Introduction
Hepatic fibrosis (HF) is a common pathological
process that occurs during the development of various chronic liver
diseases, such as liver cirrhosis, which initiates a series of
biochemical and biophysical changes in the hepatic microenvironment
(1,2). It is characterized by a radial linear
deposition of collagen around a single hepatocyte and in the space
of Disse (3). The progression and
regression of HF also relies on a complex interplay of the hepatic
microenvironment (4). Hepatic
sinusoids provide a biochemical environment for the survival and
communication of non-parenchymal hepatocytes, including liver
sinusoidal endothelial cells (LSECs), hepatic stellate cells (HSCs)
and Kupffer cells (KCs) (5).
Intercellular interactions among non-parenchymal hepatocytes are
considered to serve a pivotal role in maintaining normal liver
function and structure (6,7).
Hepatic sinusoids are specialized capillaries
located between liver plates. The wall of the hepatic sinusoid is
composed of LSECs, which are distinctive micro-vascular cells that
are key to the regulation of the liver microenvironment and
represent a permeable barrier to maintain HSC quiescence (8). Under normal circumstances,
differentiated LSECs are characterized by the presence of
fenestrations and the absence of basement membrane (BM). The
fenestration is the most characteristic structure of LSECs and
renders the cells highly permeable (9). It also enables the bidirectional
transport of metabolites between the circulation and liver
parenchyma (10). Actin is the
cytoskeletal component of LSECs, which participates in the
contraction and expansion of the fenestrae (11). The narrow space between LSECs and
liver plate is known as the space of Disse, which contains the
extracellular matrix (ECM) and separates sinusoidal cells from
parenchymal cells (5). HSCs are
specialized pericytes with lipid and retinoid droplets in the
cytoplasm that are located in the subendothelial area of hepatic
sinusoids. They directly communicate with hepatocytes and LSECs
(12). KCs are broadly located in
hepatic sinusoids and have an irregular morphology, protruding into
hepatic sinusoids or remaining unattached (13,14).
There are a number of pseudopods on the surface of KCs, which
attach to LSECs or extend into the space of Disse through the
fenestrations, and directly interact with hepatocytes and HSCs
(15).
When the normal hepatic microenvironment is
destroyed, the balance between parenchymal hepatocytes and stromal
cells is disturbed, leading to the capillarization of LSECs, which
is also known as de-differentiation (8). This process increases intrahepatic
blood flow resistance and provides a pathological basis for portal
hypertension (16). Hepatic
sinusoidal capillarization is characterized by the lack of
fenestration and formation of organized BM. It is not only a
precursor of HF, but also promotes HSC activation. Therefore, the
disordered LSEC phenotype is a key step in the process of HF
(17). In the process of
inflammation and tissue repair following chronic liver injury, HSCs
lose their droplets (18). Various
signals lead to the activation of resting HSCs and transformation
into myofibroblasts (MFBs), which increases the expression of
a-smooth muscle actin (α-SMA), thereby enhancing contractility and
hardening tissue (19,20). Activated HSCs are the main source of
stromal MFBs; they lead to an imbalance between ECM synthesis and
degradation (21). Intercellular
interactions among non-parenchymal hepatocytes results in HSC
activation and HF (22).
Additionally, activated KCs produce various cytokines, resulting in
neovascularization and an increase in intra-sinusoid pressure
(23). Autocrine and paracrine
signals among non-parenchymal hepatocytes regulate the progression
of HF in unison, as presented in Fig.
1.
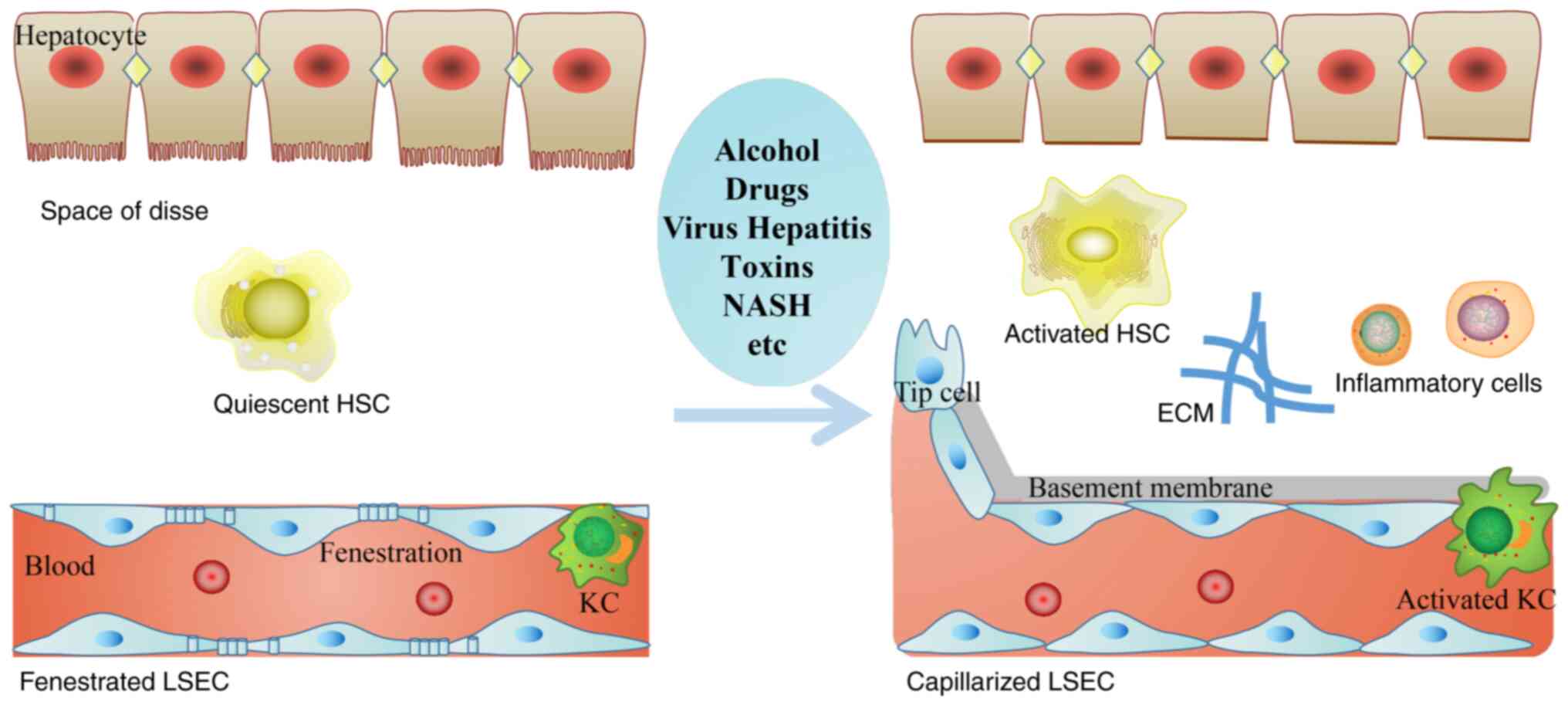 | Figure 1.Hepatic sinusoids microenvironment
alterations are induced by various chronic liver injuries. In the
process of hepatic fibrosis, the morphology and function of LSECs,
HSCs and KCs are abnormal, forming the subendothelial basement
membrane, and decreasing endothelial cell fenestrations. HSC and KC
cause a series of inflammatory responses, ECM deposition and
angiogenesis. HSCs, hepatic stellate cells; KCs, Kupffer cells;
ECM, extracellular matrix; LSEC, liver sinusoidal endothelial
cells; NASH, non-alcoholic steatohepatitis. |
LSECs regulate the hepatic microenvironment,
activate HSCs and promote HF
LSECs are the first cell type to be affected by
various etiologies in the process of HF (24). They serve a key role in hepatic
sinusoidal capillarization, angiogenesis and vasoconstriction, and
participate in the process of HF in three key ways (8,17,25).
Direct secretion of ECM by LSECs
aggravates tissue hypoxia
After liver injury, LSECs acquire a fibrogenic
phenotype and participate in HF through the direct secretion of ECM
(24). Capillarization of the
hepatic sinusoids results in a narrowing of the sinusoidal lumen
and hypoxia (26). Under hypoxic
conditions, LSECs are hypothesized to produce ECM (27). Furthermore, LSECs from fibrotic
animals have resulted in the upregulation of type I collagen mRNA,
which is 5-fold higher than that of the normal liver (28). They also secrete a large number of
components to form an organized BM, including type IV collagen,
laminin, nidogen, entactin and perlecan (27). Meanwhile, increasing studies have
revealed the pivotal role of hypoxia in the activation of HSCs.
Hypoxia induces the activation of HSCs by different mechanisms,
including the transforming growth factor (TGF)-β, AMPK-mTOR, PKCθ
and PVT1 oncogene-microRNA-152-autophagy-related protein 14
signaling pathways (29–31). Activated HSCs produce excessive
fibrotic mediators, such as α-SMA, type I collagen, matrix
metalloproteinase (MMP)-2 and TGF receptors, which promote the
abnormal accumulation of ECM (26).
During the development of HF, there is an abnormal increase in the
formation of collagen, as well as other ECM molecules, and the
total collagen volume increases nearly 10-fold (32). The massive accumulation of ECM
restricts microvascular blood flow, accentuates the narrowing and
deformation of hepatic sinusoids, and enhances hypoxia (26).
LSECs indirectly secrete cytokines to
promote HSC activation
In addition to the direct secretion of ECM
components, LSECs also indirectly contribute to HF by secreting
proinflammatory and profibrogenic factors, including TGF-β1 and
platelet-derived growth factors (PDGFs), which activate HSCs and
promote the synthesis of the ECM (11). TGF-β1 is a polypeptide growth factor
that participates in proliferation, differentiation, migration and
apoptosis of HSCs as well as other cell types, via the
TGF-β-receptor/Smads pathway (3).
TGF-β1 signaling also stimulates the transformation of HSCs into
MFBs (33). It has been reported
that TGF-β1 gene transfection can lead to the rapid development of
HF in vivo (3). The
increased expression of TGF-β1 not only recruits inflammatory cells
and fibroblasts into the injured region causing subsequent HSC
activation, but also induces the synthesis of certain BM proteins,
including laminin, type IV collagen and entactin in LSECs (34). PDGFs are potent mitogens for HSCs
(35). HSCs express PDGF receptor
(R)-α and PDGFR-β. The PDGF family, including their ligands and
receptors, serve pivotal roles in tissue repair and the formation
of ECM (36,37). PDGF-B and PDGF-D strongly promote
the migration, the conversion of HSCs to MFBs, ECM deposition and
the modulation of the tissue inhibitor of metalloproteinase
(TIMP)/MMP system (37,38). A previous study demonstrated that
PDGFR expression was increased in chronic liver disease and
overexpression of PDGF ligand induced fibrosis in mice (35). Deletion of PDGFR-β on HSCs decreased
the expression of α-SMA and type I collagen, alleviating HF
(39). Furthermore, the selective
blocking of PDGFR in HF has been found to reduce the proliferation,
survival and migration of activated HSCs (35). LSECs stimulate HSCs migration and
recruitment to sinusoids by secreting PDGF ligands and TGF-β
(40). The enhanced motility of
HSCs can facilitate their coverage around the hepatic sinusoids
(41). In addition, it has been
reported that PDGF promotes the angiogenesis phenotype of HSCs,
regulating vascular tube formation and increasing the coverage of
hepatic sinusoids, thus affecting the permeability and pressure of
hepatic sinusoids (25).
In addition to TGF-β and PDGFs, fibronectin-splice
variant containing extra domain A (Fn-EDA) induce the
trans-differentiation of HSCs to MFBs, which takes place in the
initial stages of HF (27). In
vitro studies have revealed that Fn-EDA promotes the
differentiation of proto-myofibroblasts into MFBs (27,42).
Furthermore, the increased expression of α-SMA, which is the
cellular marker of MFBs. TGF-β activation of α-SMA expression
results from the cooperation of different signal transduction
pathways raised respectively by TGF-β and Fn-EDA (43). It has been demonstrated that TGF-β
acts on LSECs to upregulate the rapid production of Fn-EDA
(43). Although HSCs produce
Fn-EDA, LSECs are the first responders and therefore may serve a
key role in the early stages of HF (27).
LSECs regulate the vascular tension of
hepatic sinusoids via paracrine action
LSECs regulate vascular tone by secreting certain
vasodilators or vasoconstrictors, such as endothelin 1 (ET-1),
thromboxane A2, cyclooxygenase 1 and nitric oxide (NO), all of
which can act on HSCs to regulate their contraction, thus
regulating hepatic sinusoidal pressure (25). For example, ET-1 belongs to a family
of potent vasoconstrictor peptides and is produced by LSECs or
activated HSCs in a paracrine or autocrine manner (44). It was originally identified from
endothelial cells (45). Elevated
serum ET-1 levels have been identified in all stages of HF
(45). In rat livers, the ET
receptor is detected in all cell types, but most abundant in HSCs.
Under normal conditions, HSCs express both endothelin A receptors
(ETARs) and endothelin B receptors (ETBRs) (45). In the process of cirrhosis, ET-1 may
have a stronger effect on HSCs by enhancing ETARs and ETBRs,
resulting in increased microvascular tone in the hepatic sinusoids
(46). ET-1 has also been revealed
to upregulate the expression of procollagen I and TGF-β1 through
ETARs (47), suggesting that the
biological effects of ET-1 on HSCs include not only increasing
intra-sinusoidal pressure via the activation of mitogen-activated
protein kinases, increased Ca2+ influx and reversible
cell contraction, but also by increasing collagen synthesis and
secretion (Fig. 2) (48).
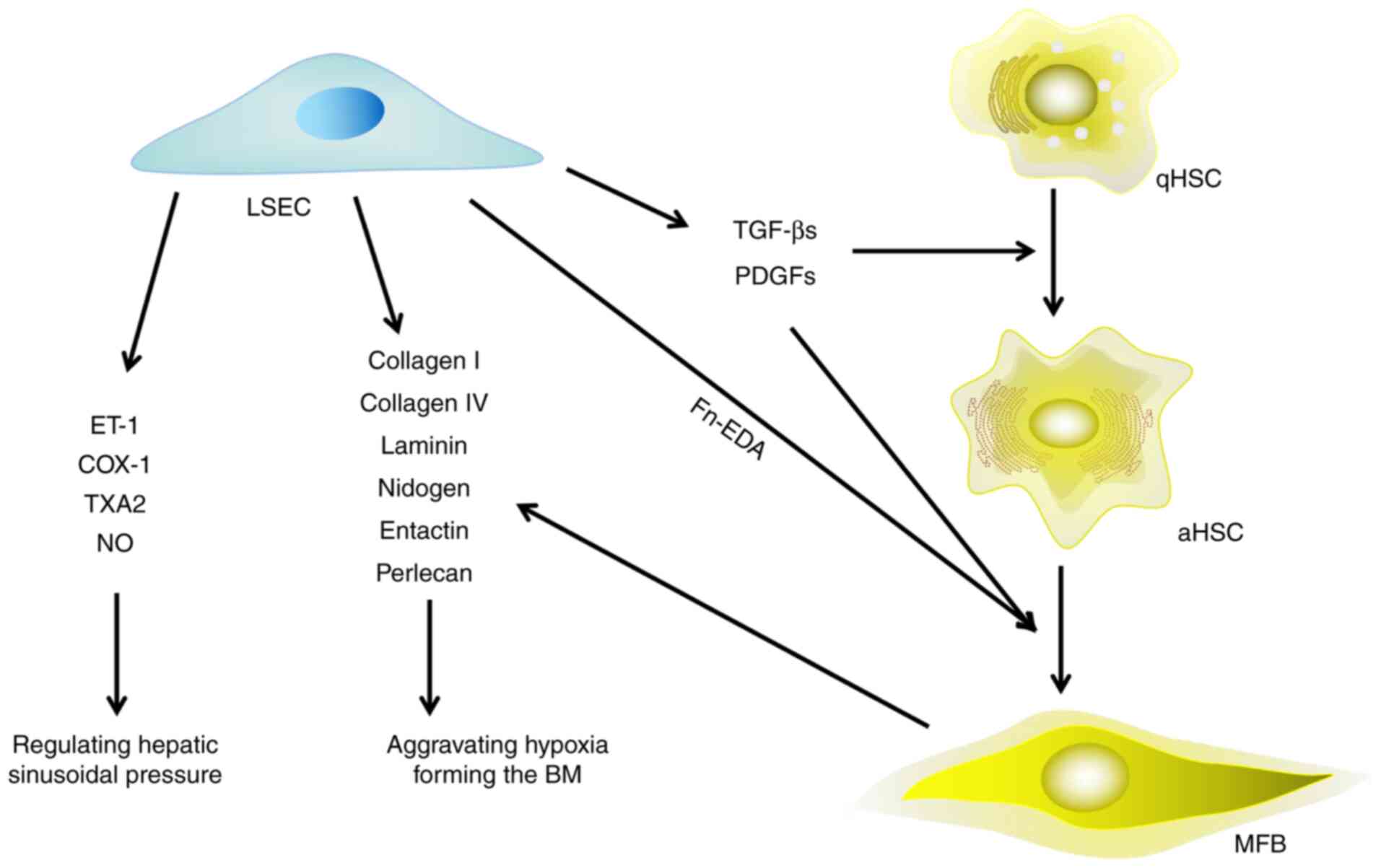 | Figure 2.Effect of LSECs on HSCs, hepatic
microenvironment and HF. LSECs are involved in the process of HF in
three aspects: i) Secreting extracellular matrix directly; ii)
secreting growth factors to promote the activation of HSCs and
their trans-differentiation to myofibroblasts; and iii) releasing
vasoconstrictors to regulate the pressure of hepatic sinusoids.
LSECs, liver sinusoidal endothelial cells; HSCs, hepatic stellate
cells; HF, hepatic fibrosis; qHSCs, quiescent hepatic stellate
cells; aHSCs, activated hepatic stellate cells; MFB, myofibroblast;
ET-1, endothelin 1; COX-1, cyclooxygenase 1; TXA2, thromboxane A2;
NO, nitric oxide; Fn-EDA, fibronectin-splice variant containing
extra domain A; BM, basement membrane; TGF-β, transforming growth
factor-β; PDGF, platelet derived growth factor. |
Activated HSCs interact with LSCSs to
regulate the hepatic microenvironment
The interaction between LSECs and HSCs is crucial
for maintaining the structure and function of hepatic sinusoids
(49). HSCs interact with LSECs in
a bidirectional manner (49). As
aforementioned, LSECs produce signals that activate HSCs (17,25),
which contributes to the capillarization of LSECs (24). HSCs promote the loss of LSEC
fenestrations and the formation of continuous BM through
inflammatory factors, cytokines, chemokines and vasoactive
mediators (50). HSCs also induce
angiogenic vascular tissue remodeling and increase vascular
resistance (51,52).
Capillarization of hepatic sinusoids
is partially associated with the accumulation of ECM
In response to liver injury, quiescent HSCs
transform into MFBs, which are the primary producers of ECM
(53). MMPs serve a crucial role in
ECM degradation, which is antagonized by TIMPs. Therefore, the
homeostasis of ECM is coordinated by its secretion and degradation,
the latter of which is partially determined by the balance between
MMPs and TIMPs (54). When HSCs are
activated, the expression of type I collagen, α-SMA and TIMP-1 are
upregulated, and the expression of MMP-2 is downregulated,
reversing the TIMP-1/MMP-2 ratio and resulting in excessive ECM
deposition and HF (55).
Under normal conditions, the hepatic sinusoids lack
BM. In chronic liver disease, activated HSCs in the space of Disse
produce various ECM components, including collagen IV, laminin and
perlecan (27). Type IV collagen,
laminin, nidogen and perlecan are the major components of BM
(56,57). The network of type IV collagen and
laminin is linked by nidogen, which is hypothesized to be the main
driving force for BM assembly (58). The co-distribution of type IV
collagen and laminin is a histochemical marker of BM formation in
liver disease (59). With the
increase of ECM deposition in hepatic sinusoids, the number of LSEC
fenestrations decreases (60).
Along with BM formation, LSECs lose their fenestrae and transform
into vascular type endothelium. These changes lead to hepatic
sinusoidal capillarization, which seriously impedes hepatic
function (59). The capillarization
of LSECs is also characterized by the decrease of LSEC defensive
function and the increase of platelet endothelial cell adhesion
molecule-1 and Laminin 1 protein surface expression (61). The physical barrier produced by the
newly formed BM impairs the intercellular exchange between
parenchymal hepatocytes and the lumen (62).
In addition to ECM oversecretion, the off switch of
certain signals that maintain hepatic microenvironment homeostasis
in activated HSCs and LSECs contribute to the capillarization of
hepatic sinusoids (63). In the
healthy liver, physiological shear stress can activate the
transcription factor Krüppel-like factor 2 in exposed LSECs,
resulting in the increase of vasodilators, including NO, and the
downregulation of vasoconstrictor molecules, including ET-1,
thereby alleviating the intra-sinusoidal pressure, reducing the
shear stress and avoiding further injury (8). Bone morphogenetic protein 9 is a
paracrine factor produced by HSCs, which maintains the quiescent
state of LSECs, as well as their fenestration and terminal
differentiation by binding to activin receptor like kinase 1
(64). In the process of HF, LSECs
lose their aforementioned defensive signals, resulting in their
impaired ability to degrade ECM, as well as the reduction of
fenestrations and the formation of a continuous BM (11).
HSCs regulate the contraction of
hepatic sinusoids
HSCs belong to hepatic sinusoidal pericytes and are
in close contact with LSECs, meaning that their contractility
regulates the contraction of hepatic sinusoids and portal vein
pressure (52,65). A single HSC can surround up to four
individual sinusoids, and when activated, can extend itself to
cover these cells (5). Previous
studies have demonstrated that junctional adhesion molecule-C
(JAM-C) expressed in HSCs interacts with junctional adhesion
molecule-B (JAM-B) expressed in LSECs (66,67).
The enhanced contractibility of HSCs, which is modulated by
JAM-C/JAM-B interaction, contributes to hepatic sinusoidal
vasoconstriction, thereby increasing hepatic sinusoidal resistance
and subsequently inducing portal hypertension (66). C-X-C chemokine receptor 4 (CXCR4) is
also expressed in activated HSCs, which combines with C-X-C motif
chemokine ligand 12 (CXCL12), inducing HSCs to enhance the
production of type I collagen and α-SMA (68). Moreover, CXCL12 promotes HSC
migration, chemotaxis, contraction and the phosphorylation of
myosin light chain in a Rho kinase-dependent manner (68). The latter is necessary for actin
stress fiber assembly, as well as HSC contraction and chemotaxis
(69). Vasodilators, including ET-1
and NO, are involved in the regulation of HSC contraction after
activation of the Rho/rock signaling pathway (65).
HSCs participate in pathological
angiogenesis
HSCs produce various biological molecules to
co-regulate LSEC capillarization and angiogenesis, including
vascular endothelial growth factor (VEGF), placental growth factor
(PLGF) and angiopoietin (Ang) (56,65,70).
VEGF is a key regulator of angiogenesis. VEGF, as
well as its receptor VEGFR1, are reported to be expressed in
hypoxia-stimulated activated HSCs (71). Overexpression of VEGF promotes HF by
secreting ECM, whereas inhibition of either VEGFR-1 or VEGFR-2
significantly attenuates HF and angiogenesis (72). VEGF disrupts the balance of
intercellular contact at the adhesion junction, enhancing
permeability and leading to BM formation (50). VEGF can also stimulate LSEC
proliferation and migration (73).
In a normal liver microenvironment, differentiated LSECs suppress
HSC activation and convert activated HSCs to the quiescence state
through enhanced NO production induced by VEGF. However, when LSECs
are de-differentiated or capillarized, they lose this effect
(74,75). Additionally, LSEC fenestration is
maintained by the paracrine secretion of VEGF by HSCs and its own
production of NO (63). Therefore,
VEGF maintains endothelial fenestrations under physiological
conditions. However, under pathological conditions, VEGF promotes
the microcirculation disorder and the capillarization of hepatic
sinusoids, resulting in dysfunction and structural changes in
hepatic sinusoids (50).
PLGF is a member of the VEGF family and a specific
ligand of VEGFR1. It was first isolated from human placenta and was
demonstrated to participate in pathological angiogenesis during
chronic liver disease (76). It has
been demonstrated that PLGF levels are increased in patients with
cirrhosis and CCl4-induced cirrhotic mice, and are
mainly distributed in activated HSCs and macrophages (77). PLGF also promotes hepatic
inflammation, angiogenesis and HF by mediating macrophage
recruitment and activation (78).
PLGF induces HSC activation and proliferation via the
phosphatidylinositol 3-kinase/Akt signaling pathway (79). Furthermore, PLGF silencing can
effectively alleviate inflammation and HF, inhibiting the
activation of HSCs (76).
Suppression of PLGF activity also alleviates the severity of HF,
inflammation and portal hypertension (80). PLGF may therefore serve as a novel
target for HF therapy.
The Ang/tyrosine kinase with immunoglobulin-like and
EGF-like domains (TIE) signaling pathway participates in vascular
homeostasis (81). Additionally,
Ang family members, including Ang-1 and Ang-2, regulates
angiogenesis. Their receptors, TIE-1 and TIE-2, are tyrosine kinase
that are expressed on LSECs. Ang-1 secreted by HSCs can bind to
TIE-2, stabilizing newly formed vessels (26). TIE-1 maintains the structural
integrity of endothelial cells, while TIE-2 is involved in
angiogenesis (26). Ang-1 and Ang-2
also modulate vascular stability and vascular permeability by
competitively binding to TIE-2 receptors (Fig. 3) (82).
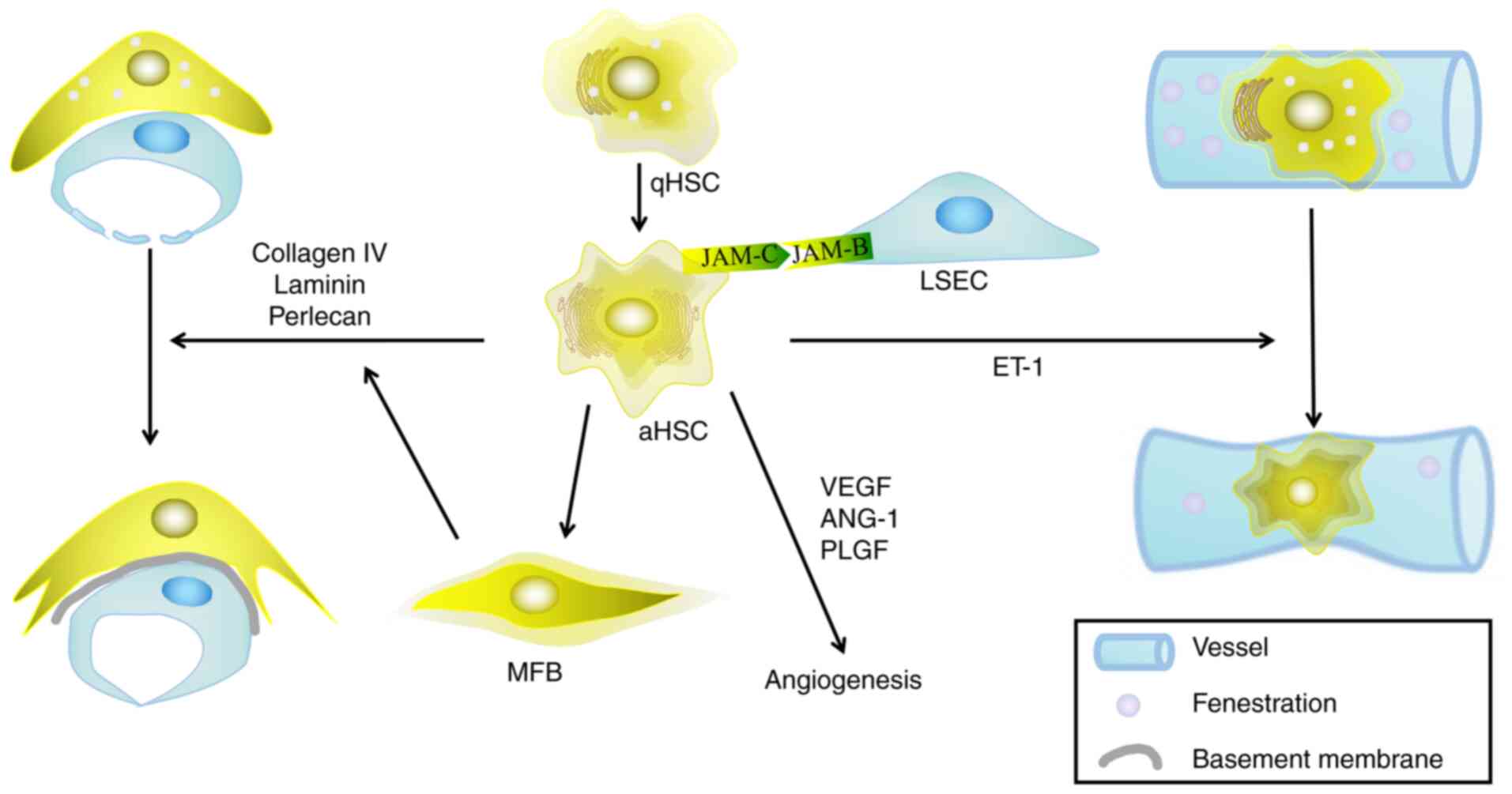 | Figure 3.Activated HSCs interact with LSECs to
regulate the hepatic microenvironment. HSCs are activated by
various liver injuries, during which stationary HSCs are
transformed into activated HSCs, leading to the remodeling of
hepatic sinusoids through a series of steps. The interaction
between HSCs and LSECs mainly manifest in the following ways: i)
The secretion of extracellular matrix reduces the number of
fenestrations in the LSECs; ii) HSCs act as pericytes to regulate
the pressure of hepatic sinusoids; and iii) HSCs secrete a series
of mediators that regulate angiogenesis. HSCs, hepatic stellate
cells; LSECs, liver sinusoidal endothelial cells; qHSC, quiescent
hepatic stellate cell; aHSC, activated hepatic stellate cell; MFB,
myofibroblast; VEGF, vascular endothelial growth factor; ANG,
angiopoietin; PLGF, placental growth factor; ET-1, endothelin 1;
JAM-, junctional adhesion molecule. |
Macrophages serve a dual role in HF and
participate in pathological angiogenesis
Previous studies have indicated the significant role
of macrophages in the regulation of HF (83–85).
Macrophages are myeloid-derived immune cells that can engulf and
degrade dead cells or foreign substances and coordinate the
inflammatory process in response (86). Macrophages not only secrete various
cytokines and growth factors, which act on HSCs and LSECs to induce
the progression and regression of hepatic fibrosis (23), but they also promote the secretion
of angiogenic factors, thus promoting neovascularization (87).
KCs interact with HSCs and other
non-parenchymal cells to aggravate HF
Intrahepatic macrophages include KCs and
infiltrating macrophages (IMs). KCs are the dominant type that
aggravate liver injury and fibrosis by mediating inflammatory
response (88).
KCs interact closely with other non-parenchymal
cells in hepatic sinusoids and mutually interact with HSCs
(89). When the liver is damaged,
the number of macrophages increases dramatically, and they rapidly
secrete various proinflammatory cytokines and chemokines, including
interleukin (IL)-1, IL-17, tumor necrosis factor-α (TNF-α), TGF-β,
C-C chemokine ligand (CCL)-2 and CCL-5, leading to the paracrine
activation of HSCs (90,91). HSCs express vascular cell adhesion
molecular-1, intercellular adhesion molecule (ICAM)-1, E-selection
and other adhesion molecules to recruit KCs, thereby infiltrating
macrophages and circulating monocytes (92).
ILs are a group of cytokines secreted by a broad
spectrum of cells, including CD4-positive T lymphocytes, monocytes,
macrophages and endothelial cells (91). Additionally, KCs and LSECs produce
ILs rapidly after injury. Previous studies have demonstrated that
mRNA levels of IL-1, IL-6 and TNF are significantly increased in
isolated KCs 2–4 weeks after CCl4 injection in rats
(93). ILs serve a complex role in
the immune response, inflammatory reaction and HF (91). They directly induce the activation
of HSCs and upregulate the production of TIMP-1, resulting in ECM
deposition and HF (90). In
addition to the direct stimulation of ECM secretion, macrophages
may promote the survival of HSC via the NF-κB signaling pathway
induced by IL-1 (94). Via NF-κB
signaling, macrophages also increase the resistance of HSCs to cell
death, thus promoting the persistence of activated HSCs and HF
(90). KCs can also convert HSCs
into MFBs by secreting IL-17 (95).
The expression of IL-17 and its receptor IL-17RA are increased in
HF mice (96). IL-17 activates
NF-κB and signal transducer and activator of transcription (STAT)3
signaling in KCs and HSCs (97).
IL-17 also induces the transformation of HSCs to MFBs through the
upregulation of TGF-β1, TNF-α and type I collagen α subunit, which
is dependent on the STAT3 signaling pathway (98).
Additionally, macrophages can release a large
quantity of VEGF and induce angiogenesis during wound healing,
inflammation and tumorigenesis (99). They secrete chemokine CXCL16 to
recruit natural killer T (NKT) cells, thus aggravating the
inflammatory response. NKT cells also promote fibrogenesis in a
CXCR6-dependent manner (89).
Macrophages contribute to the
termination of HF
Macrophages serve an important role in the
progression and resolution of HF (100). MFBs are generally considered to be
the ‘master mediators’ of fibrosis, as they are the main cell types
that synthesize ECM components (83). Additionally, previous studies have
verified that macrophages serve an important role as the ‘major
regulator’ of MFBs function (83).
Macrophages secrete TGF-β1, PDGF and other fibrogenic mediators to
recruit and activate HSCs, as well as inflammatory cells to
transform HSCs to MFBs (83,85).
Macrophages derived from Ly-6C monocytes, which accumulate during
liver injury, and mediate the transformation of HSCs into MFBs
(85). Fibrogenic macrophages can
transform into antifibrotic macrophages under low expression of
Ly-6C and high expression of anti-inflammatory mediators (23). Antifibrotic macrophages not only
remove debris directly, but also secrete factors that stimulate the
production of MMPs, including gelatinases matrix metalloproteinase
(MMP2 and MMP9), metalloelastase (MMP12), matrilysin (MMP7) and
collagenases (MMP1 and MMP13), all of which are involved in matrix
degradation, thus contributing to the resolution of fibrosis
(79,96). Macrophage-mediated changes in the
ECM also induces apoptosis and represses the survival of MFBs,
thereby reducing the ECM synthesis and contributing to the
termination and even reversion of HF (85).
It could therefore be concluded that macrophages
serve a dual role in HF and that fibrogenic macrophages contribute
to the progression of fibrosis. However, anti-inflammatory
macrophages are essential sources of MMPs. Therefore, macrophages
function differently depending on the stage of the injury, either
aggravating fibrosis or facilitating the resolution (13,101).
Macrophages stimulate LSECs during
angiogenesis
Fibrosis and angiogenesis may occur in parallel in
multiple diseases as they share a common process with ECM
accumulation and fiber formation (23). KCs are phagocytes that reside at the
sinusoidal side of the endothelium. They capture signals from the
blood and are involved in regulating blood flow (85). KCs and LSECs form the first barrier
of the portal vein system. KCs are located along the
neovascularization and secrete cytokines, ILs and growth factors,
including VEGF, PLGF and PDGF, promoting LSEC and HSC proliferation
and migration, thereby inducing pathological angiogenesis (22,83).
KCs not only regulate vascular tension and growth
through paracrine signaling during HF, but they also recruit IMs
into the surrounding spouting spots by releasing monocyte
chemoattractant protein-1 and TNF-α, enhancing their
pro-angiogenesis activity by multiplying their population (102). ICAM-1/macrophage-1 antigen
(Mac-1)-mediated IM adhesion promotes LSEC proliferation (103). Additionally, the interaction
between IMs and LSCEs though ICAM-1/Mac-1 promotes vascular
endothelial (VE)-cadherin phosphorylation at the specific sites
that control the passage of leukocytes, which delivers growth
factors to stimulate vascular sprouting (102). IMs can also activate
tyrosine-protein kinase Met or TIE-2 pathways in LSECs, which are
independent of adhesion molecules, to regulate angiogenesis
(102,104).
Angiogenesis is initiated by a single leader
endothelial cell called the ‘tip cell,’ which is followed by vessel
elongation by ‘stalk cells’ (105). Endothelial tip cells are located
in the front end of vascular branches, which are highly polarized
and extend via a large number of pseudopodia to explore the
potential environment from which they migrate, thereby stimulating
angiogenesis (106). IMs can
stimulate the proliferation of tip cells by releasing Wnt family
member 5A (Wnt5a) and stimulating the proliferation of selected
stem cells by releasing Notch-1 or Ang-1 (102). IMs co-localize with Wnt5a, Ang-1
and Notch-1 at the contact point, which corresponds to the
phosphorylation level of VE-cadherin and the degree of
adherin-mediated intercellular junction uncoupling, driving
endothelial tip cell migration and elongation (102).
To conclude, by abundant secretion of angiogenic
factors, macrophages broadly participate in angiogenesis (Fig. 4).
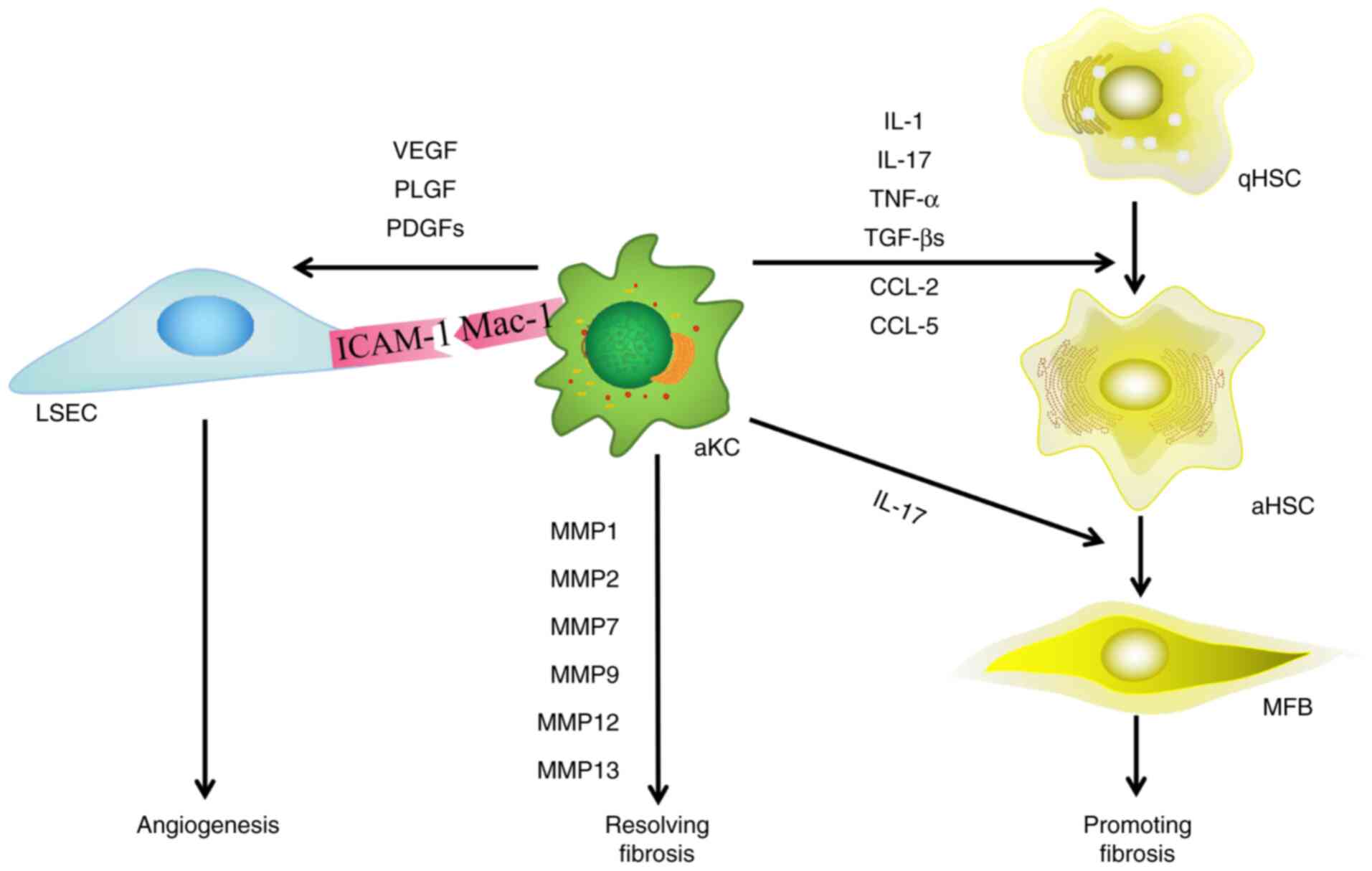 | Figure 4.Effect of macrophages on HSCs and
LSECs during HF. Macrophages serve a dual role in the progression
and resolution of HF, and participate in angiogenesis. Macrophages
activate HSCs via ILs and growth factors. When resolving HF, KCs
secrete MMPs and participate in the degradation of extracellular
matrix. Macrophages also induce pathological angiogenesis. HSCs,
hepatic stellate cells; LSECs, liver sinusoidal endothelial cells;
HF, hepatic fibrosis; VEGF, vascular endothelial growth factor;
PLGF, placental growth factor; PDGF, platelet-derived growth
factor; MFB, myofibroblast; aHSC, activated hepatic stellate cell;
qHSC, quiescent hepatic stellate cell; ICAM-1, intercellular
adhesion molecule-1; Mac-1, macrophage-1 antigen; KC, killer cells;
IL, interleukin; MMP, matrix metalloproteinase; TNF-α, tumor
necrosis factor-α; TGF-β, transforming growth factor-β; CCL-, C-C
chemokine ligand. |
Other non-parenchymal hepatocytes also
contribute to HF
Non-parenchymal hepatocytes include HSCs, LESCs and
KCs as aforementioned, in addition to natural killer (NK) cells,
NKT cells, dendritic cells (DCs) and biliary epithelial cells
(BECs), all of which have been reported to serve diverse roles in
the development of liver fibrosis (107–109).
NK cells are located in hepatic sinusoids and are in
close proximity to non-parenchymal cells of the liver (110). Previous studies have demonstrated
that NK cells inhibit the progression of fibrosis (110,111). Under various pathological
conditions, the number of NK cells increases (107). In vivo and in vitro
evidence has suggested that NK cells selectively kill early
activated HSCs, but not quiescent or fully activated HSCs (107). Activated NK cells produce IFN-γ,
which not only directly induces HSC death, but also further
enhances NK cell cytotoxicity against HSCs (112). Quiescent and fully activated HSCs
do not express elevated NK cell-activating ligands and are
resistant to NK cell killing (107). Other studies have suggested that
NK cells can kill stressed hepatocytes through natural killer group
2 member D, natural cytotoxicity triggering receptor 3 or
TNF-related apoptosis-inducing ligand-dependent pathways, and
aggravate liver damage (112,113).
In addition to NK cells, NKT cells are also enriched
among liver lymphocytes (107). NK
cells are important for killing virally infected or transformed
hepatocytes, while NKT cells are capable of rapidly aggravating
inflammatory responses under damaging conditions (94). The function of NKT cells in the
pathogenesis of liver fibrosis appear more complex than NK cells.
NKT cells not only produce pro-fibrotic cytokines [such as IL-4,
IL-13, hedgehog (Hh) ligands and osteopontin] to promote liver
fibrosis, but also produce anti-fibrotic cytokines (such as IFN-γ)
to inhibit hepatic fibrosis (107).
DCs are professional antigen-presenting cells
(92). After liver injury, DCs gain
the capacity to induce inflammation via HSCs and NK cells. Previous
studies have demonstrated that DCs induce HSC proliferation and
produce various cytokines and chemokines, such TNF-α and IL-6
(108,114). DCs have also been found to induce
NK cell-mediated cytotoxicity in vitro by producing TNF-α in
the livers of mice (108).
Additionally, DCs are involved in the regression of fibrosis after
liver injury by producing MMP9 (115).
BECs sense and respond to their surrounding
microenvironment to maintain homeostasis (109). In bile duct ligation modeled mice,
Hh ligand expression in HSCs and BECs increases, and cross-talk
between MFBs and BECs is promoted by the Hh pathway (116). Activation of the Hh pathway
stimulates BECs to specifically secrete CXCL16, which recruits NK
cells into the portal vein, further promoting the inflammatory
process (117). In addition, BECs
secrete VEGF, which can activate the production of HSCs and
collagen (109).
Conclusions
As the liver is cellularly diverse, HF is a dynamic,
highly integrated process involving molecules, cells and tissues
(118). Non-parenchymal
hepatocytes and their interactions are broadly involved in events
of the hepatic microenvironment, including hepatic sinusoid
capillarization and the inflammatory reaction of the space of Disse
in the development of HF (8,83).
Following an increased number of studies, the
reversibility of HF has been demonstrated, and effective approaches
for its reversal require urgent elucidation (119). It is broadly accepted that the
activation of HSCs is the primary mechanism of HF (120). Other studies have concluded that
the etiology of HF is the decisive factor in its development
(121). Based on those
considerations, the majority of the therapeutic strategies for HF
focus on inhibiting the activation of HSCs and removing its
etiology (24). However, it has
been revealed that other non-parenchymal hepatocytes, including
LSECs and macrophages also contribute to HF, either dependent or
independent on HSCs activation (122). Non-parenchymal hepatocytes and
their interaction are broadly involved in events of the hepatic
microenvironment, including hepatic sinusoids capillarization and
inflammatory reaction in the space of Disse during HF development.
Therefore, targeting one aspect of this complex and dynamic process
may not be enough to achieve its reversal (1).
In the future, the maintenance of the
de-differentiation phenotype of LSECs, as well the anti-fibrotic
phenotype of macrophages, is worth being considered for the
treatment of HF. This necessitates comprehensive analysis of the
interactions of various liver cell types and how they influence the
hepatic microenvironment. Further understanding of the underlying
mechanisms of these events could provide novel insight into the
discovery of effective antifibrotic therapies, thus reducing the
incidence of portal hypertension and hepatic encephalopathy, and
blocking the formation of end stage liver disease.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81670555).
Availability of data and materials
Not applicable.
Authors' contributions
QNC collected the data, wrote the manuscript and
designed the figures. YRN designed the framework of the present
review and modified the manuscript. XY and JFW assisted with
manuscript preparation. YRN and WBA conceived and designed the idea
for this study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Aydin MM and Akcali KC: Liver fibrosis.
Turk J Gastroenterol. 29:14–21. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Greuter T and Shah VH: Hepatic sinusoids
in liver injury, inflammation, and fibrosis: New pathophysiological
insights. J Gastroenterol. 51:511–519. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu X, Hu H and Yin JQ: Therapeutic
strategies against TGF-beta signaling pathway in hepatic fibrosis.
Liver Int. 26:8–22. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Higashi T, Friedman SL and Hoshida Y:
Hepatic stellate cells as key target in liver fibrosis. Adv Drug
Deliv Rev. 121:27–42. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marrone G, Shah VH and Gracia-Sancho J:
Sinusoidal communication in liver fibrosis and regeneration. J
Hepatol. 65:608–617. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Klenerman P and Ramamurthy N: Liver
sinusoidal endothelial cells: An antiviral ‘defendothelium’.
Gastroenterology. 148:288–291. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ray K: Liver: Hepatic stellate cells hold
the key to liver fibrosis. Nat Rev Gastroenterol Hepatol.
11:742014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Poisson J, Lemoinne S, Boulanger C, Durand
F, Moreau R, Valla D and Rautou PE: Liver sinusoidal endothelial
cells: Physiology and role in liver diseases. J Hepatol.
66:212–227. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shetty S, Lalor PF and Adams DH: Liver
sinusoidal endothelial cells-gatekeepers of hepatic immunity. Nat
Rev Gastroenterol Hepatol. 15:555–567. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sorensen KK, Simon-Santamaria J, McCuskey
RS and Smedsrod B: Liver Sinusoidal Endothelial Cells. Compr
Physiol. 5:1751–1774. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ni Y, Li JM, Liu MK, Zhang TT, Wang DP,
Zhou WH, Hu LZ and Lv WL: Pathological process of liver sinusoidal
endothelial cells in liver diseases. World J Gastroenterol.
23:7666–7677. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Friedman SL: Hepatic stellate cells:
Protean, multifunctional, and enigmatic cells of the liver. Physiol
Rev. 88:125–172. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dixon LJ, Barnes M, Tang H, Pritchard MT
and Nagy LE: Kupffer cells in the liver. Compr Physiol. 3:785–797.
2013.PubMed/NCBI
|
|
14
|
Dou L, Shi X, He X and Gao Y: Macrophage
phenotype and function in liver disorder. Front Immunol.
10:31122019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu HL, Lv J, Zhao ZM, Xiong AM, Tan Y,
Glenn JS, Tao YY, Weng HL and Liu CH: Fuzhenghuayu decoction
ameliorates hepatic fibrosis by attenuating experimental sinusoidal
capillarization and liver angiogenesis. Sci Rep. 9:187192019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brusilovskaya K, Konigshofer P, Schwabl P
and Reiberger T: Vascular targets for the treatment of portal
hypertension. Semin Liver Dis. 39:483–501. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
DeLeve LD: Liver sinusoidal endothelial
cells in hepatic fibrosis. Hepatology. 61:1740–1746. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tuohetahuntila M, Molenaar MR, Spee B,
Brouwers JF, Wubbolts R, Houweling M, Yan C, Du H, VanderVen BC,
Vaandrager AB and Helms JB: Lysosome-mediated degradation of a
distinct pool of lipid droplets during hepatic stellate cell
activation. J Biol Chem. 292:12436–12448. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Peterova E, Podmolikova L, Rezacova M and
Mrkvicova A: Fibroblast growth Factor-1 suppresses TGF-β-mediated
myofibroblastic differentiation of rat hepatic stellate cells. Acta
Medica (Hradec Kralove). 59:124–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schon HT, Bartneck M, Borkham-Kamphorst E,
Nattermann J, Lammers T, Tacke F and Weiskirchen R: Pharmacological
intervention in hepatic stellate cell activation and hepatic
fibrosis. Front Pharmacol. 7:332016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ezhilarasan D, Sokal E and Najimi M:
Hepatic fibrosis: It is time to go with hepatic stellate
cell-specific therapeutic targets. Hepatobiliary Pancreat Dis Int.
17:192–197. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cai X, Wang J, Wang J, Zhou Q, Yang B, He
Q and Weng Q: Intercellular crosstalk of hepatic stellate cells in
liver fibrosis: New insights into therapy. Pharmacol Res.
155:1047202020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ramirez-Pedraza M and Fernandez M:
Interplay between macrophages and angiogenesis: A double-edged
sword in liver disease. Front Immunol. 10:28822019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lafoz E, Ruart M, Anton A, Oncins A and
Hernández-Gea V: The endothelium as a driver of liver fibrosis and
regeneration. Cells. 9:9292020. View Article : Google Scholar
|
|
25
|
Soydemir S, Comella O, Abdelmottaleb D and
Pritchett J: Does mechanocrine signaling by liver sinusoidal
endothelial cells offer new opportunities for the development of
anti-fibrotics? Front Med (Lausanne). 6:3122019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kaur S and Anita K: Angiogenesis in liver
regeneration and fibrosis: ‘A double-edged sword’. Hepatol Int.
7:959–968. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wells RG: Cellular sources of
extracellular matrix in hepatic fibrosis. Clin Liver Dis.
12:759–768. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Maher JJ and McGuire RF: Extracellular
matrix gene expression increases preferentially in rat lipocytes
and sinusoidal endothelial cells during hepatic fibrosis in vivo. J
Clin Invest. 86:1641–1648. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu F, Dong B, Dong P, He Y, Zheng J and Xu
P: Hypoxia induces the activation of hepatic stellate cells through
the PVT1-miR-152-ATG14 signaling pathway. Mol Cell Biochem.
465:115–123. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shi YF, Fong CC, Zhang Q, Cheung PY, Tzang
CH, Wu RS and Yang M: Hypoxia induces the activation of human
hepatic stellate cells LX-2 through TGF-beta signaling pathway.
FEBS Lett. 581:203–210. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jin Y, Bai Y, Ni H, Qiang L, Ye L, Shan Y
and Zhou M: Activation of autophagy through calcium-dependent
AMPK/mTOR and PKCθ pathway causes activation of rat hepatic
stellate cells under hypoxic stress. FEBS Lett. 590:672–682. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen W, Rock JB, Yearsley MM, Ferrell LD
and Frankel WL: Different collagen types show distinct rates of
increase from early to late stages of hepatitis C-related liver
fibrosis. Hum Pathol. 45:160–165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ghafoory S, Varshney R, Robison T,
Kouzbari K, Woolington S, Murphy B, Xia L and Ahamed J: Platelet
TGF-β1 deficiency decreases liver fibrosis in a mouse model of
liver injury. Blood Adv. 2:470–480. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Baghy K, Iozzo RV and Kovalszky I:
Decorin-TGFβ axis in hepatic fibrosis and cirrhosis. J Histochem
Cytochem. 60:262–268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hayes BJ, Riehle KJ, Shimizu-Albergine M,
Bauer RL, Hudkins KL, Johansson F, Yeh MM, Mahoney WJ, Yeung RS and
Campbell JS: Activation of platelet-derived growth factor receptor
alpha contributes to liver fibrosis. PLoS One. 9:e929252014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ying HZ, Chen Q, Zhang WY, Zhang HH, Ma Y,
Zhang SZ, Fang J and Yu CH: PDGF signaling pathway in hepatic
fibrosis pathogenesis and therapeutics (Review). Mol Med Rep.
16:7879–7889. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Borkham-Kamphorst E and Weiskirchen R: The
PDGF system and its antagonists in liver fibrosis. Cytokine Growth
Factor Rev. 28:53–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Borkham-Kamphorst E, Meurer SK, Van de
Leur E, Haas U, Tihaa L and Weiskirchen R: PDGF-D signaling in
portal myofibroblasts and hepatic stellate cells proves identical
to PDGF-B via both PDGF receptor type alpha and β. Cell Signal.
27:1305–1314. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kocabayoglu P, Lade A, Lee YA, Dragomir A,
Sun X, Fiel MI, Thung S, Aloman C, Soriano P, Hoshida Y and
Friedman SL: β-PDGF receptor expressed by hepatic stellate cells
regulates fibrosis in murine liver injury, but not carcinogenesis.
J Hepatol. 63:141–147. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Thabut D and Shah V: Intrahepatic
angiogenesis and sinusoidal remodeling in chronic liver disease:
New targets for the treatment of portal hypertension? J Hepatol.
53:976–980. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lim BJ, Lee WK, Lee HW, Lee KS, Kim JK,
Chang HY and Lee JI: Selective deletion of hepatocyte
platelet-derived growth factor receptor α and development of liver
fibrosis in mice. Cell Commun Signal. 16:932018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Serini G, Bochaton-Piallat ML, Ropraz P,
Geinoz A, Borsi L, Zardi L and Gabbiani G: The fibronectin domain
ED-A is crucial for myofibroblastic phenotype induction by
transforming growth factor-beta1. J Cell Biol. 142:873–881. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gabbiani G: The myofibroblast in wound
healing and fibrocontractive diseases. J Pathol. 200:500–503. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bocca C, Novo E, Miglietta A and Parola M:
Angiogenesis and Fibrogenesis in chronic liver diseases. Cell Mol
Gastroenterol Hepatol. 1:477–488. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kardum D, Fabijanic D, Lukic A, Romic Z,
Petrovecki M, Bogdanovic Z, Juric K, Urek-Crncevic M and Banic M:
Correlation of endothelin-1 concentration and
angiotensin-converting enzyme activity with the staging of liver
fibrosis. Coll Antropol. 36:413–418. 2012.PubMed/NCBI
|
|
46
|
Yokomori H, Oda M, Ogi M, Kamegaya Y,
Tsukada N, Nakamura M and Ishii H: Enhanced expression of
endothelin receptor subtypes in cirrhotic rat liver. Liver.
21:114–122. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Koda M, Bauer M, Krebs A, Hahn EG,
Schuppan D and Murawaki Y: Endothelin-1 enhances fibrogenic gene
expression, but does not promote DNA synthesis or apoptosis in
hepatic stellate cells. Comp Hepatol. 5:52006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Pinzani M, Milani S, De Franco R, Grappone
C, Caligiuri A, Gentilini A, Tosti-Guerra C, Maggi M, Failli P,
Ruocco C and Gentilini P: Endothelin 1 is overexpressed in human
cirrhotic liver and exerts multiple effects on activated hepatic
stellate cells. Gastroenterology. 110:534–548. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Das A, Shergill U, Thakur L, Sinha S,
Urrutia R, Mukhopadhyay D and Shah VH: Ephrin B2/EphB4 pathway in
hepatic stellate cells stimulates Erk-dependent VEGF production and
sinusoidal endothelial cell recruitment. Am J Physiol Gastrointest
Liver Physiol. 298:G908–G915. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li G, Peng Y, Zhao T, Lin J, Duan X, Wei Y
and Ma J: Plumbagin alleviates capillarization of hepatic sinusoids
in vitro by downregulating ET-1, VEGF, LN, and type IV collagen.
Biomed Res Int. 2017:56032162017.PubMed/NCBI
|
|
51
|
Lee JS, Semela D, Iredale J and Shah VH:
Sinusoidal remodeling and angiogenesis: A new function for the
liver-specific pericyte? Hepatology. 45:817–825. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Rockey DC: Hepatic blood flow regulation
by stellate cells in normal and injured liver. Semin Liver Dis.
21:337–349. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Henderson NC and Iredale JP: Liver
fibrosis: Cellular mechanisms of progression and resolution. Clin
Sci (Lond). 112:265–280. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Knittel T, Mehde M, Kobold D, Saile B,
Dinter C and Ramadori G: Expression patterns of matrix
metalloproteinases and their inhibitors in parenchymal and
non-parenchymal cells of rat liver: Regulation by TNF-alpha and
TGF-beta1. J Hepatol. 30:48–60. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Liu T, Xu L, Wang C, Chen K, Xia Y, Li J,
Li S, Wu L, Feng J, Xu S, et al: Alleviation of hepatic fibrosis
and autophagy via inhibition of transforming growth factor-β1/Smads
pathway through shikonin. J Gastroenterol Hepatol. 34:263–276.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gupta G, Khadem F and Uzonna JE: Role of
hepatic stellate cell (HSC)-derived cytokines in hepatic
inflammation and immunity. Cytokine. 124:1545422019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Marchand M, Monnot C, Muller L and Germain
S: Extracellular matrix scaffolding in angiogenesis and capillary
homeostasis. Semin Cell Dev Biol. 89:147–156. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mokkapati S, Fleger-Weckmann A, Bechtel M,
Koch M, Breitkreutz D, Mayer U, Smyth N and Nischt R: Basement
membrane deposition of nidogen 1 but not nidogen 2 requires the
nidogen binding module of the laminin gamma1 chain. J Biol Chem.
286:1911–1918. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mak KM and Mei R: Basement membrane type
IV collagen and laminin: An overview of their biology and value as
fibrosis biomarkers of liver disease. Anat Rec (Hoboken).
300:1371–1390. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
McGuire RF, Bissell DM, Boyles J and Roll
FJ: Role of extracellular matrix in regulating fenestrations of
sinusoidal endothelial cells isolated from normal rat liver.
Hepatology. 15:989–997. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Braet F and Wisse E: Structural and
functional aspects of liver sinusoidal endothelial cell fenestrae:
A review. Comp Hepatol. 1:12002. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Natarajan V, Harris EN and Kidambi S: SECs
(Sinusoidal Endothelial Cells), liver microenvironment, and
fibrosis. Biomed Res Int. 2017:40972052017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
DeLeve LD, Wang X, Hu L, McCuskey MK and
McCuskey RS: Rat liver sinusoidal endothelial cell phenotype is
maintained by paracrine and autocrine regulation. Am J Physiol
Gastrointest Liver Physiol. 287:G757–G763. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Desroches-Castan A, Tillet E, Ricard N,
Ouarne M, Mallet C, Belmudes L, Coute Y, Boillot O, Scoazec JY,
Bailly S and Feige JJ: Bone morphogenetic protein 9 is a paracrine
factor controlling liver sinusoidal endothelial cell fenestration
and protecting against hepatic fibrosis. Hepatology. 70:1392–1408.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Soon RJ and Yee HJ: Stellate cell
contraction: Role, regulation, and potential therapeutic target.
Clin Liver Dis. 12:791–803. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hintermann E, Bayer M, Ehser J,
Aurrand-Lions M, Pfeilschifter JM, Imhof BA and Christen U: Murine
junctional adhesion molecules JAM-B and JAM-C mediate endothelial
and stellate cell interactions during hepatic fibrosis. Cell Adh
Migr. 10:419–433. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hintermann E, Bayer M, Conti CB, Fuchs S,
Fausther M, Leung PS, Aurrand-Lions M, Taubert R, Pfeilschifter JM,
Friedrich-Rust M, et al: Junctional adhesion molecules JAM-B and
JAM-C promote autoimmune-mediated liver fibrosis in mice. J
Autoimmun. 91:83–96. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Saiman Y, Agarwal R, Hickman DA, Fausther
M, El-Shamy A, Dranoff JA, Friedman SL and Bansal MB: CXCL12
induces hepatic stellate cell contraction through a
calcium-independent pathway. Am J Physiol Gastrointest Liver
Physiol. 305:G375–G382. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Sohail MA, Hashmi AZ, Hakim W, Watanabe A,
Zipprich A, Groszmann RJ, Dranoff JA, Torok NJ and Mehal WZ:
Adenosine induces loss of actin stress fibers and inhibits
contraction in hepatic stellate cells via Rho inhibition.
Hepatology. 49:185–194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhang Z, Zhang F, Lu Y and Zheng S: Update
on implications and mechanisms of angiogenesis in liver fibrosis.
Hepatol Res. 45:162–178. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Budny T, Palmes D, Stratmann U, Minin E,
Herbst H and Spiegel HU: Morphologic features in the regenerating
liver-a comparative intravital, lightmicroscopical and
ultrastructural analysis with focus on hepatic stellate cells.
Virchows Arch. 451:781–791. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Coulon S, Heindryckx F, Geerts A, Van
Steenkiste C, Colle I and Van Vlierberghe H: Angiogenesis in
chronic liver disease and its complications. Liver Int. 31:146–162.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Karkkainen MJ and Petrova TV: Vascular
endothelial growth factor receptors in the regulation of
angiogenesis and lymphangiogenesis. Oncogene. 19:5598–5605. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Xie G, Wang X, Wang L, Wang L, Atkinson
RD, Kanel GC, Gaarde WA and Deleve LD: Role of differentiation of
liver sinusoidal endothelial cells in progression and regression of
hepatic fibrosis in rats. Gastroenterology. 142:918–927. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Deleve LD, Wang X and Guo Y: Sinusoidal
endothelial cells prevent rat stellate cell activation and promote
reversion to quiescence. Hepatology. 48:920–930. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Li X, Yao Q, Liu H, Jin Q, Xu B, Zhang S
and Tu C: Placental growth factor silencing ameliorates liver
fibrosis and angiogenesis and inhibits activation of hepatic
stellate cells in a murine model of chronic liver disease. J Cell
Mol Med. 21:2370–2385. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Dewerchin M and Carmeliet P: PlGF: A
multitasking cytokine with disease-restricted activity. Cold Spring
Harb Perspect Med. 2:a0110562012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Li X, Jin Q, Yao Q, Zhou Y, Zou Y, Li Z,
Zhang S and Tu C: Placental growth factor contributes to liver
inflammation, angiogenesis, Fibrosis in mice by promoting hepatic
macrophage recruitment and activation. Front Immunol. 8:8012017.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Reif S, Lang A, Lindquist JN, Yata Y,
Gabele E, Scanga A, Brenner DA and Rippe RA: The role of focal
adhesion kinase-phosphatidylinositol 3-kinase-akt signaling in
hepatic stellate cell proliferation and type I collagen expression.
J Biol Chem. 278:8083–8090. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Van Steenkiste C, Ribera J, Geerts A,
Pauta M, Tugues S, Casteleyn C, Libbrecht L, Olievier K, Schroyen
B, Reynaert H, et al: Inhibition of placental growth factor
activity reduces the severity of fibrosis, inflammation, and portal
hypertension in cirrhotic mice. Hepatology. 53:1629–1640. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Augustin HG, Koh GY, Thurston G and
Alitalo K: Control of vascular morphogenesis and homeostasis
through the angiopoietin-Tie system. Nat Rev Mol Cell Biol.
10:165–177. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Gurnik S, Devraj K, Macas J, Yamaji M,
Starke J, Scholz A, Sommer K, Di Tacchio M, Vutukuri R, Beck H, et
al: Angiopoietin-2-induced blood-brain barrier compromise and
increased stroke size are rescued by VE-PTP-dependent restoration
of Tie2 signaling. Acta Neuropathol. 131:753–773. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Wynn TA and Barron L: Macrophages: Master
regulators of inflammation and fibrosis. Semin Liver Dis.
30:245–257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Ma PF, Gao CC, Yi J, Zhao JL, Liang SQ,
Zhao Y, Ye YC, Bai J, Zheng QJ, Dou KF, et al: Cytotherapy with
M1-polarized macrophages ameliorates liver fibrosis by modulating
immune microenvironment in mice. J Hepatol. 67:770–779. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Tacke F: Targeting hepatic macrophages to
treat liver diseases. J Hepatol. 66:1300–1312. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Varol C, Mildner A and Jung S:
Macrophages: Development and tissue specialization. Annu Rev
Immunol. 33:643–675. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
You Q, Holt M, Yin H, Li G, Hu CJ and Ju
C: Role of hepatic resident and infiltrating macrophages in liver
repair after acute injury. Biochem Pharmacol. 86:836–843. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Vollmar B, Siegmund S, Richter S and
Menger MD: Microvascular consequences of Kupffer cell modulation in
rat liver fibrogenesis. J Pathol. 189:85–91. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wehr A, Baeck C, Heymann F, Niemietz PM,
Hammerich L, Martin C, Zimmermann HW, Pack O, Gassler N, Hittatiya
K, et al: Chemokine receptor CXCR6-dependent hepatic NK T cell
accumulation promotes inflammation and liver fibrosis. J Immunol.
190:5226–5236. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Tacke F and Zimmermann HW: Macrophage
heterogeneity in liver injury and fibrosis. J Hepatol.
60:1090–1096. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Zhou WC, Zhang QB and Qiao L: Pathogenesis
of liver cirrhosis. World J Gastroenterol. 20:7312–7324. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Koyama Y and Brenner DA: Liver
inflammation and fibrosis. J Clin Invest. 127:55–64. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Luckey SW and Petersen DR: Activation of
Kupffer cells during the course of carbon tetrachloride-induced
liver injury and fibrosis in rats. Exp Mol Pathol. 71:226–240.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Weiskirchen R and Tacke F: Cellular and
molecular functions of hepatic stellate cells in inflammatory
responses and liver immunology. Hepatobiliary Surg Nutr. 3:344–363.
2014.PubMed/NCBI
|
|
95
|
Wang J, Leclercq I, Brymora JM, Xu N,
Ramezani-Moghadam M, London RM, Brigstock D and George J: Kupffer
cells mediate leptin-induced liver fibrosis. Gastroenterology.
137:713–723. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Meng F, Wang K, Aoyama T, Grivennikov SI,
Paik Y, Scholten D, Cong M, Iwaisako K, Liu X, Zhang M, et al:
Interleukin-17 signaling in inflammatory, Kupffer cells, and
hepatic stellate cells exacerbates liver fibrosis in mice.
Gastroenterology. 143:765–776. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Seki E and Brenner DA: Recent advancement
of molecular mechanisms of liver fibrosis. J Hepatobiliary Pancreat
Sci. 22:512–518. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Hara M, Kono H, Furuya S, Hirayama K,
Tsuchiya M and Fujii H: Interleukin-17A plays a pivotal role in
cholestatic liver fibrosis in mice. J Surg Res. 183:574–582. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Mochida S, Ishikawa K, Toshima K, Inao M,
Ikeda H, Matsui A, Shibuya M and Fujiwara K: The mechanisms of
hepatic sinusoidal endothelial cell regeneration: A possible
communication system associated with vascular endothelial growth
factor in liver cells. J Gastroenterol Hepatol. 13 (Suppl 1):S1–S5.
1998. View Article : Google Scholar
|
|
100
|
Zhang CY, Yuan WG, He P, Lei JH and Wang
CX: Liver fibrosis and hepatic stellate cells: Etiology,
pathological hallmarks and therapeutic targets. World J
Gastroenterol. 22:10512–10522. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Lee UE and Friedman SL: Mechanisms of
hepatic fibrogenesis. Best Pract Res Clin Gastroenterol.
25:195–206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Melgar-Lesmes P and Edelman ER:
Monocyte-endothelial cell interactions in the regulation of
vascular sprouting and liver regeneration in mouse. J Hepatol.
63:917–925. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Hoefer IE, van Royen N, Rectenwald JE,
Deindl E, Hua J, Jost M, Grundmann S, Voskuil M, Ozaki CK, Piek JJ
and Buschmann IR: Arteriogenesis proceeds via ICAM-1/Mac-1-mediated
mechanisms. Circ Res. 94:1179–1185. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Schubert SY, Benarroch A, Monter-Solans J
and Edelman ER: Primary monocytes regulate endothelial cell
survival through secretion of angiopoietin-1 and activation of
endothelial Tie2. Arterioscler Thromb Vasc Biol. 31:870–875. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Priya MK, Sahu G, Soto-Pantoja DR, Goldy
N, Sundaresan AM, Jadhav V, Barathkumar TR, Saran U, Jaffar AB,
Roberts DD, et al: Tipping off endothelial tubes: Nitric oxide
drives tip cells. Angiogenesis. 18:175–189. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
De Smet F, Segura I, De Bock K,
Hohensinner PJ and Carmeliet P: Mechanisms of vessel branching:
Filopodia on endothelial tip cells lead the way. Arterioscler
Thromb Vasc Biol. 29:639–649. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Gao B and Radaeva S: Natural killer and
natural killer T cells in liver fibrosis. Biochim Biophys Acta.
1832:1061–1069. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Connolly MK, Bedrosian AS, Mallen-St CJ,
Mitchell AP, Ibrahim J, Stroud A, Pachter HL, Bar-Sagi D, Frey AB
and Miller G: In liver fibrosis, dendritic cells govern hepatic
inflammation in mice via TNF-alpha. J Clin Invest. 119:3213–3225.
2009.PubMed/NCBI
|
|
109
|
Ehrlich L, Scrushy M, Meng F, Lairmore TC,
Alpini G and Glaser S: Biliary epithelium: A neuroendocrine
compartment in cholestatic liver disease. Clin Res Hepatol
Gastroenterol. 42:296–305. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Gao B, Radaeva S and Park O: Liver natural
killer and natural killer T cells: Immunobiology and emerging roles
in liver diseases. J Leukoc Biol. 86:513–528. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Wang H and Yin S: Natural killer T cells
in liver injury, inflammation and cancer. Expert Rev Gastroenterol
Hepatol. 9:1077–1085. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Radaeva S, Sun R, Jaruga B, Nguyen VT,
Tian Z and Gao B: Natural killer cells ameliorate liver fibrosis by
killing activated stellate cells in NKG2D-dependent and tumor
necrosis factor-related apoptosis-inducing ligand-dependent
manners. Gastroenterology. 130:435–452. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Peng Y, Yang T, Huang K, Shen L, Tao Y and
Liu C: Salvia miltiorrhiza ameliorates liver fibrosis by activating
hepatic natural killer cells in vivo and in vitro. Front Pharmacol.
9:7622018. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Cheng JT, Deng YN, Yi HM, Wang GY, Fu BS,
Chen WJ, Liu W, Tai Y, Peng YW and Zhang Q: Hepatic
carcinoma-associated fibroblasts induce IDO-producing regulatory
dendritic cells through IL-6-mediated STAT3 activation.
Oncogenesis. 5:e1982016. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Jiao J, Sastre D, Fiel MI, Lee UE,
Ghiassi-Nejad Z, Ginhoux F, Vivier E, Friedman SL, Merad M and
Aloman C: Dendritic cell regulation of carbon tetrachloride-induced
murine liver fibrosis regression. Hepatology. 55:244–255. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Sato K, Meng F, Giang T, Glaser S and
Alpini G: Mechanisms of cholangiocyte responses to injury. Biochim
Biophys Acta Mol Basis Dis. 1864:1262–1269. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Omenetti A, Syn WK, Jung Y, Francis H,
Porrello A, Witek RP, Choi SS, Yang L, Mayo MJ, Gershwin ME, et al:
Repair-related activation of hedgehog signaling promotes
cholangiocyte chemokine production. Hepatology. 50:518–527. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Parola M and Pinzani M: Liver fibrosis:
Pathophysiology, pathogenetic targets and clinical issues. Mol
Aspects Med. 65:37–55. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Sohrabpour AA, Mohamadnejad M and
Malekzadeh R: Review article: The reversibility of cirrhosis.
Aliment Pharmacol Ther. 36:824–832. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Poilil SS, George TR, Moon MJ and Jeong
YY: Nanoparticles for the treatment of liver fibrosis. Int J
Nanomedicine. 12:6997–7006. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Feng R, Yuan X, Shao C, Ding H, Liebe R
and Weng HL: Are we any closer to treating liver fibrosis (and if
no, why not)? J Dig Dis. 19:118–126. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Gracia-Sancho J, Marrone G and
Fernandez-Iglesias A: Hepatic microcirculation and mechanisms of
portal hypertension. Nat Rev Gastroenterol Hepatol. 16:221–234.
2019. View Article : Google Scholar : PubMed/NCBI
|














