Introduction
Cirrhosis is an increasing global health burden that
accounts for >100 million deaths annually worldwide (1). Liver fibrosis is the result of
wound-healing response to chronic liver impairment triggered by a
variety of causes, including hepatitis virus, ethanol, drugs and
poisons, parasites, metabolism and genetics, cholestasis and immune
deregulation (2,3). Without diagnosis and treatment,
hepatic fibrosis will ultimately progress to hepatic cirrhosis, and
even to hepatocellular carcinoma (4). Thus, it is of great importance to
actively intervene in liver fibrosis. Hepatic fibrosis is
characterized by the deposition of extracellular matrix (ECM)
proteins, which destroy the normal liver histological structure and
functions (5). Hepatic stellate
cells (HSCs) play a vital role in the development of liver
fibrosis, and are the main producers of ECM (3). In the case of liver injury, certain
cytokines and growth factors crucial for HSC activation are
released, and promote HSC activation into myofibroblasts, which are
responsible for the synthesis of ECM proteins, including α-smooth
muscle actin (α-SMA, which is encoded by Acta2), collagen type I
(Col1α1), matrix metalloproteinases and tissue inhibitor of
metalloproteinases (6). Therefore,
directly inactivating HSCs is of great importance for fibrosis
resolution, representing a therapeutic strategy for the treatment
of hepatic fibrosis.
Epigenetic modifications regulate patterns of gene
expression by modulating DNA accessibility and chromatin structure.
The epigenetic machinery, particularly certain epigenetic enzymes,
has been demonstrated to be involved in myofibroblast activation
and regulation of fibrotic gene expression (7,8).
Blocking the expression of the DNA methyltransferase DNMT3B has
been reported to significantly reduce α-SMA and Col1α1 expression
in ischemic heart disease (9). In
addition, the histone deacetylase (HDAC) inhibitors (HDACis),
MS-275 and trichostatin A have been found to reduce renal fibrosis
by diminishing the accumulation of ECM proteins (10–12).
By contrast, other studies have indicated that targeted inhibition
of certain epigenetic enzymes might aggravate fibrosis. It has been
reported that inhibition of type I protein arginine
methyltransferases can aggravate renal fibrosis by reducing
asymmetric dimethylarginine accumulation, increasing nitric oxide
concentrations and enhancing the expression of profibrotic proteins
(13). However, there is currently
no effective high-throughput screening method to identify candidate
compounds for the treatment and prevention of liver fibrotic
diseases.
Aiming to identify a novel candidate compound for
the treatment of hepatic fibrosis, the present study established a
cell-based high-throughput assay based on HSC activation, and
screened our in-house epigenetic compound library (14). The HDACi givinostat, which has been
used in phase I/II clinical trials for the treatment of Duchenne
muscular dystrophy (15), was
identified as the most potent hit. Givinostat reduced the
expression of α-SMA and collagen, which are markers of HSC
activation in vitro. Carbon tetrachloride (CCl4) has been
widely used to induce liver injury and fibrosis in mice for decades
(16), and C57BL/6J inbred mice are
frequently used for fibrosis studies in the CCl4 model of the ready
availability of genetically-modified mice (17). In a chronic CCl4-challenged mouse
model in the present study, mice developed mild liver fibrosis
after 2 weeks of CCl4 treatment, and were then treated with
givinostat for 6 weeks. Givinostat significantly ameliorated
CCl4-induced mouse liver injury and fibrosis. RNA-sequencing
(RNA-seq) analysis of the liver tissue from the givinostat
treatment and solvent groups of CCl4-challenged mice revealed genes
regulated by givinostat treatment, among which, dermokine
(Dmkn), mesothelin (Msln) and uroplakin-3b
(Upk3b) were further identified as crucial genes regulating
HSC activation. Givinostat inhibited HSC activation and alleviated
liver fibrosis in vivo and in vitro, making it a
promising tool for developing a novel therapy for the treatment of
hepatic fibrosis.
Materials and methods
Animals and treatment
Female C57BL/6J mice (8–9 weeks old, weighting 21–23
g, specific pathogen-free) were purchased from the Animal Center of
the Chinese Academy of Medical Sciences. Animal care was carried
out according to the guidelines of the Principles of Laboratory
Animal Care (18), all experimental
protocols were approved by the Institutional Animal Care and Use
Committee at the Shanghai Institute of Materia Medica (approval no.
2018-12-LC-11; Shanghai, China). The animals were allowed free
access to a standard laboratory diet and tap water. All mice were
kept in standard laboratory conditions (21±2°C, 12-h light/dark
cycle), and were fed adaptively for 1 week before starting the
experiments. A total of 24 mice were randomly divided into three
groups with 8 mice per group: i) The normal control group; ii) the
solvent group of CCl4-challenged mice; and iii) the givinostat
treatment group of CCl4-challenged mice. In the givinostat
treatment group, mice were stimulated with CCl4 for 8 weeks, and in
the last 6 weeks, the animals received an intraperitoneal (i.p.)
injection of givinostat (10 mg/kg, formulated in PBS) daily
(19). Mice were i.p. injected with
10% CCl4 dissolved in olive oil at a dose of 1 ml/kg body weight
twice a week for 8 weeks to trigger liver fibrosis (20). Givinostat or PBS solvent was i.p.
injected after CCl4 treatment for 2 weeks, when mild fibrosis was
shown. At the end of the experiment, the mice were sacrificed, and
blood as well as liver samples were harvested. Although there was a
total of 24 mice used overall, too little blood was collected
during blood collection to be used for experiments so the number of
experimental results displayed was n=8 in normal control group, n=6
in CCl4 group and n=7 in the CCl4 + givinostat group. All surgeries
(blood and liver samples were harvested) were performed under
sodium pentobarbital anesthesia (50 mg/kg), and then all mice were
euthanized by 5% isoflurane (cat. no. HR135327; Hairui Chemical).
Death of the mice was confirmed by checking whether their heartbeat
had completely stopped and whether their pupils were dilated.
Liver histopathology and
immunohistochemistry
Liver tissues were fixed in 4% paraformaldehyde for
24 h at 37°C, dehydrated and paraffin embedded. The 3–4-mm thick
liver sections were stained with hematoxylin and eosin (cat. no.
G1005; Wuhan Servicebio Technology Co., Ltd.) at 37°C for 5 min and
15 sec, respectively and Sirius Red (cat. no. G1018; Wuhan
Servicebio Technology Co., Ltd.). The liver tissue sections were
deparaffinized using xylene (Wuhan Servicebio Technology Co.,
Ltd.), rehydrated with graded alcohol, treated with 0.3% endogenous
peroxidase blocking solution (Sigma-Aldrich; Merck KGaA) for 10
min. Following high pressure heating retrieval (125°C and 103 kPa)
and blocking with 10% normal goat serum (Wuhan Servicebio
Technology Co., Ltd.) at 37°C for 30 min, the sections were
incubated overnight at 4°C with the following primary antibodies
(Wuhan Servicebio Technology Co., Ltd.): anti-α-SMA (cat. no.
GB13044; 1:100) and anti-Col1α1 (cat. no. GB11022-1; 1:100).
Following washing with PBS, goat anti-rabbit non-biotinylated
reagents (cat. no. G1213; 1:1,000; Wuhan Servicebio Technology Co.,
Ltd.) were used to react with the primary antibody for 2 h at 37°C.
Images were captured by observers who were blinded to the
experimental conditions at 6–8 non-consecutive random fields under
a light microscope (magnification, ×100), and were used to assess
the histological changes using Image-Pro Plus 6.0 software (Media
Cybernetics, Inc.). Representative views were displayed.
Cell culture
The human HSC LX-2 cell line and the rat HSC-T6 cell
line were obtained from the FuHeng Cell Center, and were cultured
in Dulbecco's modified Eagle's medium (DMEM cat. no. L110KJ;
Shanghai BasalMedia Technologies Co., Ltd.) supplemented with 10%
fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin and streptomycin (Gibco; Thermo Fisher Scientific,
Inc.), at 37°C in a 95% air humidified atmosphere containing 5%
CO2. For stimulation, the cells were starved in
serum-free DMEM for 24 h before being treated with recombinant
human TGF-β1 (10 ng/ml; cat. no. 100-21C; PeproTech, Inc.)
(21) and/or givinostat (900, 300
or 100 nM; cat. no. CSN16577; CSNpharm) for 24 h.
One-step reverse
transcription-quantitative PCR (RT-qPCR)
HSC LX-2 cells (5×105 cells/well) were
seeded in 96-well cell culture plates (Cellvis) overnight and
sequentially stimulated with TGF-β1 and compounds (2 µM) or with
TGF-β1 solely. Cells were harvested 24 h after TGF-β1 stimulation
with Total RNA extraction reagent (Vazyme Biotech Co., Ltd.).
Cytolysis was then subjected to RT-qPCR using
TransScript® Green One-Step qRT-PCR SuperMix
(cat. no. AQ211; TransGen Biotech Co., Ltd.) (14). The compounds library containing 46
molecule probes targeting epigenetic proteins was screened to find
small molecule compounds able to inhibit α-SMA
expression.
RNA-seq analysis
Total RNA was isolated from flash-frozen mice liver
tissues. Total RNA was isolated and purified using DNase I (Takara
Bio, Inc.) and Dynabeads Oligo (dT) 25 (Thermo Fisher Scientific,
Inc.). Subsequently, purified RNA (100 ng) was used for cDNA
library construction, using the NEBNext Ultra™ RNA Library Prep kit
for Illumina® (cat. no. E7530L; New England BioLabs,
Inc.). Sequencing data was collected on an Illumina HiSeq 2500
instrument. The RNA integrity number (RIN) value was used to assess
the quality of the isolated RNAs. Only RNAs with RIN ≥7.0 were used
for sequencing. The sequencing reads were located to mm10 by STAR
2.5 (22), and gene counting was
quantified using featureCounts (Subread package 2.0.0) (23). The edgeR R package (24) was used for differential gene
expression analysis. The P-value was adjusted using the Benjamini
and Hochberg method (25), and a 5%
FDR cutoff value and fold-change >1.5 were set as the threshold
of the significant gene. The differentially expressed genes were
further analyzed by gene-annotation enrichment analysis using The
Database for Annotation, Visualization and Integrated Discovery 6.8
bioinformatics platform (26).
Cytoscape was used for network analysis (27). The original data generated using
high-throughput sequencing methodologies has been submitted to the
GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE161981).
Small interfering (si)RNA
transfection
Msln siRNA (sense, 5′-GCCUUGCUUUCCAGAACAU-3′ and
antisense, 5′-AUGUUCUGGAAAGCAAGGC-3′; and sense,
5′-GGACGUCCUAAAGCAUAAA-3′ and antisense,
5′-UUUAUGCUUUAGGACGUCC-3′), Dmkn siRNA (sense,
5′-GCAGAGACGAUCAGAACUA-3′ and antisense, 5′-UAGUUCUGAUCGUCUCUGC-3′;
and sense, 5′-GCCUAUGGUGGGAAGUACU-3′ and antisense,
5′-AGUACUUCCCACCAUAGGC-3′) and Upk3b siRNA (sense,
5′-GCCCUACACACCACAGAUA-3′ and antisense, 5′-UAUCUGUGGUGUGUAGGGC-3′;
and sense, 5′-GCUACAUGACCCACCACAU-3′ and antisense,
5′-AUGUGGUGGGUCAUGUAGC-3′) for human cells were synthesized by
Shanghai GenePharma Co., Ltd. Transfection with siRNA against Msln,
Upk3b or Dmkn, or with control siRNA (sense,
5′-UUCUCCGAACGUGUCACGU-3′ and antisense, 5′-ACGUGACACGUUCGGAGAA-3′)
was performed according to the manufacturer's protocol. LX-2 cells
were seeded in a 6-well plate at 60–80% confluence. Briefly, siRNA
(20 µM, 1.5 µl) and 9 µl Lipofectamine® RNAiMAX
transfection reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
were mixed with 150 µl Opti MEM (cat. no. 31985070; Gibco; Thermo
Fisher Scientific, Inc.). Next, diluted siRNA was added to diluted
Lipofectamine RNAiMAX reagent and cultured for 5 min at room
temperature. siRNA-lipid complex was added to cells for 6–8 h at
37°C. Subsequent experiments were performed 24 h after
transfection.
RNA extraction and RT-qPCR
Total RNA was extracted from HSC LX-2 cells, HSC-T6
cells or liver tissues using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocols. Total RNA (1,000 ng) was reverse
transcribed into cDNA using a cDNA synthesis kit (Vazyme Biotech
Co., Ltd.). The reverse transcription temperature protocol was as
follows: 50°C for 15 min, followed by 80°C for 5 sec. RT-qPCR was
performed using SYBR-Green (Vazyme Biotech Co., Ltd.). The
thermocycling conditions were as follows: Pre-denaturation at 95°C
for 10 min, 40 cycles of denaturation at 95°C for 15 sec, and
annealing at 60°C for 30 sec, followed by extension at 72°C for 1
min. Subsequently, the expression values of mRNA were calculated
using the 2−ΔΔCq method (28) The expression of target genes was
normalized to GAPDH expression. The primer sequences are shown in
Table I.
 | Table I.RT-qPCR primer sequences used in the
present study. |
Table I.
RT-qPCR primer sequences used in the
present study.
| A, Primer sequences
used for mice liver tissues |
|---|
|
|---|
| Gene | Primer sequences
(5′→3′) |
|---|
| Acta2
(α-SMA) | F:
GCTGAAGTATCCGATAGAACACG |
|
| R:
GGTCTCAAACATAATCTGGGTCA |
| Col1α1 | F:
TCAGAGGCGAAGGCAACAGT |
|
| R:
CCCCAAGTTCCGGTGTGA |
| GAPDH | F:
GGTGAAGGTCGGTGTGAACGGA |
|
| R:
CCAAAGTTGTCATGGATGACCTTGG |
| Sult3a1 | F:
TATTTTGAGGGTCATCGGAACAG |
|
| R:
GGTGATGGCATTTTGGCATAGT |
| Upk3b | F:
CATCTGGCTAGTGGTGGCTTT |
|
| R:
GGTAATGTCATATAGTGGCCGTC |
| Adgrd1 | F:
GTCTGCTCTAGTGGTGGAAAGG |
|
| R:
TAAGGACCGGGAGGGTTGAAG |
| Snai3 | F:
GGTCCCCAACTACGGGAAAC |
|
| R:
CTGTAGGGGGTCACTGGGATT |
| Msln | F:
CACCGACGAGGAACTGAATG |
|
| R:
CTCCGTGGGAGTACATCCACA |
| Dmkn | F:
AGCTGACCAGTTTTCTAAGCC |
|
| R:
GCCAGTTGTAAGTAGGATTCACC |
| Upk1b | F:
AACAGGAAAATCCTCTTGGCG |
|
| R:
AAAAAGTCGCGTTGTGTTGCT |
| Cebpe | F:
ATTCGCCTATCCCTCACACAC |
|
| R:
GTAGCTGCCTCGACTGGTG |
|
Eif4ebp3 | F:
GTCCACGAGTTGCCCAATTC |
|
| R:
GGGGTAGTGGCGTATAGTGTG |
| Slc2a5 | F:
TTCCAATATGGGTACAACGTAGC |
|
| R:
GCGTCAAGGTGAAGGACTCAA |
| Ntrk1 | F:
GCCTAACCATCGTGAAGAGTG |
|
| R:
CCAACGCATTGGAGGACAGAT |
|
| B, Primer
sequences used for human HSC LX-2 cells |
|
| Gene | Primer sequences
(5′→3′) |
|
| Acta2
(α-SMA) | F:
TACGAGTAGAACGCTGTCCG |
|
| R:
CAGCACCGCCTGGATAGCC |
| Col1α1 | F:
GTGCGATGACGTGATCTGTGA |
|
| R:
CGGTGGTTTCTTGGTCGGT |
| GAPDH | F:
AGGTCGGTGTGAACGGATTTG |
|
| R:
GGGGTCGTTGATGGCAACA |
| SULT3A1 | F:
CATTATAAAAGATGAACTCGG |
|
| R:
GAAAACAATCTAGGCACA |
| UPK3B | F:
GCCTCTCCTCTGCCTTTCTGG |
|
| R:
TGGAGGTGGGGACTGGGTATC |
| ADGRD1 | F:
CTGCCTACTCAAATCTCTCTGC |
|
| R:
GAGTCCAATAGGGGCCTTCA |
| SNAI3 | F:
CCCTTCCAAGAGTCAGAGCC |
|
| R:
TGCTGCAATGGAACTAGGCA |
| MSLN | F:
CAGAGGAGGCTCAGAGAGCTA |
|
| R:
GTCCCACAGGACCCCAACAG |
| DMKN | F:
CCAAGGGACCAGAGAAGCAG |
|
| R:
CCCAGTGTTTCCCAGAGCAT |
| UPK1B | F:
GAACCTCTCAACCTGGAGGC |
|
| R:
TGGTACCCAGGAGAACCCAA |
| CEBPE | F:
CTCCGATCTCTTTGCCGTGA |
|
| R:
GTCTGGGCCGAAGGTATGTG |
|
EIF4EBP3 | F:
CCACTAGCTGCCCGATTCC |
|
| R:
GGTAGTGGCGTATAGCGTGC |
| SLC2A5 | F:
CAAGAAAGTTGAGTATGTTGGCT |
|
| R:
CAAGAAAGTTGAGTATGTTGGCT |
| NTRK1 | F:
CCATCCCTGACACTAACAGCA |
|
| R:
GCACAAGGAGCAGCGTAGAA |
Biochemical analysis
The levels of serum aspartate aminotransferase (AST;
cat. no. C010-3-1) and alanine aminotransferase (ALT; cat. no.
C009-3-1) were measured using the corresponding kits (Nanjing
Jiancheng Bioengineering Institute) and were assessed using a
Hitachi 7020 automatic analyzer (Hitachi, Ltd.).
Western blotting
Total protein was extracted from HSC LX-2 cells,
HSC-T6 cells or liver tissues with 1X SDS sample loading buffer
(250 mM Tris HCL pH 6.8, 10% SDS, 30% glycerol, 5%
β-mercaptoethanol and 0.02% bromophenol blue). Protein
concentration was determined using a BCA kit (Thermo Fisher
Scientific, Inc.). The lysates (25 µg/lane) were separated via
SDS-PAGE on 6, 10 or 12% gels, and subsequently transferred to a
nitrocellulose membrane (EMD Millipore). Following blocking with 5%
milk at room temperature for 1 h, membranes were incubated with
primary antibodies at 4°C overnight. Subsequently, the membranes
were incubated with a HRP-conjugated goat anti-Rabbit IgG secondary
antibody (1:10,000; cat. no. D110058; Sangon Biotech Co., Ltd.) for
1 h at room temperature. The bands were visualized using an
enhanced chemiluminescence kit (Thermo Fisher Scientific, Inc.)
with a ChemiScope 3400 mini imaging system (Clinx Science
Instruments Co., Ltd.). Densitometry was performed for each group
using ImageJ software (v1.50b; National Institutes of Health). The
following primary antibodies were used: Anti-α-SMA (1:1,000; cat.
no. 19245; Cell Signaling Technology, Inc.), anti-Col1α1 (1:1,000;
cat. no. 72026; Cell Signaling Technology, Inc.) and anti-GAPDH
(1:5,000; cat. no. 8884; Cell Signaling Technology, Inc.), which
was used as the loading control.
Statistical analysis
All numerical results are expressed as the mean ±
standard deviation, and represent data from a minimum of three
independent experiments. Two-tailed unpaired t-test was used to
analyze differences between two groups. One-way ANOVA or two-way
ANOVA were used to compare the means of multiple groups followed by
LSD post hoc test. All analyses were performed using GraphPad Prism
7.0 statistical software (GraphPad Software, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
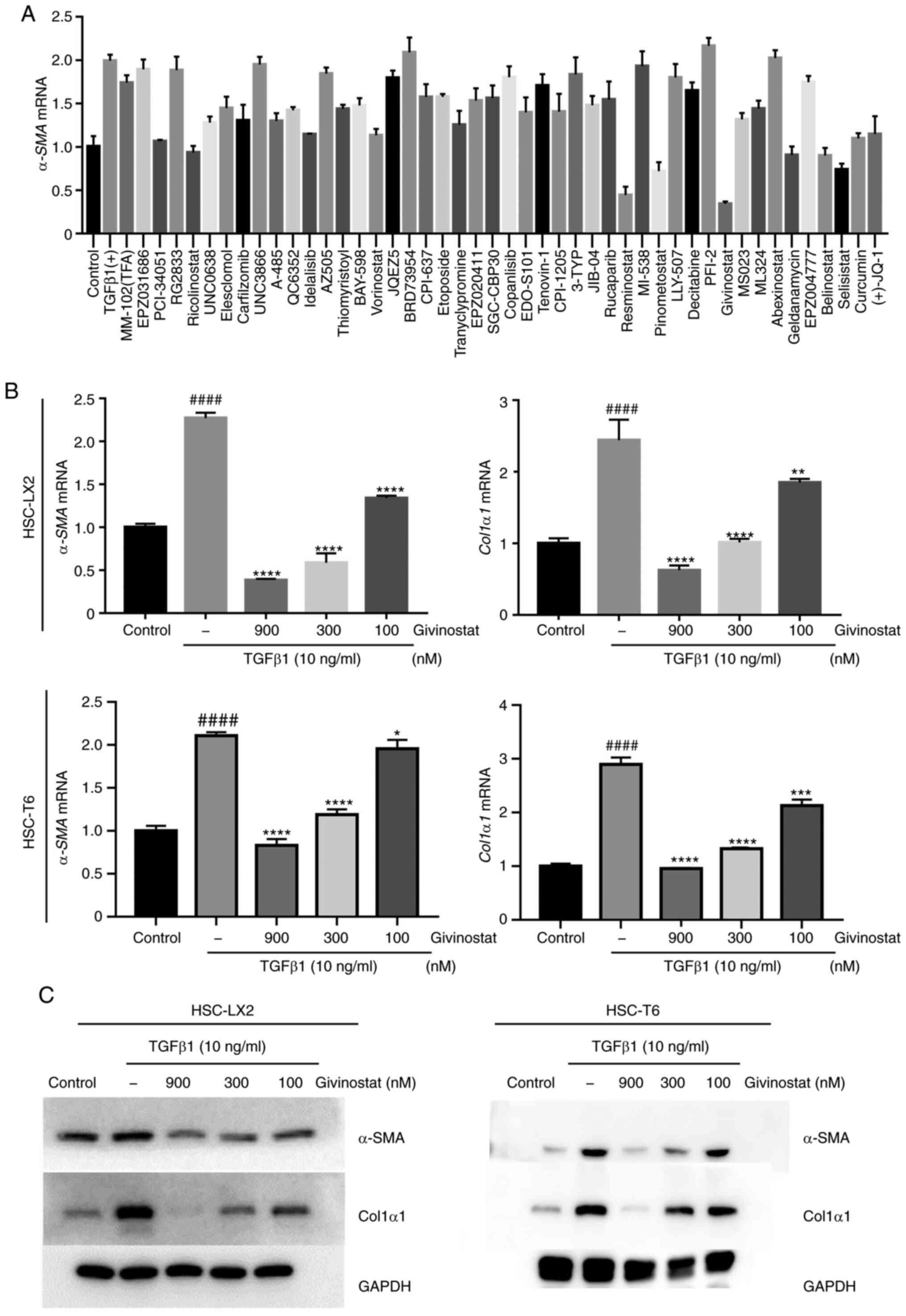
Identification of givinostat as an
inhibitor of HSC activation using cell-based high-throughput
screening
Aiming to find potential candidate compounds that
can inhibit HSC activation for the treatment of liver fibrosis, the
present study established a high-throughput screening assay based
on one-step RT-qPCR for detecting α-SMA mRNA expression as a
readout for HSC activation, since activated HSCs are characterized
by the expression of α-SMA (21). A
library of small molecule inhibitors targeting epigenetic enzymes
was screened, and givinostat was identified as the most potent
compound that inhibited TGF-β1-induced α-SMA expression
(Fig. 1A). Givinostat, a pan-HDACi
that belongs to the hydroxamic acids family with HDAC type I and
type II inhibitory activity, has been used in phase I/II clinical
trials for the treatment of Duchenne muscular dystrophy. To
validate the inhibitory effect of givinostat on HSC activation, the
mRNA and protein expression levels of Col1α1 and α-SMA after
givinostat treatment were examined. Givinostat dose-dependently
inhibited Col1α1 and α-SMA mRNA expression in the
presence of TGF-β1 (Fig. 1B).
Consistently, western blotting also confirmed that givinostat
inhibited Col1α1 and α-SMA protein expression levels in a
dose-dependent manner (Fig. 1C).
Based on these data, givinostat was identified as a potent
inhibitor of HSC activation in vitro.
Givinostat treatment alleviates
chronic liver injury and fibrosis in mice treated with CCl4
Whether givinostat could inhibit HSC activation
in vivo and alleviate liver fibrosis, which has not been
extensively studied, was further examined in the present study,
which investigated its potential role in the treatment of chronic
liver fibrotic diseases. A widely used mouse liver fibrosis model
with i.p. repeated injection of CCl4 was used to evaluate the
efficacy of givinostat for the treatment of hepatic injury and
liver fibrosis (29). C57BL/6J mice
were i.p. injected with CCl4 for 2 weeks; at that time there was
already mild liver fibrosis (30).
Mice were then treated with givinostat for the following 6 weeks
after CCl4 treatment (Fig. 2A). As
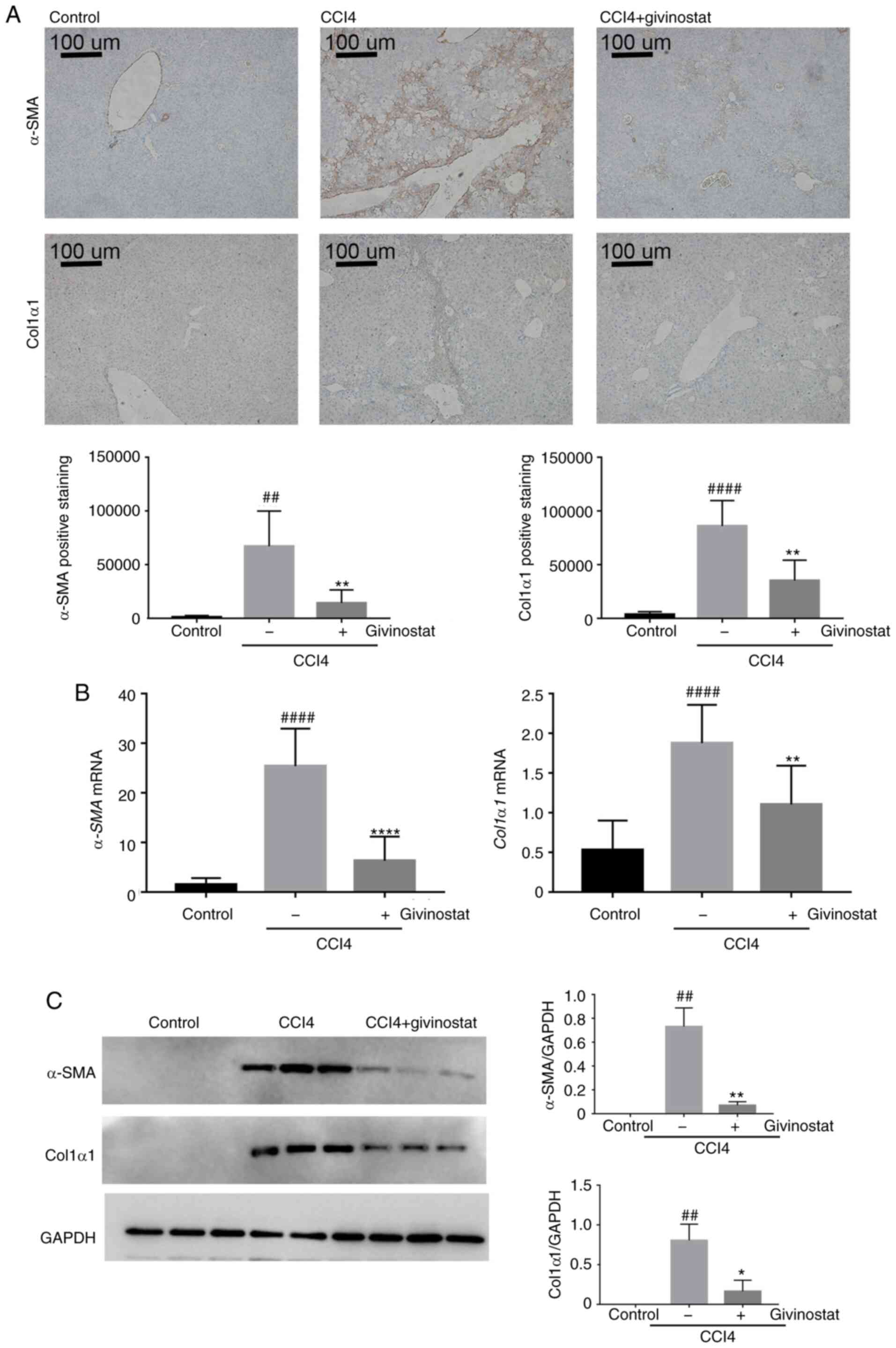
shown in Fig. 2B, persistent i.p.
injection of CCl4 for 8 weeks caused hepatocyte steatosis (Fig. 2B middle panel), varying degrees of
central venous wall thickening and collagen deposition (Fig. 2C middle panel), which are consistent
with previous reports (31,32). The results indicated that givinostat
treatment markedly ameliorated the extent of hepatocyte steatosis
(Fig. 2B right panel). Since the
deposition of ECM (primarily collagens) is the major characteristic
of liver fibrosis, the collagen contents were evaluated based on
Sirius Red staining. Images (n=6–8 images per group) showed that
hepatic lobules maintained a normal physiological structure in the
liver of control mice, while higher collagen accumulation was
observed in the liver of CCl4-treated mice (P<0.0001; Fig. 2C). In the givinostat treatment
group, the hepatic structure was improved and collagen was
decreased (P<0.0001; Fig. 2C).
There was a significant increase in the levels of serum ALT (n=6;
469±41.65; P<0.0001) and AST (n=6; 536.8±95.53; P<0.01) in
CCl4-treated mice, whereas treatment with givinostat significantly
decreased these serum markers of liver injury, ALT (n=6;
137.7±71.72; P<0.01) and AST (n=6; 156.5±71.44; P<0.01)
(Fig. 2D). Furthermore, no systemic
toxicity was observed at the dose of givinostat used in this
experiment (data not shown). Thus, givinostat treatment
significantly alleviated liver fibrosis and injury in
vivo.
 | Figure 2.Givinostat treatment alleviates
chronic liver injury and fibrosis in CCl4-induced mice. (A) Mice
were randomly divided into the following three groups (n=8/group):
Normal control, CCl4 and CCl4 + givinostat. In the CCl4 +
givinostat group, mice were stimulated with CCl4 for 8 weeks, and
in the last 6 weeks, the animals received an injection of
givinostat (10 mg/kg/day, i.p.). In the CCl4 group, mice were i.p.
injected with CCl4 twice a week for 8 weeks. (B) Representative
images of liver tissues, and microphotograph of hematoxylin and
eosin-stained paraffin-embedded sections of liver tissues. Scale
bar, 100 µm. (C) Representative microphotographs and
quantifications of Sirius Red-stained paraffin-embedded sections of
liver tissues by Image-Pro Plus 6.0 (n=6–8 images/group from normal
control, CCl4 and CCl4 + givinostat groups). Scale bar, 100 µm. (D)
Serum ALT and AST levels in mice from the normal control, CCl4 and
CCl4 + givinostat groups (n=6–8 mice/group). Data are expressed as
the mean ± SD (n=8). **P<0.01, ****P<0.0001 vs. the CCl4
group; ##P<0.01, ####P<0.0001 vs. the
normal control group. CCl4, carbon tetrachloride; i.p.,
intraperitoneally; ALT, alanine aminotransferase; AST, aspartate
aminotransferase. |
Givinostat inhibits HSC activation in
mice with CCl4-induced liver fibrosis
Quiescent HSCs are activated on account of liver
injury and considered to be the primary source of ECM yielding
during hepatic fibrosis. Since givinostat significantly inhibited
HSC activation in vitro and alleviated liver fibrosis in
vivo, the present study assessed whether it inhibited HSC
activation in vivo. α-SMA and Col1α1 are the most abundant
ECM proteins in liver tissue, and are markers of HSC activation
(33,34). Thus, α-SMA or Col1α1-positive cells
were detected by morphometric quantification to evaluate the
accumulation of activated HSCs in mouse liver tissues.
Immunohistochemical staining for α-SMA (n=6; 65.800±14.861;
P<0.01) or Col1α1 (n=6; 8.215±1.069; P<0.0001) showed that
positively stained brown-colored cells were notably increased in
the liver tissues of mice treated with CCl4 (Fig. 3A middle panel). By contrast,
immunohistochemical staining for α-SMA (n=6; 12.886±5.603;
P<0.01) or Col1α1 (n=6; 3.140±859.9; P<0.01) in the liver
tissue of givinostat-treated mice was much weaker compared with
that of solvent-treated mice, almost at a level similar to that of
the normal control group (Fig. 3A
right panel). RT-qPCR analysis confirmed that the increase in mRNA
expression of α-SMA and Col1α1 in the liver tissues
of CCl4-challenged mice was significantly reduced by givinostat
treatment (P<0.01; Fig. 3B).
Consistently, western blot analysis further confirmed that the
protein expression levels of α-SMA and Col1α1 in mouse liver
tissues were increased in the CCl4-chanlleged group, and were
reduced by givinostat treatment (P<0.05; Fig. 3C). Taken together, these results
demonstrated that givinostat alleviated liver fibrosis and
inhibited HSC activation in vivo.
Identification of crucial genes for
HSC activation that are regulated by givinostat via transcriptomic
analysis
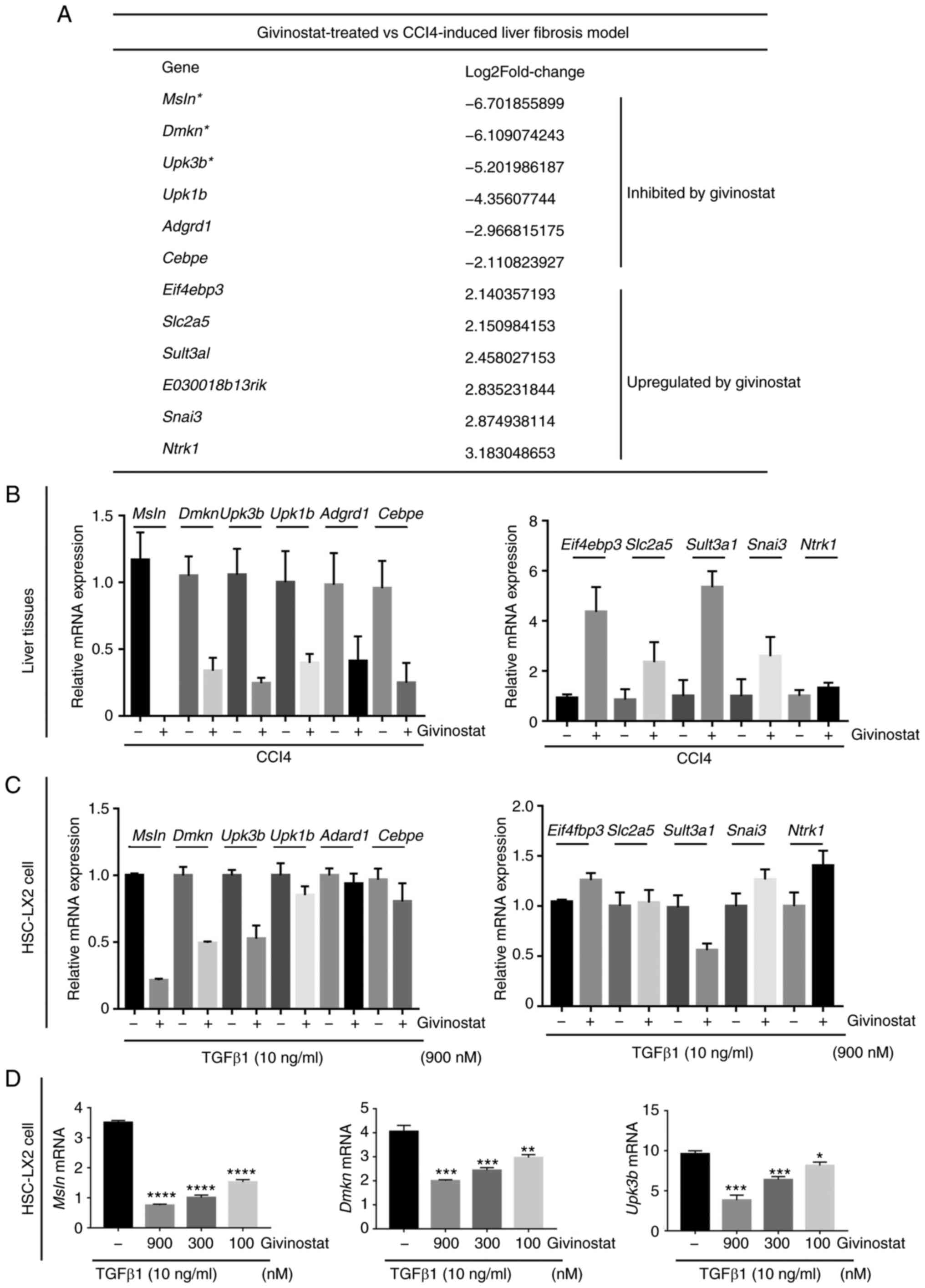
To explore the mechanism underlying the improvement
of hepatic fibrosis by givinostat treatment in CCl4-challenged
mice, RNA-seq analysis was performed to compare the gene expression
profile of liver tissues from CCl4-challenged mice with or without
givinostat treatment. Differential gene expression analysis
identified genes upregulated or downregulated in givinostat-treated
group compared with their expression in the solvent group in
CCl4-challenged mice. The most significantly regulated genes by
givinostat treatment are shown in Fig.
4A. RT-qPCR analysis confirmed that givinostat treatment
inhibited or upregulated the mRNA expression of these genes in
liver tissues (Fig. 4B), which was
consistent with the RNA-seq results.
The present study next examined whether givinostat
regulated the transcription of these genes in vitro in HSCs.
The mRNA expression of these genes was analyzed via RT-qPCR in HSC
LX-2 cells stimulated by TGF-β1 in the absence or presence of
givinostat treatment. The mRNA expression levels of Msln,
Dmkn and Upk3b were also reduced in HSC LX-2 cells
(Fig. 4C), which was consistent
with the findings in liver tissues of mice treated with givinostat.
As for the genes that were most notably upregulated by givinostat
treatment in vivo, their mRNA expression was increased in
vitro to a much lower extent (Fig.
4C). As shown in Fig. 4D,
givinostat dose-dependently inhibited Msln, Dmkn and
Upk3b mRNA expression in the presence of TGF-β1. These
results confirmed that givinostat inhibited Msln, Dmkn and
Upk3b gene expression in vivo and in
vitro.
To further validate whether these
givinostat-regulated genes play crucial roles during HSC
activation, the effect of Msln, Dmkn and Upk3b depletion on
TGF-β1-induced HSC activation was examined. RT-qPCR analysis
confirmed the knockdown efficiency of Msln, Dmkn and Upk3b by
siRNA, which resulted in >70% reduction in mRNA levels in HSC
LX-2 cells compared with that of the siControl group (P<0.0001;
Fig. 5A). RT-qPCR analysis showed
that the knockdown of Msln, Dmkn and Upk3b inhibited the mRNA
levels of α-SMA and Col1α1 in HSC-LX2 cells
stimulated with TGF-β1 compared with the siControl + TGF-β1 group
(P<0.0001; Fig. 5B and C). These
results demonstrated that givinostat inhibited Msln, Dmkn
and Upk3b expression, and these genes were shown to be
crucial for HSC activation.
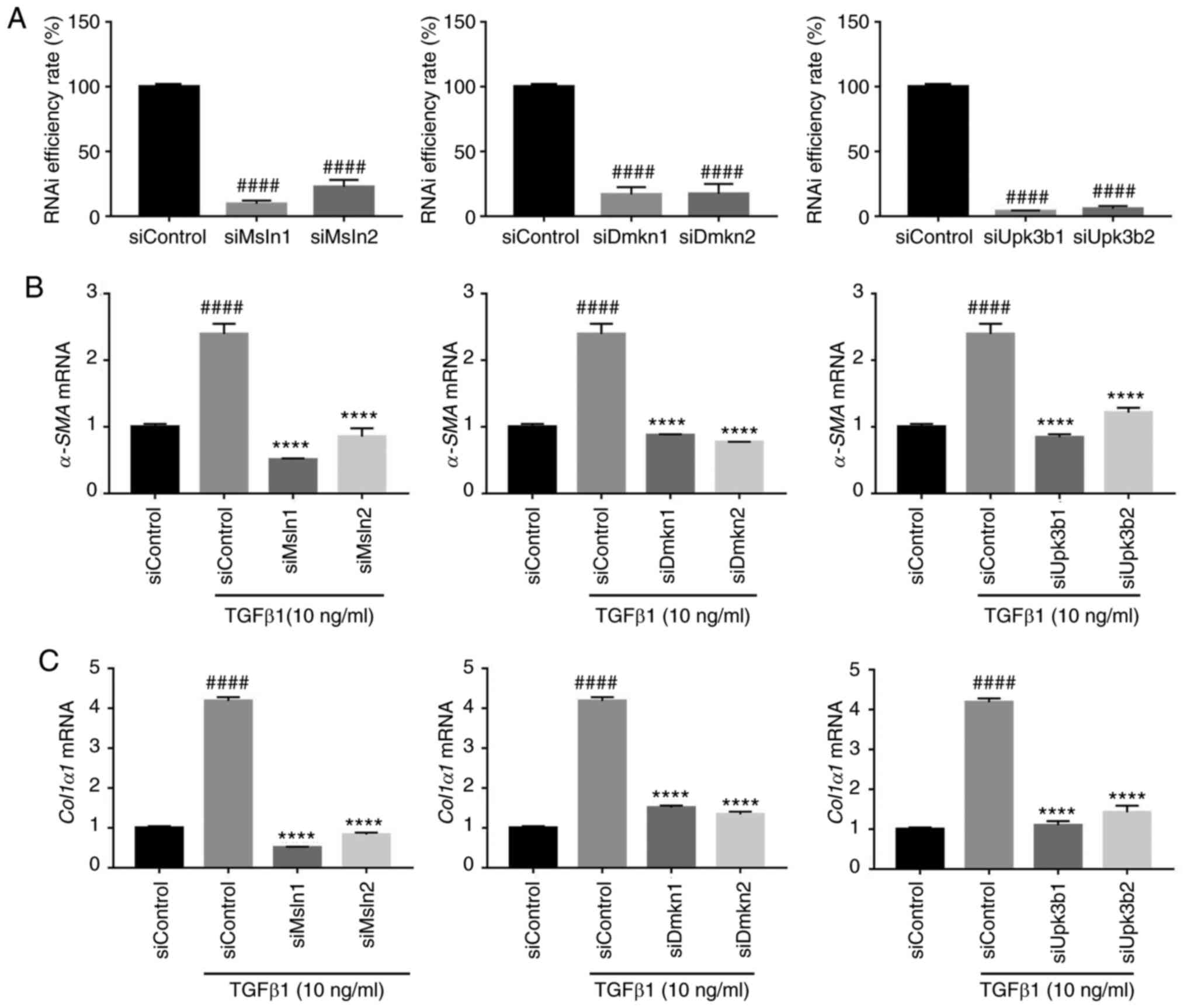 | Figure 5.Msln, Dmkn and Upk3b
knockdown significantly inhibits the activation of HSCs. (A)
Knockdown efficiency of Msln, Dmkn and Upk3b by siRNA
in HSC LX-2 cells. The mRNA expression of fibrogenic genes (B)
α-SMA and (C) Col1α1 were quantified by reverse
transcription-quantitative PCR in HSC LX-2 cells transfected with
siRNA (siControl, siMsln, siDmkn and siUpk3b). The results are
normalized to β-actin, and error bars indicate the SD of three
independent experiments. Data are expressed as the mean ± SD.
####P<0.0001 vs. the siControl group; ****P<0.0001
vs. the siControl + TGF-β1 group. HSC, hepatic stellate cell;
α-SMA, α-smooth muscle actin; Col1α1, collagen type I; Dmkn,
dermokine; Msln, mesothelin; Upk3b, uroplakin-3b; si-, small
interfering RNA; RNAi, RNA interference. |
Discussion
As a chronic hepatic disease with limited treatment
options, liver fibrosis affects millions of individuals worldwide.
Fibroblasts and myofibroblasts, which are primarily responsible for
the synthesis of ECM proteins, have been identified as pivotal
fibrotic effectors in multiple organs (35). HSCs, following activation and
transformation to a myofibroblast phenotype, as marked by
expression of α-SMA, are the dominating cell type that produce ECM
proteins during liver fibrogenesis (36,37).
Thus, the inactivation of HSCs is currently regarded as a potential
treatment strategy for the treatment of hepatic fibrosis (21).
The present study established high-throughput
screening assays to identify compounds that inhibited HSC
activation. In view of the potential regulatory role of epigenetic
mechanisms in modulating HSC function and reprogramming, an
epigenetic inhibitor library was screened. The HDACi givinostat,
which has been used in phase I/II clinical trials for Duchenne
muscular dystrophy (15), was
identified and validated as a potent inhibitor of HSC activation
in vitro and in vivo. In the screening assay,
givinostat was the most potent inhibitor of HSC activation, and it
was more potent than other HDACi drugs. It was also observed that
givinostat treatment alleviated liver fibrosis and liver injury in
CCl4-injected mice, which had shown mild liver fibrosis. The liver
organization architecture and function in mice with hepatic
fibrosis were markedly improved after givinostat treatment. A
previous study reported that givinostat can inhibit the
proliferation and induce the apoptosis of HSC cells, thereby
inhibiting liver fibrosis in mice subjected to a high-fat diet
combined with intraperitoneal injection of CCl4 (38). However, ingestion of an obesogenic
diet in that model could lead to the development of steatosis,
which might result in steatohepatitis and progressive fibrosis
(16); thus, the alleviation of
liver fibrosis might also be derived from the reduction of
steatosis by givinostat.
To evaluate the effects of givinostat on a
CCl4-induced liver fibrosis model, the present study showed a
reduction in HSC activation and alleviation of liver fibrosis by
givinostat in vivo, and also ruled out the possibility that
the alleviation of fibrosis by givinostat was due to reduced
steatosis. It has also been reported that HDAC9 knockdown can
inhibit HSC activation and decrease fibrogenic gene expression in
HSC LX-2 cells (39). Moreover, in
agreement with the anti-fibrotic effects of givinostat shown in the
present study, givinostat was reported to decrease
endothelial-to-mesenchymal transition and reduce cardiac fibrosis,
leading to improved heart performance and protection of blood
vessels from apoptosis in a mouse model of acute myocardial injury
(19). Moreover, single or repeated
oral administration of givinostat in humans has been found to be
safe, as shown in a previous clinical trial (29). In the present mouse model, no
systemic toxicity was observed at the dose of givinostat used in
this experiment. Thus, the present study suggested that givinostat
inhibited hepatic fibrosis and HSC activation in vivo, and
raised the possibility that an existing drug can be repurposed as a
new treatment of hepatic fibrosis.
Transcriptomic analysis revealed the most
significantly regulated genes in the givinostat treatment group in
comparison with the solvent group, among which, Dmkn, Msln
and Upk3b were validated in vitro in HSC LX-2 cells
as crucial genes regulating HSC activation. When Msln, Dmkn or
Upk3b expression was knocked down, the increased mRNA expression of
α-SMA and Col1α1 in response to TGF-β1 stimulation
was significantly reduced in HSC LX-2 cells, suggesting that these
three genes may play crucial roles in the activation of HSCs. To
the best of our knowledge, the role of Msln, Dmkn and Upk3b in HSC
activation was reported for the first time in the present study.
Moreover, givinostat treatment significantly reduced the mRNA
expression of Dmkn, Msln and Upk3b in both a mouse
model and HSC-LX2 cells. Certain genes that were significantly
affected by givinostat treatment in vivo were not affected
in vitro in HSC LX-2 cells, which may be unrelated to HSC
activation or could be the result of other cell types in the liver,
such as endothelial, Kupffer and bile-duct cells (40,41).
Thus, the identification of givinostat as an inhibitor of HSC
activation and its use as a chemical probe led to the
identification of novel regulators of HSC activation.
In summary, the present study established a
high-throughput cell-based assay for the identification of a
compound targeting HSC activation, and identified givinostat as a
potent inhibitor of HSC activation in vitro and in
vivo. Novel regulators of HSC activation were identified using
givinostat as a probe, and these findings illustrated the efficacy
of an epigenetic strategy that targets HSC activation for the
treatment of hepatic fibrosis.
Acknowledgements
Not applicable.
Funding
The present study was financially supported by the
National Natural Science Foundation of China (grant nos. 81070344,
81803554, 91853205, 81625022, 81821005 and 81773568), The Ministry
of Science and Technology of China (grant no. 2015CB910304), The
National Science & Technology Major Project of China (grant no.
2018ZX09711002) and Youth Innovation Promotion Association (grant
no. 2017333).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the GEO repository, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE161981.
The datasets used and/or analyzed during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
HMH, YJL, LPL, LY and JJP performed the
immunofluorescence staining, western blotting, siRNA transfection
and mouse liver fibrosis experiments, analyzed the corresponding
data and wrote the manuscript. XRZ, SJF and JH contributed to
manuscript writing and modification and analyzed the RNA-seq data.
GML, CL, CCS and YYZ conceived and supervised the project, and
revised the manuscript. The present article was conducted in
accordance with the ARRIVE guideline checklist. The authors are
accountable for all aspects of the work in ensuring that questions
related to the accuracy or integrity of any part of the work are
appropriately investigated and resolved. HMH, XRZ and LPL confirm
the authenticity of all the raw data. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Animal care was carried out according to the
guidelines of the Principles of Laboratory Animal Care, and the
protocol was approved by the Institute Animal Care and Use
Committee at the Shanghai Institute of Materia Medica (approval no.
2018-12-LC-11; Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HSCs
|
hepatic stellate cells
|
|
ECM
|
extracellular matrix
|
|
CCl4
|
carbon tetrachloride
|
|
α-SMA
|
α-smooth muscle actin
|
|
Col1α1
|
collagen type I
|
|
HDAC
|
histone deacetylase
|
|
HDACi
|
HDAC inhibitor
|
|
ALT
|
alanine aminotransferase
|
|
AST
|
aspartate aminotransferase
|
References
|
1
|
Mokdad AA, Lopez AD, Shahraz S, Lozano R,
Mokdad AH, Stanaway J, Murray CJ and Naghavi M: Liver cirrhosis
mortality in 187 countries between 1980 and 2010: A systematic
analysis. BMC Med. 12:1452014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schuppan D and Afdhal NH: Liver cirrhosis.
Lancet. 371:838–851. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bataller R and Brenner DA: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Weiskirchen R and Tacke F: Liver fibrosis:
From pathogenesis to novel therapies. Dig Dis. 34:410–422. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ginès P, Cárdenas A, Arroyo V and Rodés J:
Management of cirrhosis and ascites. N Engl J Med. 350:1646–1654.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bárcena C, Stefanovic M, Tutusaus A,
Joannas L, Menéndez A, García-Ruiz C, Sancho-Bru P, Marí M,
Caballeria J, Rothlin CV, et al: Gas6/Axl pathway is activated in
chronic liver disease and its targeting reduces fibrosis via
hepatic stellate cell inactivation. J Hepatol. 63:670–678. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Koh HB, Scruggs AM and Huang SK:
Transforming growth factor-β1 increases DNA methyltransferase 1 and
3a expression through distinct post-transcriptional mechanisms in
lung fibroblasts. J Biol Chem. 291:19287–19298. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mann J, Oakley F, Akiboye F, Elsharkawy A,
Thorne AW and Mann DA: Regulation of myofibroblast
transdifferentiation by DNA methylation and MeCP2: Implications for
wound healing and fibrogenesis. Cell Death Differ. 14:275–285.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Watson CJ, Collier P, Tea I, Neary R,
Watson JA, Robinson C, Phelan D, Ledwidge MT, McDonald KM, McCann
A, et al: Hypoxia-induced epigenetic modifications are associated
with cardiac tissue fibrosis and the development of a
myofibroblast-like phenotype. Hum Mol Genet. 23:2176–2188. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu N, He S, Ma L, Ponnusamy M, Tang J,
Tolbert E, Bayliss G, Zhao TC, Yan H and Zhuang S: Blocking the
class I histone deacetylase ameliorates renal fibrosis and inhibits
renal fibroblast activation via modulating TGF-beta and EGFR
signaling. PLoS One. 8:e540012013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marumo T, Hishikawa K, Yoshikawa M,
Hirahashi J, Kawachi S and Fujita T: Histone deacetylase modulates
the proinflammatory and -fibrotic changes in tubulointerstitial
injury. Am J Physiol Renal Physiol. 298:F133–F141. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tung CW, Hsu YC, Cai CJ, Shih YH, Wang CJ,
Chang PJ and Lin CL: Trichostatin A ameliorates renal
tubulointerstitial fibrosis through modulation of the JNK-dependent
Notch-2 signaling pathway. Sci Rep. 7:144952017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu M, Lin P, Li L, Chen D, Yang X, Xu L,
Zhou B, Wang C, Zhang Y, Luo C and Ye C: Reduced asymmetric
dimethylarginine accumulation through inhibition of the type I
protein arginine methyltransferases promotes renal fibrosis in
obstructed kidneys. FASEB J. 33:6948–6956. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Peng J, Li J, Huang J, Xu P, Huang H, Liu
Y, Yu L, Yang Y, Zhou B, Jiang H, et al: p300/CBP inhibitor A-485
alleviates acute liver injury by regulating macrophage activation
and polarization. Theranostics. 9:8344–8361. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tomaselli D, Lucidi A, Rotili D and Mai A:
Epigenetic polypharmacology: A new frontier for epi-drug discovery.
Med Res Rev. 40:190–244. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tsuchida T, Lee YA, Fujiwara N, Ybanez M,
Allen B, Martins S, Fiel MI, Goossens N, Chou HI, Hoshida Y and
Friedman SL: A simple diet- and chemical-induced murine NASH model
with rapid progression of steatohepatitis, fibrosis and liver
cancer. J Hepatol. 69:385–395. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Scholten D, Trebicka J, Liedtke C and
Weiskirchen R: The carbon tetrachloride model in mice. Lab Anim.
49((1 Suppl)): S4–S11. 2015. View Article : Google Scholar
|
|
18
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals, . Guide for the Care and Use of Laboratory Animals. 8th
edition. Washington (DC). National Academies Press; Washington
(DC): 2011
|
|
19
|
Milan M, Pace V, Maiullari F, Chirivì M,
Baci D, Maiullari S, Madaro L, Maccari S, Stati T, Marano G, et al:
Givinostat reduces adverse cardiac remodeling through regulating
fibroblasts activation. Cell Death Dis. 9:1082018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lan T, Li C, Yang G, Sun Y, Zhuang L, Ou
Y, Li H, Wang G, Kisseleva T, Brenner D and Guo J: Sphingosine
kinase 1 promotes liver fibrosis by preventing miR-19b-3p-mediated
inhibition of CCR2. Hepatology. 68:1070–1086. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang K, Han X, Zhang Z, Zheng L, Hu Z,
Yao Q, Cui H, Shu G, Si M, Li C, et al: The liver-enriched
lnc-LFAR1 promotes liver fibrosis by activating TGFβ and Notch
pathways. Nat Commun. 8:1442017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dobin A, Davis CA, Schlesinger F, Drenkow
J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR: STAR:
Ultrafast universal RNA-seq aligner. Bioinformatics. 29:15–21.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liao Y, Smyth GK and Shi W: FeatureCounts:
An efficient general-purpose program for assigning sequence reads
to genomic features. Bioinformatics. 30:923–930. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jose AF: The Benjamini-Hochberg method in
the case of discrete test statistics. Int J Biostat. 3:112007.
|
|
26
|
Huang DW, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
Bioinformatics Resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pan RL, Xiang LX, Wang P, Liu XY, Nie L,
Huang W and Shao JZ: Low-molecular-weight fibroblast growth factor
2 attenuates hepatic fibrosis by epigenetic down-regulation of
Delta-like1. Hepatology. 61:1708–1720. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Suh YG, Kim JK, Byun JS, Yi HS, Lee YS,
Eun HS, Kim SY, Han KH, Lee KS, Duester G, et al: CD11b+
Gr1+ bone marrow cells ameliorate liver fibrosis by
producing interleukin-10 in mice. Hepatology. 56:1902–1912. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Satishchandran A, Ambade A, Rao S, Hsueh
YC, Iracheta-Vellve A, Tornai D, Lowe P, Gyongyosi B, Li J,
Catalano D, et al: MicroRNA 122, regulated by GRLH2, protects
livers of mice and patients from ethanol-induced liver disease.
Gastroenterology. 154:238–252.e7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
King A, Houlihan DD, kavanagh D, Haldar D,
Luu N, Owen A, Suresh S, Than NN, Reynolds G, Penny J, et al:
Sphingosine-1-phosphate prevents egress of hematopoietic stem cells
from liver to reduce fibrosis. Gastroenterology. 153:233–248.e16.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Han CY, Koo JH, Kim SH, Gardenghi S,
Rivella S, Strnad P, Hwang SJ and Kim SG: Hepcidin inhibits Smad3
phosphorylation in hepatic stellate cells by impeding
ferroportin-mediated regulation of Akt. Nat Commun. 7:138172016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jia Y, Wang F, Guo Q, Li M, Wang L, Zhang
Z, Jiang S, Jin H, Chen A, Tan S, et al: Curcumol induces
RIPK1/RIPK3 complex-dependent necroptosis via JNK1/2-ROS signaling
in hepatic stellate cells. Redox Biol. 19:375–387. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hinz B, Phan SH, Thannickal VJ, Galli A,
Bochaton-Piallat ML and Gabbiani G: The myofibroblast: One
function, multiple origins. Am J Pathol. 170:1807–1816. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mederacke I, Hsu CC, Troeger JS, Huebener
P, Mu X, Dapito DH, Pradere JP and Schwabe RF: Fate tracing reveals
hepatic stellate cells as dominant contributors to liver fibrosis
independent of its aetiology. Nat Commun. 4:28232013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Friedman SL: Hepatic stellate cells:
Protean, multifunctional, and enigmatic cells of the liver. Physiol
Rev. 88:125–172. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang YG, Xu L, Wang T, Wei J, Meng WY,
Wang N and Shi M: Givinostat inhibition of hepatic stellate cell
proliferation and protein acetylation. World J Gastroenterol.
21:8326–8339. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang Y, Bae M, Park YK, Lee Y, Pham TX,
Rudraiah S, Manautou J, Koo SI and Lee JY: Histone deacetylase 9
plays a role in the antifibrogenic effect of astaxanthin in hepatic
stellate cells. J Nutr Biochem. 40:172–177. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Seki E, De Minicis S, Osterreicher CH,
Kluwe J, Osawa Y, Brenner DA and Schwabe RF: TLR4 enhances TGF-beta
signaling and hepatic fibrosis. Nat Med. 13:1324–1332. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ramzy MM, Abdelghany HM, Zenhom NM and
El-Tahawy NF: Effect of histone deacetylase inhibitor on
epithelial-mesenchymal transition of liver fibrosis. IUBMB Life.
70:511–518. 2018. View Article : Google Scholar : PubMed/NCBI
|