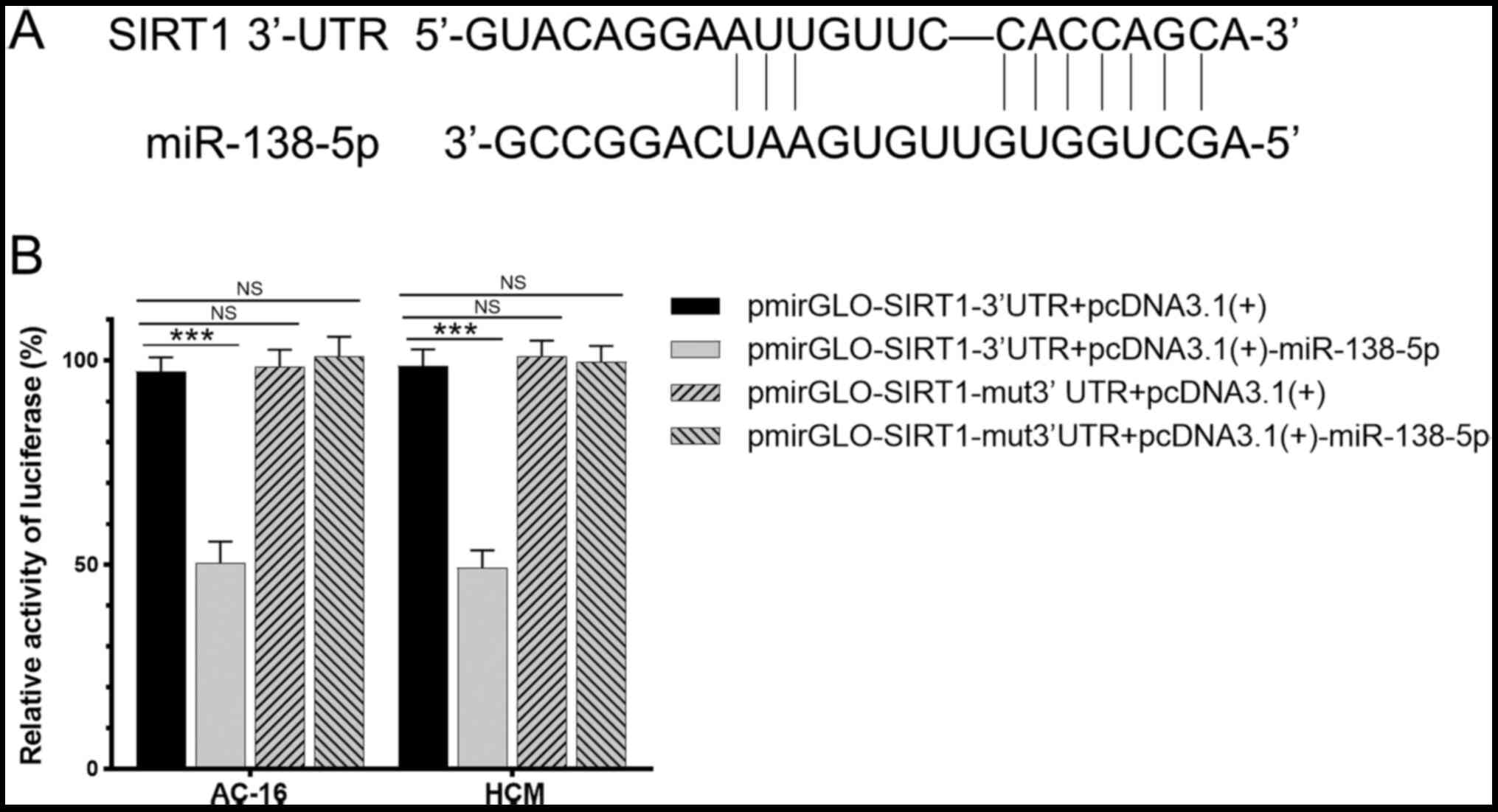
miR-138-5p targets SIRT1
Binding sites were predicted by TargetScan and the
result was shown in Fig. 2A. The
dual-luciferase reporter assay revealed that luciferase intensity
in cells co-transfected with pmirGLO-SIRT1-3′UTR and
pcDNA3.1(+)-miR-138-5p was 48% of that in the control group
(P<0.01). Notably, luciferase intensity in cells co-transfected
with pmirGLO-SIRT1-mut3′UTR and pcDNA3.1(+)-miR-138-5p was up to
99% of that in the control group, demonstrating no significant
difference (P>0.05; Fig. 2B).
This indicated that miR-138-5p may regulate SIRT1 with sequence
specificity.
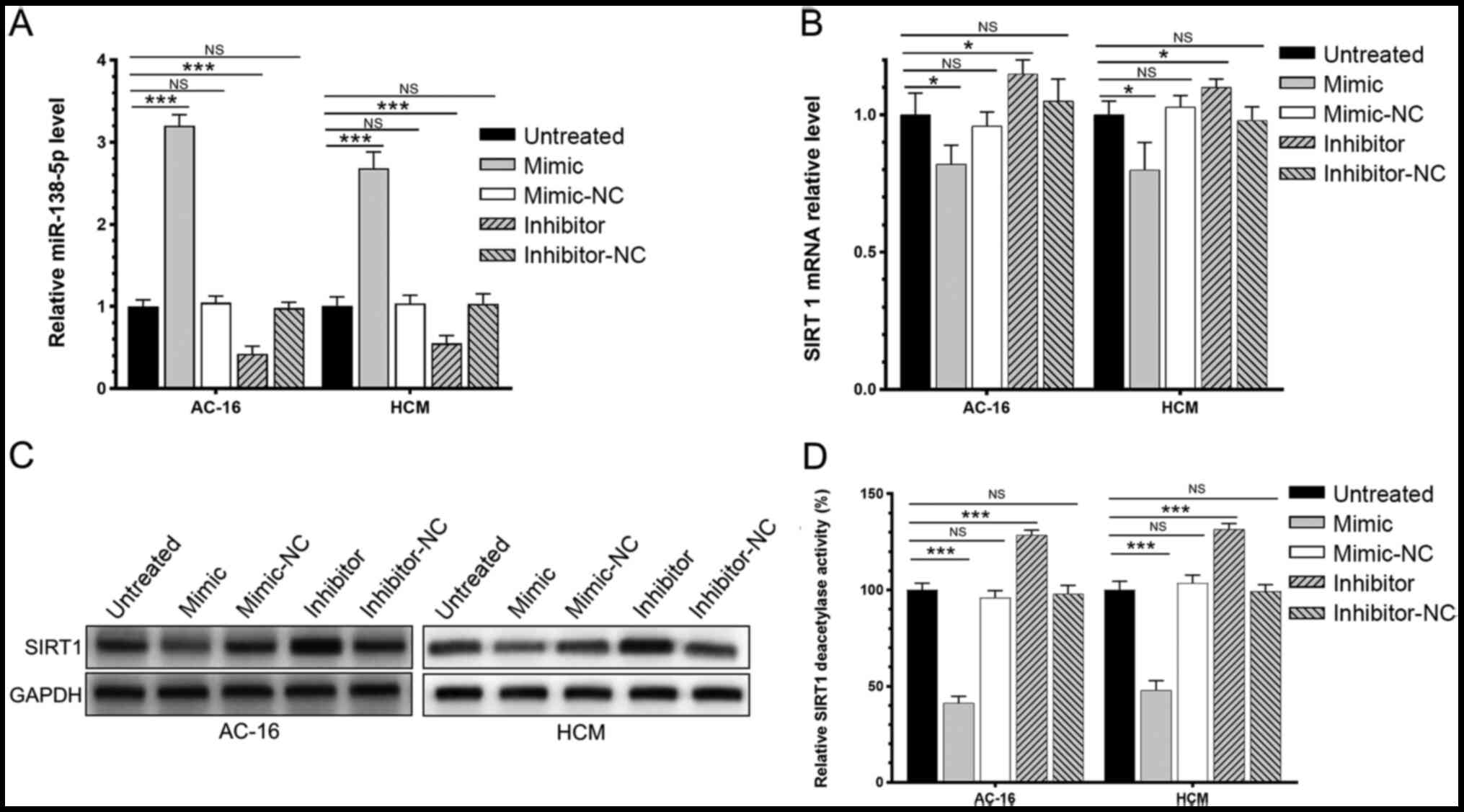
miR-138-5p inhibits the expression
level and enzyme activity of SIRT1
The respective expression level and enzymatic
activity of SIRT1 in AC-16 and HCM cells transfected with the
miR-138-5p mimic, miR-138-5p inhibitor or negative control were
investigated. It was demonstrated that the mimic significantly
increased the level of miR-138-5p while the inhibitor decreased the
level of miR-138-5p (Fig. 3A);
overexpression of miR-138-5p significantly downregulated the mRNA
and protein expression of SIRT1 in AC-16 and HCM cells (Fig. 3B and C). Furthermore, the enzymatic
activity of SIRT1 was declined in AC-16 and HCM cells
overexpressing miR-138-5p. By contrast, knockdown of miR-138-5p
significantly upregulated SIRT1 and enhanced its enzyme activity
(Fig. 3D). It was demonstrated that
miR-138-5p negatively regulated the relative level of SIRT1 and its
enzyme activity.
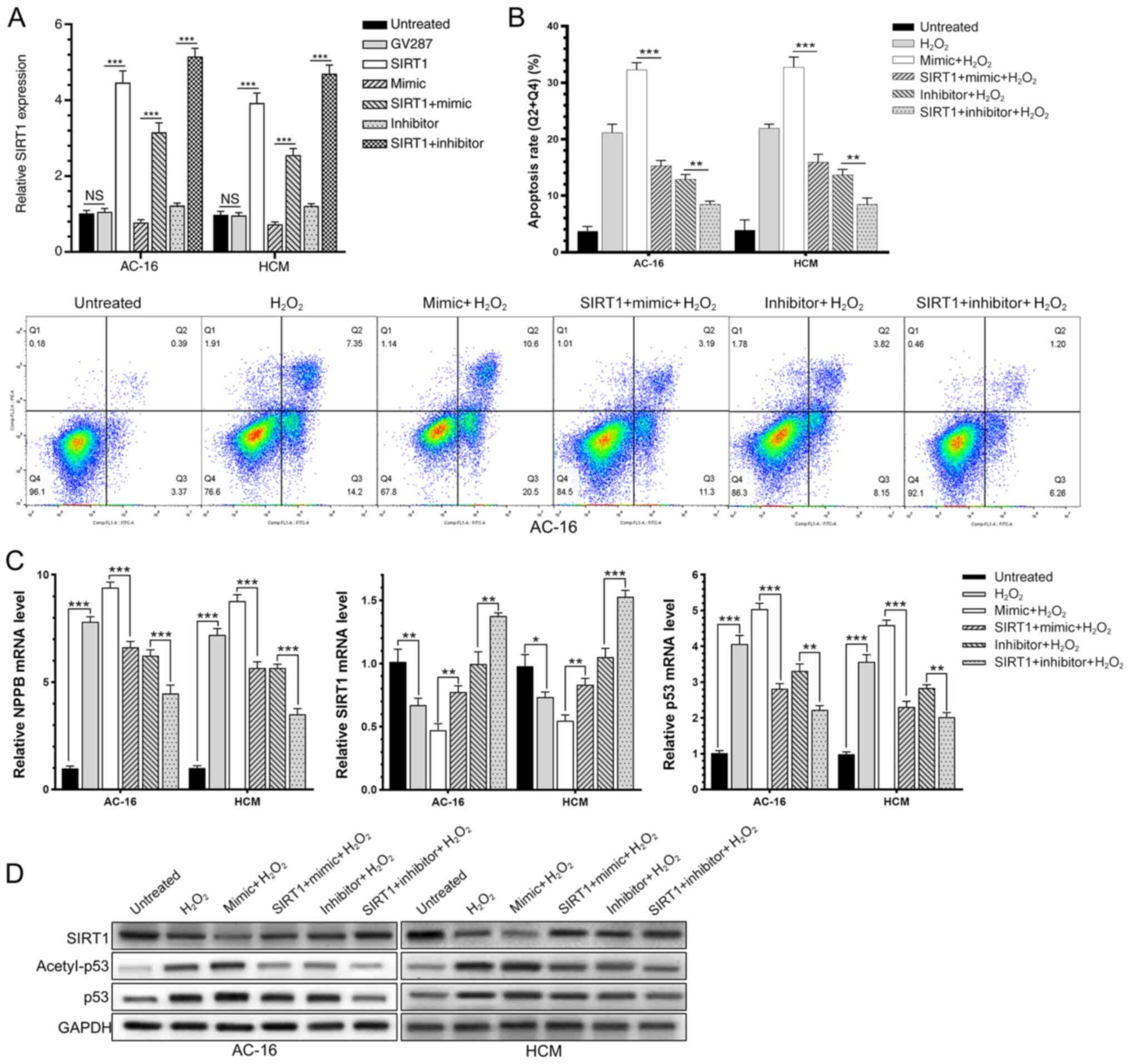
miR-138-5p regulates the HF process
via SIRT1-regulated p53 signaling
AC-16 and HCM cells were induced with 200 µM
H2O2 for 6 h, and upregulated NPPB and ROS
was detected, suggesting successful generation of in vitro
HF models (Fig. 4A and B).
Subsequently, AC-16 and HCM cells were transfected with the
miR-138-5p mimic, miR-138-5p inhibitor or negative control for 48
h, followed by H2O2 induction for a further 6
h. These results demonstrated that H2O2
induction in AC-16 and HCM cells significantly upregulated
miR-138-5p. Overexpression of miR-138-5p markedly downregulated
SIRT1 and enhanced the rate of acetylated p53 in
H2O2-induced cardiomyocytes (Fig. 4C and D). Expression changes in SIRT1
and acetylated p53 exhibited the opposite trends by knockdown of
miR-138-5p. In addition, transfection of the miR-138-5p mimic in
H2O2-induced AC-16 and HCM cells
significantly increased the rate of apoptotic cells, while
transfection of the miR-138-5p inhibitor decreased the apoptosis
rate (Fig. 4E). It was concluded
that miR-138-5p inhibited deacetylation of p53 through suppressing
the expression level and enzyme activity of SIRT1, thereby
regulating the process of HF.
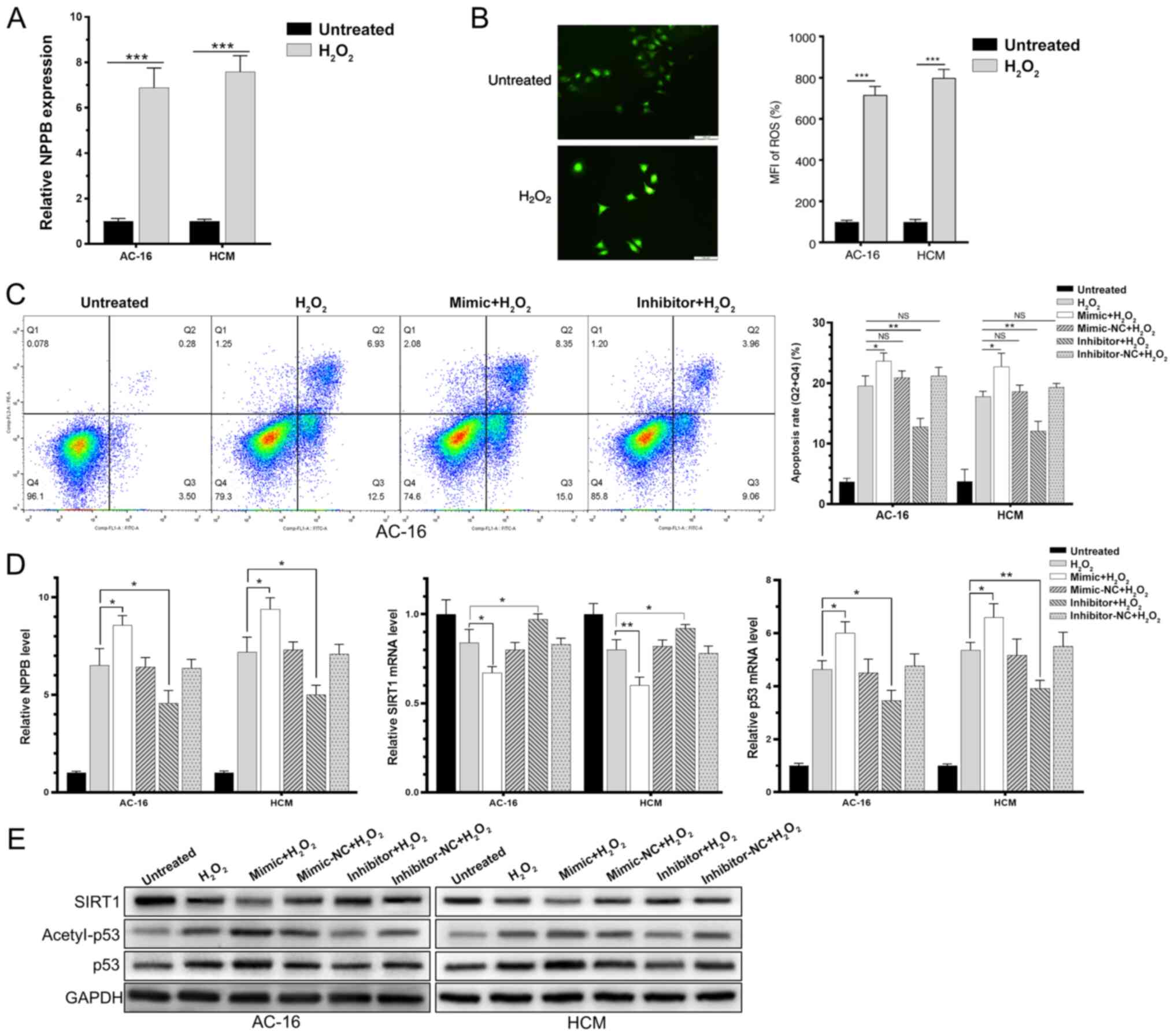 | Figure 4.miR-138-5p regulates the heart
failure process via SIRT1-regulated p53 signaling. AC-16 and HCM
were induced with H2O2 for 6 h. (A)
Upregulated NPPB level. (B) Mean fluorescence intensity of reactive
oxygen species. (C) Apoptosis proportion. (D) Relative mRNA levels
of SIRT1, p53 and NPPB. (E) Protein levels of SIRT1, p53 and
acetyl-p53. *P<0.05, **P<0.01, ***P<0.001. miR, microRNA;
NC, negative control; SIRT1, sirtuin 1; H2O2,
hydrogen peroxide; NPPB, natriuretic peptide precursor B; NS, not
significant. |
Protective effect of SIRT1 in HF
In AC-16 and HCM with lentiviral transfection
(GV287-SIRT1 or GV287), the mRNA levels of SIRT1 were significantly
upregulated (Fig. 5A). These cells
were overexpressing SIRT1 when transfected with miR-138-5p mimic or
inhibitor, and were then induced with H2O2
for 6 h to create an in vitro HF model. It was demonstrated
that the rate of acetylated p53 and the apoptosis rate were
significantly decreased in cardiomyocytes overexpressing SIRT1,
indicating improved survival of cells (Fig. 5B-D). The results demonstrated that
overexpression of SIRT1 may effectively reverse the effect of
miR-138-5p mimic or inhibitor, and decrease the acetylation of p53,
thereby protecting cardiomyocytes and decreasing apoptosis in the
H2O2-induced HF model.
Discussion
HF is the final outcome of numerous cardiovascular
diseases. The incidence and mortality of HF are high, and it
endangers the health of patients with cardiovascular and
cerebrovascular diseases (18).
Myocardial infarction is the main cause of HF (19). It is well known that cardiomyocytes
are not renewable. Necrotic cardiomyocytes in the myocardial
infarction area are recognized and cleared by the immune system,
and they are replaced by scar tissues (20). As a consequence, the structure and
function of the remaining myocardium are disrupted, eventually
leading to HF (12). The apoptosis
of cardiomyocytes often occurs throughout the process of heart
disease, and improvement of cardiac function largely relies on the
surviving cardiomyocytes (21,22).
It has been demonstrated that oxidative stress is the main cause of
hypoxic and ischemic heart diseases (23,24).
Under pathological conditions, the balance between the production
and clearance of ROS is disrupted. Excessive production of ROS
results in oxidative stress in tissues. Specifically, the
accumulation of ROS and oxygen radicals occurs during the
pathological progression of myocardial ischemia, reperfusion and
cardiac remodeling, thereby damaging the tissues and causing cell
necrosis and apoptosis (25). Piek
et al (15) reported that
NPPB may be a specific biomarker of HF and Shi et al
(16) used NPPB as a biomarker of
HF and proved that the sodium-glucose cotransporter 2 inhibitor
empagliflozin may be effective for HF. The present study generated
in vitro HF models by H2O2 induction
in AC-16 and HCM cells to mimic oxidative stress damage. The
increase in NPPB transcription and MFI of ROS was considered as
success of the generated model (15,16).
MiRNAs are a type of non-coding RNA that are 22
nucleotides in length. They are extensively expressed in eukaryotes
and participate in post-transcriptional regulation by binding to
the 3′-UTR of the target mRNA, thereby degrading them or inhibiting
their translation. miRNAs are vital regulators involved in the
onset and progression of diseases, including heart diseases. It has
been reported that knockdown of miR-21 attenuates proliferation of
cardiac fibroblasts and cardiac interstitial fibrosis by
downregulating key proteins in the ERK-MAPK signaling, thereby
improving cardiac hypertrophy and reversing cardiac remodeling
(26). Deficiency of miR-208
prevents cardiac hypertrophy, myocardial fibrosis and cardiac
remodeling in overloaded mouse cardiomyocytes (27). Through acting on the expression and
secretion of T cells and upregulating KLF13, miR-147b induces the
activation of caspase signaling and, in turn, drives cardiomyocyte
apoptosis (25). The inhibitory
effect of miR-1 on cardiomyocyte proliferation is abolished by
overexpressed miR-195. As a result, hypertrophic and disorderly
arranged cardiomyocytes trigger the onset of dilated cardiomyopathy
and HF (28). As a tumor suppressor
gene, miR-138-5p is conducive to prevent tumor progression and
metastasis (10,29,30).
Its potential biological function in cardiac diseases has been
rarely reported. A latest study revealed that SIRT1 is a potential
candidate of target gene binding of miR-138-5p by TargetScan
analysis (31). MiR-138-5p is
upregulated in insulin-resistant HepG2 cells induced by TNF-α,
which affects glucose uptake and glycogen synthesis via targeting
SIRT1. It is speculated that miR-138-5p is a promising therapeutic
target for insulin resistance in the diabetic population (32). Wang et al (10) concluded that overexpression of
miR-138-5p deteriorates ischemia-reperfusion injury in the heart by
inactivating SIRT1-PGC-1α signaling. The present study confirmed a
negative correlation between miR-138-5p and SIRT1 levels in
cardiomyocyte cell lines. Furthermore, the dual-luciferase reporter
assay confirmed the ability of miR-138-5p to regulate
post-transcriptional translation and enzymatic activity of SIRT1
via binding to its 3′-UTR.
SIRT1 belongs to Class III of the sirtuin family,
and its capacity of protein deacetylation is an important
post-translational modification. The biological functions of SIRT1
in the process of HF have been previously identified (33). For instance, SIRT1 can modify
mitochondrial function and increase ATP production by deacetylating
histone and mitochondrial proteins (i.e., UCP-2 and PGC-1α),
thereby improving cardiomyocyte metabolism and alleviating the
process of HF (7,34). Through deacetylating FOXO1, SIRT1 is
responsible for activating Rab7, and thus, it exerts its protective
effect on starvation-induced moribund myocardium via blocking
apoptosis and clearing damaged mitochondria (35,36).
The sarcoplasmic reticulum absorbs calcium through the sarcoplasmic
reticulum Ca2+-ATPase (SERCA2a). Dysfunctional SERCA2a
may cause a decrease in calcium storage of the sarcoplasmic
reticulum. The impaired systolic function of the heart further
aggravates HF. Following treatment with resveratrol (SIRT1
activator) in mice with type 1 diabetes mellitus, the abnormally
expressed SERCA2a is restored, and notably, mouse ventricular
function and ventricular dilation are restored. It has been
suggested that SIRT1 improves HF through regulating calcium ions in
cardiomyocytes (37,38). In addition, SIRT1 contributes toward
clearing mitochondria-generated ROS and relieving symptoms of
decreased ejection fraction, myocardial fibrosis and necrosis by
upregulating Mn-SOD with the involvement of HIF-2α (39). The results of the present study
illustrated that the upregulated miR-138-5p in the in vitro
HF models significantly downregulated the protein expression of
SIRT1 and its enzyme activity. Overexpression of miR-138-5p further
deteriorated the survival of cardiomyocytes by downregulating
SIRT1. By contrast, knockdown of miR-138-5p yielded the opposite
result and the survival of cardiomyocytes was significantly
improved.
p53 is the first non-histone substrate to be
deacetylated by SIRT1, and thus its pro-apoptotic activity is
suppressed. Sano et al (40)
proposed that inactivation of p53 signaling in cardiomyocytes
enhances the activity of HIF-1 and induces capillary angiogenesis,
which is beneficial in relieving compensatory cardiac hypertrophy.
Chen et al (41) identified
an interaction between HIF-1 and SIRT1. In a study conducted by
Hong et al (42),
resveratrol blocked p53 signaling by activating SIRT1 in H9C2
cells, thereby strengthening cardiac function in mice with
myocardial infarction. These findings suggest that SIRT1-induced
deacetylation of p53 is an important way of preventing HF. The
results of the present study demonstrated that in the in
vitro HF models, stimulation of apoptosis of cardiomyocytes may
be attributed to the decreased translational level of SIRT1 and
increased acetylation of p53. Overexpression of miR-138-5p
decreased the translational level and enzyme activity of SIRT1,
which further activated p53 and aggravated cardiomyocyte apoptosis.
By contrast, knockdown of miR-138-5p exerted a protective effect on
damaged cardiomyocytes through enhancing SIRT-induced deacetylation
of p53.
In conclusion, the present study identified the
upregulation of miR-138-5p at the cellular level in vitro,
clarified the targeted association between mir-138-5p and SIRT1,
and verified that mir-138-5p may inhibit the post-transcriptional
translation process of SIRT1 by targeting SIRT1 mRNA, thereby
promoting the process of HF. The present study was based on
cellular in vitro research, and more in-depth in vivo
research is required.
miR-138-5p is significantly upregulated in the in
vitro HF models. Through inhibiting the translational level and
enzyme activity of SIRT1, miR-138-5p induces worsening of HF via
activating p53 signaling. miR-138-5p and SIRT1 may be promising
diagnostic biomarkers and therapeutic targets for HF.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Shenzhen
Science and Technology Innovation Committee (grant no.
JCYJ20180302173917265).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SS, CW and JW conceived and designed the study,
performed the experiments, and analyzed and interpreted the data.
All of the authors agreed to be accountable for all aspects of the
work in ensuring that questions related to the accuracy and
integrity of any part of the work are appropriately investigated
and resolved. All authors confirm the authenticity of all the raw
data, and read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for participation
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Luscher TF: Heart failure: The
cardiovascular epidemic of the 21st century. Eur Heart J.
36:395–397. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
The L: Heart failure: The need for
improved treatment and care. Lancet. 392:4512018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Persson H, Linder-Klingsell E, Eriksson SV
and Erhardt L: Heart failure after myocardial infarction: The
importance of diastolic dysfunction. A prospective clinical and
echocardiographic study. Eur Heart J. 16:496–505. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cech TR and Steitz JA: The noncoding RNA
revolution-trashing old rules to forge new ones. Cell. 157:77–94.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
D'Onofrio N, Servillo L and Balestrieri
ML: SIRT1 and SIRT6 signaling pathways in cardiovascular disease
protection. Antioxid Redox Signal. 28:711–732. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alcendor RR, Gao S, Zhai P, Zablocki D,
Holle E, Yu X, Tian B, Wagner T, Vatner SF and Sadoshima J: Sirt1
regulates aging and resistance to oxidative stress in the heart.
Circ Res. 100:1512–1521. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Waldman M, Cohen K, Yadin D, Nudelman V,
Gorfil D, Laniado-Schwartzman M, Kornwoski R, Aravot D, Abraham NG,
Arad M and Hochhauser E: Regulation of diabetic cardiomyopathy by
caloric restriction is mediated by intracellular signaling pathways
involving ‘SIRT1 and PGC-1alpha’. Cardiovasc Diabetol. 17:1112018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang H, Bi Y, Xue L, Wang J, Lu Y, Zhang
Z, Chen X, Chu Y, Yang R, Wang R and Liu G: Multifaceted modulation
of SIRT1 in cancer and inflammation. Crit Rev Oncog. 20:49–64.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dong G, Wang B, An Y, Li J, Wang X, Jia J
and Yang Q: SIRT1 suppresses the migration and invasion of gastric
cancer by regulating ARHGAP5 expression. Cell Death Dis. 9:9772018.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang C, Sun X, Qiu Z and Chen A:
MiR-138-5p exacerbates hypoxia/reperfusion-induced heart injury
through the inactivation of SIRT1-PGC-1alpha. Inflamm Res.
68:867–876. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang J, Yang XS, Zhang Q, Zhuang X, Dong
XK, Jiang YH, Tao YN and Yang CH: Downregulated LINC01614
ameliorates Hypoxia/reoxygenation-stimulated myocardial injury by
directly sponging microRNA-138-5p. Dose Response.
18:15593258209137862020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Minicucci MF, Azevedo PS, Polegato BF,
Paiva SA and Zornoff LA: Heart failure after myocardial infarction:
Clinical implications and treatment. Clin Cardiol. 34:410–414.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang L, Wang YN, Ju JM, Shabanova A, Li
Y, Fang RN, Sun JB, Guo YY, Jin TZ, Liu YY, et al: Mzb1 protects
against myocardial infarction injury in mice via modulating
mitochondrial function and alleviating inflammation. Acta Pharmacol
Sinica. Aug 5–2020.doi: 10.1038/s41401-020-0489-0 (Epub ahead of
print).
|
|
14
|
Wei Q, Zhou HY, Shi XD, Cao HY and Qin L:
Long noncoding RNA NEAT1 promotes myocardiocyte apoptosis and
suppresses proliferation through regulation of miR-129-5p. J
Cardiovasc Pharmacol. 74:535–541. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Piek A, Suthahar N, Voors AA, de Boer RA
and Sillje HHW: A combined bioinformatics, experimental and
clinical approach to identify novel cardiac specific heart failure
biomarkers: Is Dickkopf-3 (DKK3) a possible candidate? Eur J Heart
Fail. 22:2065–2074. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shi X, Verma S, Yun J, Brand-Arzamendi K,
Singh KK, Liu X, Garg A, Quan A and Wen XY: Effect of empagliflozin
on cardiac biomarkers in a zebrafish model of heart failure: Clues
to the EMPA-REG OUTCOME trial? Mol Cell Biochem. 433:97–102. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lelonek M, Stopczynska I, Koroscik E,
Straburzynska-Migaj E and Gruchala M: Multicenter experiences with
levosimendan therapy and its safety in patients with decompensated
advanced heart failure. Adv Clin Exp Med. 29:1305–1312. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mia MM, Cibi DM, Abdul Ghani SAB, Song W,
Tee N, Ghosh S, Mao J, Olson EN and Singh MK: YAP/TAZ deficiency
reprograms macrophage phenotype and improves infarct healing and
cardiac function after myocardial infarction. PLoS Biol.
18:e30009412020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li J, Jia L, Hao Z, Xu Y, Shen J, Ma C, Wu
J, Zhao T, Zhi Y, Li P, et al: Site-specific N-glycoproteomic
analysis reveals upregulated sialylation and core fucosylation
during transient regeneration loss in neonatal mouse hearts. J
Proteome Res. 19:3191–3200. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Badreddin A, Fady Y, Attia H, Hafez M,
Khairallah A, Johar D and Bernstein L: What role does the stress
response have in congestive heart failure? J Cell Physiol.
233:2863–2870. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Szobi A, Gonçalvesová E, Varga ZV, Leszek
P, Kuśmierczyk M, Hulman M, Kyselovič J, Ferdinandy P and Adameová
A: Analysis of necroptotic proteins in failing human hearts. J
Transl Med. 15:862017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ravingerová T, Čarnická S, Nemčeková M,
Ledvényiová V, Adameová A, Khandelwal VK, Zálešák M and Kolář F:
The impact of lifestyle-related risk factors on cardiac response to
ischemia and possibilities to restore impaired ischemic tolerance.
Physiol Res. 61 (Suppl 2):S1–S10. 2012. View Article : Google Scholar
|
|
24
|
Turer AT and Hill JA: Pathogenesis of
myocardial ischemia-reperfusion injury and rationale for therapy.
Am J Cardiol. 106:360–368. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gu M, Wang J, Wang Y, Xu Y, Zhang Y, Wu W
and Liao S: miR-147b inhibits cell viability and promotes apoptosis
of rat H9c2 cardiomyocytes via down-regulating KLF13 expression.
Acta Biochim Biophys Sin (Shanghai). 50:288–297. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Adam O, Lohfelm B, Thum T, Gupta SK, Puhl
SL, Schafers HJ, Böhm M and Laufs U: Role of miR-21 in the
pathogenesis of atrial fibrosis. Basic Res Cardiol. 107:2782012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Satoh M, Minami Y, Takahashi Y, Tabuchi T
and Nakamura M: Expression of microRNA-208 is associated with
adverse clinical outcomes in human dilated cardiomyopathy. J Card
Fail. 16:404–410. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
van Rooij E, Sutherland LB, Liu N,
Williams AH, McAnally J, Gerard RD, Richardson JA and Olson EN: A
signature pattern of stress-responsive microRNAs that can evoke
cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA.
103:18255–18260. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu D, Gu L, Li Z, Jin W, Lu Q and Ren T:
miR-138-5p suppresses lung adenocarcinoma cell
epithelial-mesenchymal transition, proliferation and metastasis by
targeting ZEB2. Pathol Res Pract. 215:861–872. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang X, Zhao Y, Cao W, Wang C, Sun B, Chen
J, Li S, Chen J, Cui M, Zhang B, et al: miR-138-5p acts as a tumor
suppressor by targeting hTERT in human colorectal cancer. Int J
Clin Exp Pathol. 10:11516–11525. 2017.PubMed/NCBI
|
|
31
|
Ma J, Zhang Y, Ji H, Chen L, Chen T, Guo
C, Zhang S, Jia J and Niu P: Overexpression of miR-138-5p
suppresses MnCl2-induced autophagy by targeting SIRT1 in
SH-SY5Y cells. Environ Toxicol. 34:539–547. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Luan B and Sun C: MiR-138-5p affects
insulin resistance to regulate type 2 diabetes progression through
inducing autophagy in HepG2 cells by regulating SIRT1. Nutr Res.
59:90–98. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hsu CP, Odewale I, Alcendor RR and
Sadoshima J: Sirt1 protects the heart from aging and stress. Biol
Chem. 389:221–231. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kutsche HS, Schreckenberg R, Weber M,
Hirschhauser C, Rohrbach S, Li L, Niemann B, Schulz R and Schlüter
KD: Alterations in glucose metabolism during the transition to
heart failure: The contribution of UCP-2. Cells. 9:5522020.
View Article : Google Scholar
|
|
35
|
Hariharan N, Maejima Y, Nakae J, Paik J,
Depinho RA and Sadoshima J: Deacetylation of FoxO by Sirt1 plays an
essential role in mediating starvation-induced autophagy in cardiac
myocytes. Circ Res. 107:1470–1482. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gottlieb RA, Finley KD and Mentzer RM Jr:
Cardioprotection requires taking out the trash. Basic Res Cardiol.
104:169–180. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sulaiman M, Matta MJ, Sunderesan NR, Gupta
MP, Periasamy M and Gupta M: Resveratrol, an activator of SIRT1,
upregulates sarcoplasmic calcium ATPase and improves cardiac
function in diabetic cardiomyopathy. Am J Physiol Heart Circ
Physiol. 298:H833–H843. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen X, Zhang X, Gross S, Houser SR and
Soboloff J: Acetylation of SERCA2a, another target for heart
failure treatment? Circ Res. 124:1285–1287. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dioum EM, Chen R, Alexander MS, Zhang Q,
Hogg RT, Gerard RD and Garcia JA: Regulation of hypoxia-inducible
factor 2alpha signaling by the stress-responsive deacetylase
sirtuin 1. Science. 324:1289–1293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sano M, Minamino T, Toko H, Miyauchi H,
Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, et al:
p53-induced inhibition of Hif-1 causes cardiac dysfunction during
pressure overload. Nature. 446:444–448. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen R, Dioum EM, Hogg RT, Gerard RD and
Garcia JA: Hypoxia increases sirtuin 1 expression in a
hypoxia-inducible factor-dependent manner. J Biol Chem.
286:13869–13878. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hong W, Tatsuo S, Shou-Dong W, Qian Z,
Jian-Feng H, Jue W, Chen J, Hai-Yan Q and Yue-Jin Y: Resveratrol
upregulates cardiac SDF-1 in mice with acute myocardial infarction
through the deacetylation of cardiac p53. PLoS One.
10:e01289782015. View Article : Google Scholar : PubMed/NCBI
|