Introduction
Kidney transplantation is the best therapeutic
option for end-stage renal disease. Current available
immunosuppressive regimens have significantly improved the
short-term graft survival, yet graft survival after the first
post-transplant year has not been considerably prolonged. The
leading cause of graft loss after the first post-transplant year is
antibody-mediated rejection (ABMR), a condition without an
available effective treatment (1,2). Thus,
delineating the molecular mechanisms involved in ABMR, as well as
possible therapeutic maneuvers, is of paramount importance.
ABMR results from the de novo donor-specific
antibodies (DSA), which are specific mainly, but not exclusively,
against the human leukocyte antigens class I (HLAI) and/or class II
(HLAII) of the graft (3). Renal
endothelial cells display HLAI on their surface, and upon
activation, also upregulate HLAII expression (4). The graft endothelium is at the
forefront of the kidney transplant against the assault from the
recipient's adaptive humoral immune system and not surprisingly a
target of the latter. The effector mechanisms of DSA-mediated graft
injury include activation of the classical complement pathway,
antibody-dependent natural killer (NK) cell cytotoxicity, monocyte
cytotoxicity facilitated by antibody-FcγR binding, and not
uncommonly T-cells are also implicated, resulting in mixed
antibody- and cell-mediated rejection (5,6). In
the case of active humoral rejection, which has a worse prognosis
than acute cellular rejection, the presence of neutrophils is a
significant finding in the graft biopsies, and intra-capillary
thrombosis is also common (5,6).
The mammalian target of rapamycin (mTOR) complex I
(mTORC1) inhibitors rapamycin and everolimus are used as
immunosuppressants for kidney transplantation. Studies have shown
that these inhibitors may interfere with anti-HLA-induced
endothelial cell alterations, modifying the graft to be less
vulnerable to antibody-mediated injury (7–9). In a
model of heart ABMR, an immunosuppressive regimen containing
everolimus proved superior to an immunosuppressive regimen
containing mycophenolate (10).
mTORC1 is activated when there is a sufficient quantity of
nutrients, such as certain amino acids, and trophic factors,
promoting protein translation and cell proliferation (11,12).
Another sensor of nutrients is the general control
nonderepressible 2 kinase (GCN2K). In case of shortage of an
amino-acid, its specific tRNA remains unloaded. Unloaded tRNA
induces a conformation change in GCN2K, which is autophosphorylated
and activated. Then, GCN2K phosphorylates the eukaryotic initiator
factor 2α (eIF2α), suppressing the general translational program of
the cell and selectively enhancing the translation of proteins for
adaptation to stress (13).
Although activation of GCN2K suppresses adaptive immunity (14–16),
activators of this kinase are not still widely used as
immunosuppressants. Halofuginone, a veterinary drug against
coccidioidomycosis, which activates GCN2K by inhibiting
prolyl-tRNA synthetase, has been approved as an orphan drug for the
treatment of scleroderma (17–19).
However, the drug has not yet gained proper recognition as a
general immunosuppressant.
This study aimed to evaluate the effect of anti-HLAI
antibodies on the integrity and immunological relevant parameters
in primary human glomerular endothelial cells, and whether mTOR
inhibition or GCN2K activation may modify any anti-HLAI-induced
alterations.
Materials and methods
Cell culture conditions
Primary human glomerular endothelial cells were
purchased from Sciencell Research Laboratories. The culture medium
was Dulbecco's modified Eagle's medium (DMEM) low glucose (5.55 mM)
(Thermo Fisher Scientific, Inc.) supplemented with 20% fetal bovine
serum (Sigma-Aldrich; Merck Millipore) and antibiotic-antimycotic
solution (Sigma-Aldrich; Merck Millipore). The above primary cells
are differentiated, well-characterized passage one glomerular
endothelial cells. We expanded them in 75 cm2 flasks,
and passage two cells were used for the experiments. The cells were
cultured at 37°C in a humidified atmosphere with 5% CO2,
and cells were seeded in 6-well plates (3×105 cells per
well), 24-well plates (1×105 cells per well), or 96-well
plates (1×104 cells per well).
Cells remained untreated or treated with anti-HLAI
antibodies (Ultra-LEAF™ Purified anti-human HLA-A,B,C Antibody,
cat. no 311428, Biolegend) at a concentration of 1 µg/ml.
Anti-HLAI-treated cells were cultured in the presence or not of 10
ng/ml of the mTORC1 inhibitor everolimus (Selleck Chemicals) or 20
nM of the GCN2K activator halofuginone (Cayman Chemical) or their
combination.
The concentration of 1 µg/ml anti-HLAI was based on
the concentration administered in previous studies (7–9,20).
However, since some studies also used the concentration of 10 µg/ml
(20–22), we performed preliminary experiments
to evaluate which concentration affects mTOR and GCN2K. Western
blotting detection of phosphorylated activated mTOR and GCN2K and
their substrates' phosphorylation revealed that both concentrations
strongly activate both pathways. Thus, we continued the experiments
with a concentration of 1 µg/ml. The 10 ng/ml concentration of
everolimus was selected because it falls within this drug's
therapeutic levels (23). We also
performed preliminary experiments. In brief, glomerular endothelial
cells were cultured for 48 h in 96-well plates at a number of
10,000 cells per well with or without 10 ng/ml everolimus. Lactate
dehydrogenase (LDH) release assay revealed that the above
concentration of everolimus is not toxic for glomerular endothelial
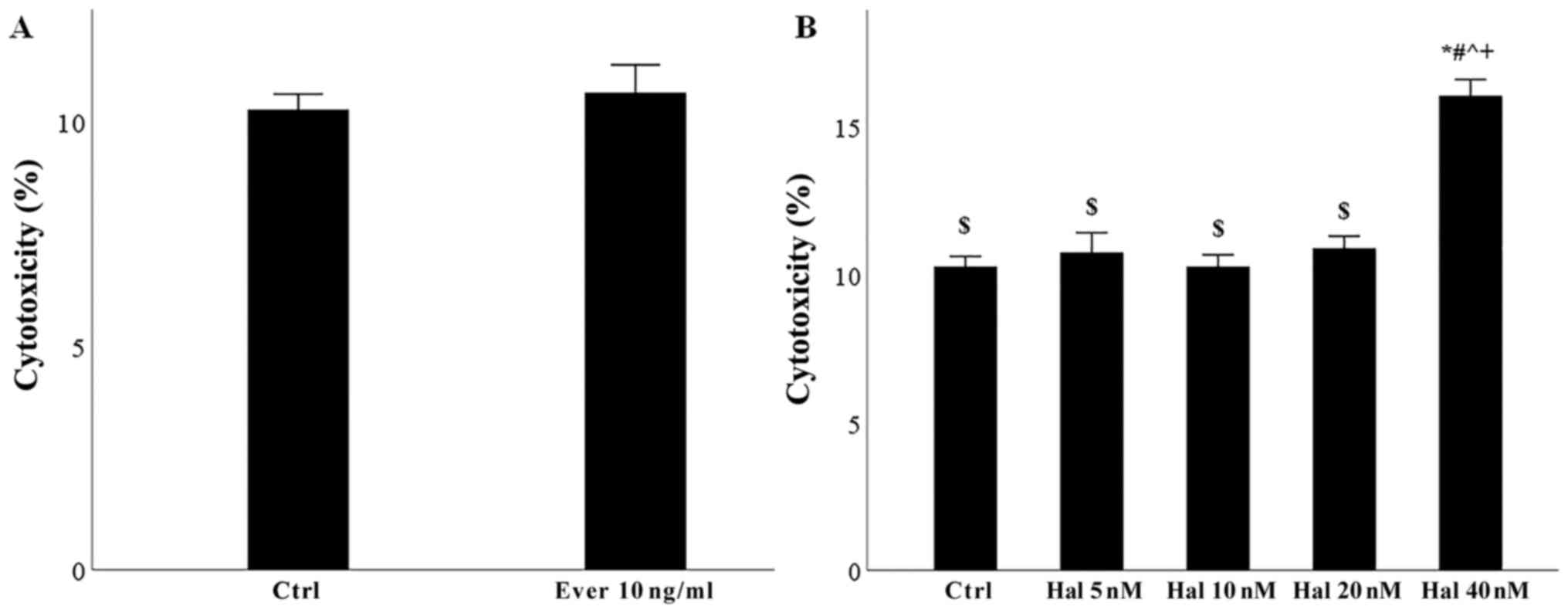
cells (Fig. 1A). Finally, since
there was no available clinical data for halofuginone, we performed
preliminary experiments culturing glomerular endothelial cells for
48 h in 96-well plates at a number of 10,000 cells per well with or
without 5, 10, 20, or 40 nM halofuginone. LDH release assay showed
that the concentration of 20 nM was the higher nontoxic
concentration for glomerular endothelial cells (Fig. 1B).
The assessment of cellular proteins involved in the
evaluated signal transduction pathways or immunological processes
was performed after 12 h of cell culture. Cytotoxicity of the
reagents, cell proliferation, and measurement of certain factors
secreted in cell culture supernatants were carried out after 48 h
of culture.
Assessment of cell necrosis and
proliferation
For evaluating the cytotoxicity of the reagents and
cell proliferation, glomerular endothelial cells were cultured in
96-well plates for 48 h under the aforementioned conditions. Six
independent experiments were performed, each one in
triplicates.
The cytotoxicity of the reagents was assessed by
evaluating cell necrosis with the LDH release assay (Cytotox
Non-Radioactive Cytotoxic Assay kit, Promega Corporation).
Cytotoxicity was calculated according to the equation Cytotoxicity
(%) = (LDH in the supernatant: Total LDH) ×100.
Bromodeoxyuridine (BrdU) labeling and
immunoenzymatic detection were used to evaluate cell proliferation
(Cell Proliferation ELISA, Roche Diagnostics). The proliferation
index was calculated according to the equation Proliferation index
= optical density of the treated cells: Optical density of the
control cells.
Assessment of cellular proteins of
interest
For assessing the level of various cellular
proteins, glomerular endothelial cells were cultured in 6-well
plates for 12 h under the aforementioned experimental conditions.
Three independent experiments were performed.
Cell proteins were extracted with the T-PER tissue
protein extraction reagent (Thermo Fisher Scientific, Inc.)
supplemented with protease and phosphatase inhibitors
(Sigma-Aldrich; Merck Millipore) and measured with Bradford Assay
(Sigma-Aldrich; Merck Millipore). For the western blotting, 10 µg
from each sample were electrophoresed in sodium dodecyl sulfate
(SDS) polyacrylamide (4–12% Bis-Tris gels, Thermo Fisher
Scientific, Inc.) and transferred on polyvinylidene fluoride (PVDF)
membranes (Thermo Fisher Scientific, Inc.).
Blots were incubated at 4°C for 16 h with the
primary antibodies specific against activated cleaved caspase-3
(cleaved caspase-3, 1:1,000, cat. no ab13847, Abcam), focal
adhesion kinase (FAK, 1:100, cat. no sc-271126, Santa Cruz
Biotechnology, Inc.), phosphorylated at Tyr397 FAK (p-FAK, 1:1,000,
cat. no 8556, Cell Signaling Technology, Inc.), mTOR (1:100, cat.
no sc-517464, Santa Cruz Biotechnology, Inc.), phosphorylated at
Ser2448 mTOR (p-mTOR, 1:100, cat. no sc-293133, Santa Cruz
Biotechnology, Inc.), p70S6 kinase (p70S6K, 1:100, cat. no sc-8418,
Santa Cruz Biotechnology, Inc.), phosphorylated at Thr389 p70S6K
(p-p70S6K, 1:1,000, cat. no 9234, Cell Signaling Technology),
protein kinase B (Akt, 1:100, cat. no sc-5298, Santa Cruz
Biotechnology, Inc.), phosphorylated at Ser474 Akt (p-Akt, 1:1,000,
cat. no 4060, Cell Signaling Technology, Inc.), GCN2 kinase (GCN2K,
1:100, cat. no sc-374609, Santa Cruz Biotechnology, Inc.),
phosphorylated at Thr899 GCN2K (p-GCN2K, 1:1,000, cat. no ab75836;
Abcam), eIF2α (1:100, cat. no sc-133132, Cell Signaling Technology,
Inc.), phosphorylated at Ser51 eIF2α (p-eIF2a, 1:1,000, cat. no
9721, Cell Signaling Technology, Inc.), intercellular adhesion
molecule 1 (ICAM-1, 1:1,000, cat. no 4915; Cell Signaling
Technology), HLA-DR (Ultra-LEAF™ Purified anti-human HLA-DR
Antibody, cat. no 307648, Biolegend), CD46 (1:1,000, cat. no
CSB-PA923298, Cusabio), CD59 (1:1,000, cat. no CSB-PA004947YA01HU,
Cusabio), and β-actin (1:5,000, cat no. 4967, Cell Signaling
Technology, Inc.). Anti-rabbit IgG, HRP-linked (1:1,000; cat. no.
7074; Cell Signaling Technology, Inc.) or anti-mouse IgG,
HRP-linked (1:1,000; cat. no. 7076; Cell Signaling Technology,
Inc.) antibodies were used as secondary antibodies and applied for
30 min at room temperature. The Restore Western Blot Stripping
Buffer (Thermo Fisher Scientific, Inc.) was used for reprobing the
PVDF blots.
Bands were visualized with the LumiSensor Plus
Chemiluminescent HRP substrate kit (GenScript Corporation), and the
ImageJ 1.51t software (National Institute of Health) was used for
the optical density (OD) measurement of the bands.
Assessment of IL-8, MCP-1, TGF-β1, and
vWF
Interleukin-8 (IL-8), monocyte chemoattractant
protein 1 (MCP-1), transforming growth factor-beta 1 (TGF-β1), and
von Willebrand factor (vWF) concentrations were measured in cell
culture supernatants of glomerular endothelial cells cultured in
24-well plates for 48 h under the aforementioned experimental
conditions. Six independent experiments were performed.
The measurements were obtained through enzyme-linked
immunosorbent assay (ELISA) using the Interleukin-8 Human ELISA kit
(Bender Medsystems), the 1/MCAF ELISA kit for MCP-1 (Cusabio), the
Human TGF-beta1 ELISA kit (AssayPro), and the Human von Willebrand
Factor ELISA kit (Cusabio).
Statistical analysis
Statistical analysis was performed with the IMB SPSS
Statistics v.20 (IBM Corp.). For comparison of means, one-way
analysis of variance (ANOVA) and Bonferroni's correction test were
used. Results were presented as mean ± standard error of mean
(SEM), and statistical significance was set at a P<0.05. Except
for the western blotting that evaluated phosphorylated protein OD
to total protein OD ratio, for readers' convenience, all the other
western blotting results were depicted after normalization of means
for the control group.
Results
The effect of anti-HLAI antibodies on
cell necrosis, apoptosis and proliferation and the impact of
halofuginone or everolimus
Neither anti-HLAI antibodies nor their combination
with halofuginone or everolimus was cytotoxic. Cytotoxicity was
11.33±0.11, 12.17±0.38, 10.67±0.64, 11.50±0.37, and 12.17±0.21% in
control, anti-HLAI-treated cells, anti-HLAI-treated cells with
halofuginone, anti-HLAI-treated cells with everolimus, and
anti-HLAI-treated cells with halofuginone and everolimus,
respectively (p n.s.) (Fig.
2A).
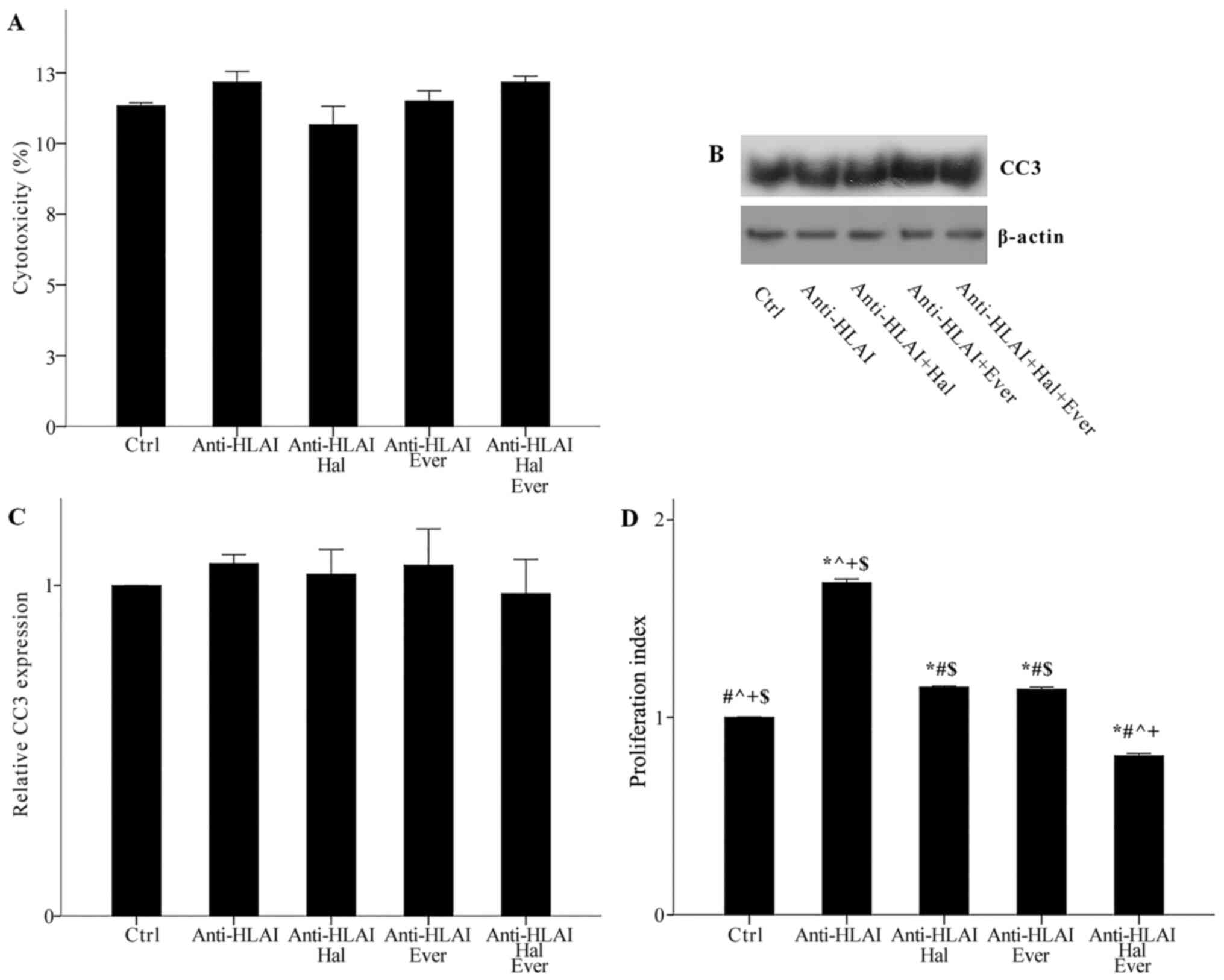 | Figure 2.Effect of anti-HLAI on cell necrosis,
apoptosis and proliferation, and the impact of halofuginone or
everolimus treatment. (A) Neither anti-HLAI antibodies nor their
combination with halofuginone or everolimus alone or in combination
induced cell necrosis. The same was confirmed for apoptosis
assessed by the level of activated cleaved caspase-3. (B) Panel B
depicts a representative experiment, while the (C) cumulative
results are presented in panel C. (D) Anti-HLAI antibodies promoted
cell proliferation, while halofuginone or everolimus inhibited
anti-HLAI-induced cell proliferation. Data are presented as the
mean ± SEM. *P<0.05 vs. control cells, #P<0.05 vs.
anti-HLAI-treated cells, ^P<0.05 vs.
anti-HLAI-treated cells administered halofuginone,
+P<0.05 vs. anti-HLAI-treated cells administered
everolimus and $P<0.05 vs. anti-HLAI-treated cells
with halofuginone and everolimus. HLAI, human leukocyte antigen
class I; Hal, halofuginone; Ever, everolimus; Ctrl, control; CC3,
cleaved caspase-3. |
Neither anti-HLAI antibodies nor their combination
with halofuginone or everolimus induced apoptosis, as it was
assessed by the level of activated cleaved caspase-3. In
anti-HLAI-treated cells, anti-HLAI-treated cells with halofuginone,
anti-HLAI-treated cells with everolimus, and anti-HLAI-treated
cells with halofuginone and everolimus cleaved caspase-3 level was
1.07±0.03, 1.03±0.07, 1.06±0.11, and 0.97±0.10 fold as found in
control cells, respectively (p n.s.) (Fig. 2B and C).
Anti-HLAI antibodies promoted cell proliferation,
whereas both halofuginone and everolimus inhibited
anti-HLAI-induced cell proliferation. In the anti-HLAI-treated
cells proliferation index was 1.68±0.02 (P<0.001, compared to
the control). Halofuginone decreased proliferation index to
1.15±0.01 (P<0.001, compared to anti-HLAI-treated cells).
Everolimus reduced proliferation index to 1.14±0.01 (P<0.001,
compared to anti-HLAI-treated cells). The combination of
halofuginone and everolimus had an even greater effect on
anti-HLAI-induced cell proliferation since, in this case, the
proliferation index was 0.80±0.01 (P<0.001 compared to control
cells, anti-HLAI-treated cells, anti-HLAI-treated cells with
halofuginone, and anti-HLAI-treated cells with everolimus (Fig. 2D).
The effect of anti-HLAI antibodies on
integrin, mTOR, and GCN2K signal transduction and the impact of
halofuginone or everolimus
The integrin, mTOR, and GCN2K signal transduction
pathways were assessed by the level of phosphorylation of proteins
that are phosphorylated once the above pathways are activated. For
this purpose, the ratio of phosphorylated to total protein was
assessed.
Anti-HLAI antibodies trigger integrin signaling as
it was assessed by the p-FAK to total FAK ratio. Compared to the
control cells, anti-HLAI antibodies increased p-FAK (P<0.001).
Halofuginone did not affect anti-HLAI-induced FAK phosphorylation
(p n.s. compared to the anti-HLAI-treated cells). On the contrary,
everolimus prevented anti-HLAI-induced FAK phosphorylation (p n.s.
compared to the control, and P<0.001 compared to the
anti-HLAI-treated cells). The combination of everolimus with
halofuginone did not alter the p-FAK level more than everolimus
alone (p n.s. compared to the control and anti-HLAI-treated cells
with everolimus) (Fig. 3A and
B).
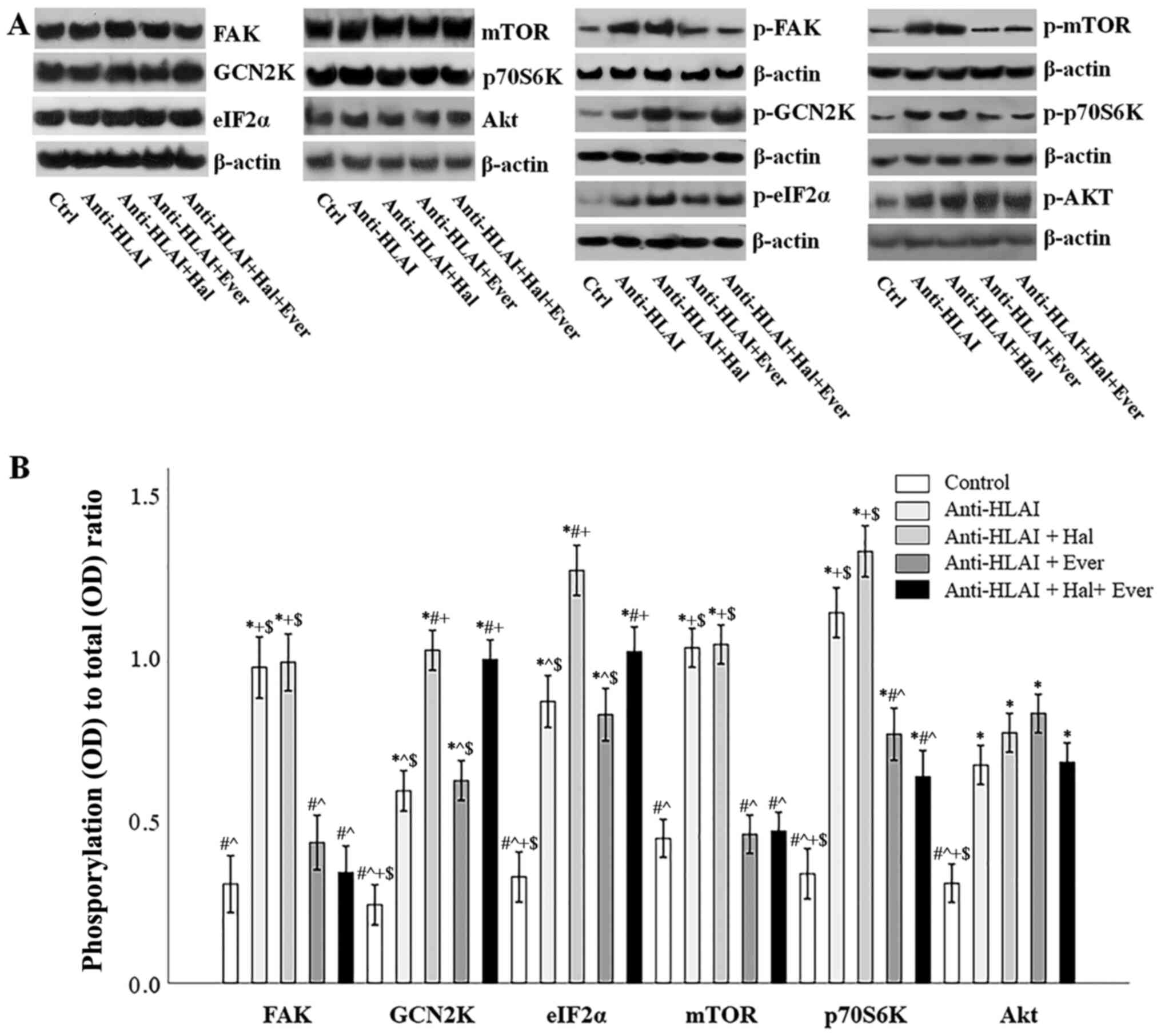 | Figure 3.Effect of anti-HLAI on integrins,
mTOR and GCN2K signal transduction and the impact of halofuginone
or everolimus. (A) Representative experiment for the expression of
total and phosphorylated FAK, GCN2K, eIF2α, mTOR, p70S6K and AKT.
(B) Cumulative results depict the phosphorylated to total protein
ratio and revealed that anti-HLAI antibodies increased p-FAK,
p-GCN2K, p-eIF2α, p-mTOR, p-p70S6K and p-AKT levels. Halofuginone
increased p-GCN2K and p-eIF2α. Everolimus treatment decreased the
anti-HLAI antibody-induced upregulation of p-FAK, p-mTOR and
p-p70S6K. Data are presented as the mean ± SEM. *P<0.05 vs.
control cells, #P<0.05 vs. anti-HLAI-treated cells,
^P<0.05 vs. anti-HLAI-treated cells administered
halofuginone, +P<0.05 vs. anti-HLAI-treated cells
administered everolimus and $P<0.05 vs.
anti-HLAI-treated cells with halofuginone and everolimus. HLAI,
human leukocyte antigen class I; mTOR, mammalian target of
rapamycin; GCN2K, general control nonderepressible 2 kinase; FAK,
focal adhesion kinase; eIF2α, eukaryotic initiator factor 2α; p,
phosphorylated; Hal, halofuginone; Ever, everolimus; Ctrl, control;
OD, optical density. |
Anti-HLAI antibodies induced GCN2K activation
assessed by the ratio of p-GCN2K to total GCN2K ratio. Compared to
the control cells, anti-HLAI antibodies increased p-GCN2K
(P<0.001). Halofuginone increased anti-HLAI-induced GCN2K
phosphorylation (P<0.001 compared to the anti-HLAI-treated
cells). On the contrary, everolimus did not affect
anti-HLAI-induced GCN2K phosphorylation (P<0.001 compared to the
control, and p n.s. compared to the anti-HLAI-treated cells). The
combination of halofuginone with everolimus did not alter the
p-GCN2K level more than halofuginone alone (P<0.001 compared to
the control, and p n.s. compared to anti-HLAI-treated cells, and
compared to anti-HLAI-treated cells with halofuginone) (Fig. 3A and B).
The anti-HLAI-induced activation of GCK2K was also
confirmed by the ratio of p-eIF2α to total eIF2α. Compared to the
control cells, anti-HLAI antibodies increased p-eIF2α (P<0.001).
Halofuginone increased anti-HLAI-induced eIF2α phosphorylation even
higher (P<0.001 compared to the anti-HLAI-treated cells). On the
contrary, everolimus did not affect anti-HLAI-induced eIF2α
phosphorylation (P<0.001 compared to the control, and p n.s.
compared to the anti-HLAI-treated cells). The combination of
halofuginone with everolimus did not alter the p-eIF2α level more
than halofuginone alone (P<0.001 compared to the control, and
anti-HLAI-treated cells, and p n.s. compared to anti-HLAI-treated
cells with halofuginone) (Fig. 3A and
B).
Anti-HLAI antibodies induced mTOR activation
assessed by the ratio of p-mTOR to total mTOR. Compared to the
control cells, anti-HLAI antibodies increased p-mTOR (P<0.001).
Halofuginone did not affect anti-HLAI-induced mTOR phosphorylation
(p n.s. compared to the anti-HLAI-treated cells). On the contrary,
everolimus prevented anti-HLAI-induced mTOR phosphorylation (p n.s.
compared to the control, and P<0.001 compared to the
anti-HLAI-treated cells). The combination of everolimus with
halofuginone did not alter the p-mTOR level more than everolimus
alone (p n.s. compared to the control and anti-HLAI-treated cells
with everolimus, and P<0.001 compared to anti-HLAI-treated
cells) (Fig. 3A and B).
Anti-HLAI-induced mTORC1 activation was also
confirmed by the p-p70S6K to total p70S6K ratio. Compared to the
control cells, anti-HLAI antibodies increased p-p70S6K
(P<0.001). Halofuginone did not affect anti-HLAI-induced p70S6K
phosphorylation (p n.s. compared to the anti-HLAI-treated cells).
On the contrary, everolimus reduced anti-HLAI-induced p70S6K
phosphorylation (P<0.001 compared to the control, and P<0.001
compared to the anti-HLAI-treated cells). The combination of
everolimus with halofuginone did not alter the p-p70S6K level more
than everolimus alone (P<0.001 compared to the control,
P<0.001 compared to anti-HLAI-treated cells and p n.s. or
anti-HLAI-treated cells with everolimus) (Fig. 3A and B).
Anti-HLAI antibodies activated mTORC2 assessed by
the p-Akt to total Akt ratio. Compared to the control cells,
anti-HLAI antibodies increased p-Akt (P<0.001). Halofuginone,
everolimus, or their combination did not affect anti-HLAI-induced
Akt phosphorylation since (p n.s compared to anti-HLAI-treated in
all cases) (Fig. 3A and B).
The effect of anti-HLAI antibodies on
ICAM-1, HLA-DR, CD46, and CD59 and the impact of halofuginone or
everolimus
Anti-HLAI antibodies upregulated the expression of
ICAM-1 to 2.91±0.13 times the control (P<0.001). Both
halofuginone and everolimus, as well as their combination,
inhibited anti-HLAI-induced ICAM-1 overexpression equally. The
ICAM-1 level was 1.55±0.18, 1.66±0.05, and 1.85±0.21 times the
control in anti-HLAI-treated cells with halofuginone,
anti-HLAI-treated cells with everolimus, and anti-HLAI-treated
cells with halofuginone and everolimus, respectively (P<0.001
compared to the control cells and also P<0.001 compared to
anti-HLAI-treated cells in all cases) (Fig. 4A and B).
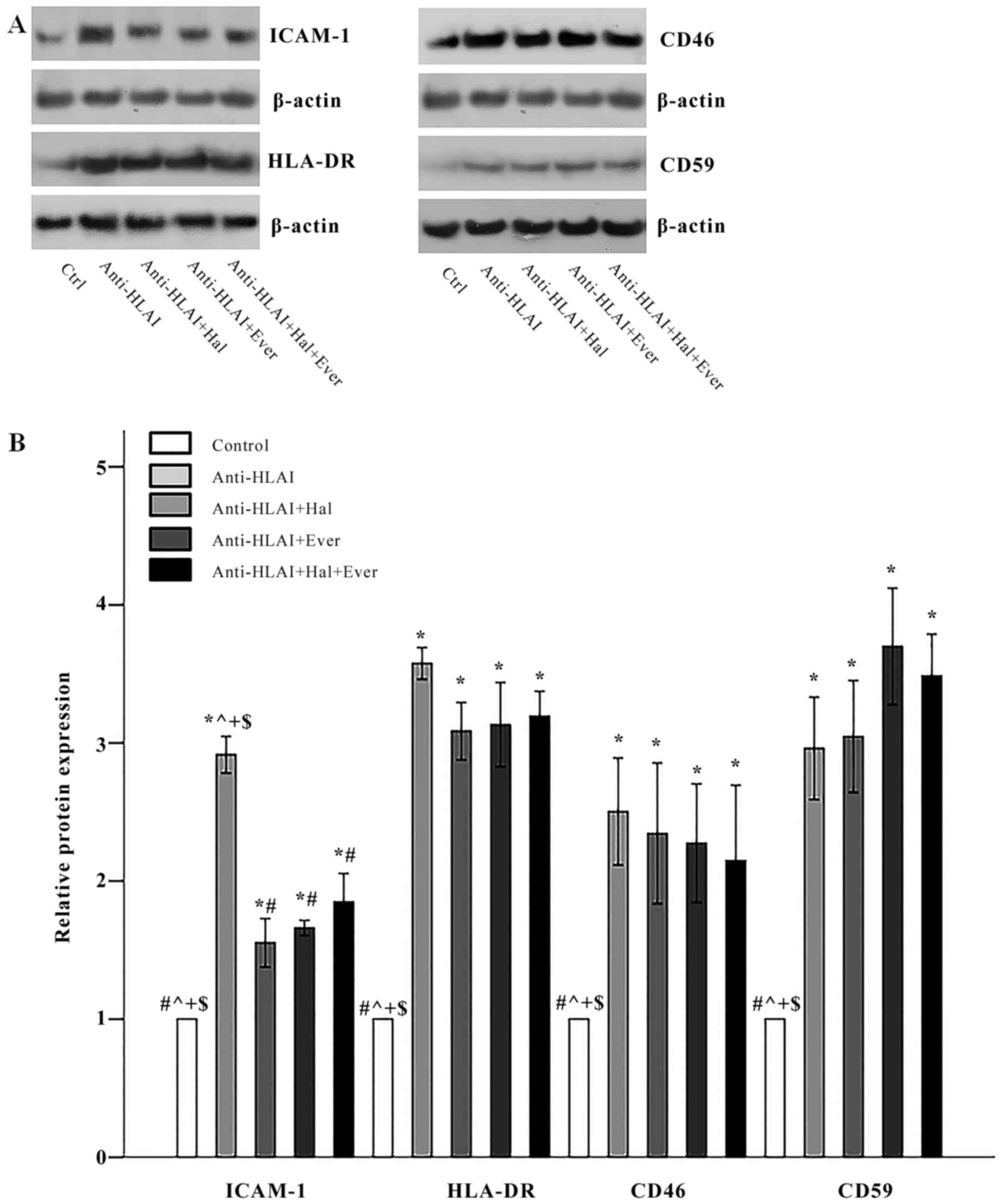 | Figure 4.Effect of anti-HLAI on ICAM-1,
HLA-DR, CD46 and CD59, and the impact of halofuginone or everolimus
treatment. (A) Representative experiment for each of the evaluated
factors. (B) Cumulative results are presented. Anti-HLAI antibodies
upregulated ICAM-1, HLA-DR, CD46 and CD59. Halofuginone or
everolimus treatment decreased ICAM-1. Data are presented as the
mean ± SEM. *P<0.05 vs. control cells, #P<0.05 vs.
anti-HLAI-treated cells, ^P<0.05 vs.
anti-HLAI-treated cells administered halofuginone,
+P<0.05 vs. anti-HLAI-treated cells administered
everolimus and $P<0.05 vs. anti-HLAI-treated cells
with halofuginone and everolimus. HLAI, human leukocyte antigen
class I; ICAM-1, intracellular adhesion molecule-1; Hal,
halofuginone; Ever, everolimus; Ctrl, control. |
Anti-HLAI antibodies elevated HLA-DR expression to
3.58±0.11 times the control (P<0.001). Halofuginone, everolimus,
or their combination did not affect anti-HLAI-induced upregulation
of HLA-DR since the HLA-DR level was 3.08±0.21, 3.13±0.31, and
3.19±0.18 times the control, respectively (P<0.001 compared to
the control cells, and p n.s. compared to anti-HLAI-treated cells)
(Fig. 4A and B).
Anti-HLAI antibodies increased CD46 expression to
2.50±0.39 times the control (P<0.001). Halofuginone, everolimus,
or their combination did not alter anti-HLAI-induced CD46
overexpression since the CD46 level was 2.35±0.51, 2.27±0.43, and
2.15±0.55 times the control, respectively (P<0.001 compared to
the control cells, and p n.s. compared to anti-HLAI-treated cells)
(Fig. 4A and B).
Anti-HLAI antibodies enhanced CD59 expression to
2.96±0.37 times the control (P<0.001). Halofuginone, everolimus,
or their combination did not change anti-HLAI-induced CD59
overexpression since CD59 level was 3.05±0.41, 3.70±0.42, and
3.49±0.30 times to the control, respectively (P<0.001 compared
to the control cells, and p n.s. compared to anti-HLAI-treated
cells) (Fig. 4A and B).
The effect of anti-HLAI antibodies on
IL-8, MCP-1, TGF-β1, and vWF and the impact of halofuginone or
everolimus
Anti-HLAI antibodies raised IL-8 concentration from
13.3±0.4 to 536.0±37.6 pg/ml (P<0.001). Halofuginone decreased
anti-HLAI-induced IL-8 increase to 291.3±17.8 pg/ml (P<0.001
compared to the control, P<0.001 compared to anti-HLAI-treated
cells). Everolimus proved a more potent inhibitor of IL-8
production than halofuginone since, in this case, IL-8 was
187.4±13.1 pg/ml (P<0.001 compared to the control, and
anti-HLAI-treated cells, and P<0.05 compared to
anti-HLAI-treated cells with halofuginone). The combination of
halofuginone and everolimus decreased IL-8 concentration further to
76.3±9.7 pg/ml (P<0.05 compared to the control and P<0.001
compared to anti-HLAI-treated cells with halofuginone and P<0.05
compared to anti-HLAI-treated cells with everolimus) (Fig. 5A).
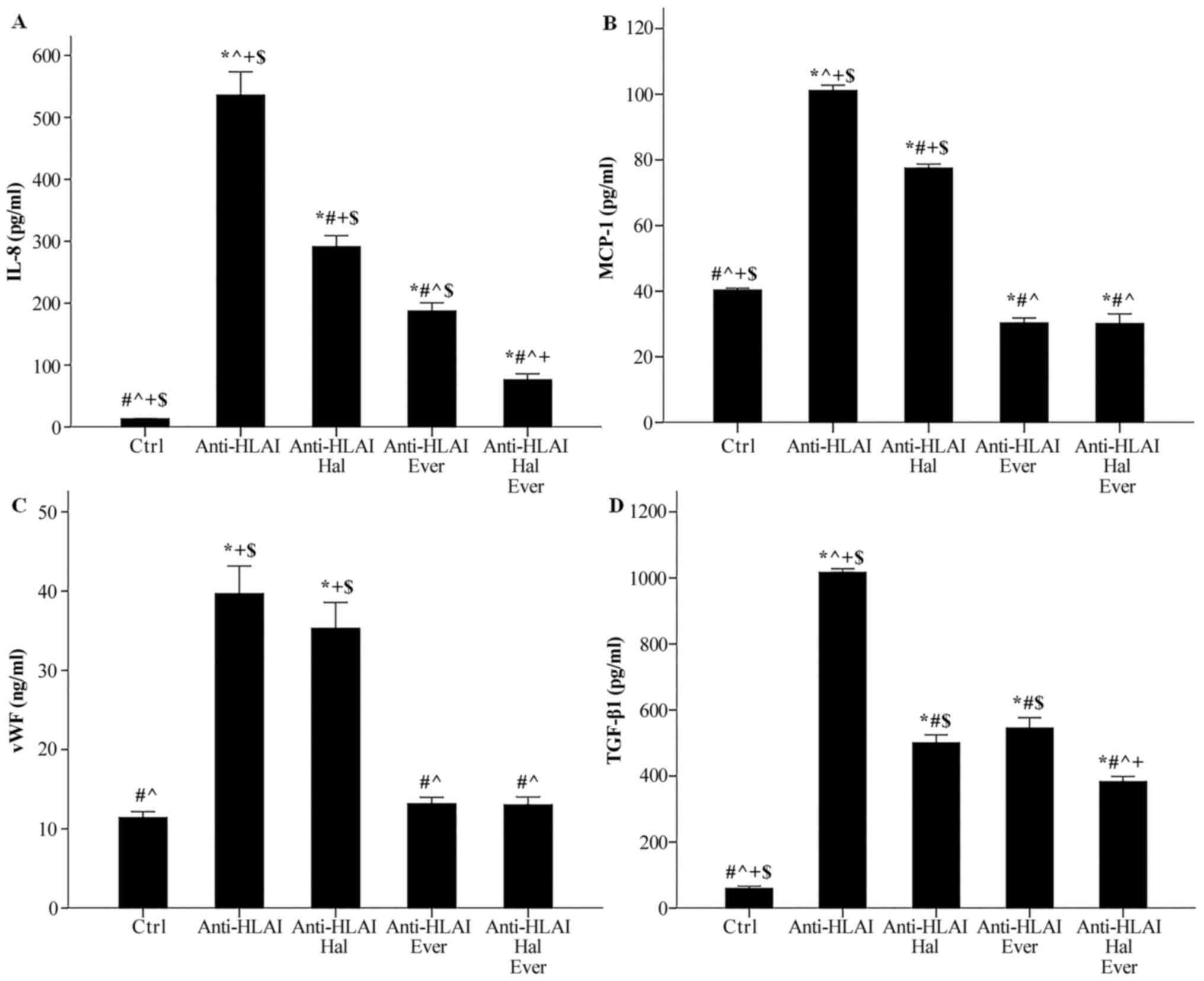 | Figure 5.Effect of anti-HLAI on IL-8, MCP-1,
vWF and anti-TGF-β1, and the impact of halofuginone or everolimus
treatment. (A) Anti-HLAI antibodies increased IL-8. Halofuginone or
everolimus decreased anti-HLAI-induced IL-8 upregulation. (B)
Anti-HLAI antibodies enhanced MCP-1. Halofuginone or everolimus
reduced anti-HLAI-induced MCP-1 production. (C) Anti-HLAI
antibodies increased vWF. Halofuginone did not affect vWF, while
everolimus prevented anti-HLAI-induced vWF upregulation. (D)
Anti-HLAI antibodies raised the TGF-β1 level. Halofuginone or
everolimus suppressed anti-HLAI-induced TGF-β1 production. Data are
presented as the mean ± SEM. The superscripts *P<0.05 vs.
control cells, #P<0.05 vs. anti-HLAI-treated cells,
^P<0.05 vs. anti-HLAI-treated cells administered
halofuginone, +P<0.05 vs. anti-HLAI-treated cells
administered everolimus and $P<0.05 vs.
anti-HLAI-treated cells with halofuginone and everolimus. HLAI,
human leukocyte antigen class I; MCP-1, monocyte chemoattractive
protein-1; vWF, von Willebrand factor; Hal, halofuginone; Ever,
everolimus; Ctrl, control. |
Anti-HLAI antibodies enhanced MCP-1 concentration
from 40.4±0.6 to 101.2±1.6 pg/ml (P<0.001). Halofuginone reduced
anti-HLAI-induced MCP-1 augmentation to 77.5±1.3 pg/ml (P<0.001
compared to the control, and P<0.001 compared to
anti-HLAI-treated cells). Everolimus decreased the MCP-1 level
further. In this case, MCP-1 concentration was 30.3±1.5 pg/ml
(P<0.05 compared to the control, and P<0.001 compared to
anti-HLAI-treated cells with halofuginone). The combination of
halofuginone and everolimus also decreased MCP-1 concentration to
30.2±2.9 pg/ml, that is no more than the inhibition induced by
everolimus alone (P<0.05 compared to the control, P<0.001
compared to anti-HLAI-treated cells with halofuginone and p n.s
compared to anti-HLAI-treated cells with everolimus) (Fig. 5B).
Anti-HLAI antibodies increased vWF concentration in
the cell culture supernatant from 11.4±0.7 to 39.7±3.5 ng/ml
(P<0.001). Halofuginone did not affect anti-HLAI-induced vWF
production since, in this case, the vWF concentration was 35.3±3.2
ng/ml (P<0.001 compared to the control and p n.s. compared to
anti-HLAI-treated cells). Everolimus inhibited anti-HLAI-induced
vWF enhancement to 13.2±0.8 ng/ml (p n.s. compared to the control
and P<0.001 compared to anti-HLAI-treated cells). The
combination of halofuginone and everolimus also reduced vWF
concentration, but no more than everolimus alone. In this case, the
level of vWF in cell culture supernatants was 13.0±1.0 ng/ml (p
n.s. compared to the control, P<0.001 compared to
anti-HLAI-treated cells, P<0.001 compared to anti-HLAI-treated
cells with halofuginone, and p n.s compared to anti-HLAI-treated
cells with everolimus) (Fig.
5C).
Anti-HLAI antibodies upregulated TGF-β1
concentration from 59.7±6.8 to 1016.3±11.2 pg/ml (P<0.001).
Halofuginone reduced anti-HLAI-induced TGF-β1 increase to
500.7±23.9 pg/ml (P<0.001 compared to the control and P<0.001
compared to anti-HLAI-treated cells). Everolimus decreased the
TGF-β1 level to the same extent as halofuginone did. In this case,
the TGF-β1 concentration was 545.0±31.6 pg/ml (P<0.001 compared
to the control, P<0.001 compared to anti-HLAI-treated cells, and
p n.s. compared to anti-HLAI-treated cells with halofuginone). The
combination of halofuginone and everolimus also decreased TGF-β1
concentration further to 383.0±15.6 pg/ml (P<0.001 compared to
the control, P<0.001 compared to anti-HLAI-treated cells,
P<0.05 compared to anti-HLAI-treated cells with halofuginone,
and P<0.001 compared to anti-HLAI-treated cells with everolimus)
(Fig. 5D).
Discussion
Graft endothelial cells are at the forefront of the
kidney transplant against the attack from the recipient's immune
system. This study evaluated the effect of anti-HLAI antibodies on
immunological relevant properties of human glomerular endothelial
cells, as well as their modification by mTOR inhibition or GCN2
kinase activation.
First, we examined whether the anti-HLAI antibodies
alone or in combination with the mTORC1 inhibitor everolimus or the
GCN2K activator halofuginone are cytotoxic or may affect cell
proliferation. None of the above reagents induced cell necrosis or
cell apoptosis as assessed by LDH release assay or by the level of
activated cleaved caspase-3, in which all the apoptotic pathways
converge (24), respectively. The
aim of our study was not to evaluate the effect of halofuginone or
everolimus on resting glomerular endothelial cells, but whether
they can modify the anti-HLAI-induced alterations. Thus, any impact
of the above substances on unstimulated cells was not assessed.
Moreover, no changes in cellular integrity were monitored when
these substances were co-administered with anti-HLAI antibodies,
allowing us to continue on our experimental approach. In accordance
with previous studies, anti-HLAI antibodies induce cell
proliferation (7,8,22,25).
Everolimus, halofuginone, or their combination prevented
anti-HLAI-induced cell proliferation. The latter is expected since
activated mTORC1 and inactivated GCN2K promote the necessary for
cell proliferation protein synthesis (11–13).
Next, we evaluated whether the above reagents exert
the expected signal transduction effects in glomerular endothelial
cells. Anti-HLAI antibodies induce integrin clustering and
autophosphorylation of FAK at Tyr397. Besides FAK activation, the
above phosphorylation turns FAK into a docking site for sarcoma
(Src) family kinases, and eventually to mTORC1 activation and cell
proliferation (7,22,25).
Our experiments confirmed the anti-HLAI-induced FAK
phosphorylation. Halofuginone did not affect FAK phosphorylation,
while everolimus decreased it significantly, indicating that
inhibition of mTORC1 suppresses the anti-HLAI-derived signal
transduction at a very early point; and consequently, may have a
considerable therapeutic potential. Albeit in different
experimental concepts, previous studies also detected that both
everolimus and rapamycin downregulate phosphorylation of FAK at
Tyr397 (26–28). Upon integrin activation and F-actin
polymerization, activated mTOR is recruited to the F-actin polymers
and induce further phosphorylation of FAK Tyr residues (29). The latter should be accomplished
indirectly through mTOR-mediated Src kinase activation since mTOR
is a serine/threonine and not a tyrosine protein kinase (11,12).
Thus, in our experiments, everolimus seems to break a positive
feedback loop consisting of anti-HLAI-induced integrin activation,
FAK phosphorylation, mTOR activation, and further mTOR-mediated FAK
phosphorylation.
Anti-HLAI antibodies induce mTORC1 activation in
endothelial cells (7–9). In its turn, activated phosphorylated
at Ser2448 mTORC1 phosphorylates other targets such as p70S6K and
4E-BP1, and promotes the necessary for cell proliferation protein
synthesis (11,12). In our experiments, anti-HLAI
antibodies induced mTORC1 activation assessed by the level of its
phosphorylation and by the level of phosphorylation of the mTORC1
target p70S6K. As expected, everolimus prevented anti-HLAI-induced
mTORC1 phosphorylation, as well as the phosphorylation of the
mTORC1 target p70S6K. On the contrary, halofuginone did not affect
the mTORC1 pathway.
According to previous studies, in endothelial cells,
besides mTORC1, anti-HLAI antibodies activate mTORC2, too (7,8).
Interestingly, although Akt activation is upstream of mTORC1
activation, it is also downstream of mTORC2 activation (12). Activated mTORC2 phosphorylates Akt
at Ser473, activating it, and promoting cell survival (30,31).
We also found that anti-HLAI antibodies activate mTORC2, assessed
by the level of phosphorylated at Ser473 Akt. However, everolimus
did not affect the anti-HLAI-induced mTORC2 activation in
glomerular endothelial cells. The latter contradicts the results of
a previous study, which has shown inhibition of anti-HLAI-induced
mTORC2 activation by everolimus (8). This discrepancy may result from
different cell types, reagent concentrations, or time-points.
Halofuginone did not affect anti-HLAI-induced mTORC2 activation as
well.
We evaluated for the first time the effect of
anti-HLAI antibodies on GCN2K activation status, and we showed that
anti-HLAI antibodies induce GCK2K activation assessed both by the
level of its activated phosphorylated at Thr899 form, as well as by
the level of phosphorylation of the GCN2K target e-IF2α at Ser51
(13). We did not evaluate the
molecular mechanisms that govern the anti-HLAI-induced GCN2K
activation, and the related literature is scarce. Interestingly,
one study has shown that perturbation of F-actin dynamics activates
GCN2K, and as already noted, integrin clustering induces F-actin
polymerization (32). Whether such
a mechanism is implicated in our model remains to be elucidated.
Everolimus did not affect the GCN2K pathway in glomerular
endothelial cells.
During ABMR, many immune cells infiltrate and injure
the graft. Monocytes, NK-cells, B-cells, and especially in the
mixed type of rejection T-cells are present, while in the case of
early active humoral rejection, neutrophils may predominate
(5,6). ICAM-1 plays a significant role in the
interaction between endothelial cells and the aforementioned immune
cell types (9,33–38).
In accordance with a previous study (21), we found anti-HLAI antibodies to
increase ICAM-1 expression in glomerular endothelial cells, a fact
that may render the graft more vulnerable to injury from the entire
above immune cell types. We also found that mTORC1 inhibition
decreases anti-HLAI-induced ICAM-1 upregulation, marking mTORC1 a
promising pharmaceutical target for amelioration of humoral
rejection. A previous study has shown that mTORC1 inhibitors
decrease ICAM-1 signal transduction and the interaction between
monocytes and endothelial cells (9). Interestingly, halofuginone also
decreased anti-HLAI-induced ICAM-1 overexpression, indicating that
the GCN2K pathway may also serve as a therapeutic strategy against
humoral rejection.
As noted, especially in the case of mixed cellular
and humoral rejection, T-cells play a significant role in graft
injury (6). We found that
glomerular endothelial cells express the HLAII HLA-DR and that
anti-HLAI antibodies enhance the HLA-DR level significantly. The
latter has never been evaluated before and indicates that anti-HLAI
antibodies may render graft endothelial cells more vulnerable to
CD4+ T-cell-mediated injury (39). Also, by increasing HLA-DR, anti-HLAI
antibodies facilitate anti-HLAII antibodies-mediated injury.
Interestingly, in the clinic, kidney graft survival is worse in the
presence of late anti-HLAII instead of anti-HLAI DSA (40). Finally, by upregulating HLA-DR,
anti-HLAI antibodies may turn glomerular endothelial cells into
effective antigen-presenting cells, enhancing both cellular and
humoral alloimmune response (39).
Unfortunately, neither everolimus nor halofuginone affected
anti-HLAI-induced HLA-DR overexpression in glomerular endothelial
cells. The latter indicates that anti-HLAI antibodies upregulate
HLADR independently of the mTORC1 or the GCN2K pathways.
At first glance, it was surprising that HLA-DR
expression was observed in cultured endothelial cells in the
absence of interferon (IFN)-γ treatment. However, unlike rats and
mice, human endothelial cells, especially those that cover the
microvasculature, express HLA-DR in vivo constantly
(4,41–45).
Primary human microvasculature endothelial cells retain HLA-DR
expression in culture for several days before losing it unless
treated with IFN-γ (4,46). We used primary human
microvasculature endothelial cells at passage two, i.e., after a
short culture period, a fact that explains the observed HLA-DR
expression. The molecular mechanisms involved in anti-HLAI-induced
HLA-DR upregulation remain to be investigated.
In the clinic, from the various anti-HLA antibodies,
the most harmful are those that activate the complement (47,48).
For the first time, we evaluated the effect of anti-HLAI antibodies
on two membrane complement regulatory proteins, the CD46 and the
CD59 (49). We found that anti-HLAI
antibodies increase the expression of both CD46 and CD59 in
glomerular endothelial cells, possibly creating a negative feedback
loop that partially protects the cells from the anti-HLAI-mediated
complement activation. Neither everolimus nor halofuginone affected
the anti-HLAI-induced upregulation of CD46 and CD59. Thus, neither
the mTORC1 nor the GCN2K pathway is responsible for
anti-HLAI-induced CD46 and CD59 overexpression in glomerular
endothelial cells.
Next, we evaluated the effect of anti-HLAI
antibodies on the levels of IL-8 and MCP-1. IL-8 attracts
neutrophils (50), whereas MCP-1
attracts monocytes, NK-cells, and T-cells (51). A previous study detected higher IL-8
production by endothelial cells treated with anti-HLAI antibodies
(21). Our research confirmed that
anti-HLAI antibodies increase IL-8 in glomerular endothelial cells.
Everolimus and halofuginone decreased the production of IL-8, with
their combination inducing an even greater decrease. Also, we found
anti-HLAI antibodies to enhance MCP-1 concentration in the
supernatants of glomerular endothelial cells culture. Halofuginone
reduced anti-HLAI-induced MCP-1 production, and everolimus exerted
the same effect to an even greater extent. Consequently, anti-HLAI
antibodies may facilitate the recruitment of immune cells into the
graft and rejection. Regarding IL-8 and MCP-1 chemokine production,
both mTOR inhibition and GCN2K activation may prove useful
therapeutic maneuvers against humoral rejection.
According to many (20,38),
but not all researchers (52),
anti-HLAI antibodies induce exocytosis of Weibel-Palade bodies in
endothelial cells. Weibel-Palade bodies contain the vWF, which,
when released, facilitates thrombus formation (53). Interestingly, intra-capillary
thrombi are not uncommon in severe active humoral rejection
(6). We found anti-HLAI antibodies
to increase vWF in the supernatants of glomerular endothelial cell
cultures, indicating that anti-HLAI antibodies may render
endothelial cell prothrombotic. Between the two tested
pharmaceutical compounds, only the mTORC1 inhibitor everolimus
prevented the anti-HLAI-induced vWF upregulation.
Similarly to many kidney diseases, the ending remark
of chronic graft failure due to ABMR is fibrosis (6). Thus, we evaluated the effect of
anti-HLAI antibodies on the production of the archetype profibrotic
cytokine TGF-β1 (54). Anti-HLAI
antibodies significantly increased TGF-β1 production by glomerular
endothelial cells. Both everolimus and halofuginone reduced TGF-β1
and their combination to an even higher degree. Therefore, both
mTOR inhibition and GCN2K activation may help in preventing chronic
and irreversible fibrosis of the graft.
Our study has many levels of novelty. The effects of
anti-HLAI antibodies on human glomerular endothelial was evaluated
for the first time. Also, some of the parameters being assessed,
such as the effect of anti-HLAI antibodies on HLA-DR, CD46, and
CD59, have never been considered before. In addition, our study
showed for the first time that the GCN2K activator halofuginone
makes the human glomerular endothelium less vulnerable to
anti-HLAI-induced injury. Finally, although mTOR inhibitors were
introduced in the kidney transplantation immunosuppressive regimen
many years ago, they are administered only in a minority of kidney
transplant recipients (55).
However, recently, the TRANSFORM study showed an immunosuppressive
regimen consisting of everolimus, low dose tacrolimus, and
prednisone is not inferior to the classic immunosuppressive regimen
tacrolimus-mycophenolate-prednisone. Also, the incidence of de
novo DSA was lower in the everolimus group, as well as the
incidence of CMV and BK-virus infection (56). Thus, although mTOR inhibitors are
available for many years, there is still significant debate and
investigation about their final place in kidney transplantation.
Clinical data about the effect of mTOR inhibitors on the glomerular
endothelium in the context of ABMR are not available. Our study
confirms previous experimental data and evaluates new parameters on
the latter topic.
The in vitro nature of our study is a
limitation since drawing direct conclusions from in vitro
studies to the in vivo model is not always safe. However,
under the strictly controlled in vitro conditions, we were
able for the first time to detect certain anti-HLAI-induced changes
in the immunological properties of human glomerular endothelial
cells and the impact of mTOR inhibition or GCN2K activation. Thus,
our study could be considered as a starting point for further
investigation of the ABMR pathophysiology in vivo, always
keeping in mind the interspecies differences.
In conclusion, anti-HLAI antibodies trigger integrin
signal transduction, activate mTOR and GCN2K, do not affect cell
integrity, and promote cell proliferation. Also, by increasing
ICAM-1, HLA-DR, IL-8, and MCP-1, anti-HLAI antibodies enhance the
ability of various immune cells to reach and interact with
glomerular endothelial cells facilitating graft rejection. On the
contrary, by upregulating CD46 and CD59, anti-HLAI antibodies may
render glomerular endothelial cells less vulnerable to
complement-mediated injury. Finally, by enhancing vWF and TGF-β1
production, anti-HLAI antibodies may render endothelium
prothrombotic, and facilitate fibrosis and graft failure,
respectively. Both mTORC1 inhibition and GCN2K activation may prove
useful pharmaceutical targets since they prevent cell
proliferation, downregulate ICAM-1, IL-8, MCP-1, and TGF-β1 induced
by anti-HLAI antibodies. Finally, mTORC1 inhibition decreases
vWF.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The analyzed datasets used and/or analyzed during
the present study are available from the corresponding author on
reasonable request.
Authors' contributions
TE designed the present study. GP and TE performed
the experiments. TE, GP, MC, NA, GF and VL analyzed the results. TE
and GP wrote the manuscript. IS contributed to the analysis and
interpretation of data. TE and GP confirm the authenticity of all
raw data. All authors approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests
References
|
1
|
Sellares J, de Freitas DG, Mengel M, Reeve
J, Einecke G, Sis B, Hidalgo LG, Famulski K, Matas A and Halloran
PF: Understanding the causes of kidney transplant failure: The
dominant role of antibody-mediated rejection and nonadherence. Am J
Transplant. 12:388–399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Abramowicz D, Oberbauer R, Heemann U,
Viklicky O, Peruzzi L, Mariat C, Crespo M, Budde K and Oniscu GC:
Recent advances in kidney transplantation: A viewpoint from the
descartes advisory board. Nephrol Dial Transplant. 33:1699–1707.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Loupy A and Lefaucheur C:
Antibody-mediated rejection of solid-organ allografts. N Engl J
Med. 379:1150–1160. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Muczynski KA, Ekle DM, Coder DM and
Anderson SK: Normal human kidney HLA-DR-expressing renal
microvascular endothelial cells: Characterization, isolation, and
regulation of MHC class II expression. J Am Soc Nephrol.
14:1336–1348. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thomas KA, Valenzuela NM and Reed EF: The
perfect storm: HLA antibodies, complement, FcγRs, and endothelium
in transplant rejection. Trends Mol Med. 21:319–329. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Katsuma A, Yamakawa T, Nakada Y, Yamamoto
I and Yokoo T: Histopathological findings in transplanted kidneys.
Renal Replacement Therapy. 3:62017. View Article : Google Scholar
|
|
7
|
Jindra PT, Jin YP, Rozengurt E and Reed
EF: HLA class I antibody-mediated endothelial cell proliferation
via the mTOR pathway. J Immunol. 180:2357–2366. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jin YP, Valenzuela NM, Ziegler ME,
Rozengurt E and Reed EF: Everolimus inhibits Anti-HLA I
antibody-mediated endothelial cell signaling, migration and
proliferation more potently than sirolimus. Am J Transplant.
14:806–819. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Salehi S, Sosa RA, Jin YP, Kageyama S,
Fishbein MC, Rozengurt E, Kupiec-Weglinski JW and Reed EF:
Outside-in HLA class I signaling regulates ICAM-1 clustering and
endothelial cell-monocyte interactions via mTOR in transplant
antibody-mediated rejection. Am J Transplant. 18:1096–1109. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li F, Rao P, Hong L, Fishbein MC, Gjertson
DW and Reed EF: OR49 effect of everolimus immunotherapy on
HLA-antibody mediated activation of endothelial cells in heart
transplantation. Hum Immunol. 78:462017. View Article : Google Scholar
|
|
11
|
Laplante M and Sabatini DM: mTOR signaling
at a glance. J Cell Sci. 122:3589–3594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Saran U, Foti M and Dufour JF: Cellular
and molecular effects of the mTOR inhibitor everolimus. Clin Sci
(Lond). 129:895–914. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Castilho BA, Shanmugam R, Silva RC, Ramesh
R, Himme BM and Sattlegger E: Keeping the eIF2 alpha kinase Gcn2 in
check. Biochim Biophys Acta. 1843:1948–1968. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Munn DH, Sharma MD, Baban B, Harding HP,
Zhang Y, Ron D and Mellor AL: GCN2 kinase in T cells mediates
proliferative arrest and anergy induction in response to
indoleamine 2,3-dioxygenase. Immunity. 22:633–642. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eleftheriadis T, Pissas G, Antoniadi G,
Spanoulis A, Liakopoulos V and Stefanidis I: Indoleamine
2,3-dioxygenase increases p53 levels in alloreactive human T cells,
and both indoleamine 2,3-dioxygenase and p53 suppress glucose
uptake, glycolysis and proliferation. Int Immunol. 26:673–684.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eleftheriadis T, Pissas G, Antoniadi G,
Liakopoulos V and Stefanidis I: Indoleamine 2,3-dioxygenase
depletes tryptophan, activates general control non-derepressible 2
kinase and down-regulates key enzymes involved in fatty acid
synthesis in primary human CD4+ T cells. Immunology.
146:292–300. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pines M and Spector I: Halofuginone-the
multifaceted molecule. Molecules. 20:573–594. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sundrud MS, Koralov SB, Feuerer M, Calado
DP, Kozhaya AE, Rhule-Smith A, Lefebvre RE, Unutmaz D, Mazitschek
R, Waldner H, et al: Halofuginone inhibits TH17 cell
differentiation by activating the amino acid starvation response.
Science. 324:1334–1338. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Carlson TJ, Pellerin A, Djuretic IM,
Trivigno C, Koralov SB, Rao A and Sundrud MS: Halofuginone-induced
amino acid starvation regulates Stat3-dependent Th17 effector
function and reduces established autoimmune inflammation. J
Immunol. 192:2167–2176. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yamakuchi M, Kirkiles-Smith NC, Ferlito M,
Cameron SJ, Bao C, Fox-Talbot K, Wasowska BA, Baldwin WM III, Pober
JS and Lowenstein CJ: Antibody to human leukocyte antigen triggers
endothelial exocytosis. Proc Natl Acad Sci USA. 104:1301–1306.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Naemi FM, Carter V, Kirby JA and Ali S:
Anti-donor HLA class I antibodies: Pathways to endothelial cell
activation and cell-mediated allograft rejection. Transplantation.
96:258–266. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jin YP, Singh RP, Du ZY, Rajasekaran AK,
Rozengurt E and Reed EF: Ligation of HLA class I molecules on
endothelial cells induces phosphorylation of Src, paxillin, and
focal adhesion kinase in an actin-dependent manner. J Immunol.
168:5415–5423. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rostaing L, Christiaans MH, Kovarik JM and
Pascual J: The pharmacokinetics of everolimus in de novo kidney
transplant patients receiving tacrolimus: An analysis from the
randomized ASSET study. Ann Transplant. 19:337–345. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fadeel B and Orrenius S: Apoptosis: A
basic biological phenomenon with wide-ranging implications in human
disease. J Intern Med. 258:479–517. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang X, Rozengurt E and Reed EF: HLA
class I molecules partner with integrin β4 to stimulate endothelial
cell proliferation and migration. Sci Signal. 3:ra852010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu L, Chen L, Chung J and Huang S:
Rapamycin inhibits F-actin reorganization and phosphorylation of
focal adhesion proteins. Oncogene. 27:4998–5010. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Drolet MC, Desbiens-Brassard V, Roussel E,
Tu V, Couet J and Arsenault M: Blockade of the acute activation of
mTOR complex 1 decreases hypertrophy development in rats with
severe aortic valve regurgitation. Springerplus. 4:4352015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiao Y, Li G, Li Q, Ali R, Qin L, Li W,
Qyang Y, Greif DM, Geirsson A, Humphrey JD and Tellides G: mTOR
(Mechanistic Target of Rapamycin) inhibition decreases
mechanosignaling, collagen accumulation, and stiffening of the
thoracic aorta in elastin-deficient mice. Arterioscler Thromb Vasc
Biol. 37:1657–1666. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee FY, Zhen YY, Yuen CM, Fan R, Chen YT,
Sheu JJ, Chen YL, Wang CJ, Sun CK and Yip HK: The mTOR-FAK
mechanotransduction signaling axis for focal adhesion maturation
and cell proliferation. Am J Transl Res. 9:1603–1617.
2017.PubMed/NCBI
|
|
30
|
Sarbassov DD: Phosphorylation and
regulation of Akt/PKB by the Rictor-mTOR complex. Science.
307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jacinto E, Facchinetti V, Liu D, Soto N,
Wei S, Jung SY, Huang Q, Qin J and Su B: SIN1/MIP1 Maintains
rictor-mTOR complex integrity and regulates Akt phosphorylation and
substrate specificity. Cell. 127:125–137. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Silva RC, Sattlegger E and Castilho BA:
Perturbations in actin dynamics reconfigure protein complexes that
modulate GCN2 activity and promote an eIF2 response. J Cell Sci.
129:4521–4533. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang L, Froio RM, Sciuto TE, Dvorak AM,
Alon R and Luscinskas FW: ICAM-1 regulates neutrophil adhesion and
transcellular migration of TNF-alpha-activated vascular endothelium
under flow. Blood. 106:584–592. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kaizuka Y, Douglass AD, Varma R, Dustin ML
and Vale RD: Mechanisms for segregating T cell receptor and
adhesion molecules during immunological synapse formation in Jurkat
T cells. Proc Natl Acad Sci USA. 104:20296–20301. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kuokkanen E, Šuštar V and Mattila PK:
Molecular control of B cell activation and immunological synapse
formation. Traffic. 16:311–326. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Barber DF, Faure M and Long EO: LFA-1
contributes an early signal for NK cell cytotoxicity. J Immunol.
173:3653–3659. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hsu HT and Orange JS: Distinct
integrin-dependent signals define requirements for lytic granule
convergence and polarization in natural killer cells. Sci Signal.
7:pe242014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Valenzuela NM, Mulder A and Reed EF: HLA
class I antibodies trigger increased adherence of monocytes to
endothelial cells by eliciting an increase in endothelial
P-selectin and, depending on subclass, by engaging FcγRs. J
Immunol. 190:6635–6650. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Neefjes J, Jongsma ML, Paul P and Bakke O:
Towards a systems understanding of MHC class I and MHC class II
antigen presentation. Nat Rev Immunol. 11:823–836. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bentall A, Cornell LD, Gloor JM, Park WD,
Gandhi MJ, Winters JL, Chedid MF, Dean PG and Stegall MD: Five-year
outcomes in living donor kidney transplants with a positive
crossmatch. Am J Transplant. 13:76–85. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mestas J and Hughes CC: Of mice and not
men: Differences between mouse and human immunology. J Immunol.
172:2731–2738. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hayry P, von Willebrand E and Andersson
LC: Expression of HLA-ABC and -DR locus antigens on human kidney,
endothelial, tubular and glomerular cells. Scand J Immunol.
11:303–310. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Evans PR, Trickett LP, Smith JL, MacIver
AG, Tate D and Slapak M: Varying expression of major
histocompatibility complex antigens on human renal endothelium and
epithelium. Br J Exp Pathol. 66:79–87. 1985.PubMed/NCBI
|
|
44
|
Daar AS, Fuggle SV, Fabre JW, Ting A and
Morris PJ: The detailed distribution of mhc class II antigens in
normal human organs. Transplantation. 38:293–298. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Muczynski KA, Cotner T and Anderson SK:
Unusual expression of human lymphocyte antigen class II in normal
renal microvascular endothelium. Kidney Int. 59:488–497. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
McDouall RM, Yacoub M and Rose ML:
Isolation, culture, and characterisation of MHC class II-positive
microvascular endothelial cells from the human heart. Microvasc
Res. 51:137–152. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Orandi BJ, Alachkar N, Kraus ES, Naqvi F,
Lonze BE, Lees L, Van Arendonk KJ, Wickliffe C, Bagnasco SM,
Zachary AA, et al: Presentation and outcomes of C4d-negative
antibody-mediated rejection after kidney transplantation. Am J
Transplant. 16:213–220. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Loupy A, Lefaucheur C, Vernerey D, Prugger
C, Duong van Huyen JP, Mooney N, Suberbielle C, Frémeaux-Bacchi V,
Méjean A, Desgrandchamps F, et al: Complement-binding anti-HLA
antibodies and kidney-allograft survival. N Engl J Med.
369:1215–1226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Merle NS, Church SE, Fremeaux-Bacchi V and
Roumenina LT: Complement System Part I-molecular mechanisms of
activation and regulation. Front Immunol. 6:2622015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Matsushima K, Baldwin ET and Mukaida N:
Interleukin-8 and MCAF: Novel leukocyte recruitment and activating
cytokines. Chem Immunol. 51:236–265. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Deshmane SL, Kremlev S, Amini S and Sawaya
BE: Monocyte chemoattractant Protein-1 (MCP-1): An overview. J
Interferon Cytokine Res. 29:313–326. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Meli A, Carter T, McCormack A, Hannah MJ
and Rose ML: Antibody alone is not a stimulator of exocytosis of
weibel-palade bodies from human endothelial cells. Transplantation.
94:794–801. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hassan MI, Saxena A and Ahmad F: Structure
and function of von Willebrand factor. Blood Coagul Fibrinolysis.
23:11–22. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Prud'homme GJ: Pathobiology of
transforming growth factor beta in cancer, fibrosis and immunologic
disease, and therapeutic considerations. Lab Invest. 87:1077–1091.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hart A, Smith JM, Skeans MA, Gustafson SK,
Stewart DE, Cherikh WS, Wainright JL, Boyle G, Snyder JJ, Kasiske
BL and Israni AK: Kidney. Am J Transplant. 16 (Suppl 2):S11–S46.
2016. View Article : Google Scholar
|
|
56
|
Berger SP, Sommerer C, Witzke O, Tedesco
H, Chadban S, Mulgaonkar S, Qazi Y, de Fijter JW, Oppenheimer F,
Cruzado JM, et al: Two-year outcomes in de novo renal transplant
recipients receiving everolimus-facilitated calcineurin inhibitor
reduction regimen from the TRANSFORM study. Am J Transplant.
19:3018–3034. 2019. View Article : Google Scholar : PubMed/NCBI
|