Introduction
Systemic lupus erythematosus (SLE) is a serious
autoimmune disease that predominantly affects women (1). SLE is characterized by widespread
inflammation and tissue damage in multiple organs, including the
blood vessels, liver, kidneys, skin, heart and joints (2). The hyperactivation of T and B
lymphocytes as well as the increased expression of specific
inflammatory cytokines directly contribute to the occurrence of SLE
(3). A previous study demonstrated
that SLE is characterized by a clear increase in IL-10, IL-2 and
IFN-γ production during exacerbation (4). In clinical practice,
hydroxychloroquine, corticosteroids and cyclophosphamide are
commonly used in the treatment of SLE (5). However, these interventions are often
accompanied by serious adverse events including blindness,
osteoporosis and infertility (6),
making the identification of novel targets for SLE treatment a high
priority.
microRNAs (miRNAs/miRs) are important regulators
involved in the pathogenesis of diverse autoimmune diseases,
including SLE (7,8). Previous studies have demonstrated that
several miRNAs, including miR-125b (9), miR-410 (10) and miR-15b (11), are downregulated in SLE and
increasing evidence suggests that these and other miRNAs serve a
pivotal regulatory role in SLE. For example, the downregulation of
miR-633 increases the expression of inflammatory chemokines in SLE
(12), while overexpression of
miR-155 attenuates autoantibody production and inflammatory
responses by increasing IL-21 signaling capacity in patients with
SLE (13). Upregulation of miR-30a
accelerates B cell proliferation and IgG antibody production in
patients with SLE (14). Previous
studies have identified miR-101-3p as an important regulator in the
progression of inflammatory diseases. miR-101-3p is a diagnostic
biomarker for several autoimmune disorders and attenuates the
inflammation and fibroblast-like synoviocyte proliferation in
rheumatoid arthritis via its interactions with
prostaglandin-endoperoxide synthase 2 (15). Sun et al (16) demonstrated that miR-101-3p
expression is decreased in SLE and that miR-101-3p overexpression
reduces the production of inflammatory cytokines (IFN-γ, IL-6 and
IL-17A) in the peripheral blood mononuclear cells (PBMCs) of SLE
patients, making it a potential novel therapeutic target for SLE.
However, the molecular mechanism underlying miR-101-3p activity in
SLE remains to be elucidated. Therefore, it is necessary to explore
the detailed molecular mechanism underlying the activities of
miR-101-3p in SLE.
MAPKs, a family of serine/threonine kinases, are
known to be closely linked to the pathogenesis of SLE (17). Blocking the activation of p38 MAPK
reduces IFN-γ and IL-6 production in SLE samples (18). Notably, miR-101 is known to regulate
the MAPK response by targeting MAPK phosphatase 1, influencing the
secretion of the downstream inflammatory cytokines in SLE (19). However, the specific regulatory
relationship between miR-101-3p and the MAPK proteins in SLE
remains to be elucidated.
The NF-κB pathway is known to be a prototypical
pro-inflammatory pathway (20),
where NF-κB modulates the transcriptional activation of diverse
genes involved in autoimmune diseases including SLE (21,22).
Previous studies have reported that the NF-κB pathway serves a
pivotal role in the pathology of SLE with the expression of several
NF-κB pathway proteins experiencing marked upregulation in patients
with SLE (23). Inhibition of NF-κB
activity in SLE-prone mice visibly reduces the incidence and
severity of the disease (18).
Notably, miR-146a has been demonstrated to attenuate the
development of SLE via its inhibition of the NF-κB pathway
(24). In addition, following
miR-101 suppression, the activation levels in the NF-κB signaling
pathway increase significantly, inducing increased neuropathic pain
(25). However, despite the ample
evidence of their interaction, the exact relationship between
miR-101-3p and the NF-κB pathway in SLE remains unclear.
The present study evaluated miR-101-3p expression in
SLE PBMCs and its specific effects on the inflammatory response of
these cells. The underlying mechanisms facilitating the
interactions between miR-101-3p and MAPK1 or NF-κB were then
determined with the aim of enhancing the understanding of their
regulatory mechanism. The findings of the present study may
identify a promising therapeutic target for SLE and elucidate the
underlying mechanism of action for miR-101-3p in SLE.
Materials and methods
Patients
A total of 40 female patients with SLE and 20
healthy controls (HC), receiving treatment at Xijing Hospital, were
enrolled in the present study between June 2016 and August 2018 and
any SLE diagnoses were based on the revised criteria issued by the
American College of Rheumatology in 1997 (26). All HC volunteers had no history of
SLE, other autoimmune inflammatory disease or cancer. Patients with
SLE and concurrent infection or additional inflammatory diseases
were excluded from the study. Disease activity was assessed using
the SLE Disease Activity Index (SLEDAI) (27). To exclude the influence of certain
therapies on gene expression, all SLE medications were withheld 24
h before sampling. The characteristics of patients with SLE and HCs
are described in Table I and no
significant differences in age or serum creatinine were identified.
The present study was approved by the Institutional Ethics
Committee of the Xijing Hospital (approval no. 20181016) and
written informed consent from both patients and volunteers were
obtained prior to sample collection.
 | Table I.The clinical characteristics of
patients with SLE and HC. |
Table I.
The clinical characteristics of
patients with SLE and HC.
| Characteristic | SLE (n=40) | HC (n=20) | P-value |
|---|
| Male:Female | 0:40 | 0:20 | – |
| Age | 34.7±11 | 34.7±8.7 | 0.99 |
| SLEDAI | 12.2±3.1 | – | – |
| Serum creatinine
(µmol/l) | 78±20 |
75±20 | 0.70 |
| Anti-dsDNA
(IU/ml) | 234±27.9 | – | – |
| ESR (mm/h) | 85.6±24.1 | 11.5±4.6 | <0.0001 |
| CRP (mg/l) | 61.5±28.4 |
6.52±1.65 | <0.0001 |
| IgG (g/l) | 23.3±10.6 | – | – |
| C3 (g/l) | 0.68±0.23 | – | – |
| Clinical features
(no. of patients) |
|
|
|
|
Arthritis | 10 | – | – |
|
Leukopenia | 13 | – | – |
|
Rash | 6 | – | – |
|
Serositis | 6 | – | – |
|
Leukopenia | 11 | – | – |
| Oral
ulcers | 8 | – | – |
| Renal
disease | 15 | – | – |
| Treatment (no. of
patients) |
|
|
|
|
Hydroxychloroquine | 19 | – | – |
|
Prednisone | 18 | – | – |
|
None | 6 | – | – |
Measurements of blood samples
Evaluations were completed using peripheral blood
samples (10 ml) collected from patients with SLE (n=40) and HC
(n=20) from the Xijing Hospital. The erythrocyte sedimentation rate
(ESR) was measured using a modified Westergren method (Excyte ESR
Non-Vacuum Tubes kit; cat. no. EX-10100; Vital Diagnostics Pty.
Ltd.). An IMMAGE 800 fully automatic protein analyzer (Beckman
Coulter, Inc.) was employed to test the levels of C-reactive
protein (CRP) and complement 3 (C3). Anti-double stranded DNA
(anti-dsDNA) and immunoglobulin G (IgG) antibodies were then
evaluated using a human anti-dsDNA ELISA kit (cat. no. 69-98345;
mskbio) or human IgG ELISA kit (cat. no. 69-99047; mskbio)
respectively.
Cell culture
Peripheral blood samples were collected from
patients with SLE and PBMCs were isolated by Ficoll-Paque density
gradient centrifugation (GE Healthcare Bio-Sciences). Briefly, 4 ml
Ficoll (GE Healthcare Bio-Sciences) was added to a centrifuge tube
with the diluted peripheral blood (4 ml), and centrifuged at 1,000
× g for 30 min at 20°C. The PBMC layer was transferred into a new
centrifuge tube, washed with 3× the volume of PBS, centrifuged at
250 × g for 10 min at 20°C, and then the supernatant was removed.
The washing step was repeated once and then PBMCs were resuspended
in PBS. PBMCs were maintained in RPMI1640 (HyClone; Cytiva)
supplemented with 10% fetal bovine serum (HyClone; Cytiva) and
grown at 37°C and 5% CO2. Logarithmic growth phase cells
were used for all subsequent assays.
Cell transfection and treatment
The miR-101-3p mimics or inhibitor were used in
either the overexpression or silencing of miR-101-3p, respectively.
The MAPK1 gene was cloned into a pcDNA3.1 vector for MAPK1
overexpression (pcDNA3.1-MAPK1). Small interfering RNAs (siRNAs)
that specifically targeted MAPK1 (si-MAPK1-1/2/3) were used to
silence MAPK1. The mimics-NC, inhibitor-NC, pcDNA3.1 (empty vector)
and si-NC served as the relevant negative controls of miR-101-3p
mimics, miR-101-3p inhibitor, pcDNA3.1-MAPK1 and si-MAPK1-1/2/3,
respectively. The sequences of oligonucleotides used were as
follows: si-MAPK1-1, sense 5′-AGUUCGAGUAUACUUCAAGTT-3′ and
antisense 5′-CUUGAUAGCGCUACGAACUTT-3′; si-MAPK1-2, sense
5′-CAUGGUAGUCACUAACAUATT-3′ and antisense
5′-UAUGUUAGUGACUACCAUGTT-3′; si-MAPK1-3, sense
5′-GAAGCGUGCAGGUUAACUTT-3′ and antisense
5′-GUACUGCAACGCACAUUUCTT-3′; si-NC, sense
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense
5′-ACGUGACACGUUCGGAGAATT-3′; miR-101-3p mimics, sense
5′-UACAGUACUGUGAUAACUGAA-3′ and antisense
5′-UUCAGUUAUCACAGUACUGUA-3′; mimics-NC, sense
5′-UUUGUACUACACAAAAGUACUG-3′ and antisense
5′-CAGUACUUUUGUGUAGUACAAA-3′; miR-101-3p inhibitor,
5′-UUCAGUUAUCACAGUACUGUA-3′; inhibitor NC,
5′-CAGUACUUUUGUGUAGUACAA-3′. All the above oligonucleotides or
plasmids were purchased from Shanghai GenePharma Co., Ltd.. When
the SLE PBMCs reached 60% confluence, they were transfected or
co-transfected with the relevant oligonucleotides (30 nM) or
plasmids (1.5 µg per well) using Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to
manufacturer's protocols. All cells were cultured for 48 h at 37°C
with 5% CO2. The subsequent experiments were performed
48 h after transfection, when these cells were used for the assays
of the regulatory mechanism of miR-101-3p/MAPK1 axis on
inflammation and NF-κB pathway in SLE. To further explore the
regulatory relationship between miR-101-3p/MAPK1 and the NF-κB
pathway, BAY11-7085, an NF-κB activator and BAY11-7082, an NF-κB
inhibitor, were used to activate or block the NF-κB pathway,
respectively. SLE PBMCs transfected with miR-101-3p mimics were
treated with 5 µmol/l BAY11-7085 and SLE PBMCs transfected with
pcDNA3.1-MAPK1 were treated with 5 µmol/l BAY11-7082 for an
additional 48 h. After the treatment, cells were used for the
assays of the regulatory mechanism of miR-101-3p/MAPK1 associated
with the NF-κB pathway in the inflammation of SLE. SLE PBMCs
without treatment were considered as the Mock group.
Bioinformatics-based prediction and
analyses
The online tools StarBase V2.0 (http://starbase.sysu.edu.cn/starbase2)
and miRDB (http://mirdb.org) were employed to
predict the possible target genes of miR-101-3p and the MAPK1 gene
was simultaneously identified by both prediction tools. Notably,
MAPK1 gene expression is upregulated in patients with SLE and
correlates with inflammatory cytokine expression. MAPK1 (ERK2) was
therefore selected as the target of miR-101-3p in the subsequent
analyses.
Reverse transcription-quantitative
(RT-q) PCR
Total RNA was extracted from PBMCS by
TRIzol® reagent (Thermo Fisher Scientific, Inc.). The
corresponding RNA was reverse transcribed into cDNA using an
ExScript RT reagent kit (Takara Biotechnology Co., Ltd.) following
DNase I (Sigma-Aldrich; Merck KGaA) treatment. Reverse
transcription conditions were 55°C for 20 min and 80°C for 10 min.
Then, RT-qPCR analysis was detected using the SYBR Green qPCR
Master Mix (5 µl; Invitrogen; Thermo Fisher Scientific, Inc.) on an
ABI 7500 real-time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.). For the PCR experiments, the following primers
were used (Table II): miR-101-3p,
forward 5′-TCCGAAAGTCAATAGTGTC-3′ and reverse
5′-GTGCAGGGTCCGAGGT-3′; U6, forward 5′-CTCGCTTCGGCAGCACA-3′ and
reverse 5′-AACGCTTCACGAATTTGCGT-3′; MAPK1 forward
5′-AGATTCCAGCCAGGATACA-3′ and reverse 5′-GCATAAAAGCCACAACTACC-3′;
β-actin forward 5′-ACACCTTCTACAATGAGCTG-3′ and reverse,
5′-CTGCTTGCTGATCCACATCT-3′. U6 and β-actin were used for the
normalization of miR-101-3p and MAPK1, respectively. The
thermocycling conditions of PCR program included an initial
denaturation step at 95°C for 3 min followed by 40 cycles at 95°C
for 15 sec, 60°C for 30 sec and 72°C for 20 sec with a final 2 min
extension step at 72°C. Relative expression levels were then
calculated using the 2−ΔΔCq method (28).
| Table II.Sequences of siRNAs and primers. |
Table II.
Sequences of siRNAs and primers.
|
| Sequences |
|---|
| siMAPK1-1 | Forward:
5′-AGUUCGAGUAUACUUCAAGTT-3′ |
|
| Reverse:
5′-CUUGAUAGCGCUACGAACUTT-3′ |
| siMAPK1-2 | Forward:
5′-CAUGGUAGUCACUAACAUATT-3′ |
|
| Reverse:
5′-UAUGUUAGUGACUACCAUGTT-3′ |
| siMAPK1-3 | Forward:
5′-GAAGCGUGCAGGUUAACUTT-3′ |
|
| Reverse:
5′-GUACUGCAACGCACAUUUCTT-3′ |
| si-NC | Forward:
5′-UUCUCCGAACGUGUCACGUTT-3′ |
|
| Reverse:
5′-ACGUGACACGUUCGGAGAATT-3′ |
| miR-101-3p | Forward:
5′-TCCGAAAGTCAATAGTGTC-3′ |
|
| Reverse:
5′-GTGCAGGGTCCGAGGT-3′ |
| U6 | Forward:
5′-CTCGCTTCGGCAGCACA-3′ |
|
| Reverse:
5′-AACGCTTCACGAATTTGCGT-3′ |
| MAPK1 | Forward:
5′-AGATTCCAGCCAGGATACA-3′ |
|
| Reverse:
5′-GCATAAAAGCCACAACTACC-3′ |
| β-actin | Forward:
5′-ACACCTTCTACAATGAGCTG-3′ |
|
| Reverse:
5′-CTGCTTGCTGATCCACATCT-3′ |
Western blotting
Proteins were isolated in RIPA lysis buffer
(Invitrogen; Thermo Fisher Scientific, Inc.) and quantified using a
BCA assay kit (Invitrogen; Thermo Fisher Scientific, Inc.). The
proteins (20 µg/lane) were then separated by 10–12% SDS-PAGE and
transferred to a polyvinylidene fluoride membrane (EMD Millipore).
The membrane was blocked using 5% skimmed milk at 37°C for 1 h. The
membranes were then incubated with the appropriate primary
antibodies, including GAPDH (1:1,000; cat. no. ab9485; Abcam), MAPK
(1:2,000; cat. no. 4695; Cell Signaling Technology, Inc.), NF-κBp65
(1:2,000; cat. no. 8242; Cell Signaling Technology, Inc.) and
phosphorylated (p-) IκBα (p-IκBα) (1:2,000; cat. no. 2859; Cell
Signaling Technology, Inc.) overnight at 4°C. After incubation with
an HRP-conjugated secondary antibody (1:5,000; cat. no. 12-348;
Sigma-Aldrich; Merck KGaA) for 1 h at 25°C, the bands were
visualized using an enhanced chemiluminescence kit (Invitrogen;
Thermo Fisher Scientific, Inc.). The relative expression of each
protein was then quantified using ImageJ version 1.46r (National
Institutes of Health), normalized to GAPDH and standardized against
the control (Mock or pcDNA3.1 + mimics-NC group).
Dual luciferase reporter gene (DLR)
assay
PBMCs were co-transfected with luciferase PsiCHECK-2
(Promega Corporation) carrying MAPK1-wildtype (MAPK1-Wt) or
MAPK1-mutant (MAPK1-Mut) (GenePharma) sequences and miR-101-3p
mimics or inhibitors. The experimental methods were as follows: The
Wt and Mut primers for the MAPK1 target fragments were designed and
synthesized by Sangon Biotech Co., Ltd.. For the luciferase
reporter assays, cells were seeded in a 48-well plate and the
luciferase reporter vectors were co-transfected with miR-101-3p
mimics, mimics-NC, miR-101-3p inhibitor or inhibitor-NC using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) and incubated at 37°C for 48 h. Relative
luciferase activity was measured using a Dual Luciferase Reporter
assay kit (Promega Corporation) following 48 h of transfection.
Firefly luciferase activity was normalized to Renilla
luciferase activity.
RNA pull-down assay
PBMCs were lysed using RIPA lysis buffer
(Invitrogen; Thermo Fisher Scientific, Inc.) and then incubated
with 3′-biotin-labeled miR-101-3p (Bio-miR-101-3p-Wt),
3′-biotin-labeled miR-101-3p with a mutation at the binding site
between MAPK1 and miR-101-3p (Bio-miR-101-3p-Mut) or
3′-biotin-labeled miR-NC (Bio-miR-NC) (Shanghai GenePharma Co.,
Ltd.) for 1 h at 37°C. These samples were then incubated with
streptavidin agarose beads (Invitrogen; Thermo Fisher Scientific,
Inc.) for another 1 h at 37°C. The beads were subsequently washed
twice with cold lysis buffer, thrice with low-salt buffer and once
with high-salt buffer at 4°C. Finally, the RNA complexes bound to
these beads were eluted and extracted for qRT-PCR analysis.
ELISA
IL-10 and IFN-γ levels in SLE PBMCs were evaluated
using Human IL-10 ELISA kit (Abcam; cat. no. ab46034) and Human
IFN-γ ELISA kit (Abcam; cat. no. ab174443), respectively. Briefly,
PBMCs were incubated with specific primary antibodies for 30 min in
coating buffer and then with peroxidase-labeled secondary antibody
for 30 min. These samples were then incubated in TMB substrate
(Sigma-Aldrich; Merck KGaA) for 10 min and the OD 450/550 nm values
were determined using a microplate reader (Molecular Devices,
LLC.).
Statistical analyses
Statistical analyses were performed using SPSS
version 22.0 (IBM Corp.) software and the data are described as the
mean ± standard deviation. The differences between two groups were
evaluated using a paired t-test while the differences among
multiple groups were assessed by one-way ANOVA followed by a
Tukey's post-hoc test. Significant correlations were identified by
Pearson correlation analysis. P<0.05 was considered to indicate
a statistically significant difference.
Results
miR-101-3p is downregulated in SLE
PBMCs
RT-qPCR confirmed that miR-101-3p is differently
expressed in SLE PBMCs, with the data clearly showing that
miR-101-3p expression was significantly downregulated in SLE PBMCs
when compared with HC PBMCs (P<0.0001; Fig. 1A). miR-101-3p expression was
negatively associated with anti-dsDNA, ESR, CRP, IgG and SLEDAI
values (Fig. 1B-G) and positively
associated with C3 in patients with SLE (P<0.0001; Fig. 1B-G). In addition, the receiver
operating characteristic (ROC) curve was plotted and used to
evaluate the diagnostic value of miR-101-3p in SLE. An area under
the curve of 0.845 was obtained, which suggests that there may be
some discriminatory power in the expression of this miRNA (Fig. 1H).
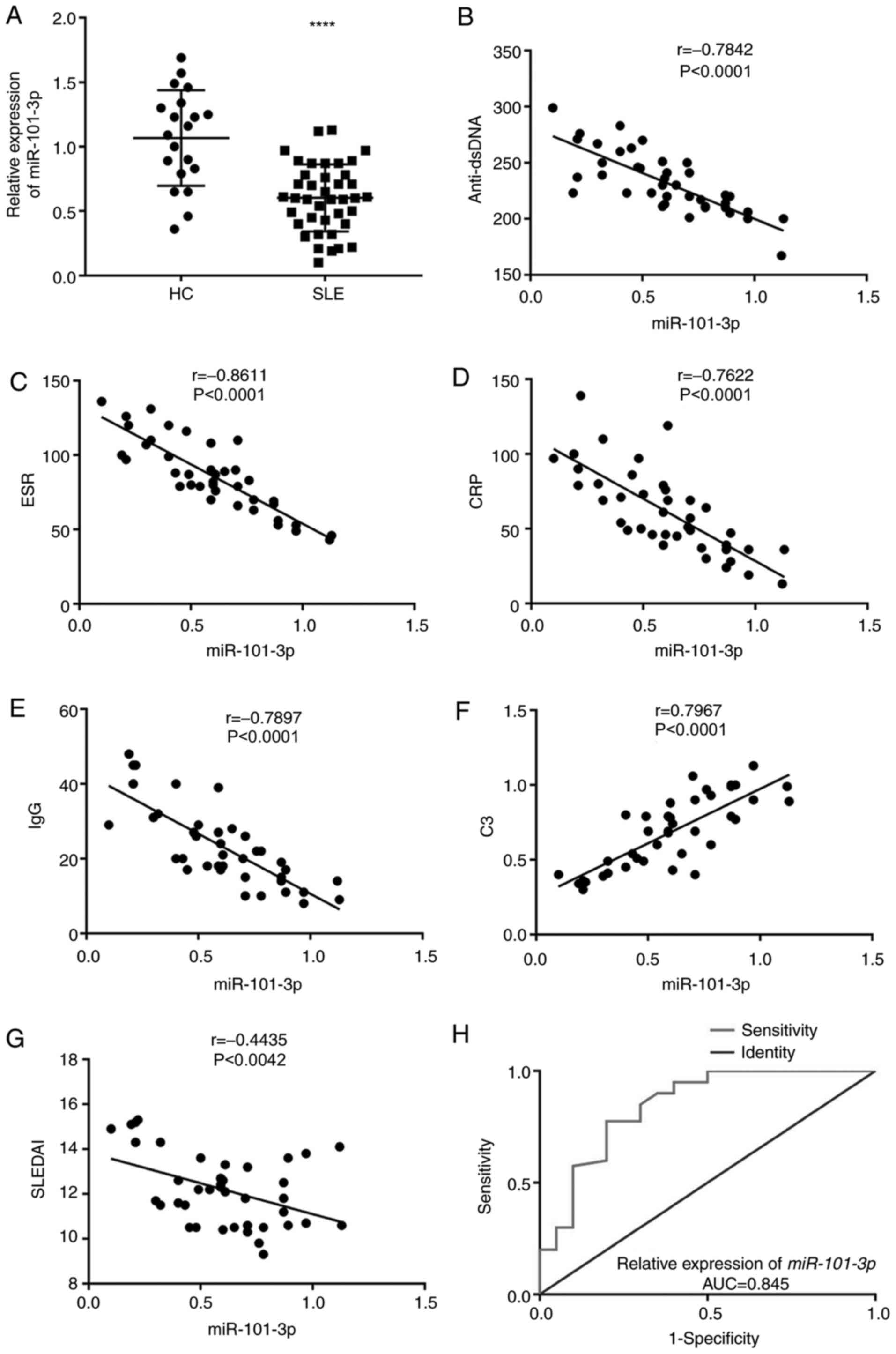 | Figure 1.miR-101-3p expression is
downregulated in SLE PBMCs. (A) Relative expression of miR-101-3p
in HC and SLE PBMCs at the mRNA level. ****P<0.0001 vs. HC. (B)
Correlation between miR-101-3p and anti-dsDNA. (C) Correlation
between miR-101-3p and ESR. (D) Correlation between miR-101-3p and
CRP. (E) Correlation between miR-101-3p and IgG. (F) Correlation
between miR-101-3p and C3 (G) correlation between miR-101-3p and
SLEDAI. (H) ROC curves of miR-101-3p in diagnosis of SLE. miR,
microRNA; SLE, systemic lupus erythematosus; PBMCs, peripheral
blood mononuclear cells; HC, healthy control; dsDNA, double
stranded DNA; ESR, erythrocyte sedimentation rate; CRP, C-reactive
protein; C3, complement 3; SLEDAI, SLE Disease Activity Index; ROC,
receiver operating characteristic; AUC, area under the curve. |
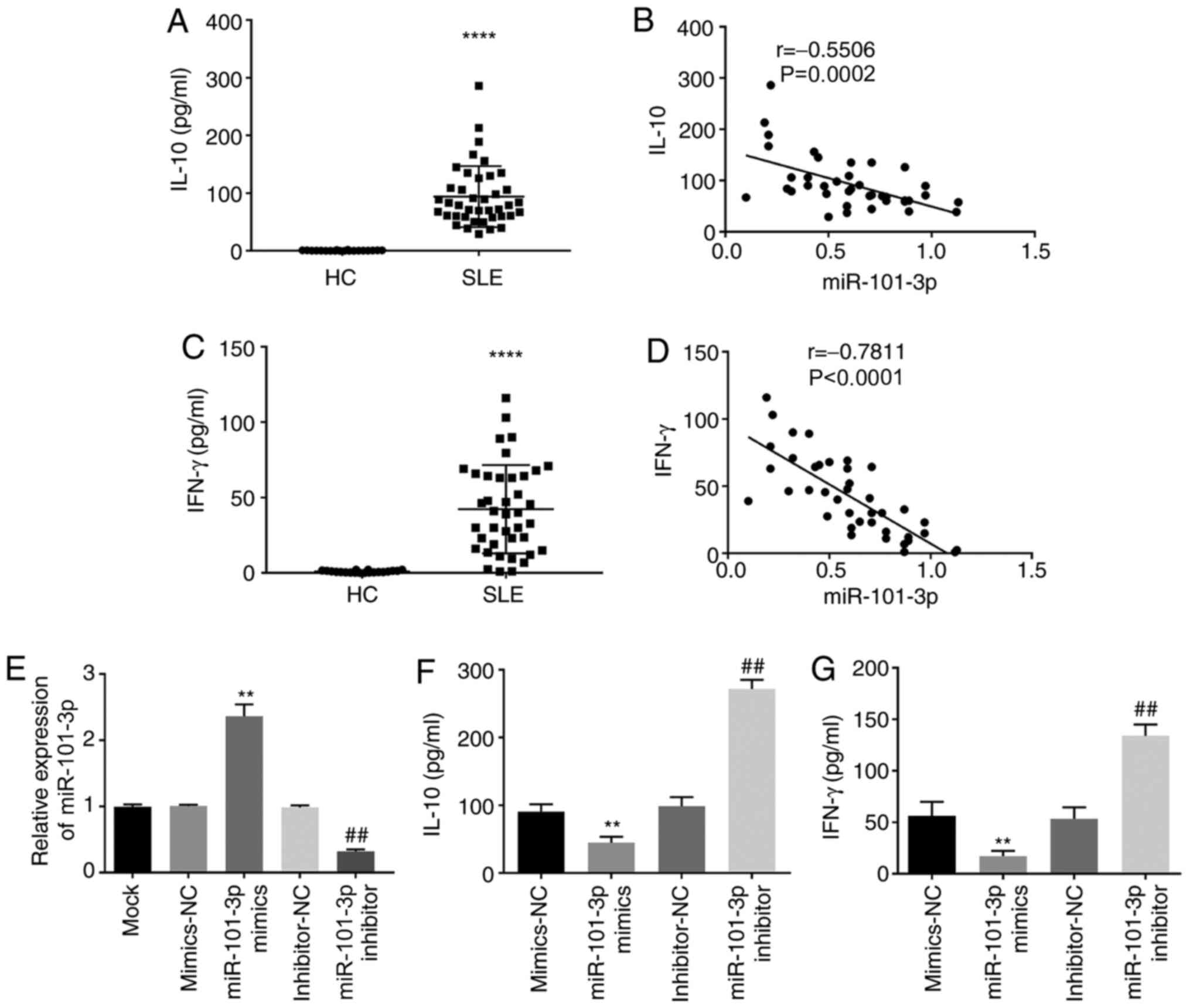
miR-101-3p decreases IL-10 and IFN-γ
expression in SLE PBMCs
To investigate the effects of miR-101-3p expression
in SLE, the levels of IL-10 and IFN-γ were evaluated using ELISA.
ELISA assays showed that IL-10 expression was significantly higher
in SLE PBMCs when compared with HC PBMCs (P<0.0001; Fig. 2A) and that miR-101-3p expression was
negatively associated with IL-10 concentration in SLE PBMCs
(P=0.0002; Fig. 2B). In addition,
IFN-γ expression increased in the SLE PBMCs when compared with the
HC PBMCs (P<0.0001; Fig. 2C) and
there was a negative correlation between IFN-γ and miR-101-3p
expression in SLE PBMCs (P<0.0001; Fig. 2D). miR-101-3p expression was then
manipulated using mimics and siRNAs, with these assays confirming
these observations (Fig. 2E). IL-10
and IFN-γ concentration decreased in the presence of miR-101-3p
mimics and increased in the presence of miR-101-3p inhibitors
(P<0.01; Fig. 2F and G).
MAPK1 is a target of miR-101-3p
RT-qPCR results suggested that MAPK1 expression was
significantly higher in SLE PBMCs compared with HC PBMCs
(P<0.001; Fig. 3A), while
miR-101-3p expression was demonstrated to be negatively associated
with MAPK1 expression in SLE PBMCs (P<0.01) (Fig. 3B). MAPK1 was identified as a
potential miR-101-3p target using the StarBase software (Fig. 3C) which was subsequently confirmed
in a DLR assay. This assay showed that the relative luciferase
activity of the reporter constructs was significantly decreased in
SLE PBMCs following co-transfection with miR-101-3p mimics and
MAPK1 Wt compared with SLE PBMCs co-transfected with mimics-NC and
MAPK1 Wt (P<0.01; Fig. 3D),
while the addition of the miR-101-3p inhibitor increased the
luciferase activity of the Wt MAPK1 reporter in SLE PBMCs
(P<0.05; Fig. 3E). RNA-pull down
assays confirmed the interaction between MAPK1 and
Bio-miR-101-3p-Wt and demonstrated that Bio-miR-101-3p-Mut with a
mutated MAPK1 binding site failed to interact with this protein
(P<0.01; Fig. 3F). Additionally,
overexpression and silencing of miR-101-3p significantly decreased
or increased the mRNA and protein expression of MAPK1 in SLE PBMCs,
respectively (P<0.01; Fig. 3G and
H).
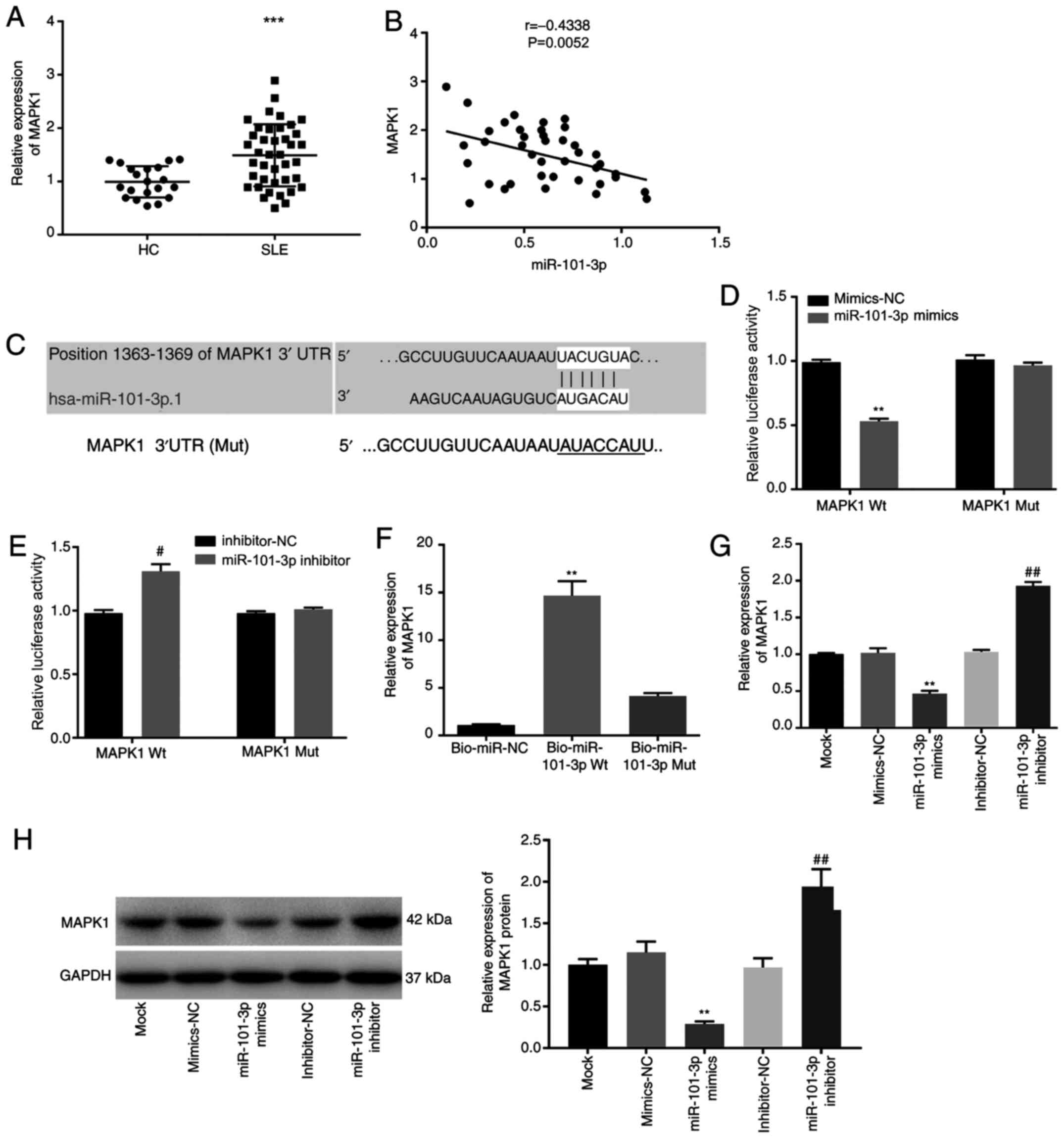 | Figure 3.MAPK1 is a target of miR-101-3p. (A)
MAPK1 expression in HC and SLE PBMCs. ***P<0.001 vs. HC. (B)
Correlation between miR-101-3p and MAPK1. (C) A binding site
predicted by an online target gene prediction software (starBase).
(D and E) Interaction analyzed by dual luciferase reporter assay.
**P<0.01 vs. mimics-NC, #P<0.05 vs. inhibitor-NC.
(F) Interaction analyzed by RNA pull-down assay. **P<0.01 vs.
Bio-NC and Bio-miR-101-3p Mut. (G and H) Relative mRNA and protein
expression of MAPK1 in SLE PBMCs transfected with miR-101-3p
mimics/inhibitor. **P<0.01 vs. mimics-NC, ##P<0.01
vs. inhibitor-NC. miR, microRNA; SLE, systemic lupus erythematosus;
PBMCs, peripheral blood mononuclear cells; HC, healthy control;
Mock, SLE PBMCs without transfection; mimics-NC, miR-101-3p mimics
negative control; inhibitor-NC, miR-101-3p inhibitor negative
control; Bio-NC, biotin RNA-labeled negative control;
Bio-miR-101-3p Wt, biotin RNA-labeled miR-101-3p-wildtype;
Bio-miR-101-3p Mut, biotin RNA-labeled miR-101-3p-mutant. |
MAPK1 overexpression eliminates
miR-101-3p inhibition of IL-10 and IFN-γ in SLE PBMCs
To explore the molecular mechanism of MAPK1
signaling in SLE, cellular signaling was evaluated when cells were
treated with an MAPK1 inhibitor, si-MAPK1-1, −2 and −3. RT-qPCR
results showed that SLE PBMC MAPK1 transcription was significantly
decreased following the addition of si-MAPK1-1, −2 and −3
(P<0.01; Fig. 4A). si-MAPK1-2
was used for subsequent assays due to its relatively high silencing
efficiency and was demonstrated to also significantly decrease the
SLE PBMC expression of MAPK1 at the protein level (P<0.01;
Fig. 4B). Notably, si-MAPK1-2 also
significantly decreased IL-10 and IFN-γ expression in SLE PBMCs
(P<0.01; Fig. 4C and D). In
addition, when MAPK1 was overexpressed in these cells, following
PBMCs transfection with pcDNA3.1-MAPK1 (P<0.01; Fig. 4E), IL-10 and IFN-γ both increased
significantly eliminating any inhibitory effects of the miR-101-3p
mimics (P<0.05; Fig. 4F and
G).
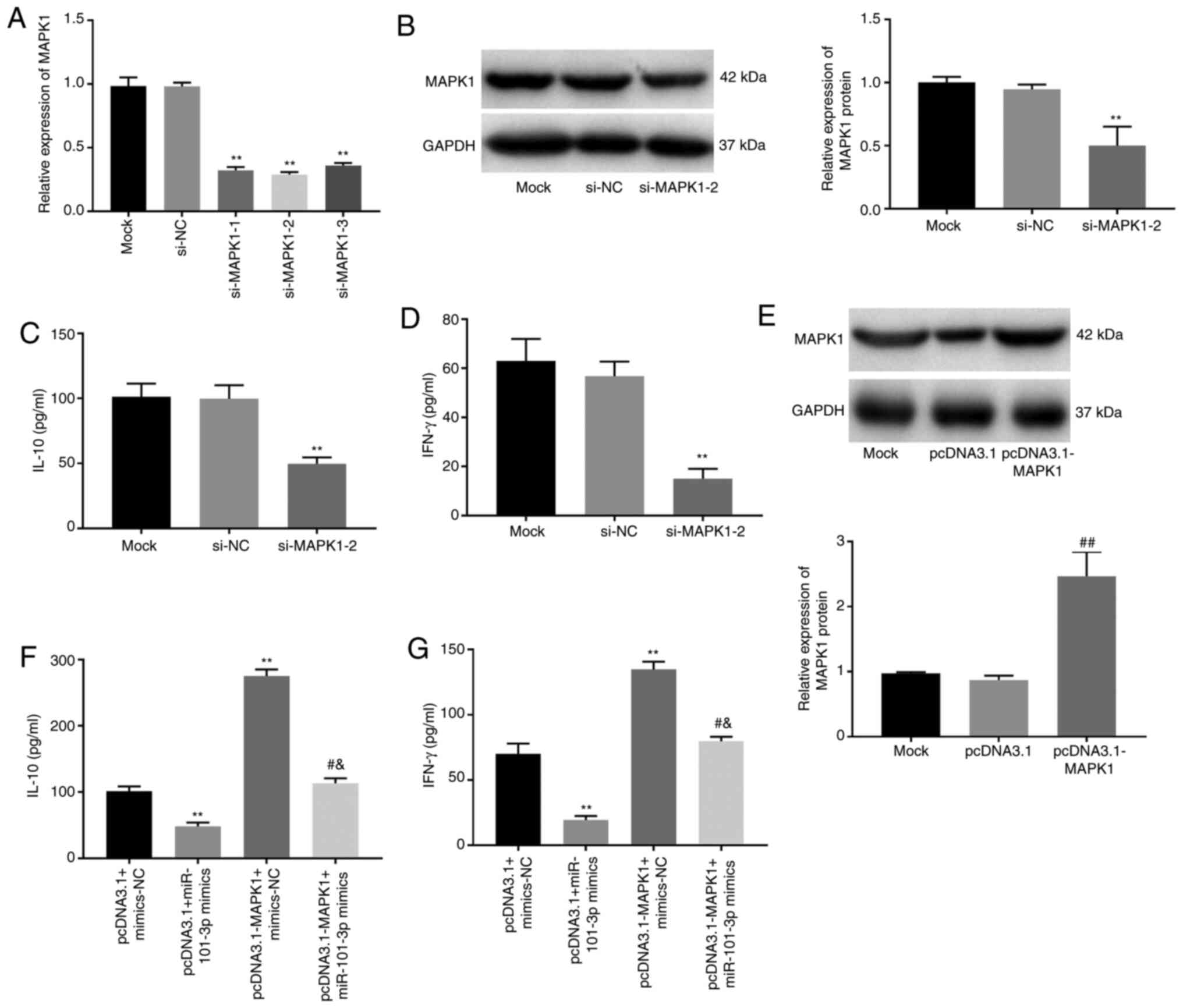 | Figure 4.MAPK1 overexpression eliminates
miR-101-3p inhibition of IL-10 and IFN-γ in SLE PBMCs. (A) Relative
expression of MAPK1 in SLE PBMCs transfected with siRNA-MAPK
(si-MAPK)1-1, −2 and −3 at the mRNA level. **P<0.01 vs. si-NC.
(B) Relative expression of MAPK1 in SLE PBMCs transfected with
si-MAPK1-2 at the protein level. **P<0.01 vs. si-NC. (C) IL-10
level in SLE PBMCs transfected with pcDNA3.1-MAPK1. **P<0.01 vs.
si-NC. (D) IFN-γ level in SLE PBMCs transfected with
pcDNA3.1-MAPK1. **P<0.01 vs. si-NC. (E) Relative expression of
MAPK1 in SLE PBMCs transfected with pcDNA3.1-MAPK1 at the protein
level. ##P<0.01 vs. pcDNA3.1. (F) IL-10 level in SLE
PBMCs transfected with pcDNA3.1-MAPK1 and/or miR-101-3p mimics.
**P<0.01, vs. pcDNA3.1+mimics-NC; #P<0.05 vs.
pcDNA3.1+miR-101-3p mimics; &P<0.05 vs.
pcDNA3.1-MAPK1+ mimics-NC. (G) IFN-γ level in SLE PBMCs transfected
with pcDNA3.1-MAPK1 and/or miR-101-3p mimics. **P<0.01 vs.
pcDNA3.1+mimics-NC; #P<0.05 vs. pcDNA3.1+miR-101-3p
mimics; &P<0.05 vs. pcDNA3.1-MAPK1+ mimics-NC.
miR, microRNA; SLE, systemic lupus erythematosus; PBMCs, peripheral
blood mononuclear cells; si-, small interfering; Mock, SLE PBMCs
without transfection; si-NC, siRNA-MAPK negative control; pcDNA3.1,
empty pcDNA3.1; mimics-NC, miR-101-3p mimics negative control. |
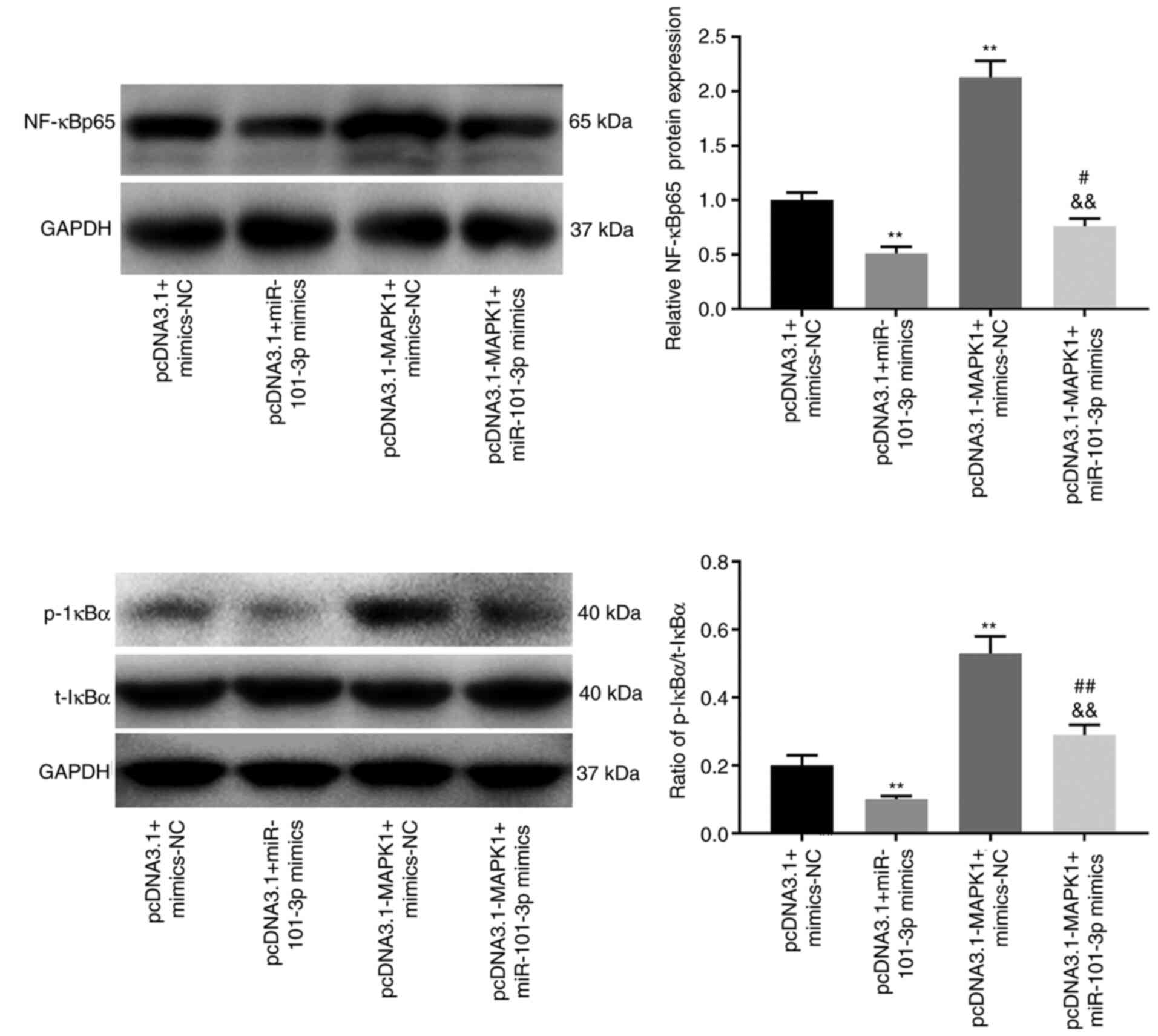
The NF-κB pathway is repressed by
miR-101-3p
Given the results of the previous assays the present
study proceeded to evaluate the regulatory relationship between
miR-101-3p and the NF-κB pathway. The protein level of NF-κBp65 is
a pivotal index to evaluate NF-κB pathway activity and the
phosphorylation of IκBα causes the activation of NF-κB pathway. As
shown in Fig. 5, the protein level
of NF-κBp65 and the ratio of p-IκBα/total (t)-IκBα were
significantly decreased in the presence of miR-101-3p mimics
(P<0.01). In addition, MAPK1 overexpression increased the
protein level of NF-κBp65 and the ratio of p-IκBα/t-IκBα in SLE
PBMCs (P<0.01), which could be reversed following the addition
of the miR-101-3p mimics (P<0.01).
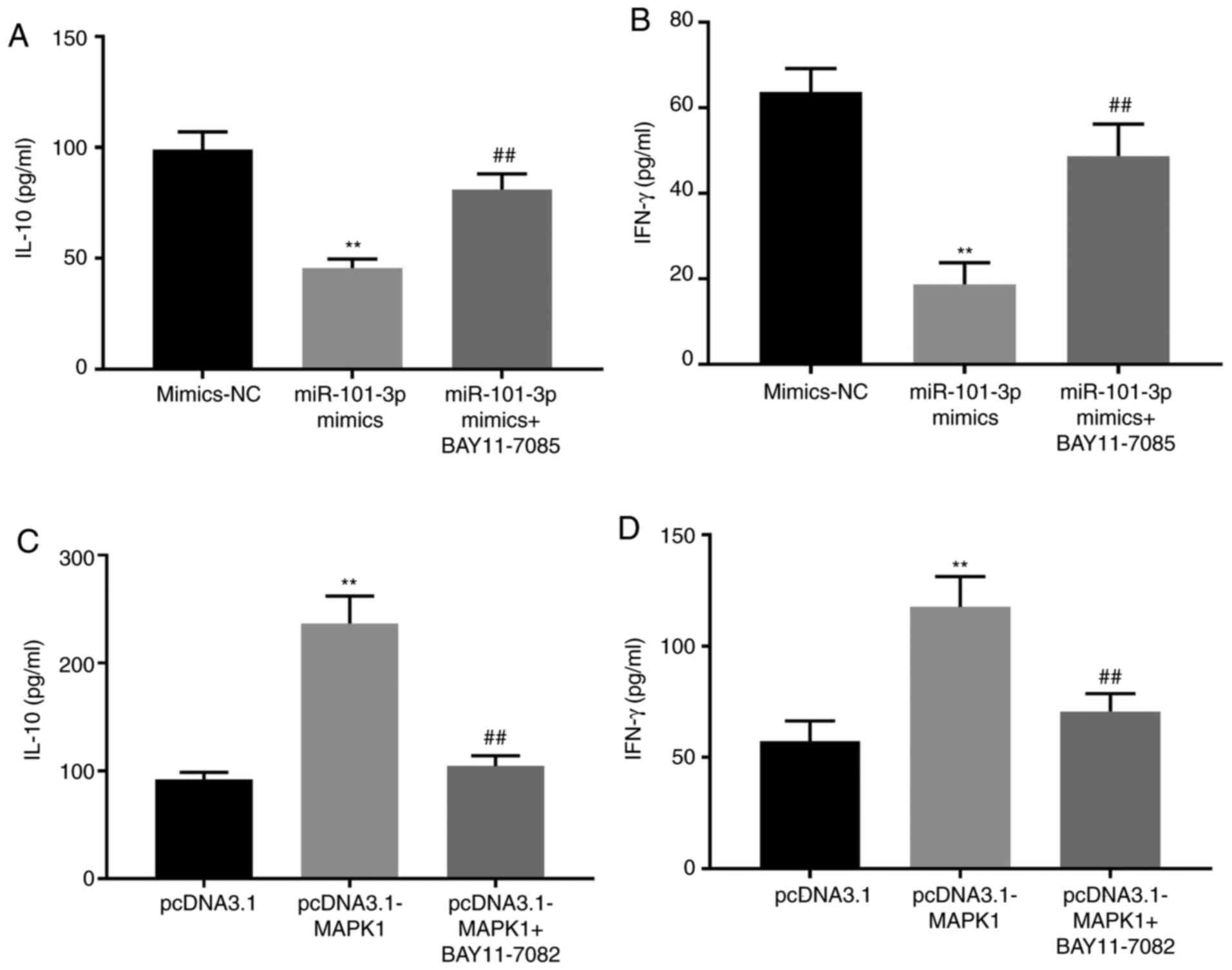
miR-101-3p decreases IL-10 and IFN-γ
expression in SLE PBMCs by inhibiting the NF-κB pathway
NF-κB pathway activities can be modulated via the
addition of BAY11-7085 (NF-κB activator) or BAY11-7082 (NF-κB
inhibitor). When cells were treated with BAY11-7085 a significant
increase in IL-10 and IFN-γ expression levels was observed, even in
the presence of miR-101-3p mimics (P<0.01). By contrast,
treatment with BAY11-7082 significantly decreased IL-10 and IFN-γ
expressions even in the presence of the pcDNA3.1-MAPK1 construct
(P<0.01; Fig. 6).
Discussion
SLE is a serious disorder of the immune system,
which results from the interaction of a wide range of risk factors,
including hormones, environmental signals and genetic factors
(29). Aberrations in miRNA
expression have been linked to SLE pathogenesis, with the
expression of some miRNAs, including miR-98 (30), miR-1246 (31) and miR-17-5p (32) demonstrated to be decreased in SLE.
Similar to the previous studies, miR-101-3p expression was markedly
reduced in SLE in the current study. To date, SLE diagnosis is
supported by a variety of indices including anti-dsDNA, ESR, CRP,
IgG and C3 (33–36). Reduced expression of miR-98 is found
to be associated with anti-dsDNA and SLEDAI in SLE PBMCs (37) while miR-203 expression is negatively
associated with ESR and CRP in patients with SLE (38). The plasma levels of miR-145 and
miR-183 are positively associated with complement C3 in patients
with SLE (39) and the results of
the present study confirm this. In the present study, miR-101-3p
expression was negatively associated with anti-dsDNA, ESR, CRP, IgG
and SLEDAI and positively associated with C3. In addition, ROC
curve analysis demonstrated that miR-101-3p may possess some value
as a diagnostic biomarker for SLE. As patients with SLE and
concurrent infections or inflammatory diseases were excluded from
the study, this application may be limited, but it does suggest
that miR-101-3p may be used as an additional diagnostic marker for
SLE.
The overproduction of pro-inflammatory cytokines is
an important characteristic of SLE (40) and the production of IL-10 and IFN-γ,
which act as regulators of this inflammatory response have been
identified as critical mediators of SLE pathogenesis (41–44).
In the present study, IL-10 and IFN-γ concentrations were
demonstrated to be significantly higher in SLE PBMCs when compared
with HC PBMCs, which was consistent with previous studies (45,46)
and further supported the link between the inflammatory response
and SLE pathogenesis. Previous studies have demonstrated that
various miRNAs can attenuate the inflammatory response in SLE. Yang
et al (19) demonstrated
that miR-101 affects the secretion of inflammatory factors such as
TNF-α, IFN, IL-1 and IL-10 via its regulation of the MAPK proteins
in SLE. Liu et al (10)
demonstrated that the overexpression of miR-410 significantly
reduces IL-10 expression in T cells from patients with SLE and Sun
et al (16) confirmed that
the expression of inflammatory cytokines IL-17, IL-6 and IFN-γ
increases in SLE PBMCs when miR-101-3p expression was reduced. The
findings of the present study suggested that overexpression of
miR-101-3p reduced IL-10 and IFN-γ expression in SLE PBMCs. Taken
together, these data suggest that the overexpression of miR-101-3p
may alleviate SLE progression by modulating the inflammatory
response.
MAPK1 is involved in the regulation of cell
proliferation, adhesion and differentiation (47,48).
The production of inflammatory cytokines is responsible for the
MAPKs (49) and increasing evidence
suggests that there is a strong correlation between MAPK1 and miRNA
expression during the inflammatory response. For instance, miR-320a
is decreased in patients with myasthenia gravis and attenuates the
expression of inflammatory cytokines via its targeting of MAPK1
(50). In acute lung injuries
miR-342 suppresses lipopolysaccharide (LPS)-induced inflammatory
responses by downregulating MAPK1 (51). In addition, miR-212-3p suppresses
inflammatory cytokine production induced by the HBeAg via its
targeted regulation of MAPK1 in the liver following HBV infection
(52). The present study showed
that MAPK1 was a direct target of miR-101-3p and that miR-101-3p
could negatively regulate MAPK1 expression in SLE PBMCs. Given
this, it was hypothesized that miR-101-3p participated in the
development of SLE via its regulation of MAPK1 and that its
underlying mechanism will be similar to those described above.
Various studies have linked MAPK and the inflammation process.
Shalini et al (53) reported
that the specific p38 MAPK inhibitors prevent LPS-induced
production of IFN-γ and TNF-α in atherosclerosis. Matsuzawa et
al (54) demonstrated that p38
MAPK-mediated autophagy supports IFN-γ-mediated innate immunity and
that p38 MAPK inhibitors attenuate IFN-γ-mediated bactericidal
activity. Notably, Garcia-Rodriguez et al (55) demonstrated that MAPK1 gene
expression is upregulated in patients with SLE and that this
expression is strongly associated with IL-10 expression. Similarly,
the present study found that silencing MAPK1 significantly
increased IL-10 and IFN-γ expression in SLE PBMCs and eliminated
the inhibitory effects of miR-101-3p mimics. These results further
support the hypothesis that miR-101-3p relieved SLE-associated
inflammation via its downregulation of MAPK1.
It has been documented that the protein level of
NF-κBp65 and the ratio of p-IκBα/t-IκBα can be used to evaluate the
activity of the NF-κB pathway in SLE (49,56,57).
The present study explored the regulatory relationship between
miR-101-3p and NF-κB pathway on the inflammation of SLE. Therefore,
NF-κBp65 and the ratio of p-IκBα/t-IκBα were chosen to evaluate the
activity of the NF-κB pathway. The NF-κB pathway is involved in the
regulation of the inflammatory and immune responses (58) and miR-101 regulates the NF-κB
pathway in several diseases. For example, miR-101 attenuates
cisplatin chemoresistance by reducing NF-κB signaling in
hepatocarcinoma (59) and miR-101
silencing facilitates the pathogenesis of neuropathic pain by
promoting NF-κB activity (60). In
the present study, the expression of NF-κB-related proteins in SLE
PBMCs was markedly decreased following miR-101-3p overexpression,
indicating that miR-101-3p may be involved in the development of
SLE via its inhibition of the NF-κB pathway. Additionally, MAPK1
overexpression increased the NF-κB-related protein expression in
SLE PBMCs, suggesting that MAPK1 promoted NF-κB signaling in these
cells. Considering the interactions between miR-101-3p and MAPK1,
it was hypothesized that miR-101-3p reduced MAPK1 expression
inhibiting NF-κB signaling in SLE PBMCs. However, the specific
regulatory mechanisms underlying these interactions remain to be
elucidated.
Previous studies have demonstrated that inhibition
of the NF-κB pathway attenuates the inflammatory response in
autoimmune diseases. For instance, demethylzeylasteral alleviates
the inflammatory response in lupus nephritis by restricting NF-κB
signaling (61) and miR-146a
inhibits inflammation in the kidney tissues during SLE via its
regulation of the NF-κB pathway (62). In the present study, BAY11-7085 and
BAY11-7082 were demonstrated to effectively reverse the inhibitory
effects of miR-101-3p overexpression or promote the effects of
MAPK1 overexpression in SLE PBMCs. These feedback experiments
demonstrated that miR-101-3p reduces inflammation by inhibiting
NF-κB signaling in SLE PBMCs. Taken together the results of the
present study suggested that the overexpression of miR-101-3p could
alleviate SLE via the inhibition of MAPK1 expression and the
prevention of NF-κB signal transduction.
In conclusion, miR-101-3p is downregulated in SLE
PBMCs and may be used as an additional diagnostic marker for SLE.
miR-101-3p inhibited inflammation in SLE PBMCs via downregulation
of MAPK1 and indirect prevention of NF-κB signaling. These results
suggested that both miR-101-3p and its target MAPK1 may be
promising therapeutic targets for SLE. However, there were some
limitations to the present study. First, it was limited to the
cellular level and further in vivo experiments are required
to clarify the regulatory mechanism controlling miR-101-3p-mediated
changes to the inflammatory response in SLE. Second, there are a
number of other downstream targets and pathways of miR-101-3p that
have not yet been evaluated in SLE, suggesting that future
evaluations should consider using more high throughput technologies
such as microarrays to evaluate more targets rapidly.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (grant no. 81401337).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ, YF and SL made substantial contributions to the
conception and design of the study, acquisition of data and
analysis and interpretation of data. ZW and NB took part in
drafting the article and revising it critically for important
intellectual content. ZW and NB were also responsible for
performing the experiments and the analysis of the experimental
data, and for the management of the whole project. XZ, SL, ZW and
NB confirm the authenticity of all the raw data. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Ethics Committee of the Xijing Hospital (approval no. 20181016) and
written informed consent from both patients and volunteers were
obtained prior to sample collection.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zucchi D, Elefante E, Calabresi E,
Signorini V, Bortoluzzi A and Tani C: One year in review 2019:
Systemic lupus erythematosus. Clin Exp Rheumatol. 37:715–722.
2019.PubMed/NCBI
|
|
2
|
Tsang-A-Sjoe M and Bultink IE: Systemic
lupus erythematosus: Review of synthetic drugs. Expert Opin
Pharmacother. 16:2793–2806. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Aringer M: Inflammatory markers in
systemic lupus erythematosus. J Autoimmun. 110:1023742020.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Uzrail AH, Assaf AM and Abdalla SS:
Correlations of expression levels of a panel of genes (IRF5, STAT4,
TNFSF4, MECP2 and TLR7) and cytokine levels (IL-2, IL-6, IL-10,
IL-12, IFN-γ and TNF-α) with systemic lupus erythematosus outcomes
in Jordanian patients. Biomed Res Int. 2019:17038422019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Di Battista M, Marcucci E, Elefante E,
Tripoli A, Governato G, Zucchi D, Tani C and Alunno A: One year in
review 2018: Systemic lupus erythematosus. Clin Exp Rheumatol.
36:763–777. 2018.PubMed/NCBI
|
|
6
|
Ali M, Firoz CK, Jabir NR, Rehan M, Khan
MS and Tabrez S: An insight on the pathogenesis and treatment of
systemic lupus erythematosus. Endocr Metab Immune Disord Drug
Targets. 18:110–123. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Katsuyama E, Yan M, Watanabe KS, Narazaki
M, Matsushima S, Yamamura Y, Hiramatsu S, Ohashi K, Watanabe H,
Katsuyama T, et al: Downregulation of miR-200a-3p, targeting CtBP2
complex, is involved in the hypoproduction of IL-2 in systemic
lupus erythematosus-derived T cells. J Immunol. 198:4268–4276.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zheng B, Zhang P, Yuan L, Chhetri RK, Guo
Y and Deng D: Effects of human umbilical cord mesenchymal stem
cells on inflammatory factors and miR-181a in T lymphocytes from
patients with systemic lupus erythematosus. Lupus. 29:126–135.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cao W, Qian G, Luo W, Liu X, Pu Y, Hu G,
Han L, Yuan L, A X and Deng D: miR-125b is downregulated in
systemic lupus erythematosus patients and inhibits autophagy by
targeting UVRAG. Biomed Pharmacother. 99:791–797. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu D, Zhang N, Zhang X, Qin M, Dong Y and
Jin L: MiR-410 down-regulates the expression of interleukin-10 by
targeting STAT3 in the pathogenesis of systemic lupus
erythematosus. Cell Physiol Biochem. 39:303–315. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ren D, Liu F, Dong G, You M, Ji J, Huang
Y, Hou Y and Fan H: Activation of TLR7 increases CCND3 expression
via the downregulation of miR-15b in B cells of systemic lupus
erythematosus. Cell Mol Immunol. 13:764–775. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen S, Wang Y, Qin H, Lin J, Xie L, Chen
S, Liang J and Xu J: Downregulation of miR-633 activated AKT/mTOR
pathway by targeting AKT1 in lupus CD4+ T cells. Lupus.
28:510–519. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rasmussen TK, Andersen T, Bak RO, Yiu G,
Sørensen CM, Stengaard-Pedersen K, Mikkelsen JG, Utz PJ, Holm CK
and Deleuran B: Overexpression of microRNA-155 increases IL-21
mediated STAT3 signaling and IL-21 production in systemic lupus
erythematosus. Arthritis Res Ther. 17:1542015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu Y, Dong J, Mu R, Gao Y, Tan X, Li Y,
Li Z and Yang G: MicroRNA-30a promotes B cell hyperactivity in
patients with systemic lupus erythematosus by direct interaction
with Lyn. Arthritis Rheum. 65:1603–1611. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wei Q, Lv F, Zhang H, Wang X, Geng Q,
Zhang X, Li T, Wang S, Wang Y and Cui Y: MicroRNA-101-3p inhibits
fibroblast-like synoviocyte proliferation and inflammation in
rheumatoid arthritis by targeting PTGS2. Biosci Rep.
40:BSR201911362020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun H, Guo F and Xu L: Downregulation of
microRNA-101-3p participates in systemic lupus erythematosus
progression via negatively regulating HDAC9. J Cell Biochem.
121:4310–4320. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gang C, Jiahui Y, Huaizhou W, Qing C,
Dongbao Z and Qian S: Defects of mitogen-activated protein kinase
in ICOS signaling pathway lead to CD4(+) and CD8(+) T-cell
dysfunction in patients with active SLE. Cell Immunol. 258:83–89.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu Y, Deng W, Meng Q, Qiu X, Sun D and
Dai C: CD8+iTregs attenuate glomerular endothelial cell injury in
lupus-prone mice through blocking the activation of p38 MAPK and
NF-κB. Mol Immunol. 103:133–143. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang J, Lu YW, Lu MM, Leng RX, Pan HF and
Ye DQ: MicroRNA-101, mitogen-activated protein kinases and
mitogen-activated protein kinases phosphatase-1 in systemic lupus
erythematosus. Lupus. 22:115–120. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hellweg CE: The nuclear factor κB pathway:
A link to the immune system in the radiation response. Cancer Lett.
368:275–289. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cao HY, Li D, Wang YP, Lu HX, Sun J and Li
HB: The protection of NF-κB inhibition on kidney injury of systemic
lupus erythematosus mice may be correlated with lncRNA TUG1.
Kaohsiung J Med Sci. 36:354–362. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu J, Huang X, Hao S, Wang Y, Liu M, Xu
J, Zhang X, Yu T, Gan S, Dai D, et al: Peli1 negatively regulates
noncanonical NF-κB signaling to restrain systemic lupus
erythematosus. Nat Commun. 9:11362018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ji L, Hou X, Liu W, Deng X, Jiang Z, Huang
K and Li R: Paeoniflorin inhibits activation of the IRAK1-NF-κB
signaling pathway in peritoneal macrophages from lupus-prone
MRL/lpr mice. Microb Pathog. 124:223–229. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou C, Zhao L, Wang K, Qi Q, Wang M, Yang
L, Sun P and Mu H: MicroRNA-146a inhibits NF-κB activation and
pro-inflammatory cytokine production by regulating IRAK1 expression
in THP-1 cells. Exp Ther Med. 18:3078–3084. 2019.PubMed/NCBI
|
|
25
|
Zhang W, Yu T, Cui X, Yu H and Li X:
Analgesic effect of dexmedetomidine in rats after chronic
constriction injury by mediating microRNA-101 expression and the
E2F2-TLR4-NF-κB axis. Exp Physiol. 105:1588–1597. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hochberg MC: Updating the American college
of rheumatology revised criteria for the classification of systemic
lupus erythematosus. Arthritis Rheum. 40:17251997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bombardier C, Gladman DD, Urowitz MB,
Caron D and Chang CH: Derivation of the SLEDAI. A disease activity
index for lupus patients. The committee on prognosis studies in
SLE. Arthritis Rheum. 35:630–640. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fava A and Petri M: Systemic lupus
erythematosus: Diagnosis and clinical management. J Autoimmun.
96:1–13. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xie L and Xu J: Role of MiR-98 and its
underlying mechanisms in systemic lupus erythematosus. J Rheumatol.
45:1397–1405. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Luo S, Liu Y, Liang G, Zhao M, Wu H, Liang
Y, Qiu X, Tan Y, Dai Y, Yung S, et al: The role of microRNA-1246 in
the regulation of B cell activation and the pathogenesis of
systemic lupus erythematosus. Clin Epigenetics. 7:242015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sarhan RA, Aboelenein HR, Sourour SK,
Fawzy IO, Salah S and Abdelaziz AI: Targeting E2F1 and c-Myc
expression by microRNA-17-5p represses interferon-stimulated gene
MxA in peripheral blood mononuclear cells of pediatric systemic
lupus erythematosus patients. Discov Med. 19:419–425.
2015.PubMed/NCBI
|
|
33
|
Motawi TK, Mohsen DA, El-Maraghy SA and
Kortam MA: MicroRNA-21, microRNA-181a and microRNA-196a as
potential biomarkers in adult Egyptian patients with systemic lupus
erythematosus. Chem Biol Interact. 260:110–116. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Clancy R, El Bannoudi H, Rasmussen SE,
Bornkamp N, Allen N, Dann R, Reynolds H, Buyon JP and Berger JS:
Human low-affinity IgG receptor FcγRIIA polymorphism H131R
associates with subclinical atherosclerosis and increased platelet
activity in systemic lupus erythematosus. J Thromb Haemost.
17:532–537. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yuan W, Cao H, Wan P, Shi R, Zhou S and
Zheng J: Clinical evaluation of total and high-avidity anti-dsDNA
antibody assays for the diagnosis of systemic lupus erythematosus.
Lupus. 28:1387–1396. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sun J, Li X, Zhou H, Liu X, Jia J, Xie Q,
Peng S, Sun X, Wang Q and Yi L: Anti-GAPDH autoantibody is
associated with increased disease activity and intracranial
pressure in systemic lupus erythematosus. J Immunol Res.
2019:74307802019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yuan S, Tang C, Chen D, Li F, Huang M, Ye
J, He Z, Li W, Chen Y, Lin X, et al: miR-98 modulates cytokine
production from human PBMCs in systemic lupus erythematosus by
targeting IL-6 mRNA. J Immunol Res. 2019:98275742019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li HS, Ning Y, Li SB, Shao PY, Chen SJ, Ye
Q and Heng X: Expression and clinical significance of miR-181a and
miR-203 in systemic lupus erythematosus patients. Eur Rev Med
Pharmacol Sci. 21:4790–4796. 2017.PubMed/NCBI
|
|
39
|
Lin LJ, Mai LJ, Chen G, Zhao EN, Xue M and
Su XD: Expression and diagnostic value of plasma miR-145 and
miR-183 in children with lupus nephritis. Zhongguo Dang Dai Er Ke
Za Zhi. 22:632–637. 2020.(In Chinese). PubMed/NCBI
|
|
40
|
Yao Y, Wang JB, Xin MM, Li H, Liu B, Wang
LL, Wang LQ and Zhao L: Balance between inflammatory and regulatory
cytokines in systemic lupus erythematosus. Genet Mol Res. 15:1–8.
2016. View Article : Google Scholar
|
|
41
|
Godsell J, Rudloff I, Kandane-Rathnayake
R, Hoi A, Nold MF, Morand EF and Harris J: Clinical associations of
IL-10 and IL-37 in systemic lupus erythematosus. Sci Rep.
6:346042016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yuan Y, Wang X, Ren L, Kong Y, Bai J and
Yan Y: Associations between interleukin-10 gene polymorphisms and
systemic lupus erythematosus risk: A meta-analysis with trial
sequential analysis. Clin Exp Rheumatol. 37:242–253.
2019.PubMed/NCBI
|
|
43
|
Liu M, Liu J, Hao S, Wu P, Zhang X, Xiao
Y, Jiang G and Huang X: Higher activation of the interferon-gamma
signaling pathway in systemic lupus erythematosus patients with a
high type I IFN score: Relation to disease activity. Clin
Rheumatol. 37:2675–2684. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kokic V, Martinovic Kaliterna D, Radic M,
Perkovic D, Cvek M and Capkun V: Relationship between vitamin D,
IFN-γ, and E2 levels in systemic lupus erythematosus. Lupus.
25:282–288. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jiao Q, Qian Q, Zhao Z, Fang F, Hu X, An
J, Wu J and Liu C: Expression of human T cell immunoglobulin domain
and mucin-3 (TIM-3) and TIM-3 ligands in peripheral blood from
patients with systemic lupus erythematosus. Arch Dermatol Res.
308:553–561. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dahal LN, Basu N, Youssef H, Khanolkar RC,
Barker RN, Erwig LP and Ward FJ: Immunoregulatory soluble CTLA-4
modifies effector T-cell responses in systemic lupus erythematosus.
Arthritis Res Ther. 18:1802016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bashanfer SAA, Saleem M, Heidenreich O,
Moses EJ and Yusoff NM: Disruption of MAPK1 expression in the ERK
signalling pathway and the RUNX1-RUNX1T1 fusion gene attenuate the
differentiation and proliferation and induces the growth arrest in
t(8;21) leukaemia cells. Oncol Rep. 41:2027–2040. 2019.PubMed/NCBI
|
|
48
|
Zhu Y, Yang T, Duan J, Mu N and Zhang T:
MALAT1/miR-15b-5p/MAPK1 mediates endothelial progenitor cells
autophagy and affects coronary atherosclerotic heart disease via
mTOR signaling pathway. Aging (Albany NY). 11:1089–1109. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Aparicio-Soto M, Sánchez-Hidalgo M,
Cárdeno A, Rosillo MÁ, Sánchez-Fidalgo S, Utrilla J, Martín-Lacave
I and Alarcón-de-la-Lastra C: Dietary extra virgin olive oil
attenuates kidney injury in pristane-induced SLE model via
activation of HO-1/Nrf-2 antioxidant pathway and suppression of
JAK/STAT, NF-κB and MAPK activation. J Nutr Biochem. 27:278–288.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cheng Z, Qiu S, Jiang L, Zhang A, Bao W,
Liu P and Liu J: MiR-320a is downregulated in patients with
myasthenia gravis and modulates inflammatory cytokines production
by targeting mitogen-activated protein kinase 1. J Clin Immunol.
33:567–576. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhu S, Song W, Sun Y, Zhou Y and Kong F:
MiR-342 attenuates lipopolysaccharide-induced acute lung injury via
inhibiting MAPK1 expression. Clin Exp Pharmacol Physiol.
47:1448–1454. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chen W, Bian H, Xie X, Yang X, Bi B, Li C,
Zhang Y, Zhu Q, Song J, Qin C and Qi J: Negative feedback loop of
ERK/CREB/miR-212-3p inhibits HBeAg-induced macrophage activation. J
Cell Mol Med. 24:10935–10945. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shalini V, Pushpan CK, G S, A J and A H:
Tricin, flavonoid from Njavara reduces inflammatory responses in
hPBMCs by modulating the p38MAPK and PI3K/Akt pathways and prevents
inflammation associated endothelial dysfunction in HUVECs.
Immunobiology. 221:137–144. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Matsuzawa T, Fujiwara E and Washi Y:
Autophagy activation by interferon-γ via the p38 mitogen-activated
protein kinase signalling pathway is involved in macrophage
bactericidal activity. Immunology. 141:61–69. 2013. View Article : Google Scholar
|
|
55
|
Garcia-Rodriguez S, Callejas-Rubio JL,
Ortego-Centeno N, Zumaquero E, Ríos-Fernandez R, Arias-Santiago S,
Navarro P, Sancho J and Zubiaur M: Altered AKT1 and MAPK1 gene
expression on peripheral blood mononuclear cells and correlation
with T-helper-transcription factors in systemic lupus erythematosus
patients. Mediators Inflamm. 2012:4959342012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Shi X, Qian T, Li M, Chen F, Chen Y and
Hao F: Aberrant low expression of A20 in tumor necrosis
factor-α-stimulated SLE monocytes mediates sustained NF-κB
inflammatory response. Immunol Invest. 44:497–508. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kong X, Zhang Z, Fu T, Ji J, Yang J and Gu
Z: TNF-α regulates microglial activation via the NF-κB signaling
pathway in systemic lupus erythematosus with depression. Int J Biol
Macromol. 125:892–900. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Sun SC: The non-canonical NF-κB pathway in
immunity and inflammation. Nat Rev Immunol. 17:545–558. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Chai Z, Yin X, Chen J, Shi J, Sun J, Liu
C, Liu F and Cheng S: MicroRNA-101 modulates cisplatin
chemoresistance in liver cancer cells via the DNA-PKcs signaling
pathway. Oncol Lett. 18:3655–3663. 2019.PubMed/NCBI
|
|
60
|
Liu JC, Xue DF, Wang XQ, Ai DB and Qin PJ:
MiR-101 relates to chronic peripheral neuropathic pain through
targeting KPNB1 and regulating NF-κB signaling. Kaohsiung J Med
Sci. 35:139–145. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hu Q, Yang C, Wang Q, Zeng H and Qin W:
Demethylzeylasteral (T-96) treatment ameliorates mice lupus
nephritis accompanied by inhibiting activation of NF-κB pathway.
PLoS One. 10:e01337242015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Fu HX, Fan XP, Li M, Liu MJ and Sun QL:
MiR-146a relieves kidney injury in mice with systemic lupus
erythematosus through regulating NF-κB pathway. Eur Rev Med
Pharmacol Sci. 23:7024–7032. 2019.PubMed/NCBI
|