Introduction
Autism spectrum disorder (ASD) is characterized by
persistent deficits in language communication and social
interaction, as well as the occurrence of stereotyped and
repetitive behaviors (1). It is a
group of neurodevelopmental disorders with high clinical and
etiologic heterogeneity (1,2). The prevalence rate of autism is 1–2%
worldwide according to the Autism KnowledgeBase (AutismKB 2.0)
database (3). Autism is usually
caused by genetic and environmental factors, of which genetic
factors account for a larger proportion (1,3,4). Over
the last decade, the genetic etiology of autism has been studied
using classic cytogenetics, whole-genome linkage analysis,
candidate gene resequencing and genetic association analysis,
whole-genome association analysis, whole-genome copy number
variation, whole-exome and whole-genome sequencing approaches
(2,3). Marked progress has been made in the
study of candidate genes associated with autism. For instance, 228
highly credible autism-related genes (30 syndrome genes and 198
non-syndrome genes) were identified and deposited in the Autism
KnowledgeBase (AutismKB) database (3,4),
including PTEN, NRXN 1, RELN, SHANK3, POGZ and CHD8.
However, there is a lack of repeatability in different genetic
studies (5–8) and it is not clear how many genes are
associated with autism. General studies have shown that the genetic
basis of autism has obvious heterogeneity (1,9). A
number of autism-related genes can lead to a variety of phenotypes
and diseases and the number of genes involved in autism is quite
large, which brings great challenges to the genetic research of
autism (6). Except for a small
number of autistic patients with single-gene defects or dominant
chromosomal abnormalities, most of the patients have not undergone
prenatal diagnosis. Thus, it is necessary to understand the complex
polygenic background of autism and to implement prenatal diagnosis
of this disease (10,11).
Whole-exome sequencing (WES) is an advanced genomic
technique that sequences all exonic regions in the entire genome
captured through target enrichment. Comprehensive analysis of the
whole exome aids in the identification of disease-related coding
region mutations to determine the impact of these DNA mutations on
the pathogenesis of a disease (12). There are a few reports on how to
preliminarily screen for gene mutations of familial autism based on
a large number of mutations annotated from the WES results and how
to further identify highly credible autism-related genes (13–18).
However, the screening conditions and parameters used are not
consistent. WES has not been applied in the prenatal diagnosis of
ASD.
In the present study, two autistic children were
subjected to WES and reasonable conditions were established for the
preliminary screening of variant annotations and the identification
of credible autism-related genes. The present study aimed to
compare and evaluate the genetic heterogeneity of credible
autism-related genes between two autistic children in the same
pedigree to provide more evidence for the genetic risk and complex
polygenic background of familiar ASD.
Materials and methods
Autism pedigree and peripheral blood
samples
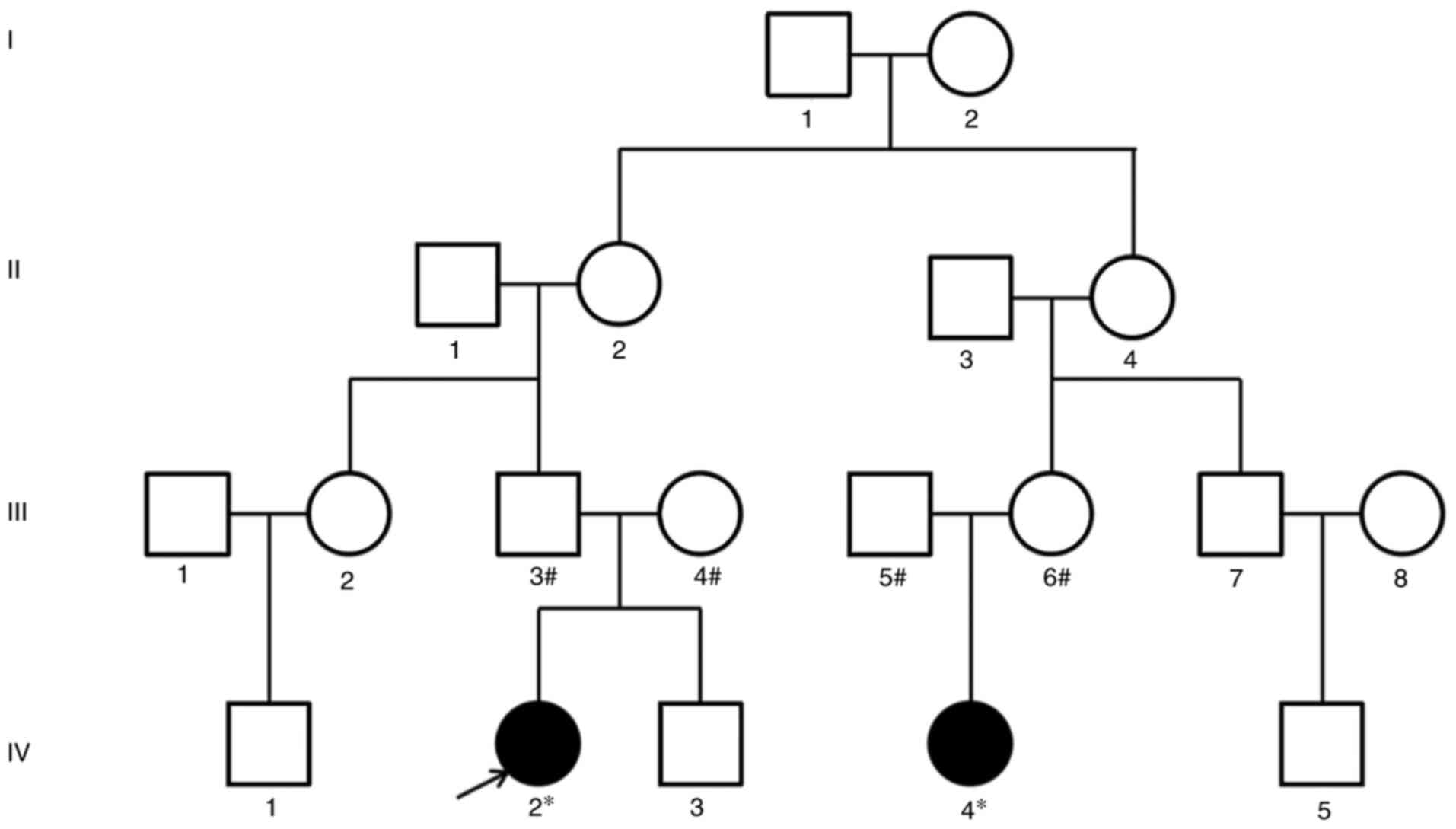
A total of two autistic children (IV2 and IV4) in a
family pedigree were recruited between January 2019 and December
2020 for the present study (Fig.
1). They were diagnosed at the Second Affiliated Hospital of
Wenzhou Medical University (Wenzhou, China) and identified as ASD
according to Wenzhou Medical Identification Committee of Sick and
Disabled Children. The diagnosis of ASD was confirmed by the Autism
Diagnostic Observation Schedule (ADOS) (19) and expert clinical judgment. IV2 and
IV4 children were found to have ASD characteristics at age 2 and 3
and diagnosed as autistic children at age 3 and 4, respectively.
The scores of ADOS (model 1) were 10 and 8, respectively. IV2 was
associated with mild cerebral palsy and mild intellectual
disability, whereas IV4 was associated with epilepsy, brain
development delay and mild intellectual disability. The parents of
IV2 and IV4 were without clinical autism symptoms and the marriage
type was not consanguineous. Psychological training, such as
training in language communication and social interaction, was
performed at the Mental Health Center, Wenzhou Medical University
(Wenzhou, China), which is one of the author-affiliated institutes
associated with this study. The present study was approved by the
Ethics Committee of Wenzhou Hospital of Integrated Traditional
Chinese and Western Medicine (approval number: 2018-57; Wenzhou,
China); the Ethics Committee of this hospital was shared with the
Wenzhou Maternal and Child Health Guidance Center (Wenzhou, China).
Written informed consent forms was obtained from the parents of the
children, for both their participation and the participation of the
children. The clinical data and 2–3 ml/each peripheral blood
samples of two autistic children and their parents were provided by
Wenzhou Maternal and Child Health Guidance Center. The peripheral
blood samples of IV2 and IV4 autistic children were collected at
the age of 7 and 5 years, respectively.
WES
WES of the autism samples (2 ml) was performed by
iGeneTech Biotech (Beijing) Co. Ltd. Genomic DNA was extracted
using a Blood Genomic DNA Isolation kit (magnetic bead method;
Kangwei Biologic Co. Ltd.) in accordance with the manufacturer's
protocols. The quality and quantity of DNA extracts were evaluated.
DNA quality was examined using 1% agarose gel electrophoresis. The
DNA concentrations were determined using a Qubit dsDNA BR assay kit
(Thermo Fisher Scientific, Inc.). The whole exome liquid-phase
capture technique was used to efficiently and specifically enrich
all the human exonic regions. Briefly, fragmentation was carried
out using a hydrodynamic shearing system (Covaris, Inc.) to
generate 150–200 bp fragments. Remaining overhangs were converted
into blunt ends via exonuclease/polymerase activities using the
Fast Library Prep Kit [iGeneTech Biotech (Beijing) Co. Ltd.]. After
adenylation of 3′ ends of DNA fragments, adapter oligonucleotides
were ligated using the Fast Library Prep Kit. The Fast Library Prep
Kit was used in accordance with the manufacturer's protocols. DNA
fragments with ligated adapter molecules on both ends were
selectively enriched by PCR. Subsequently, libraries were
hybridized in liquid phase with a biotin-labeled probe, and
magnetic beads with a streptomycin monolayer were employed to
capture the exons of genes using the TargetSeq One Kit [iGeneTech
Biotech (Beijing) Co. Ltd.], in accordance with the manufacturer's
protocols. Captured libraries were enriched by PCR to prepare for
sequencing. DNA library construction was performed using a Fast
Library Preparation kit [iGeneTech Biotech (Beijing) Co. Ltd.].
Library quantification and quality assessment were then performed.
Qubit 3.0 (Thermo Fisher Scientific, Inc.) was used for library
quantification; library concentration >25 ng/µl was considered a
qualified library. DNA quality was examined using Bioptic Qsep 100
DNA fragment analyzer (BiOptic, Inc.). The length of library
fragments was generally between 270 and 350 bp. The Illumina
NovaSeq6000 platform (Illumina, Inc.) was used for PE150
high-throughput double-terminal sequencing. The loading
concentration of the final library was 450–600 pmol/l; library
quantification was performed by qPCR. The raw WES data were
obtained and mapped to the human reference genome (UCSC
GRCh37/hg19, 2009). The type of analysis was single sample
whole-exome analysis, which included quality evaluation of
sequencing data as well as the detection and screening of
variations. Based on the BAM results of alignment with the genome
reference sequences, Samtools (http://www.htslib.org/) and GATK (https://gatk.broadinstitute.org/hc/en-us) were used to
find single nucleotide polymorphisms (SNPs) and small insertions
and deletions (InDel) of <50 bp. ANNOVAR (http://www.openbioinformatics.org/annovar/)
software was used to annotate the SNP and InDel loci to determine
the corresponding gene information, functional data and
harmfulness. The annotations on SNP and InDel were carried out by
iGeneTech Biotech (Beijing) Co. Ltd.
Screening for SNP and InDel
annotations
A large number of genetic variants were detected. To
identify the variants with significant functions, the following
conditions were set: i) The variants could be divided into high
level, possible high level, intermediate level and low level
according to the priority of harmfulness. In the present study, the
screening for the priority of variants was limited to high level
only. The high priority of the variant indicated that it was
located in the exonic region of the gene, had a minor allele
frequency (MAF) of <0.01 in East Asian populations [i.e. the
1000 Genomes Project database (1000G), the Exome Aggregation
Consortium (ExAC) and the Genome Aggregation Database (gnomAD)]
(20) and could lead to an altered
amino acid sequence. In addition, at least 2 of the 10 prediction
software packages [including functional prediction software SIFT,
Polyphen2_HDIV, Polyphen2_HVAR, LRT, MutationTaster,
MutationAssessor, FATHMM, fathmm-MKL, total effect prediction
software CADD, conservative prediction software GERP; all from
dbNSFP version 3.5a (annotated using Annovar; updated on September
21, 2018)] predicted it as harmful. ii) The quality evaluation of
the variants could be divided into high, medium and low quality. In
the present study, the quality of the variant was limited to high
or medium quality only. The high quality of the variant is denoted
by its depth of ≥50 layers, genotype quality value of >30 and
frequency deviation of <0.1 (heterozygous 0.4–0.6; homozygous
0.9–1.0) (20). The medium quality
of the variant is denoted by its depth of ~20 layers, genotype
quality value of >20 and frequency deviation of <0.2
(heterozygous 0.3–0.7; homozygous 0.8–1.0) (20). iii) The mutant genes were required
to be associated with autism in the human gene mutation database
(HGMD) (http://www.hgmd.cf.ac.uk/ac/index.php).
Sanger sequencing
All the preliminary screening variants of IV2 and
IV4 were verified by Sanger sequencing. In addition, the parents of
IV2 and IV4 were sequenced for these variants to determine the
variant source. Conventional PCR amplification and Sanger
sequencing were performed on all the exons where the variant loci
of the corresponding gene were located; PCR primer synthesis and
Sanger sequencing were conducted by Qingke Biology Co., Ltd. The
primer sequences are provided in Table
SI.
Screening of autism-related genes
through autism databases
The AutDB autism database (http://www.mindspec.org/autdb.html; also known as
SFARI Gene, http://gene.sfari.org) is updated in
real time (21). By September 2020,
it contained 1,237 genes related to autism, with an evidence score
of 0–5. In the present study, the genes with evidence score of ≥3
in the AutDB database were selected as candidate autism-related
genes. AutismKB Database is the world's largest database for the
genetic evidence of ASD (3,4), with 1,379 genes and 11,669 single
nucleotide variants or small InDels integrated in version 2.0
(2018). According to AutismKB, in addition to 99 syndrome genes,
1,280 non-syndrome genes with an evidence score of >4 were
designated as candidate autism-related genes, of which 30 syndrome
genes and 198 non-syndrome genes with an evidence score of >16
were designated as substantially credible autism-related genes. The
criteria set by Yang et al (3) used for calculating the ‘evidence
score’ of AutismKB v2.0 database were as follows: Next-generation
sequencing (NGS) de novo mutation studies or low-scale gene
studies, weight 10; NGS other studies, weight 8; genome-wide
association studies, weight 4; Copy-number variation/structural
variation studies, linkage analyses or low-scale genetic
association studies, weight 2; and expression profiling or NGS
mosaic mutation studies, weight 1.
Results
Preliminary screening of the WES
results of autistic child IV2
Among all the detected variants in autistic child
IV2, there were 21 variants (in 21 genes) with high harmful effects
and high or medium quality, and their mutant genes were associated
with autism in HGMD; further details are provided in Table I. Notably, the variant types of all
genes were missense (nonsynonymous), except that GPR152 and
OAS3 genes contained deletion (frameshift, InDel below 50
bp) and nonsense (stopgain) variants. All the variants were located
in exons. The genotypes were all heterozygous and the variant
frequency of each gene in this sample ranged from 0.32–0.59; that
is, the minimum variant frequency (that of gene NRP2) was
0.32 and the maximum variant frequency (that of gene OAS3)
was 0.59 in this sample. All of the mutant genes were located on
autosomal chromosomes, except that ARHGEF6 was on the X
chromosome.
 | Table I.Preliminary screening of whole-exome
sequencing results of autistic child IV2. |
Table I.
Preliminary screening of whole-exome
sequencing results of autistic child IV2.
| Gene | Location | Transcript/Exon
(cDNA change/AA change) | dbSNP150 ID | Variant frequency
in IV2 |
|---|
| DNAH7 | 2q32.3 |
NM018897/exon18 | rs769106832 | 0.52 |
|
|
|
(c.G2323A/p.E775K) |
|
|
| CAPN10 | 2q37.3 | NM023083/exon7 | rs552159586 | 0.48 |
|
|
|
(c.T1145G/p.L382R) |
|
|
| LPA | 6q25.3 | NM005577/exon34
(c.T5395A/p.C1799S) | rs189946882 | 0.42 |
| ABCB5 | 7p21.1 |
NM178559/exon12 | rs80123476 | 0.47 |
|
|
|
(c.G1234A/p.A412T) |
|
|
| LAMB1 | 7q31.1 | NM002291/exon3 | N/A | 0.4 |
|
|
|
(c.C38T/p.A13V) |
|
|
| CTTNBP2 | 7q31.31 | NM033427/exon5 | N/A | 0.45 |
|
|
|
(c.C2188G/p.L730V) |
|
|
| ZNF79 | 9q33.3 |
NM001286696/exon5 | rs182450731 | 0.56 |
|
|
|
(c.T259C/p.W87R) |
|
|
| LAMC3 | 9q34.12 |
NM006059/exon15 | rs200121474 | 0.44 |
|
|
|
(c.G2687A/p.R896Q) |
|
|
| JMJD1C | 10q21.3 |
NM001322254/exon6 | rs201464655 | 0.46 |
|
|
|
(c.A1046C/p.D349A) |
|
|
| SCUBE2 | 11p15.4 |
NM001170690/exon7 | rs991815964 | 0.41 |
|
|
|
(c.C838T/p.R280W) |
|
|
| GPR152 | 11q13.2 | NM206997/exon1 | rs776403683 | 0.48 |
|
|
|
(c.1149delG/p.Q383fs) |
|
|
| OAS3 | 12q24.13 | NM006187/exon9 | N/A | 0.59 |
|
|
|
(c.C1996T/p.Q666X) |
|
|
| MYCBP2 | 13q22.3 |
NM015057/exon50 | N/A | 0.41 |
|
|
|
(c.C7394G/p.P2465R) |
|
|
| CACNA1H | 16p13.3 |
NM001005407/exon25 | N/A | 0.46 |
|
|
|
(c.A4643G/p.N1548S) |
|
|
| CHD3 | 17p13.1 |
NM001005271/exon11 | rs747768901 | 0.49 |
|
|
|
(c.G1969A/p.G657S) |
|
|
| MYH4 | 17p13.1 | NM017533/exon4 | rs201896271 | 0.48 |
|
|
|
(c.G331A/p.A111T |
|
|
| ARHGEF6 | Xq26.3 |
NM001306177/exon12 | rs201896882 | 0.44 |
|
|
|
(c.G944A/p.R315Q) |
|
|
| ATP2B4 | 1q32.1 |
NM001001396/exon12 | rs138521935 | 0.4 |
|
|
|
(c.G1996A/p.A666T) |
|
|
| NRP2 | 2q33.3 | NM003872/exon9 | rs201900948 | 0.32 |
|
|
|
(c.A1333C/p.I445L) |
|
|
| CSMD1 | 8p23.2 |
NM033225/exon41 | rs201454449 | 0.4 |
|
|
|
(c.A6236T/p.Y2079F) |
|
|
| SORCS1 | 10q25.1 |
NM001013031/exon1 | rs118055903 | 0.38 |
|
|
|
′(c.C503T/p.T168M) |
|
|
Preliminary screening of the WES
results in autistic child IV4
Among all the detected variants in autistic child
IV4, there were 22 variants (in 21 genes) with high harmful effects
and high or medium quality, and their mutant genes were associated
with autism in HGMD; further details are given in Table II. All of the variants were
missense type and were located in exonic regions. The genotypes
were all heterozygous, and the variant frequency of each gene in
this sample ranged from 0.32–0.57; that is, the minimum variant
frequency (that of gene MTMR12) was 0.32 and the maximum
variant frequency (that of gene SMARCC2) was 0.57 in this
sample. The mutant genes were all located on autosomal chromosomes.
Based on the comparative assessment of WES results in Tables I and II, the two autistic children IV2 and IV4
shared a rare variant (ATP2B4 c.G1996A).
| Table II.Preliminary screening of whole-exome
sequencing results of autistic child IV4. |
Table II.
Preliminary screening of whole-exome
sequencing results of autistic child IV4.
| Gene | Location | Transcript/Exon
(cDNA change/AA change) | dbSNP150 ID | Variant frequency
in IV4 |
|---|
| USH2A | 1q41 |
NM007123/exon13 | rs145383772 | 0.45 |
|
|
|
(c.G2792A/p.C931Y) |
|
|
| SNTG2 | 2p25.3 | NM018968/exon3 | rs192264442 | 0.47 |
|
|
|
(c.C229A/p.R77S) |
|
|
| SCN1A | 2q24.3 |
NM001165963/exon10 | rs200176684 | 0.45 |
|
|
|
(c.G1499A/p.R500Q) |
|
|
| SETD5 | 3p25.3 |
NM001080517/exon20 | rs527600519 | 0.45 |
|
|
|
c.G3325A/p.D1109N) |
|
|
| CCDC14 | 3q21.1 |
NM001308317/exon5 | rs116934420 | 0.51 |
|
|
|
(c.A116G/p.H39R) |
|
|
| ROS1 | 6q22.1 |
NM002944/exon43 | rs538891848 | 0.47 |
|
|
|
(c.C6764T/p.S2255L) |
|
|
| ROS1 | 6q22.1 |
NM002944/exon43 | rs547573917 | 0.47 |
|
|
|
(c.T6763C/p.S2255P) |
|
|
| SYNE1 | 6q25.2 |
NM033071/exon72 | rs375752705 | 0.42 |
|
|
|
(c.C11786A/p.A3929E) |
|
|
| THSD7A | 7p21.3 |
NM015204/exon24 | N/A | 0.56 |
|
|
|
(c.G4540T/p.D1514Y) |
|
|
| PLXNA4 | 7q32.3 |
NM020911/exon31 | rs200763664 | 0.49 |
|
|
|
(c.C5564T/p.S1855F) |
|
|
| CHD7 | 8q12.2 | NM017780/exon3 | N/A | 0.57 |
|
|
|
(c.G1892A/p.R631K) |
|
|
| PKHD1L1 | 8q23.2 |
NM177531/exon77 | rs146897796 | 0.41 |
|
|
|
(c.C12673G/p.L4225V) |
|
|
| KCNMA1 | 10q22.3 |
NM001014797/exon1 | rs77602559 | 0.44 |
|
|
|
(c.A34G/p.S12G) |
|
|
| SLK | 10q24.33 |
NM001304743/exon12 | rs554070501 | 0.47 |
|
|
|
(c.G2774A/p.R925Q) |
|
|
| NAV2 | 11p15.1 |
NM001111018/exon5 | N/A | 0.46 |
|
|
|
(c.T362C/p.F121S) |
|
|
| NCAPD2 | 12p13.31 |
NM014865/exon20 | rs200921592 | 0.49 |
|
|
|
(c.C2548T/p.R850W) |
|
|
| SMARCC2 | 12q13.2 |
NM003075/exon26 | rs200833916 | 0.57 |
|
|
|
(c.C2920A/p.P974T) |
|
|
| GOLGA3 | 12q24.33 |
NM001172557/exon12 | N/A | 0.5 |
|
|
|
(c.C2492T/p.T831I) |
|
|
| SPATA13 | 13q12.12 |
NM001166271/exon2 | N/A | 0.44 |
|
|
|
(c.T959C/p.V320A) |
|
|
| ATP2B4 | 1q32.1 |
NM001001396/exon12 | rs138521935 | 0.36 |
|
|
|
(c.G1996A/p.A666T) |
|
|
| MTMR12 | 5p13.3 |
NM001040446/exon5 | rs768253146 | 0.32 |
|
|
|
(c.C424T/p.H142Y) |
|
|
| DNAH9 | 17p12 |
NM004662/exon15 | N/A | 0.36 |
|
|
|
(c.C2209T/p.P737S) |
|
|
Sanger sequencing to verify the
preliminary screening variants in autistic children and to verify
the sources of these variants
The 21 and 22 preliminary screening variants in
autistic children IV2 and IV4, respectively, were verified by
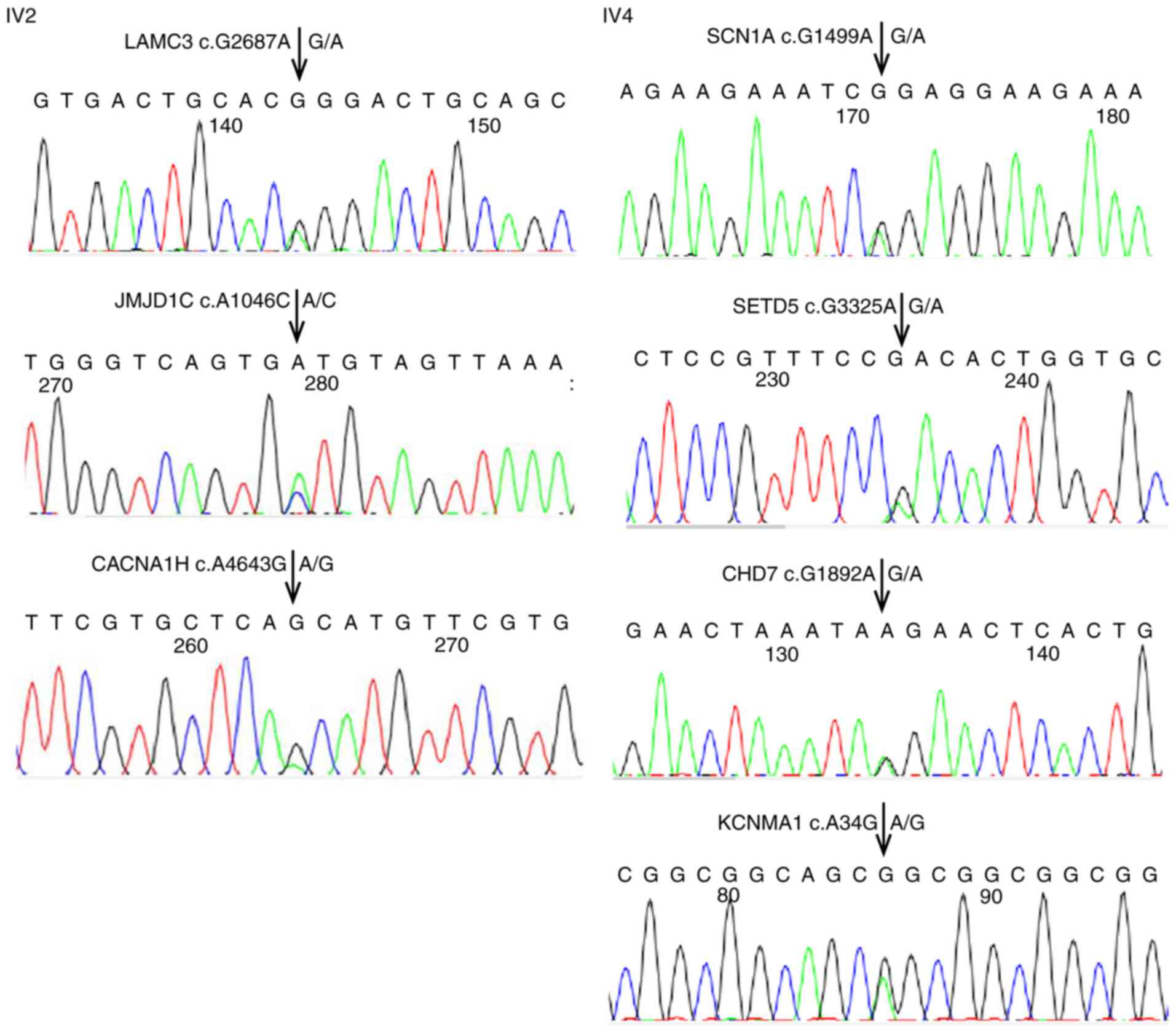
Sanger sequencing. The sequencing maps of three variants in IV2 and
four variants in IV4 are provided in Fig. 2. As shown in Tables III and IV, these variants were ultimately
identified as highly credible autism-related variants. The
sequencing maps of the remaining variants will be provided upon
reasonable request (see Availability of data and materials
below). To determine the source of these preliminary screening
variants, the sequencing data of the parents were analyzed. The
results indicated that all the preliminary screening variants in
the two autistic children were inherited from their parents, who
were heterozygous for all of these variants, and there were no
de novo variants (Tables
III and IV). The sequencing
maps of the parents will be provided upon reasonable request (see
Availability of data and materials below).
| Table III.Further bioinformatics analysis of
preliminary screening gene variations in autistic child IV2. |
Table III.
Further bioinformatics analysis of
preliminary screening gene variations in autistic child IV2.
| Gene | Variation
harmfulness | Variation
quality | Variation
source | Included in
AutismKB 2.0/Evidence score | Included in
AuthB/Evidence score |
|---|
| DNAH7 | High | High | Father | Yes/2 | No |
| CAPN10 | High | High | Mother | Yes/0 | No |
| LPA | High | High | Father | Yes/0 | No |
| ABCB5 | High | High | Father | No | No |
|
LAMB1a | High | High | Mother | Yes/16 | Yes/3 |
|
CTTNBP2a | High | High | Mother | Yes/2 | Yes/3 |
| ZNF79 | High | High | Father | Yes/2 | No |
|
LAMC3a | High | High | Father | Yes/38 | Yes/3 |
|
JMJD1Ca | High | High | Father | Yes/20 | Yes/3 |
| SCUBE2 | High | High | Father | Yes/0 | No |
| GPR152 | High | High | Father | Yes/0 | No |
| OAS3 | High | High | Mother | Yes/0 | No |
|
MYCBP2a | High | High | Mother | Yes/12 | No |
|
CACNA1Ha | High | High | Mother | Yes/22 | Yes/3 |
|
CHD3a | High | High | Mother | Yes/12 | Yes/3 |
|
MYH4a | High | High | Mother | Yes/12 | Yes/2 |
|
ARHGEF6a | High | High | Father | Yes/syndromic | No |
| ATP2B4 | High | Medium | Father | Yes/0 | No |
| NRP2 | High | Medium | Father | Yes/4 | Yes/0 |
|
CSMD1a | High | Medium | Father | Yes/14 | Yes/3 |
|
SORCS1a | High | Medium | Mother | Yes/15 | No |
| Table IV.Further bioinformatics analysis of
preliminary screening gene variations in autistic child IV4. |
Table IV.
Further bioinformatics analysis of
preliminary screening gene variations in autistic child IV4.
| Gene | Variation
harmfulness | Variation
quality | Variation
source | Included in
AutismKB 2.0/Evidence score | Included in
AuthB/Evidence score |
|---|
|
USH2Aa | High | High | Father | Yes/10 | Yes/3 |
|
SNTG2a | High | High | Father | Yes/14 | Yes/2 |
|
SCN1Aa | High | High | Mother |
Yes/20/syndromic | Yes/5 |
|
SETD5a | High | High | Mother | Yes/22 | Yes/5 |
| CCDC14 | High | High | Father | Yes/0 | No |
| ROS1 | High | High | Mother | Yes/0 | No |
| ROS1 | High | High | Mother | Yes/0 | No |
|
SYNE1a | High | High | Father | Yes/11 | Yes/3 |
| THSD7A | High | High | Father | Yes/0 | No |
|
PLXNA4a | High | High | Father | Yes/11 | Yes/2 |
|
CHD7a | High | High | Father |
Yes/18/syndromic | Yes/3 |
| PKHD1L1 | High | High | Mother | Yes/0 | No |
|
KCNMA1a | High | High | Mother | Yes/34 | Yes/3 |
| SLK | High | High | Mother | Yes/0 | No |
|
NAV2a | High | High | Father | Yes/12 | Yes/3 |
| NCAPD2 | High | High | Mother | Yes/2 | No |
|
SMARCC2a | High | High | Mother | Yes/0 | Yes/4 |
| GOLGA3 | High | High | Mother | Yes/2 | No |
| SPATA13 | High | High | Father | Yes/3 | No |
| ATP2B4 | High | Medium | Mother | Yes/0 | No |
| MTMR12 | High | Medium | Father | Yes/0 | No |
| DNAH9 | High | Medium | Father | Yes/0 | No |
Bioinformatics analysis of preliminary
screening variants in autistic child IV2
A total of 11 candidate autism-related genes
(indicated with a in Table
III) in autistic child IV2 were screened according to the
evidence score of >4 in AutismKB or ≥3 in AutDB. The candidate
genes with an evidence score of >16 in AutismKB were regarded as
highly credible autism-related genes, which included LAMC3,
JMJD1C and CACNA1H (indicated in bold in Table III).
Based on the possible autism-related gene functions
in Gene Ontology (GO; updated on January 1, 2018) database
(https://www.uniprot.org/help/gene_ontology),
LAMC3 was associated with astrocyte development,
JMJD1C was related to transcriptional regulation and histone
demethylase activity (that is, chromatin remodeling) and
CACNA1H was involved in neuronal action potential and
membrane potential regulation (that is, synaptic function).
The specific missense variant loci of these three
genes were reported for the first time in the present study, to the
best of our knowledge. None of the loci were found in AutismKB 2.0
autism database (version 2018), AutDB autism database (version
2020) or HGMD database (version 2019). The three gene variants were
located outside the repeat region. The MAFs of LAMC3
c.G2687A were 0.001, 0.01 and 0.008 in East Asian populations
(1000G, ExAC and gnomAD, respectively). However, CACNA1H
c.A1046C and JMJD1C c.A4643G were not detected in these
populations. According to the HGMD database, 40.91% (9/22), 92.00%
(23/25) and 92.73% (51/55) of the missense variants in LAMC3,
JMJD1C and CACNA1H genes, respectively, were pathogenic.
The functional effects of the variants in these three genes, as
predicted by 10 software packages are listed in Table V. Notably, eight of the 10 packages
predicted LAMC3 c.G2687A as harmful (i.e. very high
harmfulness), eight of the 10 packages are predicted CACNA1H
c.A1046C as harmful (i.e. very high harmfulness) and two of 10
software predicted JMJD1C c.A4643G as harmful (i.e. high
harmfulness).
| Table V.Functional impact of highly credible
autism-related gene mutations in autistic children IV2 and IV4 as
predicted by 10 software programs. |
Table V.
Functional impact of highly credible
autism-related gene mutations in autistic children IV2 and IV4 as
predicted by 10 software programs.
|
|
Software |
|---|
|
|
|
|---|
| Gene | SIFTa | Polyphen2
HDIVb | Polyphen2
HVARc | LRTd | Mutation
Tastere | Mutation
Assessorf | FATHMMg | Fathmm
MKLh | CADDi | GERPj |
|---|
| LAMC3 | D | D | P | D | D | L | T | D | 25.3 | 5.12 |
| JMJD1C | D | B | B | N | N | L | T | D | 4.9 | 4.91 |
| CACNA1H | D | D | D | D | D | N/A | D | D | 25.6 | 4 |
| SCN1A | D | B | B | N | D | M | D | D | 23.8 | 5.28 |
| SETD5 | D | D | D | N | D | M | D | D | 23.3 | 6.07 |
| CHD7 | T | P | P | D | D | L | T | D | 17.7 | 5.23 |
| KCNMA1 | D | B | B | N/A | N | N | D | T | 12.1 | −1.58 |
In addition to LAMC3 and JMJD1C
inherited from the father III3 (descended from I1 and I2), there
was also the cumulative effect of CACNA1H inherited from the
mother III4 (not descended from I1 and I2), i.e. there was a
cumulative effect of CACNA1H with LAMC3 and
JMJD1C. The results of the present study could provide
guidance for the prenatal diagnosis of ASD. If the father III3 and
mother III4 of autistic child IV2 give birth again, prenatal
genetic diagnosis should be conducted on these three highly
credible autism-related genes.
Bioinformatics analysis of preliminary
screening variants in autistic child IV4
A total of 10 candidate autism-related genes
(indicated with a in Table
IV) in autistic child IV4 were screened according to the
evidence score of >4 in the AutismKB or at ≥3 in the AutDB. The
candidate genes with an evidence score of >16 in the AutismKB
were considered as highly credible autism-related genes, which
included SCN1A, SETD5, CHD7 and KCNMA1 (indicated in
bold in Table IV).
Based on the possible autism-related gene functions
in GO database, SCN1A was related to axon initiation segment
and neuronal action potential transmission (synaptic function),
SETD5 was involved in transcriptional regulation and histone
acetylation regulation (i.e. chromatin remodeling), CHD7 was
associated with central nervous system development (i.e. synaptic
function) and transcriptional positive regulation, and
KCNMA1 was responsible for the regulation of membrane
potential (i.e. synaptic function).
SCN1A c.G1499A has been reported to be
associated with Dravet syndrome in the HGMD database (updated in
2019), whereas the missense variant loci of the remaining three
genes were first discovered in the present study, to the best of
our knowledge, and were not found in the AutismKB 2.0 database
(version 2018), the AutDB database (version 2020) or the HGMD
database (version 2019).
All variants in these four genes were located
outside the repeat region. The MAFs of SCN1A c.G1499A,
SETD5 c.G3325A and KCNMA1 c.A34G were (0.003, 0 and
0.0016), (0.001, 0 and 0.0032) and (0.001, 0 and 0.0014) in East
Asian populations (1000G, ExAC and gnomAD), respectively; whereas
CHD7 c.G1892A was not found in these populations. According
to the HGMD database, 51.62% (860/1666), 18.75% (6/32), 20.14%
(171/849) and 66.67% (6/9) of the missense variants in SCN1A,
SETD5, CHD7 and KCNMA1 genes, respectively, were
pathogenic. The functional effects of the variants in these four
genes predicted by 10 software are listed in Table V. Notably, seven of the 10 software
packages predicted that SCN1A c.G1499A was harmful (i.e.
very high harmfulness), nine of the 10 software packages predicted
that SETD5 c.G3325A was harmful (i.e. very high
harmfulness), seven of the 10 software packages predicted that
CHD7 c.G1892A was harmful (i.e. very high harmfulness) and
two of the 10 software packages predicted that KCNMA1 c.A34G
was harmful (i.e. high harmfulness).
In addition to SCN1A, SETD5 and KCNMA1
inherited from the mother III6 (descended from I1 and I2), there
was also the cumulative effect of CHD7 inherited from the
father III5 (not descended from I1 and I2), i.e. there was a
cumulative effect of CHD7 with SCN1A, SETD5 and
KCNMA1. The results of the present study may provide
guidance for the prenatal diagnosis of ASD. If the father III5 and
the mother III6 of child IV4 give birth again, prenatal genetic
diagnosis should be performed on these four highly credible
autism-related genes.
The filtering procedure and number of the remaining
variants in IV2 and IV4 autistic children are summarized in
Table VI.
| Table VI.Filtering procedure and number of
remaining variants of autistic children IV2 and IV4 for each
step. |
Table VI.
Filtering procedure and number of
remaining variants of autistic children IV2 and IV4 for each
step.
|
| Variant |
|---|
|
|
|
|---|
| Filtering
procedure | IV2 | IV4 |
|---|
| Total variants | 252,419 | 176,044 |
| Excluding variants
if QD<2.0 or FS >60.0 or MQ<40.0 or DP<4 | 135,315 | 121,156 |
| Excluding variants
if harmfulness of variant (priority) was low |
3,237 |
2,105 |
| Retaining variants
if genes involved in autism in HGMD | 145 | 84 |
| Retaining variants
if harmfulness of variant was high and if quality of variant was
high or medium (preliminary screening gene variants in Tables I and II) | 21 | 22 |
| Following
confirmation by Sanger sequencing; retaining variants if evidence
score of gene was >4 in AutismKB or at ≥3 in AutDB (candidate
autism–related gene variants in Tables III and IV) | 11 | 10 |
| Retaining variants
if evidence score of gene was >16 in AutismKB (highly credible
autism-related gene variants in Tables III and IV) | 3 | 4 |
Discussion
De novo mutations and genetic variations both
have important effects on autism. In recent years, remarkable
progress has been made towards understanding the genetic risks of
autism using WES (22,23). For example, some de novo
mutations were found by comparing the WESs of sporadic cases with
those of their parents (17,24–29).
It has been suggested that these de novo mutations can
increase the risk of autism but may not lead to autism. Therefore,
it is necessary to perform a WES study based on the family pedigree
of ASD patients to explore possible hereditary mutations (14–17).
The present study set the reasonable conditions of
high or medium variant quality, such as depth, genotype quality
value and frequency deviation, to ensure the reliability of the WES
results. Additionally, to ensure that the selected variants were of
high pathogenicity it set the conditions that the harmfulness of
the variant was high, the variant was located in the exonic region,
the MAF value was low in three East Asian populations, the variant
resulted in amino acid changes and that various software packages
predicted the variant as harmful. The present study also set
reasonable conditions for the preliminary screening of mutant genes
related to autism in the HGMD database and further selected the
genes with high evidence score in the AutismKB and the AutDB autism
databases to ensure that the identified autism-related genes were
highly credible.
The results of the present study showed that all the
preliminary screening genes, except for ATP2B4, were
different between two autistic children in the same pedigree and
the highly credible autism-related genes were completely different
between two autistic children, including LAMC3, JMJD1C and
CACNA1H in autistic child IV2 and SCN1A, SETD5, CHD7
and KCNMA1 in autistic child IV4. These data indicated that
familial ASD patients exhibited a high degree of genetic
heterogeneity. The two autistic children in this pedigree were
fourth-degree relatives, which might contribute to a high degree of
genetic heterogeneity. A previous study showed that genetic
heterogeneity exists in the first-degree relatives of autistic
patients and even identical twins (1). High clinical heterogeneity might also
be the main cause of high genetic heterogeneity observed in the
present study (30). Apart from the
core symptoms, the vast majority of autism cases are accompanied by
other concurrent symptoms, including abnormal head circumference,
sleep disorders, epilepsy, gastrointestinal diseases, immune
disorders and metabolic disorders (31). Autistic child IV2 in the present
study pedigree was accompanied by mild cerebral palsy and mild
intellectual disability, while IV4 was accompanied by epilepsy,
brain growth delay and mild intellectual disability, which may be
partly attributed to the heterogeneous variations of autism risk
genes.
Regarding the functions of seven highly credible
autism-related genes in autistic children, JMJD1C and
SETD5 were associated with transcriptional regulation and
chromatin remodeling, CACNA1H, SCN1A and KCNMA1 were
related to synaptic function, CHD7 was involved in synaptic
function and transcriptional regulation and LAMC3 was
related to astrocytes. There are three key pathways by which gene
mutations affect nerve development in patients with ASD: i)
Chromatin remodeling, ii) transcriptional regulation and splicing
regulation and iii) synapse formation and function (32). Chromatin remodeling controls
potential events in the formation of neural connections, including
neurogenesis and neural differentiation and is dependent on
epigenetic mechanisms (32). Among
the top autism candidate genes, some are related to transcriptional
regulation and alternative splicing (32). Mutations in synapse-related genes
can affect multiple components in the synaptic network, from
receptors, ion channels to scaffold proteins, thus resulting in an
impaired synaptic function (32).
Synaptic defects and neuroelectrophysiological impairments have
been reported in experimental ASD models (32). Furthermore, astrocytes serve an
important role in regulating synaptic connections (33).
Genetic studies on autism have focused on both ends
of the frequency spectrum of genetic variation; namely, rare and
common variations (34). All of the
seven highly credible autism-related gene variants reported in the
present study belonged to rare variations and the corresponding
genes were included in the AutismKB and AutDB autism databases.
According to the AutDB database, CHD7, SCN1A and
SETD5 genes not only belong to rare variations but are also
associated with CHARGE, Dravet and MRD23 syndrome (OMIM#214800,
607208 and 615761, respectively). It has been suggested that the
etiology of autism can be caused by: i) Single gene defects, ii) a
combination of two or more rare mutations, each with high
pathogenic effect, iii) a rare variant with high pathogenic effect,
on the background of common variants with low pathogenic effects,
iv) multiple common variants and v) complex genetic mechanism,
including common susceptibility variants, environmental risk
factors and epigenetic variations (2,30). In
the present study, the highly credible autism-related genes of
autistic child IV2 (or IV4) were inherited not only from III3 (or
III6) (immediate family members) but also from III4 (or III5)
(extended family members), which could provide more evidence for
the irregular inheritance pattern of familial ASD. The results
demonstrated that a number of autism-related genes may be involved
in the pathogenesis of familial ASD, and each gene probably has
different degrees of contribution to this disease.
In summary, the present study provided a reasonable
and feasible method for screening highly reliable autism-related
genes from WES results, which has high application value in
clinical practice. The analysis and comparison of the WES results
indicated that several highly credible autism-related genes were
identified such as LAMC3, JMJD1C and CACNA1H in
autistic child IV2 and SCN1A, SETD5, CHD7 and KCNMA1
in autistic child IV4. These candidate genes were completely
different between the two autistic children, suggesting a high
degree of genetic heterogeneity. Except for SCN1A c.G1499A,
the specific missense variant loci of the remaining six highly
credible autism-related genes were reported for the first time in
the present study. Some of the highly credible autism-related genes
were originated from the extended family members of the pedigree.
The findings provided strong evidence for the complex polygenic
architecture of ASD, which may help in the early detection of this
disease.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from The
Wenzhou Science and Technology Bureau of China (grant no.
Y20180787), The Natural Science Foundation of Zhejiang Province
(grant no. LY19C070001) and Horizontal Scientific Research Project
of Wenzhou Medical University (grant no. KJHX1710).
Availability of data and materials
The datasets generated during the current study are
available in the NCBI sequence Read Archive (SRA) repository,
https://www.ncbi.nlm.nih.gov/sra/PRJNA733596;
BioProject accession: PRJNA733596. Other datasets used and analyzed
during the current study are available from the corresponding
author on reasonable request.
Authors' contributions
LS collected and analyzed the WES data, and wrote
and revised the paper. PL, TZ, ML, SZ, YTH and YWH collected the
samples, and clinical, WES and Sanger sequencing data, and analyzed
the data. PL, TZ and ML confirm the authenticity of all the raw
data. HL designed the research, analyzed the data, and wrote and
revised the paper. All of the authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was given ethics approval by the
Ethics Committee of Wenzhou Hospital of Integrated Traditional
Chinese and Western Medicine (Wenzhou, China; approval no.
2018-57); the Ethics Committee of this hospital was shared with the
Wenzhou Maternal and Child Health Guidance Center. Written informed
consent was obtained from the parents of the children, for both
their participation and the participation of the children.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jeste SS and Geschwind DH: Disentangling
the heterogeneity of autism spectrum disorder through genetic
findings. Nat Rev Neurol. 10:74–81. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yoon SH, Choi J, Lee WJ and Do JT: Genetic
and epigenetic etiology underlying autism spectrum disorder. J Clin
Med. 9:9662020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang C, Li J, Wu Q, Yang X, Huang AY,
Zhang J, Ye AY, Dou Y, Yan L, Zhou WZ, et al: AutismKB 2.0: A
knowledgebase for the genetic evidence of autism spectrum disorder.
Database. 2018:bay1062018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xu LM, Li JR, Huang Y, Zhao M, Tang X and
Wei L: AutismKB: An evidence-based knowledgebase of autism
genetics. Nucleic Acids Res. 40:D1016–D1022. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Betancur C: Etiological heterogeneity in
autism spectrum disorders: More than 100 genetic and genomic
disorders and still counting. Brain Res. 1380:42–77. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
De Rubeis S and Buxbaum JD: Genetics and
genomics of autism spectrum disorder: Embracing complexity. Hum Mol
Genet. 24:R24–R31. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
O'Roak BJ, Vives L, Girirajan S, Karakoc
E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al: Sporadic
autism exomes reveal a highly interconnected protein network of de
novo mutations. Nature. 485:246–250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang T, Guo H, Xiong B, Stessman HA, Wu H,
Coe BP, Turner TN, Liu Y, Zhao W, Hoekzema K, et al: De novo genic
mutations among a Chinese autism spectrum disorder cohort. Nat
Commun. 7:133162016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Devlin B and Scherer SW: Genetic
architecture in autism spectrum disorder. Curr Opin Genet Dev.
22:229–237. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vanya M, Szucs S, Szili K, Vetro A and
Bartfai G: The possibility of prenatal diagnosis of autism spectrum
disorder. Psychiatr Hung. 30:303–307. 2015.(In Hu). PubMed/NCBI
|
|
11
|
Jiang YH, Wang Y, Xiu X, Choy KW, Pursley
AN and Cheung SW: Genetic diagnosis of autism spectrum disorders:
The opportunity and challenge in the genomics era. Crit Rev Clin
Lab Sci. 51:249–262. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sener EF, Canatan H and Ozkul Y: Recent
Advances in autism spectrum disorders: Applications of whole exome
sequencing technology. Psychiatry Investig. 13:255–264. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chapman NH, Nato AQ Jr, Bernier R,
Ankenman K, Sohi H, Munson J, Patowary A, Archer M, Blue EM, Webb
SJ, et al: Whole exome sequencing in extended families with autism
spectrum disorder implicates four candidate genes. Hum Genet.
134:1055–1068. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Egawa J, Watanabe Y, Wang C, Inoue E,
Sugimoto A, Sugiyama T, Igeta H, Nunokawa A, Shibuya M, Kushima I,
et al: Novel rare missense variations and risk of autism spectrum
disorder: Whole-exome sequencing in two families with affected
siblings and a two-stage follow-up study in a Japanese population.
PLoS One. 10:e01194132015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Inoue E, Watanabe Y, Egawa J, Sugimoto A,
Nunokawa A, Shibuya M, Igeta H and Someya T: Rare heterozygous
truncating variations and risk of autism spectrum disorder:
Whole-exome sequencing of a multiplex family and follow-up study in
a Japanese population. Psychiatry Clin Neurosci. 69:472–476. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Toma C, Torrico B, Hervás A, Valdés-Mas R,
Tristán-Noguero A, Padillo V, Maristany M, Salgado M, Arenas C,
Puente XS, et al: Exome sequencing in multiplex autism families
suggests a major role for heterozygous truncating mutations. Mol
Psychiatry. 19:784–790. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thongnak C, Hnoonual A, Tangviriyapaiboon
D, Silvilairat S, Puangpetch A, Pasomsub E, Chantratita W,
Limprasert P and Sukasem C: Whole-exome sequencing identifies one
de novo variant in the FGD6 Gene in a Thai family with autism
spectrum disorder. Int J Genomics. 2018:82315472018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Al-Mubarak B, Abouelhoda M, Omar A,
AlDhalaan H, Aldosari M, Nester M, Alshamrani HA, El-Kalioby M,
Goljan E, Albar R, et al: Whole exome sequencing reveals inherited
and de novo variants in autism spectrum disorder: A trio study from
Saudi families. Sci Rep. 7:56792017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou B, Xu Q, Lu P and Xu X: Evaluation on
reliability and validity of Chinese Version Autism Diagnostic
Observation Schedule Module-1 and clinical application. Chin J Evid
Based Pediatr. 8:257–261. 2013.(In Chinese).
|
|
20
|
Do R, Stitziel NO, Won H, Jørgensen AB,
Duga S, Merlini PA, Kiezun A, Farrall M, Goel A, Zuk O, et al:
Multiple rare alleles at LDLR and APOA5 confer risk for earlyonset
myocardial infarction. Nature. 518:102–106. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Basu SN, Kollu R and Banerjee-Basu S:
AutDB: A gene reference resource for autism research. Nucleic Acids
Res. 37:D832–D836. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Satterstrom FK, Kosmicki JA, Wang J, Breen
MS, De Rubeis S, An JY, Peng M, Collins R, Grove J, Klei L, et al:
Large-scale exome sequencing study implicates both developmental
and functional changes in the neurobiology of autism. Cell.
180:568–584.e23. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Feliciano P, Zhou X, Astrovskaya I, Turner
TN, Wang T, Brueggeman L, Barnard R, Hsieh A, Snyder LG, Muzny DM,
et al: Exome sequencing of 457 autism families recruited online
provides evidence for autism risk genes. NPJ Genom Med. 4:192019.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
O'Roak BJ, Deriziotis P, Lee C, Vives L,
Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C,
et al: Exome sequencing in sporadic autism spectrum disorders
identifies severe de novo mutations. Nat Genet. 43:585–589. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Iossifov I, Ronemus M, Levy D, Wang Z,
Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et
al: De novo gene disruptions in children on the autistic spectrum.
Neuron. 74:285–299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sanders SJ, Murtha MT, Gupta AR, Murdoch
JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM,
Parikshak NN, Stein JL, et al: De novo mutations revealed by
whole-exome sequencing are strongly associated with autism. Nature.
485:237–241. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha
KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al: Patterns
and rates of exonic de novo mutations in autism spectrum disorders.
Nature. 485:242–245. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Iossifov I, O'Roak BJ, Sanders SJ, Ronemus
M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson
KE, et al: The contribution of de novo coding mutations to autism
spectrum disorder. Nature. 515:216–221. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sjaarda CP, Wood S, McNaughton AJM, Taylor
S, Hudson ML, Liu X, Guerin A and Ayub M: Exome sequencing
identifies de novo splicing variant in XRCC6 in sporadic case of
autism. J Hum Genet. 65:287–296. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo H, Wang T, Wu H, Long M, Coe BP, Li H,
Xun G, Ou J, Chen B, Duan G, et al: Inherited and multiple de novo
mutations in autism/developmental delay risk genes suggest a
multifactorial model. Mol Autism. 9:642018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lai MC, Lombardo MV and Baron-Cohen S:
Autism. Lancet. 383:896–910. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
De Rubeis S, He X, Goldberg AP, Poultney
CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al:
Synaptic, transcriptional and chromatin genes disrupted in autism.
Nature. 515:209–215. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Petrelli F and Bezzi P: mGlu5-mediated
signalling in developing astrocyte and the pathogenesis of autism
spectrum disorders. Curr Opin Neurobiol. 48:139–145. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Manolio TA, Collins FS, Cox NJ, Goldstein
DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR,
Chakravarti A, et al: Finding the missing heritability of complex
diseases. Nature. 461:747–753. 2009. View Article : Google Scholar : PubMed/NCBI
|