Introduction
Alzheimer's disease (AD) is an age-related,
progressive neurodegenerative disease, with clinical symptoms of
cognitive decline and changes in behavior and personality (1,2). AD is
the commonest type of dementia in people aged >65 (3). With the increase in human lifespan AD
is becoming, at present, a major health concern for the elderly.
Worldwide >50 million people are suffering from AD (3). However, there is no effective
treatment for delaying the disease progression of patients with AD
(4). The disease is characterized
by two hallmark lesions: Senile plaques (SP) and neurofibrillary
tangles (NFTs). SP and NFTs mainly result from the deposition of
amyloid β (Aβ) peptides and hyperphosphorylated tau, respectively
(3). Aβ fibrils have been reported
to compose SP and to be the main cause of the massive
neurodegeneration observed in AD brains (5). Along with NFTs and SP, additional
neuropathological characteristics of this disease include synapse
loss and neuronal death (6).
Synapses are a unique architecture formed by nerve cells and are
considered to be the critical sites for pathogenesis in
neurodegenerative diseases associated with aging (7,8). The
cognitive impairment in AD mainly results from synaptic loss in
neurons and the deterioration of synapses usually begins at the
level of dendritic spines (9,10).
Dendritic spines are tiny, bulbous structures protruding from the
dendrites of neurons, which receive fast excitatory synaptic input
in the brain. These structures compartmentalize the postsynaptic
machinery and biochemical signaling molecules needed to respond to
input from single presynaptic terminals (11). Recently, it has been reported that
Aβ1–42 can decrease dendritic spine density in rat
primary hippocampal neuron cultures (12). Moreover, some studies conducted in
AD mouse models have shown that the trajectories of axons and
dendrites were altered in the proximity of amyloid plaques, which
affected synaptic integration of signals (13,14).
Among Aβ peptides, Aβ1–42 is known to be
the most neurotoxic (15). Its
soluble oligomers may disrupt intracellular calcium homeostasis,
leading to the activation of Cdk5. This proline-directed
serine/threonine kinase regulates neuronal migration during
development and maintains the survival and synaptic functions of
mature neurons (16). It has been
shown that the aberrant activity of Cdk5 induces the
hyperphosphorylation of the neurofilament and
microtubule-associated protein tau and serves an important role in
neurodegeneration in AD (17). In
addition, Cdk5 has been reported to act as an upstream regulator of
mitochondrial fission during neuronal apoptosis. Inhibition of
dynamin-related protein 1 (Drp1)-dependent mitochondrial fission
alleviates neuronal apoptosis induced by aberrant Cdk5 expression
(18). However, the underlying
mechanism remains unknown. In our recent study, the mitochondrial
fission protein Drp1 was identified as a direct substrate for Cdk5
(19). Aβ1–42 stimulates
Cdk5-mediated phosphorylation of Drp1 at Ser579 in cortical
neurons, thereby regulating mitochondrial fission-mediated neuronal
apoptosis. However, whether Cdk5-mediated Drp1 phosphorylation is
also involved in Aβ1–42-induced neurodegeneration is yet
to be fully elucidated.
In the present study, we hypothesized that
phosphorylation of Drp1 at Ser579 may be involved in the
pathogenesis of neurodegeneration. To this end, a
phosphorylation-defective (phosphor-defect) mutant lentiviral
vector (Lenti-Drp1-S579A) was constructed to block Drp1
phosphorylation at Ser579. After infection, the expression level of
Drp1-S579A was first confirmed in primary cultures of cortical
neurons. The neurite outgrowth and synapse density of cortical
neurons were observed under microscope. Consistent with our
previous findings (19), blockage
of Drp1 phosphorylation also prevented Aβ1–42-induced
cleavage of caspase-3 and neuronal apoptosis. Taken together, the
present findings demonstrated that phosphorylation of Drp1 at
Ser579 served an important role in Aβ1–42-induced
neurodegeneration, suggesting that this may be an effective
strategy for the protection of neurons in this context.
Materials and methods
Experimental animals
A total of 26 C57BL/6 mice (6~8 weeks, 90–110 g)
were purchased from Hunan SJA Laboratory Animal Co., Ltd. Mice had
free access to water and food at 22–25°C with a 12-h light/dark
cycle. The humidity was ~60%. All animal handling was performed in
accordance with the guidelines of Animal Research Committee of
Nanchang University (20). All
protocols described in this article were approved by the Ethics
Committee for Animal Experimentation of Nanchang University
(approval no. 2018-035). All surgical procedures involving
experimental animals were performed under anesthesia with 1.0%
pentobarbital sodium (50 mg/kg body weight) by intraperitoneal
injection and the suffering of animals was minimized to the best of
our ability.
Primary cortical neuronal
cultures
As previously described (19), primary cortical neuronal cultures
were derived from embryonic day 14–15 fetal C57BL/6 mouse brains.
In brief, the cortex isolated from embryonic mouse brains was
placed in DMEM (HyClone; Cytiva) and treated with 0.125% trypsin
(Beijing Solarbio Science & Technology Co., Ltd.) and 0.004%
DNase-I (Sigma-Aldrich; Merck KGaA) at 37°C for 15 and 10 min,
respectively. Neurons were mechanically dissociated by pipetting
and were seeded on poly-L-lysine (Sigma-Aldrich; Merck KGaA)-coated
glass- or plastic-bottom 35-mm culture dishes (cell density was
~25,000-30,000/35-mm dish for microscopic observation, or
~45,000-50,000/35-mm dish for western blotting). Cells were first
cultured in neurobasal plating medium [neurobasal medium (Thermo
Fisher Scientific, Inc.), 2% B27 supplement, 0.5 mM L-glutamine, 25
µM L-glutamic acid, 1% penicillin-streptomycin (P/S), 10 mM HEPES,
10% FBS (Biological Industries)] and incubated at 37°C in a
humidified incubator with 95% air and 5% CO2. On the
second day, neuronal cells were cultured in neurobasal feeding
medium (neurobasal medium, 2% B27 supplement, 0.5 mM L-glutamine,
1% P/S, 10 mM HEPES). Half the volume of media was replaced with
the same volume of fresh neurobasal feeding media every 4 days.
Construction of Lenti-Drp1-S579A and
infection procedure
Lenti-Drp1-S579A was constructed by Cyagen
Biosciences, Inc. Briefly, Drp1-S579A site-directed mutagenesis was
performed using a QuickChange kit (Agilent Technologies, Inc.)
according to the manufacturer's instructions. Drp1-S579A was
further subcloned into the pLV [Exp]-Puro-EF1A vector (Cyagen
Biosciences, Inc.) and fused with a 6X His tag. The expression
vector and package vectors (2 µg of each vector) were
co-transfected into 293T cells (the American Type Culture
Collection) using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) at 37°C for 6 h. After 48 h of
culture, the supernatants containing the lentivirus were harvested.
Purification was then performed using ultracentrifugation at 80,000
× g for 2 h at 4°C and the lentiviral titer was determined.
To evaluate the infection efficiency, neurons at 3
days in vitro (DIV) were infected with the empty lentiviral
vector pLV [Exp]-Puro-EF1A-mCherry at a multiplicity of infection
(MOI) of 1–5. Then, 3 days after infection, the fluorescence of
mCherry was used to monitor and visualize the lentiviral infection
under fluorescence microscope (IX71; Olympus Corporation). The
formula for calculating the infection efficiency was as follows:
(The number of infected cells/total cells in the field) ×100%. To
examine the expression level of the exogenous mutant Drp1-S579A,
neurons at 3 DIV were infected with Lenti-Drp1-S579A or empty
lentiviral vector at MOI of 1, 2 and 5 at 37°C for 8 h. At day 5
after infection, western blot analysis probed with anti-6His was
carried out to examine the expression of phospho-defect Drp1-S579A
in neurons.
Western blot analysis
Cultured neurons for western blot analysis were
washed twice with PBS and lysed in RIPA lysis buffer (Beyotime
Institute of Biotechnology) for 30 min on ice. The whole cell
lysate was harvested via sonication (20 KHz; 20 sec; 4°C) in 4X
sample buffer and the protein concentration was measured using a
BCA protein assay reagent kit (Beyotime Institute of
Biotechnology). Proteins (20 µg per lane) were separated by 10%
SDS-PAGE gel and further transferred onto PVDF membranes
(MilliporeSigma). After blocking with 5% skim milk in TBS-0.1%
Tween 20 (TBST) buffer for 30 min at room temperature, the
membranes were probed overnight at 4°C with the following primary
antibodies: Rabbit anti-Drp1 (cat. no. 8570, 1:1,000; Cell
Signaling Technology, Inc.), phospho-Drp1-Ser616 (cat. no. 3455,
1:1,000; Cell Signaling Technology, Inc.), anti-6His (cat. no.
CW0083S; CWBIO), cleaved caspase-3 (cat. no. 9644, 1:1,000; Cell
Signaling Technology, Inc.), rabbit anti-microtubule associated
protein 2 (MAP2; cat. no. 8707, 1:1,000, Cell Signaling Technology,
Inc.) and β-actin (cat. no. 4967, 1:5,000; Cell Signaling
Technology, Inc.). After three washes in TBST, the membranes were
incubated with HRP-conjugated secondary antibodies (1:5,000; CWBIO)
for 1 h at room temperature. Protein bands were detected using an
ECL solution (CWBIO). Densitometric analysis was performed using
ImageJ software (version 1.48; National Institutes of Health).
Transfection procedures
To observe the spines on neuronal axons, the pEGFP
plasmid (Clontech; 4 µg per 35 mm-well) was transfected into
primary cortical neurons at 6 DIV using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). According to the
manufacturer's instructions, a mixture of 4 µg plasmid DNA, 5 µl
Lipofectamine 2000 and 200 µl Opti-MEM (Invitrogen; Thermo Fisher
Scientific, Inc.) was incubated at room temperature for 15 min,
then added to neuronal cultures for transfection at 37°C. After 6 h
of incubation, the medium containing Lipofectamine was replaced
with normal culture medium. Axonal spines were assessed under a
fluorescence microscope (IX71; Olympus Corporation) after 18–24 h
after transfection. To quantify the spines, ~20 neurons in each
group were randomly selected and the number of spines along 10 µm
of the axon was calculated using ImageJ software (version 1.48;
National Institutes of Health). All experiments were repeated at
least three times and the number of spines/10 µm of an axon was
used to evaluate spine density in each group.
Immunofluorescence staining
The cortical neurons were fixed in 4% ice-cold
paraformaldehyde (PFA; Beijing Solarbio Science & Technology
Co., Ltd.) at 4°C for 10 min. After fixation, cells were washed
three times with 1X PBS and permeabilized with 0.5% Triton-X 100
and 0.5% BSA (cat. no. A2058; Sigma-Aldrich; Merck KGaA) in PBS at
room temperature for 30 min. Then, the cells were incubated with
monoclonal rabbit anti-MAP2 (cat. no. 8707, 1:200; Cell Signaling
Technology, Inc.) or monoclonal rabbit anti-synapsin-1 (cat. no.
6710, 1:200; Cell Signaling Technology, Inc.) antibodies at room
temperature for 2 h and washed three times with 0.5% Triton-X 100
in 1X PBS. For immunofluorescence staining, cells were further
incubated with Alexa Fluor 594-conjugated goat-rabbit IgG (1:200;
Abcam) or Alexa Fluor 488-conjugated AffiniPure goat anti-rabbit
IgG (1:200; ProteinTech Group, Inc.) antibodies in the dark at room
temperature for 2 h. The primary and secondary antibodies were
diluted in 1X PBS with 0.5% Triton-X 100 and 0.5% BSA.
Immunofluorescence signals were observed using an inverted
fluorescence microscope (IX71; Olympus Corporation; magnification,
×40). To measure neurite length, ~30 neurons were randomly selected
and captured using fluorescence or DIC images. Neurites longer than
the diameter of the soma were defined as neurites. The fluorescent
signals from MAP2 and synapsin-1 immunofluorescent staining were
measured to evaluate neurite length and synapse density using
ImageJ software. The neurites originating from the soma were
calculated as the number of primary dendrites per neuron.
Preparation of Aβ1–42
Aβ1–42 was purchased from Sigma-Aldrich
(Merck KGaA). According to the manufacturer's instructions, the
Aβ1–42 peptide was resuspended in DMSO (Beijing Solarbio
Science & Technology Co., Ltd.) to a concentration of 5 mM and
then diluted to 100 µM in sterile 1X PBS (pH 7.4). The suspension
was allowed to oligomerize for 5 days at 37°C and diluted to the 10
µM of Aβ1–42 immediately before addition to the neuron
culture medium.
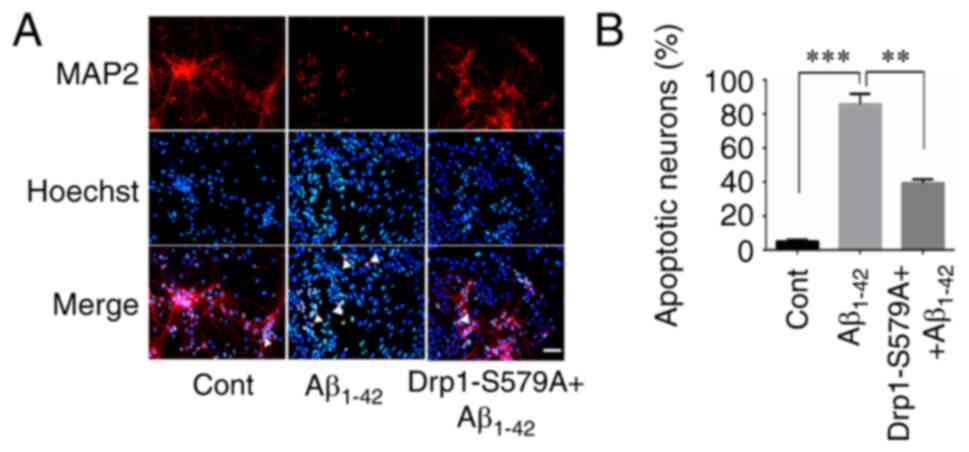
Neuronal apoptosis assays
As described previously (19), mouse cortical neuronal cultures were
treated with or without 10 µM Aβ1–42 at 37°C for 24 h.
To examine the effect of Cdk5-mediated Drp1 phosphorylation on
Aβ1–42-induced neuronal apoptosis, neurons were infected
with Lenti-Drp1-S579A 3 days prior to Aβ1–42 treatment.
Then, 24 h after Aβ1–42 incubation, the cells were fixed
with 4% ice-cold PFA at 4°C for 10 min. Immunofluorescence staining
of MAP2 was performed to label cortical neurons. Hoechst 33258
(cat. no. 94403, Sigma-Aldrich; Merck KGaA) was used for nuclear
DNA staining to evaluate chromosomal condensation and its
morphological changes in neurons. After MAP2 immunofluorescence
staining, the cells were further stained with Hoechst 33258 at room
temperature for 5 min, according to the manufacturer's protocol.
Then, the fluorescence of MAP2 and Hoechst 33258 was detected under
a fluorescence microscope (IX71; Olympus Corporation;
magnification, ×40). Normal neuronal nuclei were stained blue,
whereas apoptotic nuclei with decreased volume and condensed
chromatin were stained shiny white. Neurons with condensed and
fragmented Hoechst 33258 staining were counted as dead cells.
Statistical analysis
Statistical analysis was performed using SPSS
software (version 17.0; SPSS, Inc.). Data are presented as the mean
± SEM and multiple comparisons between groups were performed using
one-way ANOVA followed by post hoc Tukey's test. The comparisons
between two groups were analyzed via unpaired Student's t-test. All
experiments were repeated at least three times. P<0.05 was
considered to indicate a statistically significant difference.
Results
Expression level of phospho-defective
Drp1-S579A in primary cortical neurons
To examine the role of Drp1 phosphorylation at
Ser579 in Aβ1–42-induced neurodegeneration and
apoptosis, a lentiviral vector carrying the phospho-defective
mutant Drp1-S579A was constructed. As shown in Fig. 1A, cells were infected with a
lentiviral vector carrying mCherry at different multiplicity of
infections (MOIs; varying from 0–5) to evaluate infection
efficiency. The percentage of mCherry fluorescence-positive cells
reached 50–70% at an MOI of 2. Thus, in subsequent experiments,
neuronal cultures were infected with Lenti-Drp1-S579A at an MOI of
2. As the phospho-defective Drp1-S579A is fused with a 6His tag in
the lentiviral vector, the expression of Drp1-S579A in neurons
after infection could be immunoblotted using 6His antibody. As
presented in Fig. 1B, the
expression of phospho-defective Drp1-S579A was detected in neurons
infected with Lenti-Drp1-S579A. The exogenous mutant Drp1-S579A
effectively downregulated the phosphorylation of Drp1 at Ser579 in
neurons.
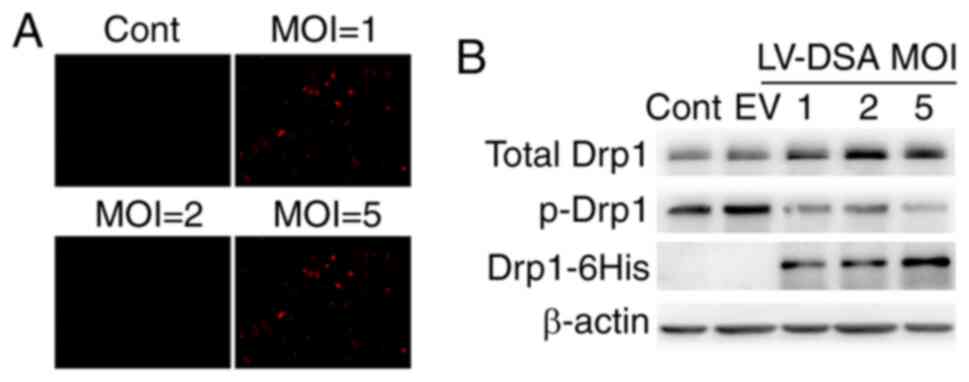 | Figure 1.Lentiviral infection efficiency and
expression level of Drp1-S579A in primary cortical neurons. (A) The
lentiviral infection efficiency in neurons at different MOI.
Primary cortical neurons were infected with lentiviral vector
carrying mCherry at a MOI of 0–5. Infection efficiency was
monitored via mCherry fluorescence in neurons (magnification, ×10).
Scale bar, 50 µm. (B) Expression of phosphorylation-defect Drp1 in
neurons infected with Lenti-Drp1-S579A. Neurons were infected with
Lenti-Drp1-S579A at a MOI of 0–5 or empty lentiviral vector at MOI
of 5. Then, 3 days later, cells were harvested for western blot
analysis to detect the expression level of 6His-tagged Drp1-S579A,
phosphorylation of Drp1 at Ser579 and total-Drp1. β-actin was used
as an endogenous control. EV, empty lentiviral vector; LV-DSA,
lenti-Drp1-S579A; Drp1, dynamin-related protein 1; MOI,
multiplicity of infection; Cont, control; p-, phosphorylated. |
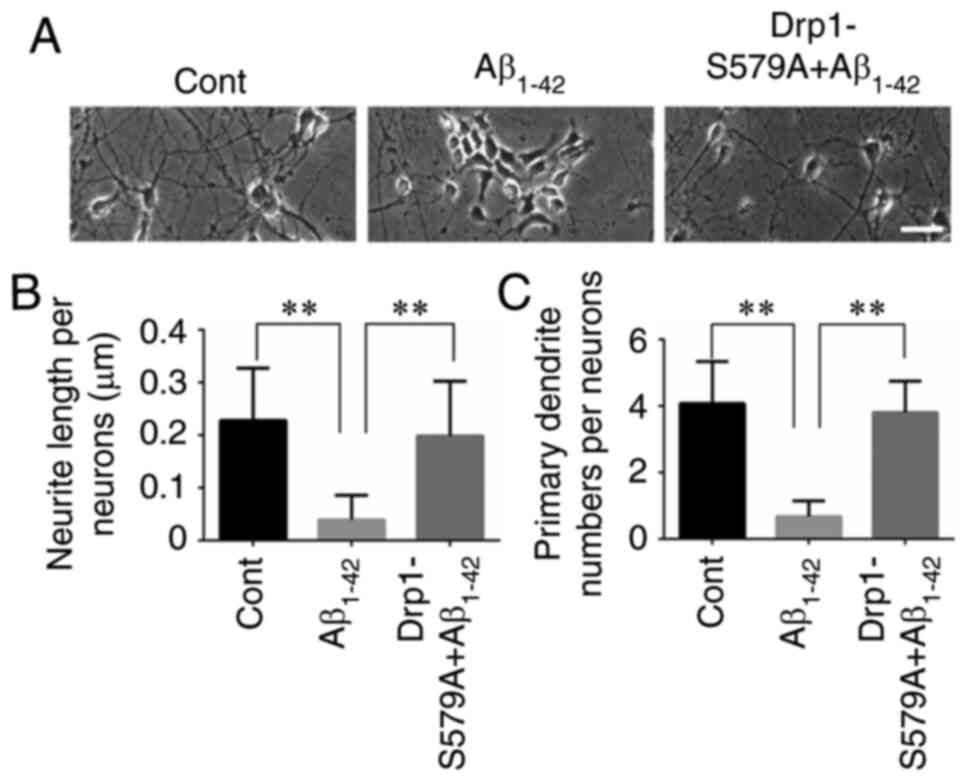
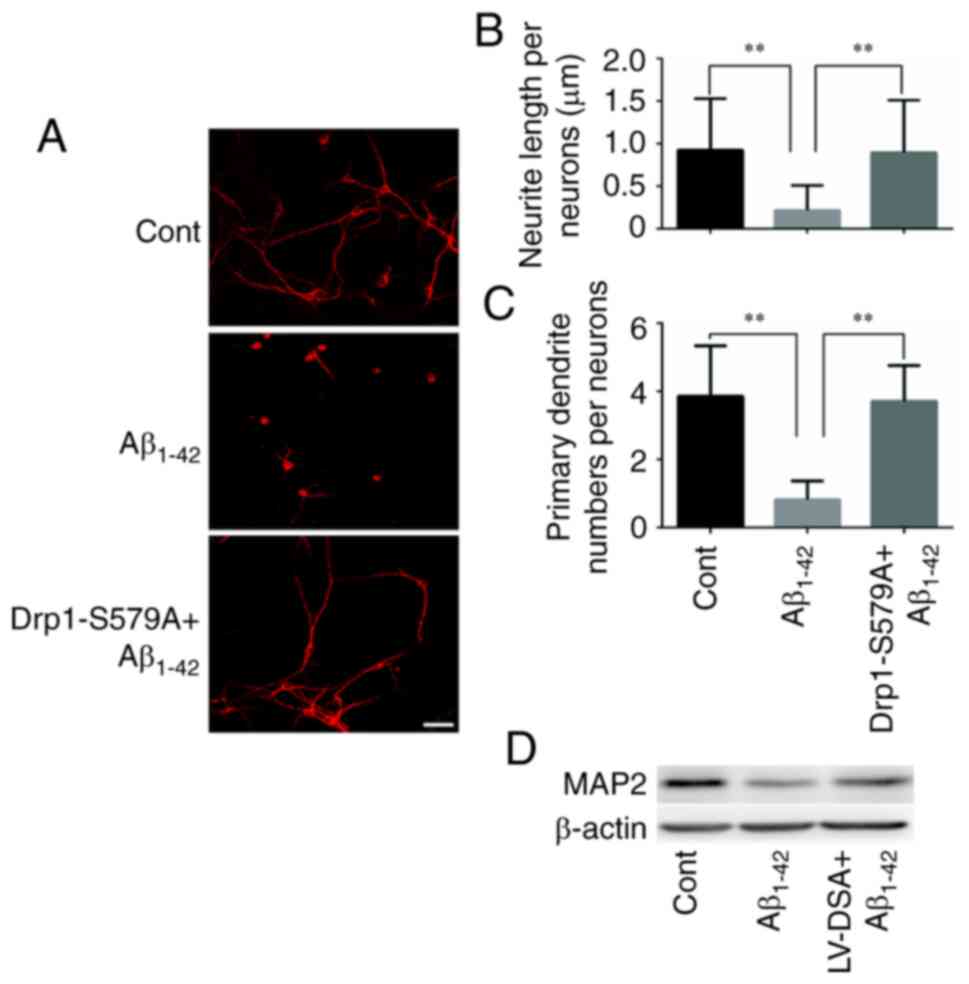
Lenti-Drp1-S579A protects neurons
against Aβ1–42-induced reduction in neurite length and
dendritic loss
Our previous study reported that Aβ1–42
exerted an inhibitory effect on neurite outgrowth (19). The present results confirmed that
primary cortical neurons at 9 DIV had long axons and well-developed
dendrites. By contrast, neurons treated with 10 µM
Aβ1–42 had shortened atrophic axons and dendrites
(Fig. 2A). When neurons were
infected with Lenti-Drp1-S579A, the deleterious effect of
Aβ1–42 on neurites was markedly weakened (Fig. 2B and C). Moreover,
immunofluorescence staining of MAP2 was used to label the neuronal
processes (Fig. 3A). It was found
that Lenti-Drp1-S579A prevented Aβ1–42-induced axonal
and dendritic atrophy in cortical neurons. Aβ1–42
significantly decreased neurite lengths and primary dendrite
numbers in neurons, which was effectively alleviated by blockage of
Drp1 phosphorylation via Lenti-Drp1-S579A (Fig. 3B and C). In addition,
Lenti-Drp1-S579A prevented the downregulation of MAP2 in neurons
following Aβ1–42 stimulation (Fig. 3D).
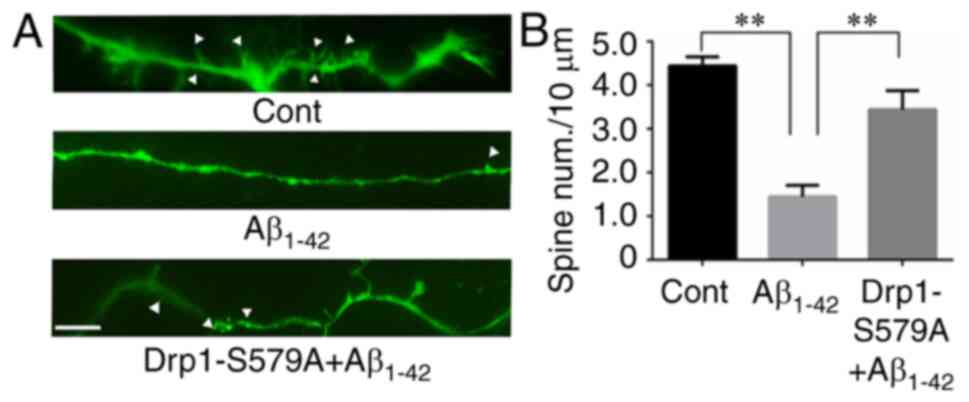
Lenti-Drp1-S579A attenuates the
inhibitory effect of Aβ1–42 on dendritic spine
growth
It has been reported that Aβ1–42 has a
deleterious effect on neurite outgrowth, including dendritic spines
(21). For visualization of spines,
primary cortical neurons were transfected with the pEGFP plasmid at
5 DIV. GFP allowed for the identification of neurons and
quantification of spines under an inverted fluorescence microscope.
As shown in Fig. 4A, mature
dendritic spines were observed in control neurons. Compared with
control cells, neurons subjected to 10 µM Aβ1–42
exhibited a significant decrease in mature dendritic spines
(4.44±0.21 vs. 1.45±0.26 spines/10 µm for control vs.
Aβ1–42, respectively; Fig.
4B). To investigate the role of Drp1 phosphorylation in
Aβ1–42-induced dendritic spine shrinkage, cortical
neurons were infected with Lenti-Drp1-S579A at 3 DIV.
Lenti-Drp1-S579A significantly restored dendritic spines in neurons
after Aβ1–42 treatment (1.45±0.26 vs. 3.44±0.43
spines/10 µm for Aβ1–42 vs. Lenti-Drp1-S579A,
respectively; Fig. 4B).
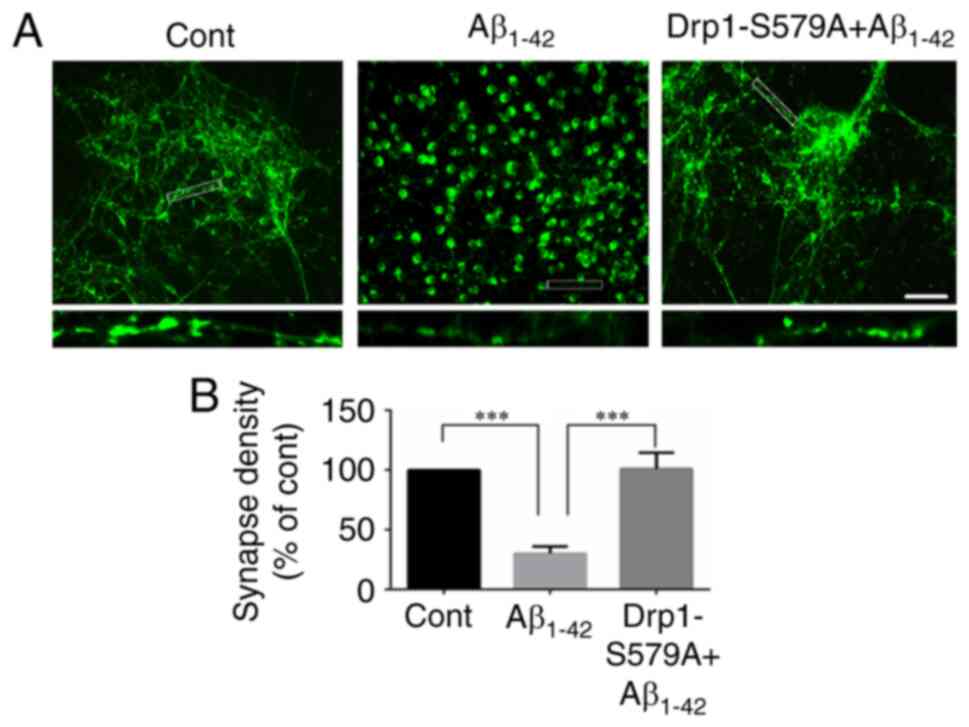
Lenti-Drp1-S579A suppresses
Aβ1–42-induced synaptic loss
It is well recognized that synapses serve an
important role in interneuronal communication and memory formation
(3). Progressive synapse loss is
one of the major hallmarks of AD and is the main cause of memory
impairment in patients with AD (14). Several studies have shown that
incubation of cortical neurons with Aβ1–42 decreases
synapse number (22,23). Immunofluorescence staining of
synapsin was conducted to detect synapses in neurons. As shown in
Fig. 5A, synapsin-1-positive puncta
were prevalent in neuronal processes in the control group. By
contrast, synapsin-1 staining displayed a localization in the soma
and decreased along neuronal processes after Aβ1–42
incubation, indicating reduced synapse density (1.00 vs.
30.11±5.80% for control vs. Aβ1–42, respectively;
Fig. 5B). Moreover, infection with
Lenti-Drp1-S579A restored synapsin-1 staining along neuronal
processes (30.11±5.80 vs. 100.90±13.66% for Aβ1–42 vs.
Lenti-Drp1-S579A, respectively; Fig.
5B).
Lenti-Drp1-S579A protects neurons
against Aβ1–42-induced apoptosis
Exposure of cultured neurons to submicromolar
concentrations of Aβ1–42 may induce direct neurotoxicity
(24). Here, the effect of
Lenti-Drp1-S579A on Aβ1–42-induced apoptosis was
examined. To detect neuronal apoptosis, the cells were stained with
Hoechst 33258 and MAP2 antibody. As presented in Fig. 6A, MAP2-positive cells with condensed
Hoechst 33258 staining were counted as apoptotic neurons. Compared
with the control group, Aβ1–42 stimulation significantly
increased the number of apoptotic neurons (5.00±1.08 vs.
85.75±6.14% for control vs. Aβ1–42, respectively).
Furthermore, infection with Lenti-Drp1-S579A efficiently alleviated
Aβ1–42-induced apoptosis (85.75±6.14 vs. 39.25±2.25% for
Aβ1–42 vs. Lenti-Drp1-S579A, respectively; Fig. 6B).
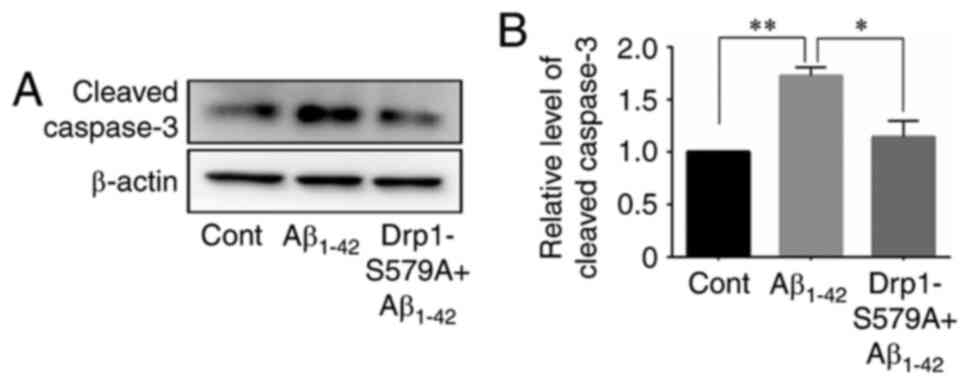
Next, the cleavage of caspase-3 was examined using
western blot analysis in neurons after Aβ1–42 exposure.
The results demonstrated that Aβ1–42 significantly
stimulated the cleavage of caspase-3 in neurons (1.00 vs. 1.73±0.08
for control vs. Aβ1–42, respectively), which was
efficiently prevented by Lenti-Drp1-S579A (1.73±0.08 vs. 1.14±0.15
for Aβ1–42 vs. Lenti-Drp1-S579A, respectively) (Fig. 7).
Discussion
AD is the most common neurodegenerative disease,
with Aβ plaques as one of the major pathological hallmarks
(25). In several AD mouse models,
Aβ peptide deposition in the brain has been reported to be
associated with various neuronal abnormalities, including the
dystrophic neurites (26),
dendritic spine loss (27),
development of synaptic dysfunction (28) and abnormal neuronal firing (29). The variety of neuronal deficits
associated with the deposition of Aβ peptides likely contributes to
cognitive decline and memory loss in patients with AD (8,30). It
is considered that the neurotoxicity of Aβ1–42 is
responsible for neurodegeneration in the AD brain (31). In addition, aberrant activity of
Cdk5 is also involved in the Aβ-evoked neurotoxic cascade (32). Cdk5 serves a vital role in the
development of the central nervous system, maintenance of synaptic
plasticity and neuronal apoptosis in response to stress (33). Moreover, it acts as an upstream
regulator of Drp1-dependent mitochondrial fission during neuronal
apoptosis (18). However, the
underlying mechanism remains to be elucidated.
In our previous study, the mitochondrial fission
protein Drp1 was identified as the direct substrate of Cdk5 and Aβ
1–42 effectively induced Cdk5-meidated Drp1
phosphorylation at Ser579. Furthermore, it was observed that
Cdk5-mediated Drp1 phosphorylation at Ser579 was involved in
Aβ1–42-induced mitochondrial fission and neuronal
apoptosis (19), indicating that
blockage of this process may be a possible strategy to prevent
Aβ1–42-induced neurodegeneration. To prove this
hypothesis, the current study constructed a lentiviral vector
carrying phospho-defective Drp1-S579A. The expression level of
total Drp1 and exogenous mutant Drp1-S579A in neurons were detected
using western blotting with Drp1 antibody and anti-6His antibody,
respectively. Since the affinity of different antibodies is
distinct, it is inappropriate to compare the expression of
endogenous and exogenous mutant Drp1 by the density of these blot
bands. Notably, the mutant Drp1-S579A effectively decreased the
level of Drp1 phosphorylation at Ser579 in neurons. This result was
consistent with that of a previous study (34), in which the phosphorylation site at
Ser579 was reported to be necessary for the GTPase activity of
Drp1. Although the phospho-defective Drp1-S579A may have the same
ability to interact with Fis1 as wild-type Drp1, mitochondrial
fission cannot be mediated by the mutant Drp1 without GTPase
activity (34). In the present
study, it remains unclear how the exogenous phospho-defective
Drp1-S579A had an inhibitory effect on the phosphorylation of
Drp1-Ser579 in neurons. Whether the mutant Drp1-S579A may
competitively inhibit Cdk5-mediated wild-type Drp1 phosphorylation
remains to be determined.
Next, the effect of Lenti-Drp1-S579A on
Aβ1–42-induced neurite atrophy, synapse loss was
examined. The results demonstrated that the
Aβ1–42-mediated decrease in neurite length, synapse
number and density of dendrites and dendritic spines was mostly
prevented by the blockage of Drp1 phosphorylation at Ser579, but it
does not suggest that Lenti-Drp1-S579A completely blocked all of
the Aβ1–42-induced neuronal injury. Moreover, the
Aβ1–42-induced neuronal apoptosis was still detected in
Drp1-S579A + Aβ1–42 group. Neuronal apoptosis might be a
later event in the Aβ1–42-induced degeneration of
dendritic growth and loss of spines and synapses and the inhibitory
effect of Lenti-Drp1-S579A on Aβ1–42-induced
neurodegeneration is greater than neuronal apoptosis. Taken
together, the current data support the important role of
Cdk5-mediated Drp1 phosphorylation at Ser579 in
Aβ1–42-induced neurodegeneration. In addition, the
decrease in neurite length, synapse number and density of dendrite
and dendrite spine is the common characteristic of
neurodegeneration. Therefore, the present study indicates that
blockage of Drp1 phosphorylation at Ser579 efficiently protects
neurons against Aβ1–42-induced neurodegeneration.
On the other hand, it has been well documented that
mitochondrial dysfunction serves a crucial role in various
neurodegenerative diseases, including AD, Parkinson's disease and
Huntington disease (35–37). Mitochondria are highly dynamic
organelles, with their morphology changing frequently via fission
and fusion events. Some large GTPases have been identified as
regulators of mitochondrial fission and fusion. Mitochondrial outer
membrane fission is mediated by Drp1 and mitochondrial fission
protein (Fis1) (38). Optic atrophy
and mitofusins regulate mitochondrial inner or outer membrane
fusion, respectively (39). Several
studies have reported abnormalities in mitochondrial function and
dynamics in neurodegenerative diseases (14,40,41).
Impaired balance of mitochondrial fusion and fission has been
observed in the hippocampal tissue of patients with AD (41). Furthermore, inhibition of
Drp1-mediated mitochondrial fission protects dopaminergic neurons
against neurite loss and apoptosis following mitochondrial stress
(42). Consistent with previous
studies, the mutant Drp1-S579A alleviated Aβ1–42-induced
cleavage of caspase-3 and neuronal apoptosis. In addition, the post
translational modifications of Drp1 are closely associated with its
activity and mitochondrial fission, including phosphorylation,
SUMOylation and nitrosylation (43–48).
Calmodulin-dependent protein kinase Ia phosphorylates Drp1 at
Ser616 and triggers mitochondrial fission by promoting the
interaction between Drp1 and Fis1 (43). GSK-3-mediated phosphorylation of
Drp1 at Ser40 and Ser44 enhances the GTPase activity of Drp1 and
induces mitochondrial fragmentation (45). In addition, S-nitrosylation of Drp1
also bridges excessive mitochondrial fission with neuronal injury
during neurodegeneration (48).
Therefore, it would be useful to further examine whether there is a
crosstalk between Cdk5-mediated Drp1 phosphorylation and
S-nitrosylation of the same protein in neurodegeneration.
The present study indicated that blockage of Drp1
phosphorylation at Ser579 protected cortical neurons against
Aβ1–42-induced degeneration and apoptosis. However,
there are some limitations in the present study. First, the effect
of lenti-Drp1-S579A on neurodegeneration was only examined in
primary cultured cortical neurons. It is still unclear whether
blockage of Drp1 phosphorylation at Ser579 prevents
neurodegeneration in vivo. Therefore, it is necessary to
examine the effect of lenti-Drp1-S579A on neurodegeneration and
neuronal apoptosis in AD animal model. Second, the functional
consequences of Cdk5-mediated Drp1 phosphorylation remain
controversial. It has been reported that Cdk5-mediated
phosphorylation of Drp1 at Ser616 inhibits mitochondrial fission
during neuronal maturation (47).
By contrast, Jahani-Asl et al (47) and our previous study (19) revealed that Cdk5-mediated
phosphorylation of Drp1 at Ser616 or Ser579 (the same conserved
serine residue as Ser585 in different Drp1 isoforms) stimulates
mitochondrial fission in neurons after exposure to
N-methyl-d-aspartate or Aβ1–42, respectively. Although
it is possible that the opposite effect of Cdk5-mediated Drp1
phosphorylation at the same conserved serine residue may be
explained by the level of maturity of neurons, it would be useful
to further investigate the underlying mechanism.
In conclusion, it was suggested that the atrophy of
neuronal processes, synapse loss and neuronal apoptosis are the
characteristic alterations in AD, a common neurodegenerative
disease and that mitochondrial dysfunction is involved in the
pathogenesis of AD. In our previous study, the Cdk5-mediated
phosphorylation of Drp1 at Ser579 was found to regulate
Aβ1–42 induced mitochondrial fission and neuronal
apoptosis (19). Thus, it was
necessary to further investigate the role of Drp1 phosphorylation
at Ser579 in neurodegeneration. The current study constructed a
lentiviral vector carrying phospho-defect Drp1-S579A. It was found
that inhibition of Drp1 phosphorylation at Ser579 by
Lenti-Drp1-S579A efficiently attenuated the
Aβ1–42-mediated decrease in neurite length, synapse
number, density of dendrites and dendritic spines and neuronal
apoptosis. This suggests the involvement of Drp1 phosphorylation at
Ser579 in neurodegeneration in AD and corroborates the potential of
blocking this process to prevent neurodegeneration.
Acknowledgements
Not applicable.
Funding
This work was supported by National Natural Science
Foundation of China (grant nos. 81660159, 81660607 and 82060177),
Jiangxi Province Natural Science Foundation (grant no.
20192BAB205050), Jiangxi Province Major discipline and academic
leaders training Program Project (grant no. 20172BCB22028), the
Research Fund for Jiangxi Geriatric Clinical Medical Research
Center (grant no. 2020BCG74003) and Jiangxi Province Key Laboratory
of Experimental Animals (grant no. 20192BCD40003), Key Research and
Development Program of Jiangxi Province (grant no. 20192BBG70049)
and the Research Fund from Jiangxi Administration of Traditional
Chinese Medicine (grant no. 2019A084).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XJH and XHQ conceived and designed the research. DX
and ZJY conducted the cellular and molecular biological
experiments. PY, LPJ and YTO raised C57BL/6 mice and conducted
neuronal primary culture. TY, JHS, QGL and YYW performed
fluorescent microscopy. DX, PY and QGL analysed and interpreted the
data. XJH and XHQ confirm the authenticity of all the raw data. XJH
and LPJ wrote the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The animal experiments in the present study were
approved by the Animal Research Ethics Committee of Affiliated
People's Hospital of Nanchang University (approval no.
2018-035).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Reddy PH, Manczak M, Mao P, Calkins MJ,
Reddy AP and Shirendeb U: Amyloid-beta and mitochondria in aging
and Alzheimer's disease: Implications for synaptic damage and
cognitive decline. J Alzheimers Dis. 20 (Suppl 2):S499–S512. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mattson MP: Pathways towards and away from
Alzheimer's disease. Nature. 430:631–639. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vaz M and Silvestre S: Alzheimer's
disease: Recent treatment strategies. Eur J Pharmacol.
887:1735542020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reddy PH, Manczak M and Yin X:
Mitochondria-division inhibitor 1 protects against amyloid-β
induced mitochondrial fragmentation and synaptic damage in
Alzheimer's disease. J Alzheimers Dis. 58:147–162. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pozueta J, Lefort R and Shelanski ML:
Synaptic changes in Alzheimer's disease and its models.
Neuroscience. 251:51–65. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Koffie RM, Hyman BT and Spires-Jones TL:
Alzheimer's disease: Synapses gone cold. Mol Neurodegener.
6:632011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
DeKosky ST and Scheff SW: Synapse loss in
frontal cortex biopsies in Alzheimer's disease: Correlation with
cognitive severity. Ann Neurol. 27:457–464. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Terry RD, Masliah E, Salmon DP, Butters N,
DeTeresa R, Hill R, Hansen LA and Katzman R: Physical basis of
cognitive alterations in Alzheimer's disease: Synapse loss is the
major correlate of cognitive impairment. Ann Neurol. 30:572–580.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Harris KM and Kater SB: Dendritic spines:
Cellular specializations impartingboth stability and flexibility to
synaptic function. Annu Rev Neurosci. 17:341–371. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Carlisle HJ and Kennedy MB: Spine
architecture and synaptic plasticity. Trends Neurosci. 28:182–187.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Serrano-Pozo A, Frosch MP, Masliah E and
Hyman BT: Neuropathological alterations in Alzheimer disease. Cold
Spring Harb Perspect Med. 1:a0061892011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ryu J, Hong BH, Kim YJ, Yang EJ, Choi M,
Kim H, Ahn S, Baik TK, Woo RS and Kim HS: Neuregulin-1 attenuates
cognitive function impairments in a transgenic mouse model of
Alzheimer's disease. Cell Death Dis. 7:e21172016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Colgan LA and Yasuda R: Plasticity of
dendritic spines: Subcompartmentalization of signaling. Annu Rev
Physiol. 76:365–385. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jankowsky JL and Zheng H: Practical
considerations for choosing a mouse model of Alzheimer's disease.
Mol Neurodegener. 12:892017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barage SH and Sonawane KD: Amyloid cascade
hypothesis: Pathogenesis and therapeutic strategies in Alzheimer's
disease. Neuropeptides. 52:1–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shah K and Lahiri DK: Cdk5 activity in the
brain-multiple paths of regulation. J Cell Sci. 127:2391–2400.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nguyen MD, Larivière RC and Julien JP:
Deregulation of Cdk5 in a mouse model of ALS: Toxicity alleviated
by perikaryal neurofilament inclusions. Neuron. 30:135–147. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Meuer K, Suppanz IE, Lingor P, Planchamp
V, Göricke B, Fichtner L, Braus GH, Dietz GP, Jakobs S, Bähr M and
Weishaupt JH: Cyclin-dependent kinase 5 is an upstream regulator of
mitochondrial fission during neuronal apoptosis. Cell Death Differ.
14:651–661. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo MY, Shang L, Hu YY, Jiang LP, Wan YY,
Zhou QQ, Zhang K, Liao HF, Yi JL and Han XJ: The role of
Cdk5-mediated Drp1 phosphorylation in Aβ1–42 induced
mitochondrial fission and neuronal apoptosis. J Cell Biochem.
119:4815–4825. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yan M, Guo A, Chen P, Jing H, Ren D, Zhong
Y, Wu Y, Fei E, Lai X, Zou S and Wang S: LRP4 LDLα repeats of
astrocyte enhance dendrite arborization of the neuron. Mol Brain.
13:1662020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kandimalla R, Manczak M, Yin X, Wang R and
Reddy PH: Hippocampal phosphorylated tau induced cognitive decline,
dendritic spine loss and mitochondrial abnormalities in a mouse
model of Alzheimer's disease. Hum Mol Genet. 27:30–40. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ingelsson M, Fukumoto H, Newell KL,
Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT and
Irizarry MC: Early Abeta accumulation and progressive synaptic
loss, gliosis and tangle formation in AD brain. Neurology.
62:925–931. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lista S and Hampel H: Synaptic
degeneration and neurogranin in the pathophysiology of Alzheimer's
disease. Expert Rev Neurother. 17:47–57. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Deshpande A, Mina E, Glabe C and Busciglio
J: Different conformations of amyloid beta induce neurotoxicity by
distinct mechanisms in human cortical neurons. J Neurosci.
26:6011–6018. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cummings BJ, Su JH, Geddes JW, Van
Nostrand WE, Wagner SL, Cunningham DD and Cotman CW: Aggregation of
the amyloid precursor protein within degenerating neurons and
dystrophic neurites in Alzheimer's disease. Neuroscience.
48:763–777. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grutzendler J, Helmin K, Tsai J and Gan
WB: Various dendritic abnormalities are associated with fibrillar
amyloid deposits in Alzheimer's disease. Ann N Y Acad Sci.
1097:30–39. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tsai J, Grutzendler J, Duff K and Gan WB:
Fibrillar amyloid deposition leads to local synaptic abnormalities
and breakage of neuronal branches. Nat Neurosci. 7:1181–1183. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Walsh DM, Klyubin I, Fadeeva JV, Cullen
WK, Anwyl R, Wolfe MS, Rowan MJ and Selkoe DJ: Naturally secreted
oligomers of amyloid beta protein potently inhibit hippocampal
long-term potentiation in vivo. Nature. 416:535–539. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Busche MA, Eichhoff G, Adelsberger H,
Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A and
Garaschuk O: Clusters of hyperactive neurons near amyloid plaques
in a mouse model of Alzheimer's disease. Science. 321:1686–1689.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Masliah E, Ellisman M, Carragher B,
Mallory M, Young S, Hansen L, DeTeresa R and Terry RD:
Three-dimensional analysis of the relationship between synaptic
pathology and neuropil threads in Alzheimer disease. J Neuropathol
Exp Neurol. 51:404–414. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Postuma RB, He W, Nunan J, Beyreuther K,
Masters CL, Barrow CJ and Small DH: Substrate-bound beta-amyloid
peptides inhibit cell adhesion and neurite outgrowth in primary
neuronal cultures. J Neurochem. 74:1122–1130. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wilkaniec A, Gąssowska-Dobrowolska M,
Strawski M, Adamczyk A and Czapski GA: Inhibition of
cyclin-dependent kinase 5 affects early neuroinflammatory
signalling in murine model of amyloid beta toxicity. J
Neuroinflammation. 15:12018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huang E, Qu D, Zhang Y, Venderova K, Haque
ME, Rousseaux MW, Slack RS, Woulfe JM and Park DS: The role of
Cdk5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation
in neuronal death. Nat Cell Biol. 12:563–571. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Taguchi N, Ishihara N, Jofuku A, Oka T and
Mihara K: Mitotic phosphorylation of dynamin-related GTPase Drp1
participates in mitochondrial fission. J Biol Chem.
282:11521–11529. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang WY, Gu ZL, Liang ZQ and Qin ZH:
Mitochondrial dysfunction and Huntington disease. Neurosci Bull.
22:129–136. 2006.PubMed/NCBI
|
|
36
|
Baloyannis SJ: Mitochondrial alterations
in Alzheimer's disease. J Alzheimers Dis. 9:119–126. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cho DH, Nakamura T, Fang J, Cieplak P,
Godzik A, Gu Z and Lipton SA: S-nitrosylation of Drp1 mediates
beta-amyloid-related mitochondrial fission and neuronal injury.
Science. 324:102–105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim H, Scimia MC, Wilkinson D, Trelles RD,
Wood MR, Bowtell D, Dillin A, Mercola M and Ronai ZA: Fine-tuning
of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial
adaptation to hypoxia. Mol Cell. 44:532–544. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chandhok G, Lazarou M and Neumann B:
Structure, function and regulation of mitofusin-2 in health and
disease. Biol Rev Camb Philos Soc. 93:933–949. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yan QW, Zhao N, Xia J, Li BX and Yin LY:
Effects of treadmill exercise on mitochondrial fusion and fission
in the hippocampus of APP/PS1 mice. Neurosci Lett. 701:84–91. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang X, Su B, Lee HG, Li X, Perry G, Smith
MA and Zhu X: Impaired balance of mitochondrial fission and fusion
in Alzheimer's disease. J Neurosci. 29:9090–9103. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Qi X, Qvit N, Su YC and Mochly-Rosen D: A
novel Drp1 inhibitor diminishes aberrant mitochondrial fission and
neurotoxicity. J Cell Sci. 126:789–802. 2013.PubMed/NCBI
|
|
43
|
Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y,
Tomizawa K, Nairn AC, Takei K, Matsui H and Matsushita M: CaM
kinase I alpha-induced phosphorylation of Drp1 regulates
mitochondrial morphology. J Cell Biol. 182:573–585. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cereghetti GM, Stangherlin A, Martins de
Brito O, Chang CR, Blackstone C, Bernardi P and Scorrano L:
Dephosphorylation by calcineurin regulates translocation of Drp1 to
mitochondria. Proc Natl Acad Sci USA. 105:15803–15808. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yan J, Liu XH, Han MZ, Wang YM, Sun XL, Yu
N, Li T, Su B and Chen ZY: Blockage of GSK3β-mediated Drp1
phosphorylation provides neuroprotection in neuronal and mouse
models of Alzheimer's disease. Neurobiol Aging. 36:211–227. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cho B, Cho HM, Kim HJ, Jeong J, Park SK,
Hwang EM, Park JY, Kim WR, Kim H and Sun W: CDK5-dependent
inhibitory phosphorylation of Drp1 during neuronal maturation. Exp
Mol Med. 46:e1052014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jahani-Asl A, Huang E, Irrcher I,
Rashidian J, Ishihara N, Lagace DC, Slack RS and Park DS: CDK5
phosphorylates DRP1 and drives mitochondrial defects in
NMDA-induced neuronal death. Hum Mol Genet. 24:4573–4583. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nakamura T, Cieplak P, Cho DH, Godzik A
and Lipton SA: S-nitrosylation of Drp1 links excessive
mitochondrial fission to neuronal injury in neurodegeneration.
Mitochondrion. 10:573–578. 2010. View Article : Google Scholar : PubMed/NCBI
|