Introduction
As a chronic vascular inflammatory disease, which is
associated with oxidative stress and endothelial dysfunction,
atherosclerosis mainly affects the walls of large and medium
arteries, such as the aorta, carotid and coronary arteries
(1,2). The characteristics of atherosclerosis
include lipid accumulation, inflammatory response, cell death and
arterial wall sclerosis, which forms the pathological basis of
ischemic heart disease (3).
Therefore, it is important to identify novel therapeutic strategies
for the treatment of atherosclerosis.
Homocysteine (HCY) is a sulfhydryl-containing amino
acid produced via the demethylation of dietary methionine, which is
rich in animal proteins (1).
Previously, increased plasma HCY was confirmed to be an independent
risk factor for atherosclerosis, and HCY may aggravate vascular
endothelial inflammation and injure the endothelial cells of major
vessels, such as the carotid artery and the aorta (1,2).
Therefore, HCY was used to establish an endothelial injury model in
the present study.
Propofol (2, 6-diisopropyl phenol) is an intravenous
general anesthetic, which is used clinically in an emulsion
formulation. It is extensively used in the induction and
maintenance of anesthetization and procedural sedation (3,4). The
functions of propofol include anti-inflammation, inhibition of the
production of pro-inflammatory cytokines (5), conversion of the production of nitric
oxide, suppression of neutrophil functions and anti-oxidation
(5). Furthermore, propofol has been
reported to upregulate the expression levels of
phospholipid-transporting ATPase ABCA1, ATP-binding cassette
sub-family G member 1 and scavenger receptor class B member 1 via
the peroxisome proliferator-activated receptor γ/oxysterols
receptor LXR-α signaling pathway in THP-1 macrophage-derived foam
cells. Several studies have also shown the protective role of
propofol in myocardial ischemia reperfusion injury in type 2
diabetic rats and its alleviating effect on the injury and
apoptosis in endothelial cells (6–8).
The endoplasmic reticulum (ER) is an organelle
covered by an extensive membrane network in eukaryotic cells. It
plays an important role in protein synthesis, folding and
transport, calcium homeostasis, lipid and steroid synthesis. A
variety of pathological factors, such as hyperlipidemia, oxidative
stress, viral infection and calcium imbalance can disturb the
homeostasis of ER and cause the accumulation of misfolded or
unfolded proteins in the ER cavity, which is called ER stress (ERS)
(9). To alleviate this stress, the
cell initiates the unfolded protein response (UPR) (10). Studies have demonstrated that ERS is
associated with atherosclerosis, and plays an important role in the
initiation and progression of atherosclerosis (11,12).
Long-term ERS can lead to apoptosis and activation of inflammatory
response pathways. In cardiovascular diseases, C/EBP-homologous
protein (CHOP) is the most widely studied biomarker in the
ERS-related apoptosis signaling pathway (13). The activation of the apoptotic
signaling pathway mediated by CHOP and ER chaperone BiP (Bip),
upregulates the expression levels of pro-apoptotic members of the
Bcl-2 family and induces apoptosis (13). As atherosclerosis progresses, the
UPR cannot control ERS, and the expression of CHOP increases,
eventually activating its induced apoptosis signaling pathway. ERS
can also activate the NF-κB pathway and the NACHT LRR and PYD
domains-containing protein 3 inflammasome, and increase the
expression levels of a large number of inflammatory molecules, such
as TNF-α and IL-1β; therefore, it can trigger inflammatory
responses (12). It has been
confirmed that ERS-mediated apoptosis and inflammation are widely
involved in all stages of atherosclerotic development (14).
The aim of the present study was to investigate
whether propofol could inhibit the injury of endothelial cells
induced by HCY and the potential mechanism involved.
Materials and methods
Cell culture
Human umbilical vein endothelial cells (HUVECs,
PCS-100-010™) were purchased from the American Type Culture
Collection, thawed at 37°C for 2 min, then transferred into a tube
containing 5 ml RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc.), followed by centrifugation at 1,000 × g and 4°C for 5 min.
The supernatant was removed, then the cells were re-suspended in
RPMI-1640 medium containing 100 U/ml penicillin, 100 µg/ml
streptomycin and 10% FBS (Gibco; Thermo Fisher Scientific, Inc.).
Subsequently, they were transferred into a 25-cm flask and
incubated at 37°C in an humidified incubator with 5%
CO2. The medium was replaced every 2 days. When the
cells reached 80% confluence, they were washed with PBS twice, then
0.25% trypsin (1 ml) was added. After attachment, cells (70–80%
confluence) were exposed to 2.5 mmol/l HCY (Sigma-Aldrich; Merck
KGaA) at 37°C for 48 h to construct the endothelial cell injury
model (2). For HCY and propofol
(Sigma-Aldrich; Merck KGaA) co-treatment, adherent cells were
pre-treated with indicated concentrations (12.5, 25, 50, 100 and
200 µM) of propofol at 37°C for 2 h, followed by exposure to 2.5
mmol/l HCY for 48 h. Untreated cells were used as the control
group. Then, cells were collected for the following analysis.
Cell transfection
For Bip overexpression, overexpression plasmids
(pcDNA3.1-Bip; ov-Bip) were purchased from Shanghai GenePharma Co.,
Ltd. The pcDNA3.1 empty vector (Shanghai GenePharma Co., Ltd.) was
used as a negative control (NC). Briefly, Lipofectamine®
2000 (Invitrogen; Thermo Fisher Scientific. Inc.) was mixed with 20
µg plasmids, which was then added to the cells at 70–80% confluence
and incubated for 6 h at 37°C. Subsequently, cells were cultured in
RPMI-1640 medium and at 48-h post-transfection, the cell samples
were used for subsequent experimentation.
Cell Counting Kit-8 (CCK-8) assay
Briefly, the HVUECs (2×103 cells per
well) were seeded into a 96-well plate and incubated at 37°C. After
attachment, cells were pre-treated with or without 12.5, 25, 50,
100 and 200 µM propofol at 37°C for 2 h, followed by exposure to
2.5 mmol/l HCY for 48 h. Subsequently 10 µl CCK-8 reagent
(Dojindo Molecular Technologies, Inc.) was added to each well and
incubated for 2 h at 37°C. Then, the absorbance of the cells was
measured at 450 nm using a microplate reader (BioTek Instruments,
Inc.).
ELISA
To determine the concentrations of TNF-α (cat. no.
F02810), IL-1β (cat. no. F01220) and IL-6 (cat. no. F01310) in the
supernatant of the HUVECs, corresponding ELISA kits (Shanghai
Xitang Biotechnology Co., Ltd.) were used according to the
manufacturer's instructions. The optical density at 450 nm was
measured using a microplate reader (Bio-Rad Laboratories, Inc.),
then a standard curve was created.
Western blot analysis
Total cell protein was extracted using RIPA lysis
(Beyotime Institute of Biotechnology), then the concentration was
determined using the BCA method. Protein (50 µg) was added to the
SDS loading buffer, and after the protein samples were heated in a
water bath for 5 min, they were separated via SDS-PAGE on a 10% gel
at 60 V for 40 min and 110 V for 60 min. Separated proteins were
then transferred onto a PVDF membrane. After washing with PBS, the
PVDF membrane was blocked with 50 g/l skimmed milk at 25°C for 1 h.
Following which, the membrane was incubated overnight at 4°C with
primary antibodies against NF-κB-p65 (cat. no. ab16502; 1:2,000;
Abcam), phosphorylated (p)-IκBα (cat. no. ab92700; 1:1,000; Abcam),
Bax (cat. no. ab32503; 1:1,000; Abcam), cleaved caspase-3 (cat. no.
ab2302; 1:1,000; Abcam), Bcl-2 (cat. no. ab32124; 1:1,000; Abcam),
Bip (cat. no. 3177; 1:1,000; Cell Signaling Technology, Inc.), CHOP
(cat. no. 2895; 1:1,000; Cell Signaling Technology, Inc.),
p-protein kinase R (PKR)-like ER kinase (PERK; cat. no. 3179;
1:1,000; Cell Signaling Technology, Inc.), PERK (cat. no. 5683;
1:1,000; Cell Signaling Technology, Inc.) and inositol-requiring 1α
(IRE1α; cat. no. 3294; 1:1,000; Cell Signaling Technology, Inc.).
After the membrane was washed with TBS with 5% Tween-20 (TBST)
three times, a goat anti-rabbit IgG (cat. no. ab205718; 1:10,000;
Abcam) and goat anti-mouse IgG (cat. no. ab6789; 1:10,000; Abcam)
secondary antibodies were added to the PVDF membrane at 37°C for 2
h. After TBST was used to wash the membrane, ECL agent (Thermo
Fisher Scientific, Inc.) was added and the images were captured
using a Bio-Rad chemiluminescence imager (Bio-Rad Laboratories,
Inc.). Protein expression levels were semi-quantified using
Image-Pro Plus software version 6.0 (Roper Technologies, Inc.).
Flow cytometry
A total of 1.5×106 cells were incubated
with 0.25% trypsin, harvested and rinsed twice with pre-chilled
PBS. Then, the apoptotic rate was evaluated with the Annexin V-FITC
Apoptosis Detection kit (BioLegend, Inc.). Briefly, the transfected
cells were resuspended in 100 µl binding buffer prior to
counterstaining with 5 µl Annexin V-FITC and 5 µl PI solution at
room temperature in the dark for 15 min. Then, the stained cells
were analyzed using a flow cytometer (FACScan; BD Biosciences). The
data was analyzed using CellQuest™ software v.5.1 (BD Biosciences)
to assess early + late apoptosis.
Statistical analysis
All data are presented as the mean ± standard
deviation from three independent repeated experiments. Statistical
data analysis was performed with SPSS v23.0 (IBM Corp.) and
GraphPad Prism v5.0 (GraphPad Software, Inc.) software. Statistical
differences between multiple groups were analyzed using one-way
ANOVA followed by Tukey's post hoc test. P<0.05 was considered
to indicate a statistically significant difference.
Results
Propofol ameliorates HCY-induced HUVEC
cell injury
To investigate the dysfunction caused by HCY in
HUVECs, the cell viability of HUVECs was detected. As shown in
Fig. 1A, the cell viability of
cells treated with HCY was significantly decreased compared with
that in the control group; however, the addition of 50–200 µmol/l
propofol enhanced cell viability in a dose-dependent manner.
Therefore, propofol at 50, 100 and 200 µmol/l was chosen for
further experimentation.
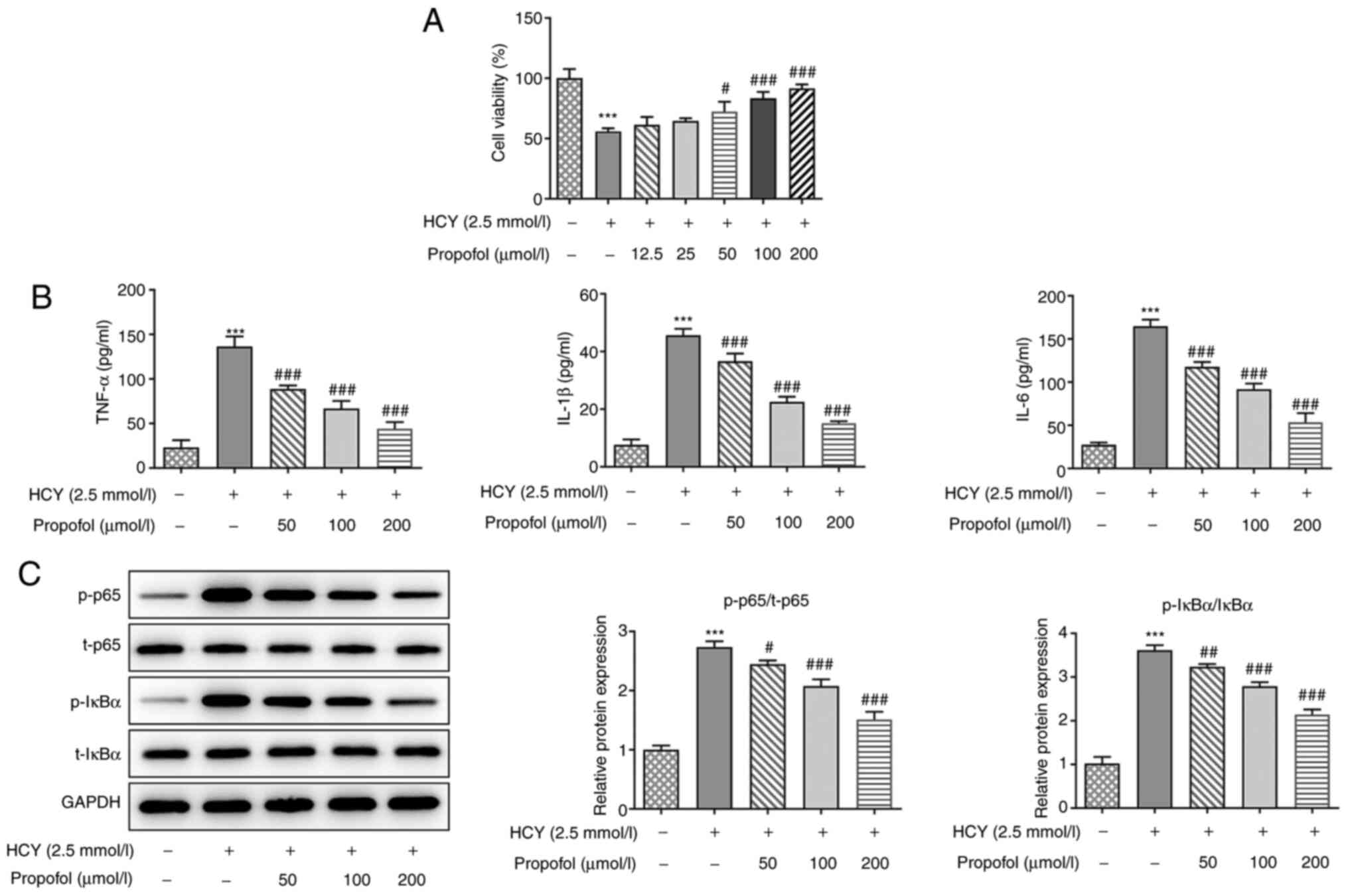 | Figure 1.Effect of propofol on HCY-induced
NF-κB signaling activation and inflammation in HUVECs. (A) HUVECs
were pretreated with different concentrations of propofol for 2 h
and stimulated with 2.5 mmol/l HCY for 48 h, then the cell
viability was measured using a Cell Counting Kit-8 assay. (B)
HUVECs were pretreated with different concentrations of propofol
for 2 h and stimulated with 2.5 mmol/l HCY for 48 h, then the
concentrations of TNF-α, IL-1β and IL-6 in the culture medium was
measured using ELISA kits. (C) HUVECs were pretreated with
different concentrations of propofol for 2 h and stimulated with
2.5 mmol/l HCY for 48 h, then the expression levels of proteins
involved in NF-κB signaling, including p65 and IκBα, were
determined via western blotting. ***P<0.001 vs. control group;
#P<0.05, ##P<0.01 and
###P<0.001 vs. HCY group. HCY, homocysteine; HUVECs,
human umbilical vein endothelial cells; p-, phosphorylated; t-,
total. |
Propofol decreases the concentration
of inflammatory factors in HCY-induced HUVECs
As NF-κB-mediated endothelial cell activation and
vascular inflammation are central to the initiation and progression
of atherosclerosis (15), the
expression levels and release of pro-inflammatory factors were
investigated. Following propofol pretreatment for 2 h and the
induction of cell injury with 2.5 mmol/l HCY for 4 h, the
concentration of TNF-α, IL-1β and IL-6 in the HUVECs were detected
using ELISA, and the protein expression levels of p-NF-κB-p65 and
p-IκBα were detected using western blot analysis. Compared with
those in the control group, the concentrations of TNF-α, IL-1β and
IL-6 were decreased following pretreatment with propofol, in a
dose-dependent manner (Fig. 1B).
Furthermore, as shown in Fig. 1C,
it was found that HCY significantly upregulated the ratio of p-p65
and p-IκBα, while increasing doses of propofol reversed the effects
of HCY. The results suggested that propofol could decrease the
production of inflammatory cytokines in HCY-induced HUVECs by
inactivating the NF-κB signaling pathway.
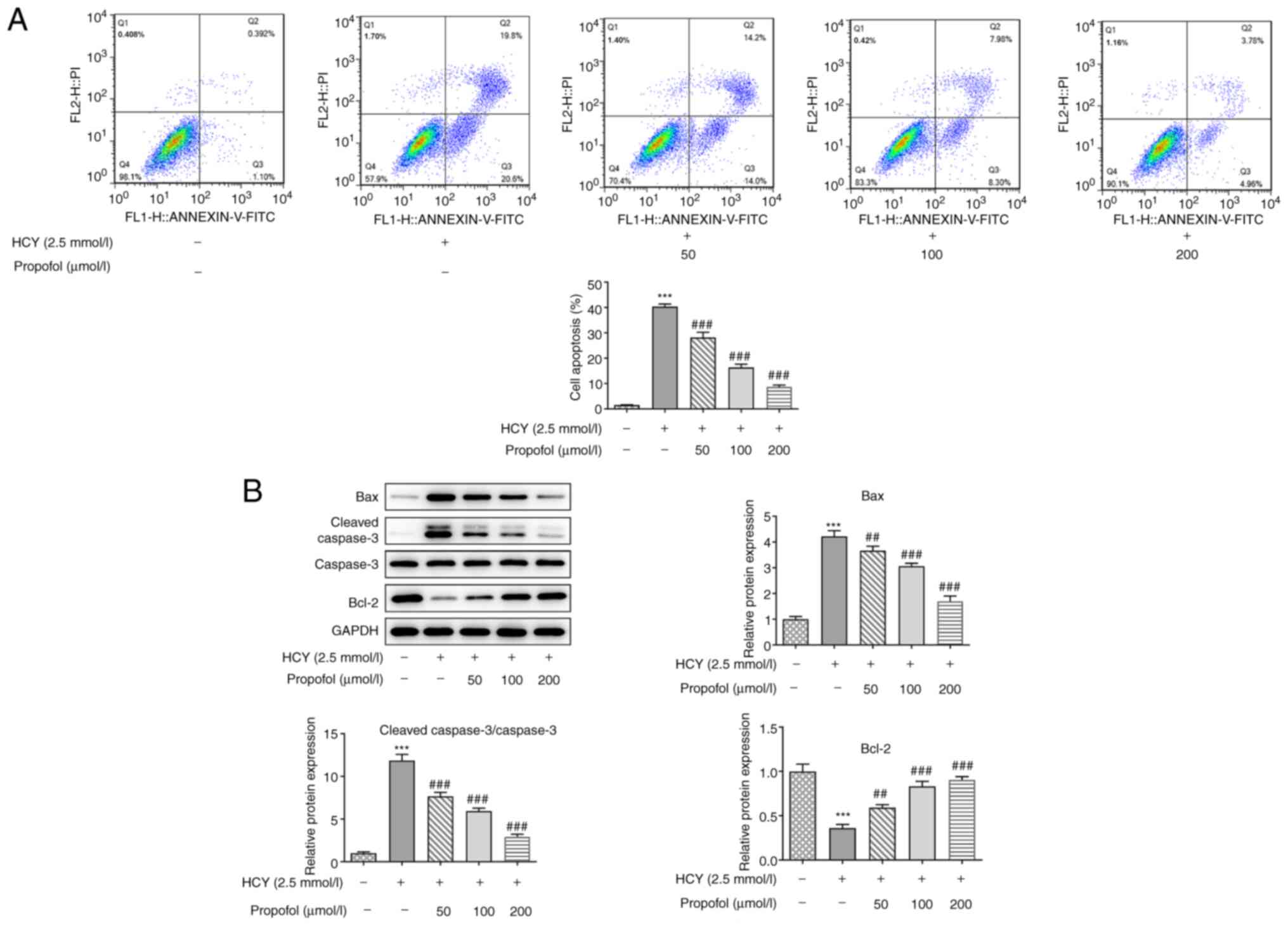
Propofol decreases cell apoptosis in
HCY-induced HUVECs
Following pretreatment with propofol for 2 h and
induction of cell injury with 2.5 mmol/l HCY in the HUVECs for 48
h, apoptosis was measured using flow cytometry. As shown in
Fig. 2A, cell apoptosis was
increased in the HCY group compared with that in the control group;
however, apoptosis gradually decreased when the cells, induced by
HCY, were pretreated with increasing concentrations of propofol.
Then, western blotting was performed to detect the expression
levels of the apoptosis-related proteins, Bax, cleaved caspase-3
and Bcl-2. As shown in Fig. 2B, the
protein expression levels of Bax and cleaved caspase-3 were
downregulated, while that of Bcl-2 was upregulated, as a result of
increasing concentrations of propofol pretreatment. These results
indicated that propofol could decrease HUVEC apoptosis induced by
HCY.
Propofol decreases ERS in HCY-induced
HUVECs
Western blot analysis was utilized to assess the
effect of propofol on the expression levels of ERS-related
proteins. HCY increased the protein expression levels of Bip and
CHOP in HUVECs, indicating that HCY increased ERS (Fig. 3A). However, pretreatment of HUVECs
with increasing concentrations of propofol alleviated the increased
protein expression levels of Bip and CHOP in a dose-dependent
manner. Next, the protein expression levels of p-PERK and p-IREα
were detected, and the results showed that their expression levels
in HCY-induced HUVECs were increasingly attenuated by propofol
(Fig. 3B). These results suggested
that propofol could decrease the expression level of proteins
involved in ERS in HUVECs induced by HCY.
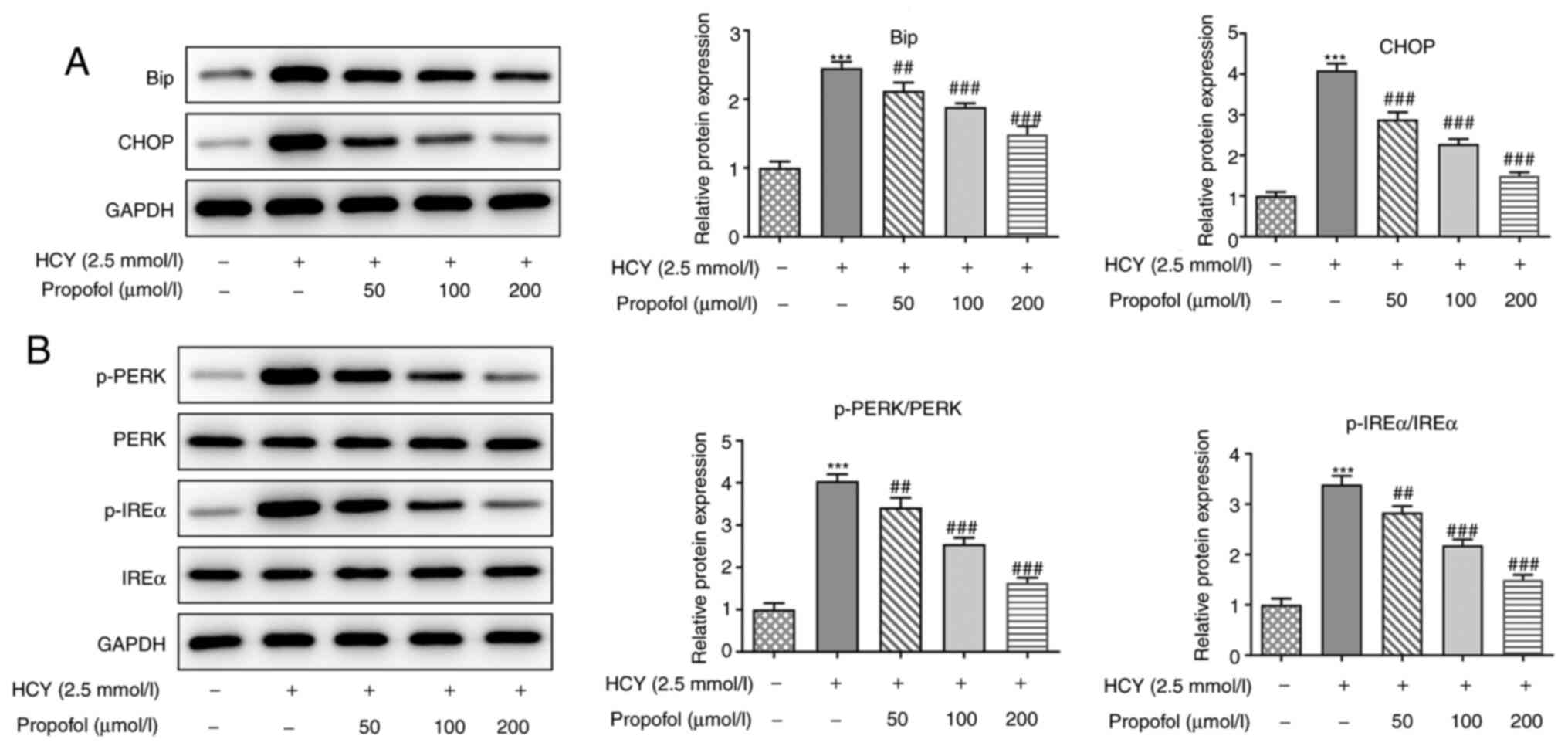 | Figure 3.Effect of propofol on the expression
levels of proteins related to ERS in HCY-treated HUVECs. HUVECs
were pretreated with different concentrations of propofol for 2 h
and stimulated with 2.5 mmol/l HCY for 48 h. Then, the expression
levels of proteins involved in ERS were detected via western
blotting, including (A) Bip and CHOP, and (B) p-PERK and p-IREα.
***P<0.001 vs. control group; ##P<0.01 and
###P<0.001 vs. HCY group. HCY, homocysteine; HUVECs,
human umbilical vein endothelial cells; ERS, endoplasmic reticulum
stress; p-, phosphorylated; Bip, ER chaperone BiP; CHOP,
C/EBP-homologous protein; PERK, protein kinase R-like ER kinase;
IREα, inositol-requiring 1α. |
Overexpression of Bip reverses the
inhibitory effect of propofol on HCY-induced HUVEC ERS
Then, the Bip overexpression plasmid was constructed
and western blotting confirmed its transfection efficiency compared
with the NC (Fig. 4A).
Subsequently, propofol, at 200 µmol/l, was selected for the next
experiments to examine the effect of Bip overexpression on the
functions of propofol. As shown in Fig.
4B, the cell viability of HCY-induced HUVECs was enhanced by
pretreatment with propofol; however, it decreased when ov-Bip was
transfected into the cells. Next, the expression levels of
ERS-related proteins were determined, as shown in Fig. 4C, the protein expression levels of
p-PERK and p-IREα in HCY-induced HUVECs were downregulated
following pretreatment with propofol. However, transfection with
ov-Bip, in these cells, increased the protein expression levels of
p-PERK and p-IREα. These results suggested that overexpression of
Bip could reverse the inhibitory effect of propofol on HCY-induced
cell viability impairment and ERS in HUVECs.
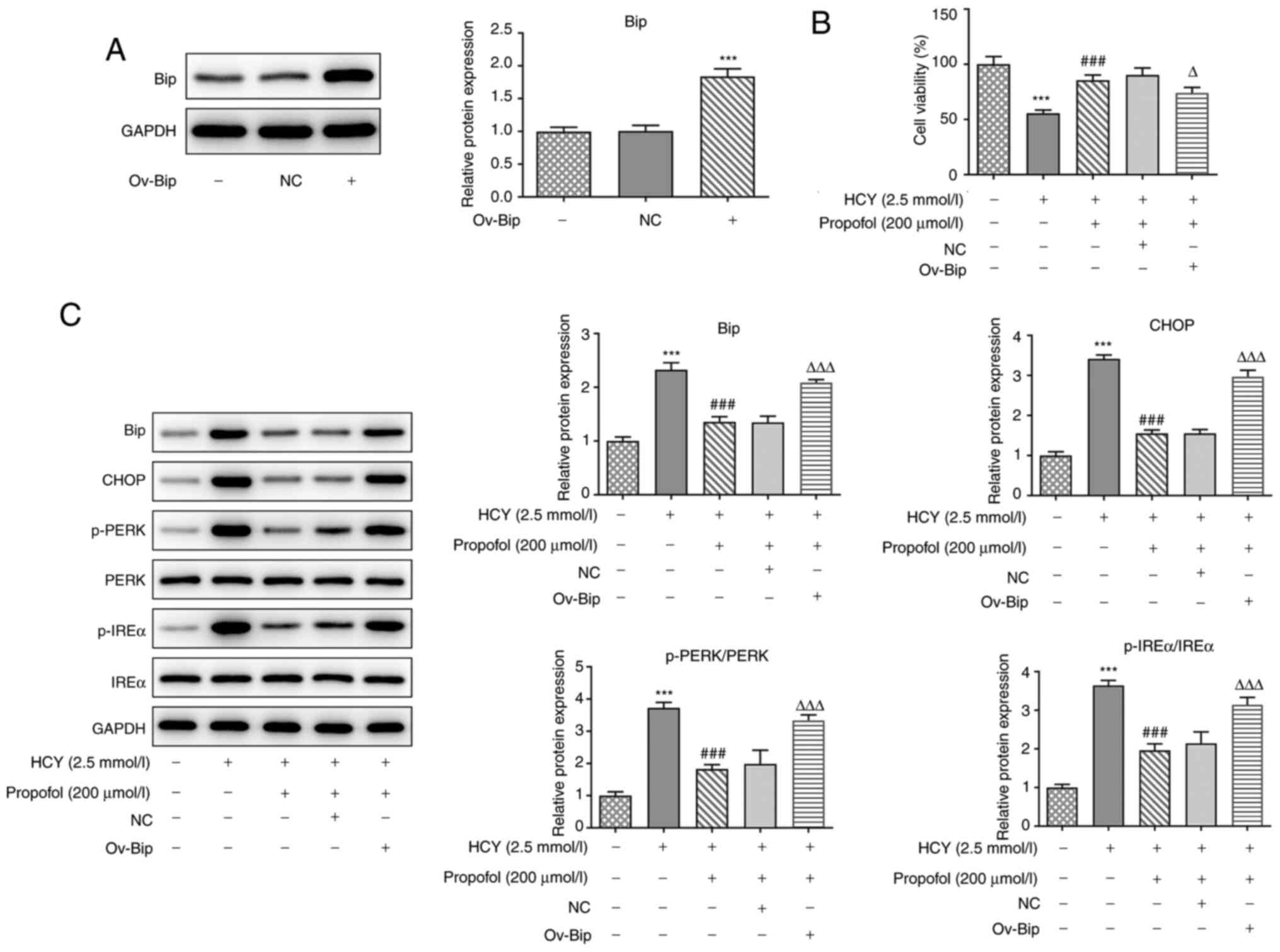 | Figure 4.Effect of Bip overexpression on the
propofol-induced reduction in the expression of ERS-related
proteins in HCY-treated HUVECs. (A) HUVECs were transfected with
ov-Bip or NC vector, then the protein expression of Bip was
detected by western blotting. (B) HUVECs transfected with ov-Bip or
NC vectors were pretreated with 200 µmol/l propofol for 2 h and
stimulated with 2.5 mmol/l HCY for 48 h, then cell viability was
detected using a Cell Counting Kit-8 assay. (C) HUVECs transfected
with ov-Bip or NC vectors were pretreated with 200 µmol/l propofol
for 2 h and stimulated with 2.5 mmol/l HCY for 48 h, then the
protein expression levels of Bip, CHOP, p-PERK/PERK and p-IREα/IREα
were detected via western blotting. ***P<0.001 vs. control
group; ###P<0.001 vs. HCY group;
ΔP<0.05 and ΔΔΔP<0.001 vs. propofol
group. HCY, homocysteine; HUVECs, human umbilical vein endothelial
cells; ov, overexpression vector; NC, negative control; p-,
phosphorylated; Bip, ER chaperone BiP; CHOP, C/EBP-homologous
protein; PERK, protein kinase R-like ER kinase; IREα,
inositol-requiring 1α. |
Overexpression of Bip reverses the
inhibitory effect of propofol on NF-κB signaling-mediated
inflammatory responses in HUVECs induced by HCY
ELISA was performed to detect the concentrations of
TNF-α, IL-1β and IL-6. Compared with that in the HCY group,
pretreatment with propofol in HCY-induced HUVECs could downregulate
the concentrations of inflammatory cytokines, which could be
reversed by the overexpression of Bip (Fig. 5A). Next, the pro-inflammatory
proteins in the NF-κB signaling pathway were investigated, and
western blot analysis showed that the ratio of p-p65 and p-IκBα was
higher in the HCY group compared with that in the control group,
and pretreatment with propofol downregulated their expression
levels (Fig. 5B). However,
transfection with ov-Bip could reverse these effects. Thus,
overexpression of Bip could reverse the inhibitory effect of
propofol on the expression of NF-κB signaling-mediated inflammatory
factors in HUVECs induced by HCY.
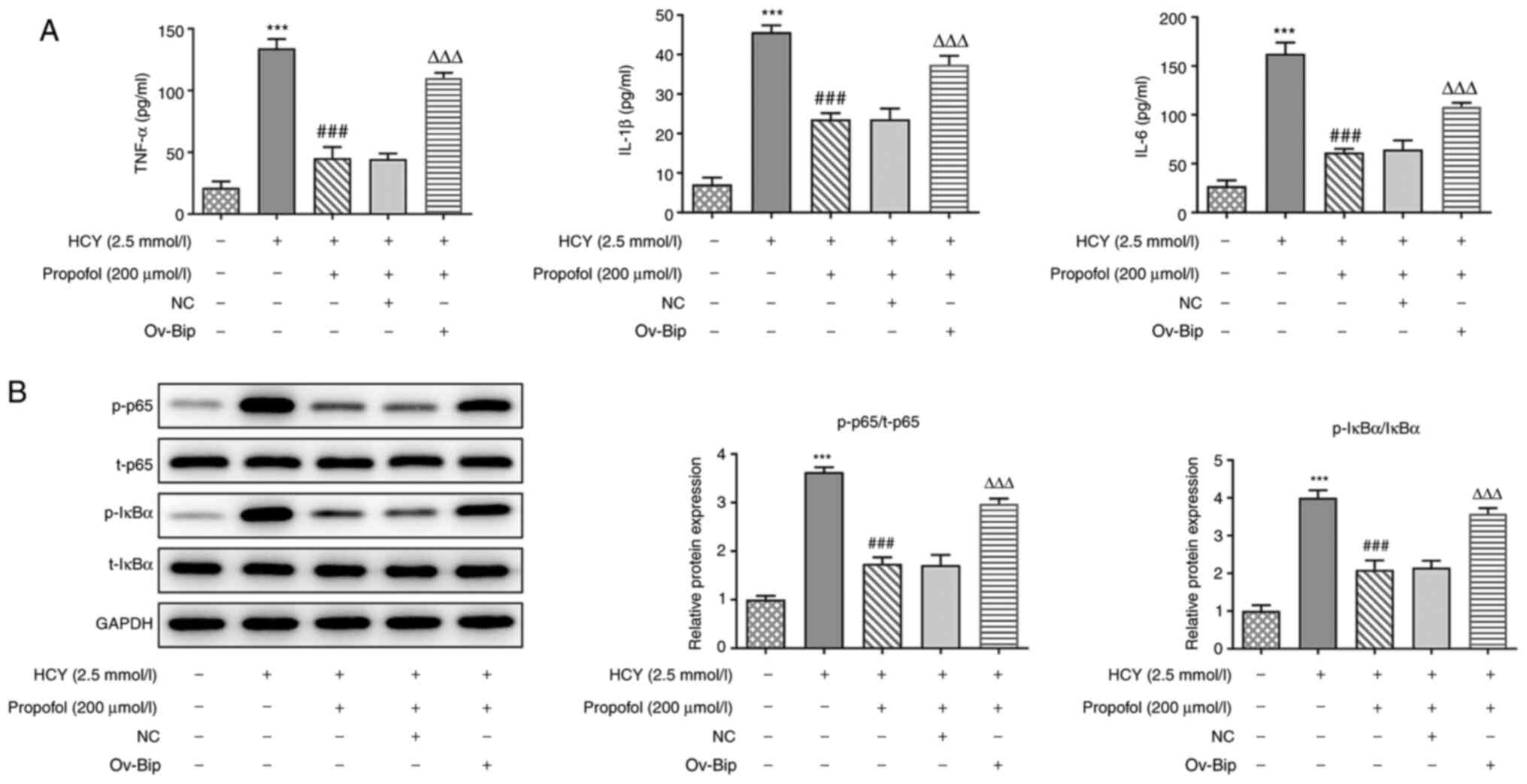 | Figure 5.Effect of Bip overexpression on the
propofol-induced suppression of inflammation and NF-κB signaling
activation in HCY-treated HUVECs. (A) HUVECs transfected with
ov-Bip or NC vectors were pretreated with 200 µmol/l propofol for 2
h and stimulated with 2.5 mmol/l HCY for 48 h, then the
concentrations of TNF-α, IL-1β and IL-6 in the culture medium was
measured using ELISA kits. (B) HUVECs transfected with ov-Bip or NC
vectors were pretreated with 200 µmol/l propofol for 2 h and
stimulated with 2.5 mmol/l HCY for 48 h, then the expression levels
of proteins involved in NF-κB signaling, including p65 and IκBα,
were determined via western blotting. ***P<0.001 vs. control
group; ###P<0.001 vs. HCY group;
ΔΔΔP<0.001 vs. propofol group. HCY, homocysteine;
HUVECs, human umbilical vein endothelial cells; ov, overexpression
vector; NC, negative control; p-, phosphorylated; t-, total; Bip,
ER chaperone BiP. |
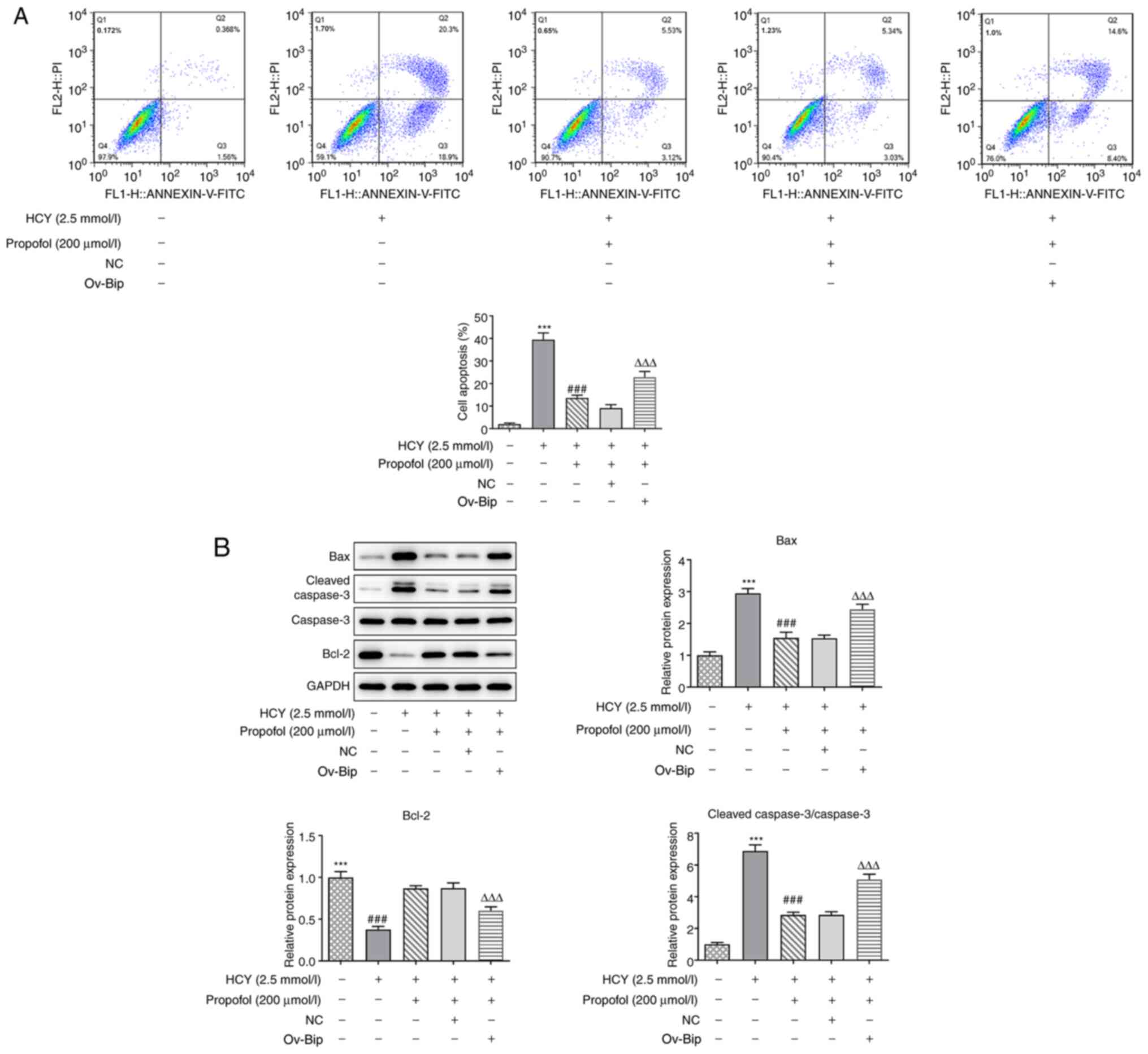
Overexpression of Bip reverses the
inhibitory effects of propofol on cell apoptosis in HCY-induced
HUVECs
To further investigate whether propofol could reduce
HCY-induced cell apoptosis by regulating ERS, flow cytometry was
utilized. The results showed that compared with that in the HCY
group, pretreatment with propofol inhibited cell apoptosis in
HCY-induced HUVECs, but transfection with ov-Bip promoted cell
apoptosis of these cells (Fig. 6A).
Furthermore, pretreatment with propofol downregulated the protein
expression levels of Bax and cleaved caspase-3 and upregulated the
expression of Bcl-2, which could be reversed by the overexpression
of Bip (Fig. 6B). The results
showed that overexpression of Bip could reverse the inhibitory
effects of propofol on cell apoptosis in HCY-induced HUVECs.
Discussion
The dysfunction of the endothelium, which is an
emergent and complex system, is involved in the pathogenesis of all
types of cardiovascular diseases and it occurs in response to
cardiovascular risk factors (16).
Previous investigations have shown that endothelial dysfunction
occurs prior to and throughout the development of atherosclerosis
(17). Furthermore, dysfunction of
the endothelial cells, which occurs in the process of lesion
formation, at the earliest time, could stimulate the development of
atherosclerosis, demonstrating that endothelial dysfunction is a
biomarker to predict the onset of atherosclerosis (18–20).
HCY is the byproduct of a high number of biological
processes and an independent risk factor for cardiovascular
disease, which was previously confirmed by some clinical trials
(21). Furthermore, some studies
identified its role in inducing inflammation, apoptosis in
endothelial cell culture and triggering endothelial desquamation
(21). Therefore, it was used as an
elicitor to induce endothelial dysfunction in the present study and
the results showed that HCY decreased cell viability, elevated the
concentration of inflammatory cytokines and promoted cell apoptosis
of HUVECs.
Inflammation is important in the occurrence and
development of arteriosclerosis and arteriosclerosis-related
complications. NF-κB is a key transcriptional regulator at the
onset of inflammation, and activation of NF-κB is extensively
involved in different inflammatory diseases, including asthma and
rheumatoid arthritis (22–25). Furthermore, NF-κB activation has
been found in atherosclerotic plaques, and specific silencing or
inhibition of the NF-κB signaling pathway has been shown to reduce
the area of the atherosclerotic plaques and decrease the incidence
of atherosclerosis-related complications (26). The results from the present study
also suggested that HCY notably increased the protein expressions
levels of p-p65 and p-IκBα, suggesting that the release of
pro-inflammatory cytokines may be associated with the activation of
the NF-κB signaling pathway.
Propofol is widely used as an anesthetic and it has
been reported to possess anti-inflammatory properties (27). A previous study identified its role
in alleviating inflammation via the inhibition of the NF-κB
signaling pathway in allergic asthmatic mice; however, whether it
could play a central role in suppressing the NF-κB signaling
pathway to reduce inflammation and cell apoptosis remains unclear
(28). In the present study,
pretreatment with propofol increased cell viability, reduced
apoptosis and ameliorated inflammation by inactivating the NF-κB
signaling pathway.
The ER is a membranous network of branching tubules
and flattened sacs in all eukaryotic cells. It is known as a
factory where proteins are synthesized, folded, assembled and
modified. ERS occurs when there is an imbalance between the
protein-folding load and the capacity of ER, as a result of an
increasing demand for folding proteins or stimuli that obstruct the
proteins to fold (29). Once ERS
occurs, UPR is activated through the evolutionally conserved
signaling pathways, including IRE1α and PERK (30). These are two ER-localized protein
sensors, both of which possess an ER-luminal domain that senses
unfolded proteins, and a cytosolic domain that transmits signals to
the transcriptional or translational apparatus. IRE1α has
protein-kinase activity and site-specific endoribonuclease (RNase)
activity (31,32). PERK also has protein-kinase activity
(33). In response to ERS, IRE1α is
autophosphorylated, thereby activating its RNase activity. As
aforementioned by some studies, HCY has been demonstrated to induce
ERS in endothelial cells, and propofol can inhibit ERS in the
retinal pigment epithelial cells, thus the present study focused on
the specific role of propofol in the regulation of ERS in
endothelial cells (34,35). The results showed that HCY elevated
the protein expression levels of Bip and CHOP, which are
ER-specific chaperones and transcriptional factors contributing to
apoptosis, respectively. Furthermore, HCY promoted PERK and IRE1α
phosphorylation in HUVECs, thereby leading to ERS. However,
increasing doses of propofol could gradually reverse these
trends.
In a previous study, hypertensive mouse models were
administrated with ERS inhibitors, which led to improved
endothelial function, suggesting that ERS might be associated with
endothelial dysfunction (36). In
addition, the association between ERS and atherosclerosis was also
investigated previously and it was confirmed in atheroma
endothelial cells and macrophages (37). Thus, in the present study it was
hypothesized that propofol could alleviate inflammation and
apoptosis of endothelial cells by regulating ERS. To further
confirm this hypothesis, ov-Bip was constructed and transfected
into the HCY-induced HUVECs, which were pretreated with propofol.
The results showed that overexpression of Bip could reverse the
inhibitory effects of propofol on ERS and decrease cell viability.
Several reports have indicated that inflammatory cytokines can
cause ERS and therefore activate the UPR (10,11,38).
Thus, the concentration of inflammatory factors was investigated in
the present study, and it was found that transfection with ov-Bip
could upregulate their concentrations. Not surprisingly, it also
reactivated the NF-κB signaling pathway. The interaction between
IRE1α and PERK can assist with the converge of the distinct
signaling pathways to produce an effective response to reduce
damage (39). However, if
overwhelmed, these signaling proteins can also induce apoptosis
(31). Correspondingly,
reactivation of these proteins, by the overexpression of Bip, in
the present study, could promote cell apoptosis. In addition,
propofol has also been reported to inhibit inflammation via other
pathways besides the NF-κB signaling pathway (40–42).
Whether propofol may relieve HUVEC injury or arteriosclerosis via
other pathways will be investigated in our future studies.
In conclusion, propofol could ameliorate
inflammation and cell apoptosis in HCY-induced HUVECs by inhibiting
ERS, which may provide a novel insight into the treatment of
atherosclerosis. However, this study lacks evidence from in
vivo experiments, so in vivo studies that utilize animal
models will be performed in our future research to determine the
protective effect of propofol against arteriosclerosis.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
MZ and CJ contributed to the study conception and
design. CJ, HY and JH contributed to the acquisition of the data.
WZ contributed to the analysis and interpretation of the data. CJ
drafted the initial manuscript and MZ revised it critically for
important intellectual content. MZ and CJ confirm the authenticity
of all the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pang X, Liu J, Zhao J, Mao J, Zhang X,
Feng L, Han C, Li M, Wang S and Wu D: Homocysteine induces the
expression of C-reactive protein via NMDAr-ROS-MAPK-NF-κB signal
pathway in rat vascular smooth muscle cells. Atherosclerosis.
236:73–81. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li J, Luo M, Xie N, Wang J and Chen L:
Curcumin protects endothelial cells against homocysteine induced
injury through inhibiting inflammation. Am J Transl Res.
8:4598–4604. 2016.PubMed/NCBI
|
|
3
|
Hales TG and Lambert JJ: The actions of
propofol on inhibitory amino acid receptors of bovine
adrenomedullary chromaffin cells and rodent central neurones. Br J
Pharmacol. 104:619–628. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mickey BJ, White AT, Arp AM, Leonardi K,
Torres MM, Larson AL, Odell DH, Whittingham SA, Beck MM, Jessop JE,
et al: Propofol for treatment-resistant depression: A pilot study.
Int J Neuropsychopharmacol. 21:1079–1089. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Allaouchiche B, Debon R, Goudable J,
Chassard D and Duflo F: Oxidative stress status during exposure to
propofol, sevoflurane and desflurane. Anesth Analg. 93:981–985.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nesseler N, Launey Y, Isslame S, Flécher
E, Lebouvier T, Mallédant Y and Seguin P: Is extracorporeal
membrane oxygenation for severe acute respiratory distress syndrome
related to intra-abdominal sepsis beneficial? Intensive Care Med.
41:943–945. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin C, Sui H, Gu J, Yang X, Deng L, Li W,
Ding W, Li D and Yang Y: Effect and mechanism of propofol on
myocardial ischemia reperfusion injury in type 2 diabetic rats.
Microvasc Res. 90:162–168. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang S and Smith JD: ABCA1 and nascent HDL
biogenesis. Biofactors. 40:547–554. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cao SS, Luo KL and Shi L: Endoplasmic
reticulum stress interacts with inflammation in human diseases. J
Cell Physiol. 231:288–294. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oakes SA and Papa FR: The role of
endoplasmic reticulum stress in human pathology. Annu Rev Pathol.
10:173–194. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang X, Xu H, Hao Y, Zhao L, Cai X, Tian
J, Zhang M, Han X, Ma S, Cao J and Jiang Y: Endoplasmic reticulum
oxidoreductin 1α mediates hepatic endoplasmic reticulum stress in
homocysteine-induced atherosclerosis. Acta Biochim Biophys Sin
(Shanghai). 46:902–910. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang S, Wu M, Li X, Zhao R, Zhao Y, Liu L
and Wang S: Role of endoplasmic reticulum stress in atherosclerosis
and its potential as a therapeutic target. Oxid Med Cell Longev.
2020:92701072020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ivanova EA and Orekhov AN: The role of
endoplasmic reticulum stress and unfolded protein response in
atherosclerosis. Int J Mol Sci. 17:1932016. View Article : Google Scholar
|
|
14
|
Hamczyk MR, Villa-Bellosta R, Quesada V,
Gonzalo P, Vidak S, Nevado RM, Andrés-Manzano MJ, Misteli T,
López-Otín C and Andrés V: Progerin accelerates atherosclerosis by
inducing endoplasmic reticulum stress in vascular smooth muscle
cells. EMBO Mol Med. 11:e97362019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Alberts-Grill N, Denning TL, Rezvan A and
Jo H: The role of the vascular dendritic cell network in
atherosclerosis. Am J Physiol Cell Physiol. 305:C1–C21. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suwaidi JA, Hamasaki S, Higano ST,
Nishimura RA, Holmes DR Jr and Lerman A: Long-term follow-up of
patients with mild coronary artery disease and endothelial
dysfunction. Circulation. 101:948–954. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Reddy KG, Nair RN, Sheehan HM and Hodgson
JM: Evidence that selective endothelial dysfunction may occur in
the absence of angiographic or ultrasound atherosclerosis in
patients with risk factors for atherosclerosis. J Am Coll Cardiol.
23:833–843. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Esper RJ, Nordaby RA, Vilariño JO,
Paragano A, Cacharron JL and Machado RA: Endothelial dysfunction: A
comprehensive appraisal. Cardiovasc Diabetol. 5:42006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Verma S, Buchanan MR and Anderson TJ:
Endothelial function testing as a biomarker of vascular disease.
Circulation. 108:2054–2059. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Canpolat U, Kocyigit D and Yildirim A:
Role of endothelial dysfunction and endocan in atherosclerosis:
Point of origin or end point? Angiology. 71:4772020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lai WK and Kan MY: Homocysteine-induced
endothelial dysfunction. Ann Nutr Metab. 67:1–12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rius J, Guma M, Schachtrup C, Akassoglou
K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG and Karin M:
NF-kappaB links innate immunity to the hypoxic response through
transcriptional regulation of HIF-1alpha. Nature. 453:807–811.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tak PP and Firestein GS: NF-kappaB: A key
role in inflammatory diseases. J Clin Invest. 107:7–11. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Holgate ST: Cytokine and anti-cytokine
therapy for the treatment of asthma and allergic disease. Cytokine.
28:152–157. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Williams RO, Paleolog E and Feldmann M:
Cytokine inhibitors in rheumatoid arthritis and other autoimmune
diseases. Curr Opin Pharmacol. 7:412–417. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Baldwin AS Jr: The NF-kappa B and I kappa
B proteins: New discoveries and insights. Annu Rev Immunol.
14:649–683. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Du QH, Xu YB, Zhang MY, Yun P and He CY:
Propofol induces apoptosis and increases gemcitabine sensitivity in
pancreatic cancer cells in vitro by inhibition of nuclear factor-κB
activity. World J Gastroenterol. 19:5485–5492. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Peng X, Li C, Yu W, Liu S, Cong Y, Fan G
and Qi S: Propofol attenuates hypoxia-induced inflammation in BV2
microglia by inhibiting oxidative stress and NF-κB/Hif-1α
signaling. Biomed Res Int. 2020:89787042020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang K and Kaufman RJ: From
endoplasmic-reticulum stress to the inflammatory response. Nature.
454:455–462. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shamu CE, Cox JS and Walter P: The
unfolded-protein-response pathway in yeast. Trends Cell Biol.
4:56–60. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cox JS, Shamu CE and Walter P:
Transcriptional induction of genes encoding endoplasmic reticulum
resident proteins requires a transmembrane protein kinase. Cell.
73:1197–1206. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shi Y, Vattem KM, Sood R, An J, Liang J,
Stramm L and Wek RC: Identification and characterization of
pancreatic eukaryotic initiation factor 2 alpha-subunit kinase,
PEK, involved in translational control. Mol Cell Biol.
18:7499–7509. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhu L, Jia F, Wei J, Yu Y, Yu T, Wang Y,
Sun J and Luo G: Salidroside protects against homocysteine-induced
injury in human umbilical vein endothelial cells via the regulation
of endoplasmic reticulum stress. Cardiovasc Ther. 35:33–39. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou X, Wei Y, Qiu S, Xu Y, Zhang T and
Zhang S: Propofol decreases endoplasmic reticulum stress-mediated
apoptosis in retinal pigment epithelial cells. PLoS One.
11:e01575902016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kassan M, Galán M, Partyka M, Saifudeen Z,
Henrion D, Trebak M and Matrougui K: Endoplasmic reticulum stress
is involved in cardiac damage and vascular endothelial dysfunction
in hypertensive mice. Arterioscler Thromb Vasc Biol. 32:1652–1661.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tabas I: The role of endoplasmic reticulum
stress in the progression of atherosclerosis. Circ Res.
107:839–850. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xue X, Piao JH, Nakajima A, Sakon-Komazawa
S, Kojima Y, Mori K, Yagita H, Okumura K, Harding H and Nakano H:
Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein
response (UPR) in a reactive oxygen species (ROS)-dependent
fashion, and the UPR counteracts ROS accumulation by TNFalpha. J
Biol Chem. 280:33917–33925. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sprenkle NT, Sims SG, Sánchez CL and
Meares GP: Endoplasmic reticulum stress and inflammation in the
central nervous system. Mol Neurodegener. 12:422017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu GJ, Lin YW, Chuang CY, Tsai HC and Chen
RM: Liver nitrosation and inflammation in septic rats were
suppressed by propofol via downregulating TLR4/NF-κB-mediated iNOS
and IL-6 gene expressions. Life Sci. 195:25–32. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ding XW, Sun X, Shen XF, Lu Y, Wang JQ,
Sun ZR, Miao CH and Chen JW: Propofol attenuates TNF-α-induced
MMP-9 expression in human cerebral microvascular endothelial cells
by inhibiting Ca2+/CAMK II/ERK/NF-κB signaling pathway.
Acta Pharmacol Sin. 40:1303–1313. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cheng L, Chen Z, Wang L, Lan Y, Zheng L
and Wu F: Propofol partially attenuates complete freund's
adjuvant-induced neuroinflammation through inhibition of the
ERK1/2/NF-κB pathway. J Cell Biochem. 120:9400–9408. 2019.
View Article : Google Scholar : PubMed/NCBI
|