Introduction
Spontaneous intracerebral hemorrhage (ICH) has the
highest mortality rate of ~40% worldwide among stroke subtypes
(1), accounts for 15–20% of all
stroke cases, and is more common in elderly patients (2–4). Acute
ICH due to large intracranial hematoma is associated with high
morbidity and mortality as it can lead to primary brain injury via
destruction of brain tissue and high intracranial pressure (ICP)
(5–7). Previous studies revealed that
craniotomy for hematoma evacuation is an effective therapy for
limiting primary brain damage and decreasing ICP following ICH
(5,8,9).
However, long-term outcomes were found to not be altered, and it
rarely affects neurological recovery (10). Increasing numbers of studies have
found that red blood cell debris (hemoglobin and its degradation
products) and other blood components trigger secondary brain injury
following ICH and contribute to a series of damaging events,
including neuroinflammation, brain edema, oxidative stress,
blood-brain barrier (BBB) damage and neuronal death (11–16).
An increasing number of studies has been investigated the
mechanisms underlying ICH-induced secondary injury to identify
improved therapeutic targets for ICH; the potential mechanisms
underlying EBI include autophagy, apoptosis, direct neuronal death
and necroptosis (13,17,18).
Numerous factors, including hypertension,
obstructive sleep apnea (OSA), smoking, obesity and hyperlipidemia,
can induce intracranial aneurysms, promote aneurysm rupture,
aggravate early brain injury (EBI) and worsen the overall outcome
of patients with vascular aneurysm following hemorrhagic stroke
(19–21). Geer et al (22) also reported that the incidence rate
of hypertension, heart disease and hyperlipidemia, as well as body
mass index, were higher in patients with OSA than in those without.
A multivariable logistic regression model showed that OSA is
associated with an increased risk of ICH in a recent study
(23). Pontes-Neto et al
(24) also found that the incidence
of OSA is higher in patients with ICH and may aggravate
perihematomal edema.
The mechanism by which OSA aggravates brain injury
is unclear. Orrù et al (25)
reported that OSA induces oxidative stress and inflammation and
disrupts vascular function by releasing excessive levels of NO and
its derivatives. Apoptosis and neuroinflammation are involved in
hypoxia-induced cell death and tissue injury, especially in OSA and
hypoxia-associated disease, such as intermittent hypoxia (26) and cerebral ischemia (27). Previous studies have reported that
activating transcription factor (ATF)4 is a transcriptional
regulation factor that serves an important role in ICH and that the
ATF4/CHOP signaling pathway regulates cell death via aggravated
neuroinflammation and apoptosis (28,29).
To the best of our knowledge, however, the effect of OSA in ICH has
not been investigated and the specific mechanism is unclear. The
present study aimed to investigate the neuronal damage induced by
OSA and the potential molecular mechanisms by which ICH-induced EBI
regulates neural apoptosis.
Materials and methods
Animals
All animal experiments complied with the National
Institutes of Health guidelines (30) for the handling of laboratory animals
and were approved by the Ethics Committee of the Wuxi Clinical
College of Anhui Medical University (approval no. YXLL-2020-112;
Wuxi, China). All experiments were performed on 159 healthy adult
male C57BL/6J mice (age, 6–8 weeks; weight, 22–25 g; Anhui Medical
University). Mice were divided into the following groups
(n=15/group): Sham, OSA, ICH, ICH + OSA, ICH + small interfering
(si)-control (Con), ICH + si-ATF4, ICH + OSA + si-Con and ICH + OSA
+ si-ATF4. Overall, there were 16 mice in the Sham group (one
dead), 16 mice in the OSA group (one dead), 19 mice in the ICH
group (four dead), 23 mice in the ICH + OSA group (eight dead), 20
mice in the ICH + si-Con (five dead), 18 mice in the ICH + si-ATF4
(three dead), 25 mice in the ICH + OSA + si-Con (10 dead), 22 mice
in the ICH + OSA + si-ATF4 (seven dead). The mice were housed in a
climate-controlled environment at 25±2°C and 55±5% humidity with
12-h light/dark cycles, and had free access to food and water.
ICH animal model
The ICH mouse model was constructed via autologous
blood injection, as previously described (31). Briefly, male C57BL6/J mice were
anesthetized by intraperitoneal (i.p.) injection of 50 mg/kg
pentobarbital sodium and placed in a prone position with a
stereotactic head frame. The rectal temperature was maintained at
37.0±0.5°C during the operation with a heating pad. An artificial
tear ointment was used to protect the eye from injury during
surgery. A midline scalp incision was made and a cranial burr hole
(1 mm diameter) was made at the following coordinates relative to
bregma: 0.2 posterior, 2.2 lateral and 3.5 mm below the dura. A
total of 30 µl autologous blood without anticoagulation was
collected from the caudal artery and rapidly injected into the
basal ganglia via the burr hole using the 26 gauge needle of a
10-µl Hamilton syringe. First, 5 µl arterial blood was injected at
a depth of 2.8 mm from the dura (injection speed, 3 µl/min). After
5 min, the remaining 25 µl blood was injected at a depth of 3.5 mm
(injection speed, 3 µl/min). Following the injection of autologous
blood, the needle was left in the brain for 10 min to prevent blood
backflow along the needle tract. Finally, the hole was covered with
medical bone wax. The animals in the Sham group underwent the same
surgical procedures but were injected at the same sites with an
equal volume of 0.9% sterile saline instead of blood.
Intermittent hypoxia (IH) model
An IH model was used to construct the OSA model as
previously described (32). At 30
min after recovery from anesthesia, mice in the OSA group were
exposed to air for 90 sec, followed by 90 sec of progressive
hypoxia to a nadir of 5% inhaled O2 to model moderate
SA. The mice were exposed to IH for 7 h/day during the light period
(10 a.m. to 5 p.m.) for 3 consecutive days. During the hypoxia
phase, the O2 concentration in the chamber was decreased
to 5% within 20 sec by infusion of N2 and remained at
that concentration for 90 sec. Then, the O2
concentration was rapidly increased to 21% within 10 sec by
flushing the chamber with compressed clean air, which was sustained
for 90 sec. Following IH induction, the mice were transferred to
the normal housing environment with room air, neurological score
was measured and mice were euthanized for brain tissue
collection.
siRNA treatment and transfection
The mice were anesthetized with pentobarbital sodium
(50 mg/kg) and placed on stereotaxic apparatus (Narishige
International Ltd.). Then, a burr hole was made in the left
hemisphere at the following coordinates: 0.2 posterior, 1.0 lateral
and 2.2 mm below the horizontal plane of the bregma. A total of 5
µl siRNA was injected into the left lateral ventricle at a rate of
0.5 µl/min. Lipofectamine™ RNAiMax reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) was used in Opti-MEM medium, according to
the manufacturer's instructions. To enhance the silencing effect,
the injection was performed 48 h before ICH. Targeted and control
siRNAs were synthesized by Shanghai GeneChem Co., Ltd., as follows:
si-ATF4 forward, 5′-GUGAGAAACUGGAUAAGAATT−3′ and reverse,
5′-UUCUUAUCCAGUUUCUCACTT−3′ and negative control siRNA forward,
5′-UUCUCCGAACGUGUCACGUTT−3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT−3′.
Neurobehavioral and mortality
assessment
The severity of brain injury was evaluated by
determining the neurological function 72 h after ICH as previously
described (17). The scoring system
consisted of six tests, and specific standards are shown in
Table SI. The final neurological
score ranged from 3 to 18 and included spontaneous activity (0–3),
movement symmetry of all limbs (0–3), forelimb outstretching (0–3),
body proprioception (1–3), response to vibrissae touch (1–3) and
climbing (1–3). A total of 10 mice in all groups
underwent neurobehavioral assessment, and a higher score
represented improved neurological function. Following the
establishment of the ICH/OSA model and the neurobehavioral
assessment, mortality assessment was performed and death was
confirmed. Dead animals were replaced to ensure 15 mice in each
group. The mortality was defined as the ratio of dead mice to the
total number in each group.
Brain water content measurement
At 72 h post-ICH, the mice were sacrificed with 100
mg/kg sodium pentobarbital via i.p. injection. Death was confirmed
by cessation of breathing and corneal reflex, then brain tissue
samples were collected. The severity of brain edema was evaluated
by measuring the brain water content using the standard wet-dry
method, as previously reported (17,27,33).
The entire brain was harvested and separated into the ipsilateral
and contralateral cortex and basal ganglia and cerebellum (wet
weight). Then, brain specimens from each group were dehydrated at
105°C for 24 h to acquire the dry weight. The percentage of brain
water content was calculated as follows: (Wet weight-dry
weight)/wet weight ×100%.
Evans blue extravasation
Evans blue extravasation was performed as previously
described (34). Briefly, all mice
were anesthetized with intraperitoneal (i.p.) pentobarbital sodium
(50 mg/kg) injection. Evans blue dye (2%; 5 ml/kg; Sigma-Aldrich;
Merck KGaA) was injected into the left femoral vein for >2 min
and allowed to circulate for 60 min. Then, mice were sacrificed
with 100 mg/kg sodium pentobarbital via i.p. injection followed by
phosphate-buffered saline (PBS) intracardial perfusion. Death was
confirmed by cessation of breathing and corneal reflex. The brains
were removed and divided into the left and right cerebral
hemispheres, weighed, homogenized in saline and centrifuged at
15,000 x g for 30 min at room temperature. The supernatant was
added to an equal volume of trichloroacetic acid, incubated
overnight at 4°C and centrifuged at 15,000 x g for 30 min at room
temperature. The supernatant was collected and
spectrophotometrically quantified at 610 nm to measure the amount
of Evans blue dye.
TUNEL staining
TUNEL and neuronal nuclei (NeuN) co-staining were
performed to assess neuronal death in the brain cortex and were
fixed with 4% paraformaldehyde for 1 h at 25°C. Paraffin-embedded
sections (10 µm) were cut from formalin-fixed tissue and stained
with TUNEL and NeuN stain. TUNEL reaction mixture (50 µl) was added
to each sample and slides were incubated in a humidified dark
chamber for 60 min at 37°C. Then, a primary antibody against NeuN
(1:200; rabbit polyclonal; cat. no. ab128886; Abcam) diluted in PBS
was added, followed by incubation overnight at 4°C. The slides were
then incubated with DAPI (0.1 mg/ml) for 5 min at room temperature
in the dark to stain the nuclei, followed by imaging with a
fluorescence microscope (magnification, ×400). The procedure was
performed according to the manufacturer's instructions with a TUNEL
staining kit (cat. no. 1684817; Roche Diagnostics GmbH). A negative
control (without the TUNEL reaction mixture) was used. The
apoptotic index (%) was calculated as follows: Number of
TUNEL-positive cells/total number of cells ×400. The cell count was
confirmed in four randomly selected high-power fields and the data
were averaged.
Cytokine measurement
Hippocampal levels of cytokines were detected by
ELISA. Briefly, hippocampal samples were collected and dissolved
using RIPA buffer (CoWin Biosciences), then cell lysate was
centrifuged for 3–5 min at 12,500 x g at room temperature and the
supernatant was collected. IL-1β (cat. no. ab197742), IL-6 (cat.
no. ab222503), TNF-α (cat. no. ab208348) and NF-κB (cat. no.
ab176663) were measured by ELISA according to the manufacturer's
instructions (all Abcam).
Reverse transcription-quantitative
(RT-q)PCR
RT-qPCR analysis was performed as described
previously (35). Total RNA was
extracted from hippocampal brain samples using TRIzol (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. Then, the RNA was reverse-transcribed into
complementary DNA using the RevertAid First Strand cDNA Synthesis
kit (cat. no. K1622; Thermo Fisher Scientific Inc.). The ATF4 and
CHOP mRNA levels in each sample were measured by qPCR using
SYBR-Green Master Mix (Toyobo Life Science). GAPDH was used as an
internal control. The qPCR thermocycling conditions were as
follows: Initial denaturation at 45°C (2 min) and 95°C (10 min),
followed by 40 cycles of denaturation at 95°C (15 sec), annealing
at 60°C (1 min) and extension at 72°C (1 min). The
2−ΔΔCq method was used to assess the relative mRNA
expression levels (36). All
samples were analyzed in triplicate. The primers used are listed as
follows: ATF4 forward, 5′-ATGACCGAAATGAGCTTCCTG−3′ and reverse,
5′-GCTGGAGAACCCATGAGGT-3′; CHOP forward,
5′-GGAAACAGAGTGGTCATTCCC−3′ and reverse,
5′-CTGCTTGAGCCGTTCATTCTC−3′; and GAPDH forward,
5′-ATGGGTGTGAACCACGAGA-3′ and reverse,
5′-CAGGGATGATGTTCTGGGCA-3′.
Western blot analysis
Western blot analysis was performed as described
previously (33). Briefly, cerebral
cortex samples were collected, homogenized and total protein was
extracted using RIPA buffer (CoWin Biosciences). A BCA Protein
Assay kit (Beyotime Institute of Biotechnology) was used to measure
the protein concentration. Total protein (30 µg) was separated via
12% SDS-PAGE and transferred onto PVDF membranes. The membranes
were blocked at room temperature for 1 h with 5% non-fat milk and
incubated with primary antibodies (all Abcam) overnight at 4°C as
follows: Rabbit anti-β-actin (1:1,000; cat. no. ab8227),
anti-Caspase-3 (1:2,000; cat. no. ab184787), anti-Bax (1:2,000;
cat. no. ab182733), anti-Bcl2 (1:2,000; cat. no. ab182858),
anti-ATF4 (1:1,000; cat. no. ab216839) and mouse anti-CHOP (5
µg/ml; cat. no. ab11419). After washing the membranes with TBS with
0.5% Tween-20 three times, horseradish peroxidase-conjugated
anti-rabbit (1:2,000; cat. no. 7074S; Cell Signaling Technology,
Inc.) or anti-mouse IgG (1:2,000; cat. no. 7076S; Cell Signaling
Technology, Inc.) secondary antibodies were applied and incubated
at room temperature for 1.5 h. The signals were developed using an
enhanced chemiluminescence reagent (MilliporeSigma) according to
the manufacturer's instructions. The protein bands were detected
using a Bio-Rad imaging system (Bio-Rad Laboratories, Inc.) and
quantified with ImageJ (version 1.52; National Institutes of
Health).
Statistical analysis
All experiments were repeated >3 times and data
are presented as the mean ± SEM. SPSS 14.0 (SPSS, Inc.) and
GraphPad Prism 6 (GraphPad Software, Inc.) were used for
statistical analysis. Student's t test (unpaired, two-tailed) was
used to analyze the statistical differences between two groups.
Differences between multiple groups were analyzed using one-way
ANOVA followed by post hoc Tukey's test. P<0.05 was considered
to indicate a statistically significant difference.
Results
Mortality and neurological function in
ICH/OSA model mice
Previous clinical studies reported that OSA
aggravates EBI, increases mortality rate and worsens overall
outcomes of patients with ICH (22,37).
Thus, an ICH model was constructed and chronic (C)IH was used to
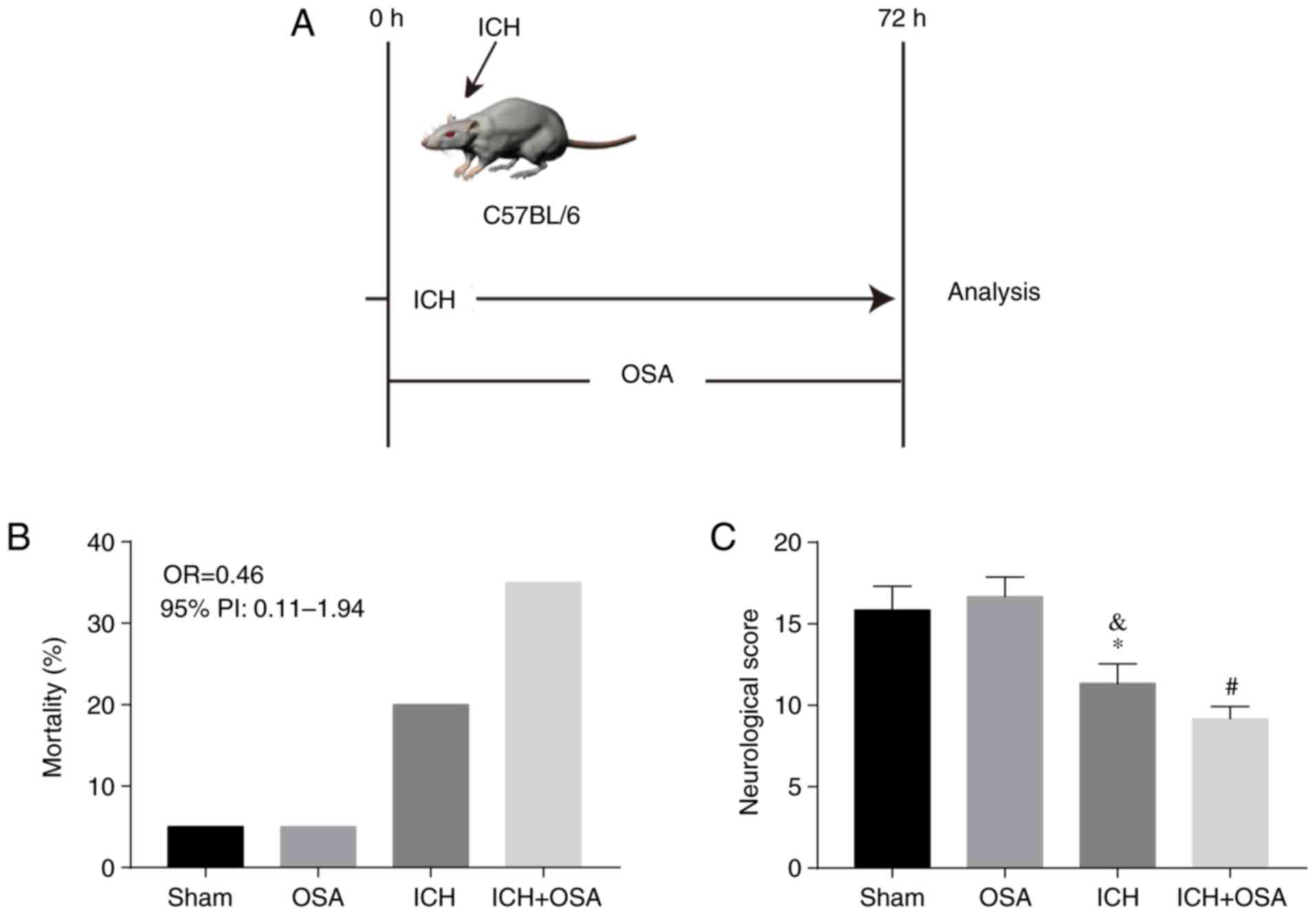
construct the OSA model (Fig. 1A).
The effect of OSA on mortality rate and neurological score was
assessed. There were 16 mice in the Sham group (one dead), 16 mice
in the OSA group (one dead), 19 mice in the ICH group (4 dead) and
23 mice in the ICH + OSA group (8 dead). Mortality rate (Fig. 1B) increased in the subarachnoid
hemorrhage (ICH) + OSA group but there was no significant
difference compared with the ICH group (OR=0.46; 95% PI,
0.11–1.94). Neurological score decreased significantly following
ICH and was further aggravated by OSA (Fig. 1C).
OSA aggravates EBI and BBB
permeability following ICH
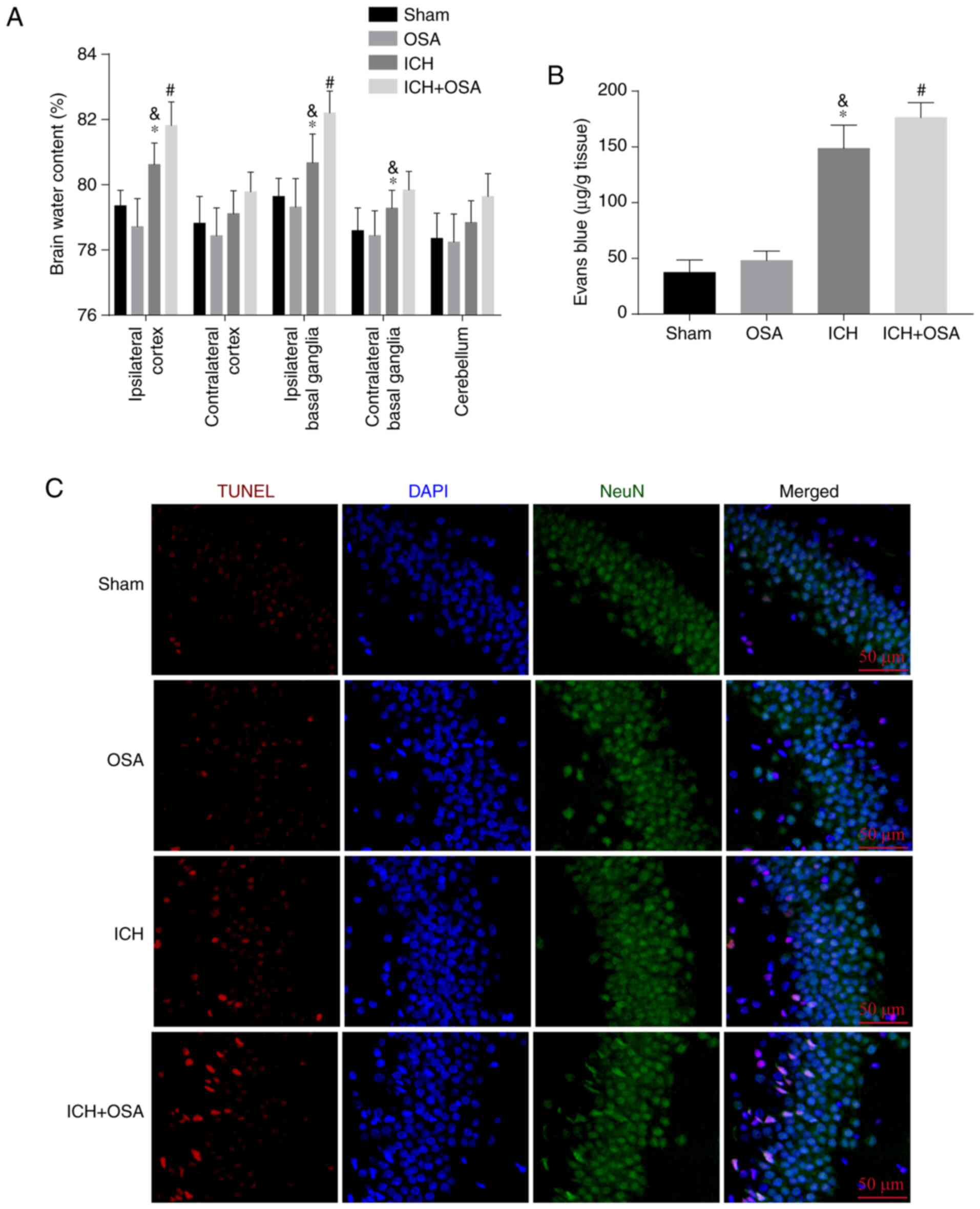
To assess EBI following ICH and OSA, brain water
content was measured by the wet-dry method at 72 h post-ICH to
evaluate the degree of brain damage. The results showed that ICH
significantly increased the brain water content in the ipsilateral
cortex and both ipsilateral and contralateral basal ganglia; OSA
significantly aggravated this in the ipsilateral cortex and basal
ganglia (Fig. 2A). Similar results
were observed for BBB permeability, which increased significantly
following ICH and was significantly aggravated by OSA (Fig. 2B). To assess hippocampal neuronal
death following ICH and OSA, TUNEL assay was performed. As
expected, more hippocampal neuron death was observed in the ICH +
OSA group compared with in the ICH group (Fig. 2C). Hence, these data showed that OSA
aggravates EBI following ICH.
OSA aggravates neuroinflammation
following ICH
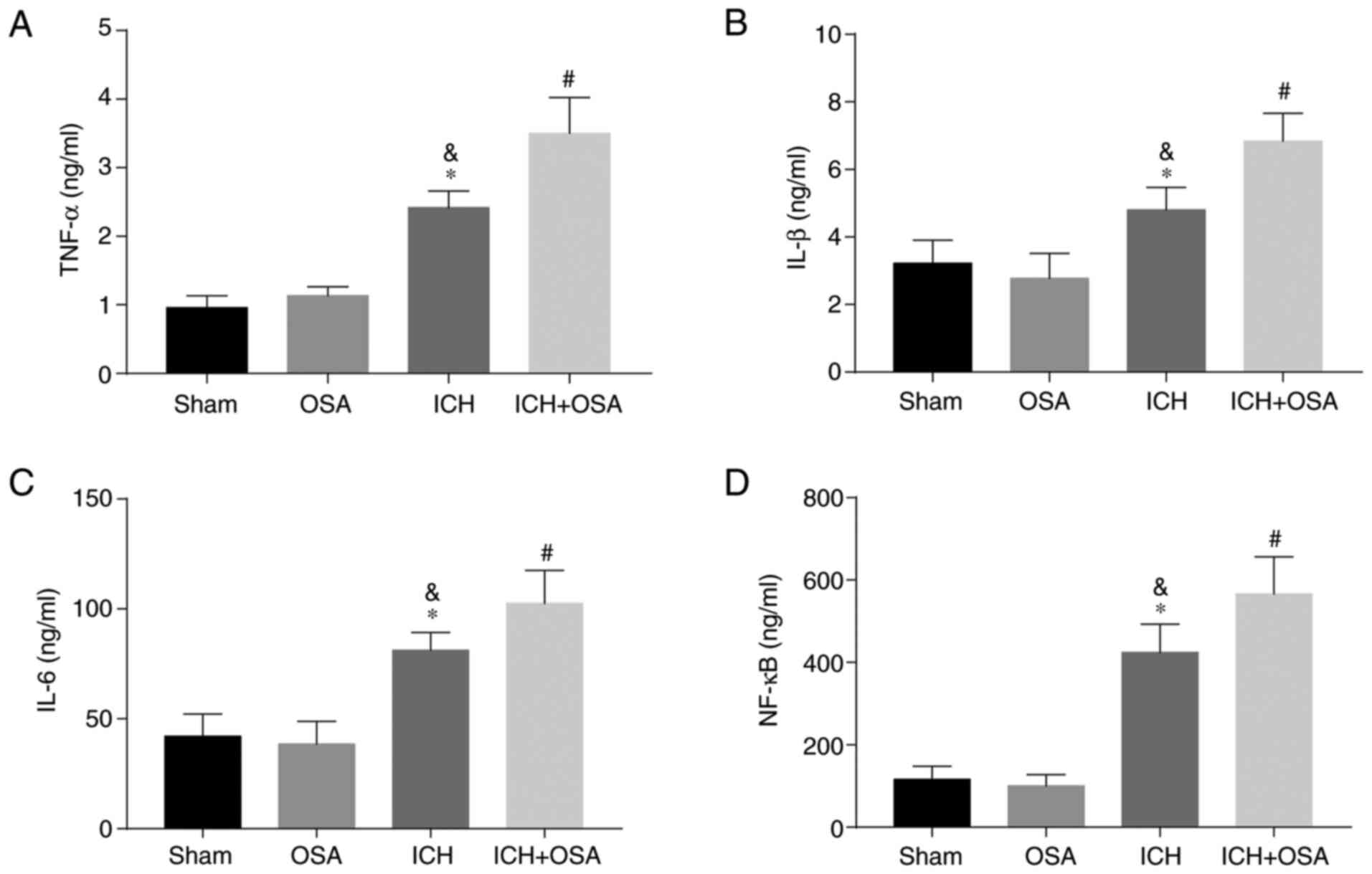
Neuroinflammation serves a key role in EBI following
ICH; increased neuroinflammation aggravates EBI (27,38).
The inflammatory complex induces secretion of pro-inflammatory
cytokines, including IL-1β, IL-6, TNF-α and NF-κB (27). Therefore, hippocampal levels of
IL-1β, IL-6, TNF-α and NF-κB were assessed by ELISA. The results
showed that expression levels of pro-inflammatory cytokines
increased significantly following ICH and were further increased in
the OSA + ICH group (Fig. 3A-D).
The results showed that OSA aggravated EBI in a mouse model of ICH
and that the mechanism may be partly associated with the activation
of neuroinflammation.
OSA further increases expression
levels of apoptosis-associated proteins following ICH
Previous studies have indicated that apoptosis is an
important form of cell death in central nervous system (CNS)
disease and serves a key role in SAH and OSA (26,27).
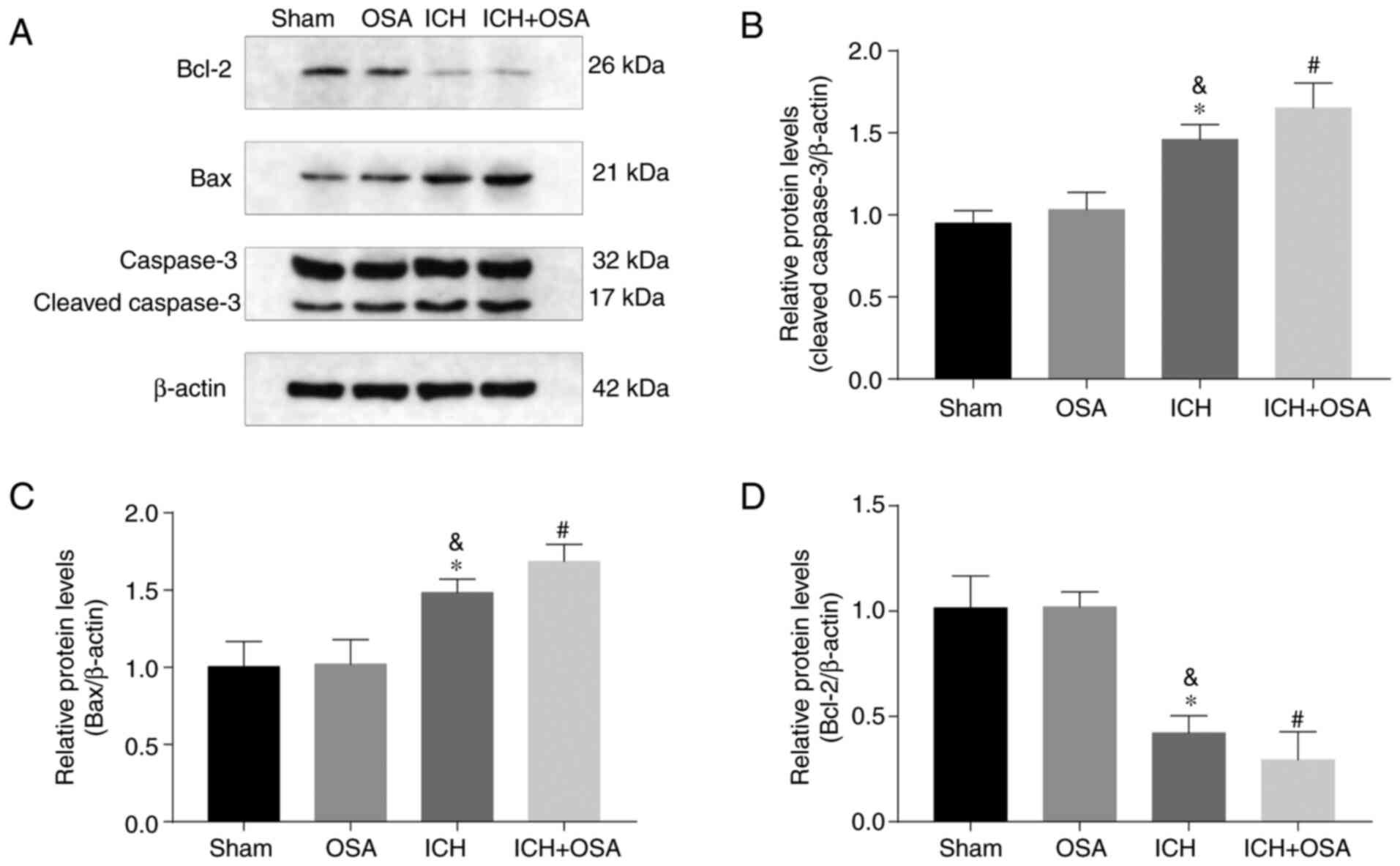
Expression levels of apoptosis-associated proteins Caspase-3, Bax,
and Bcl-2 following ICH and OSA were evaluated by western blotting
(Fig. 4A). The results showed that
expression levels of Caspase-3 and Bax increased significantly
following ICH and were further increased by subsequent OSA. Protein
expression levels of anti-apoptotic Bcl-2 were significantly
decreased by ICH and further decreased in the ICH + OSA group
(Fig. 4B-D). The results showed
that OSA aggravated EBI in a mouse model of ICH and that the
mechanism may be partly associated with the activation of
apoptosis.
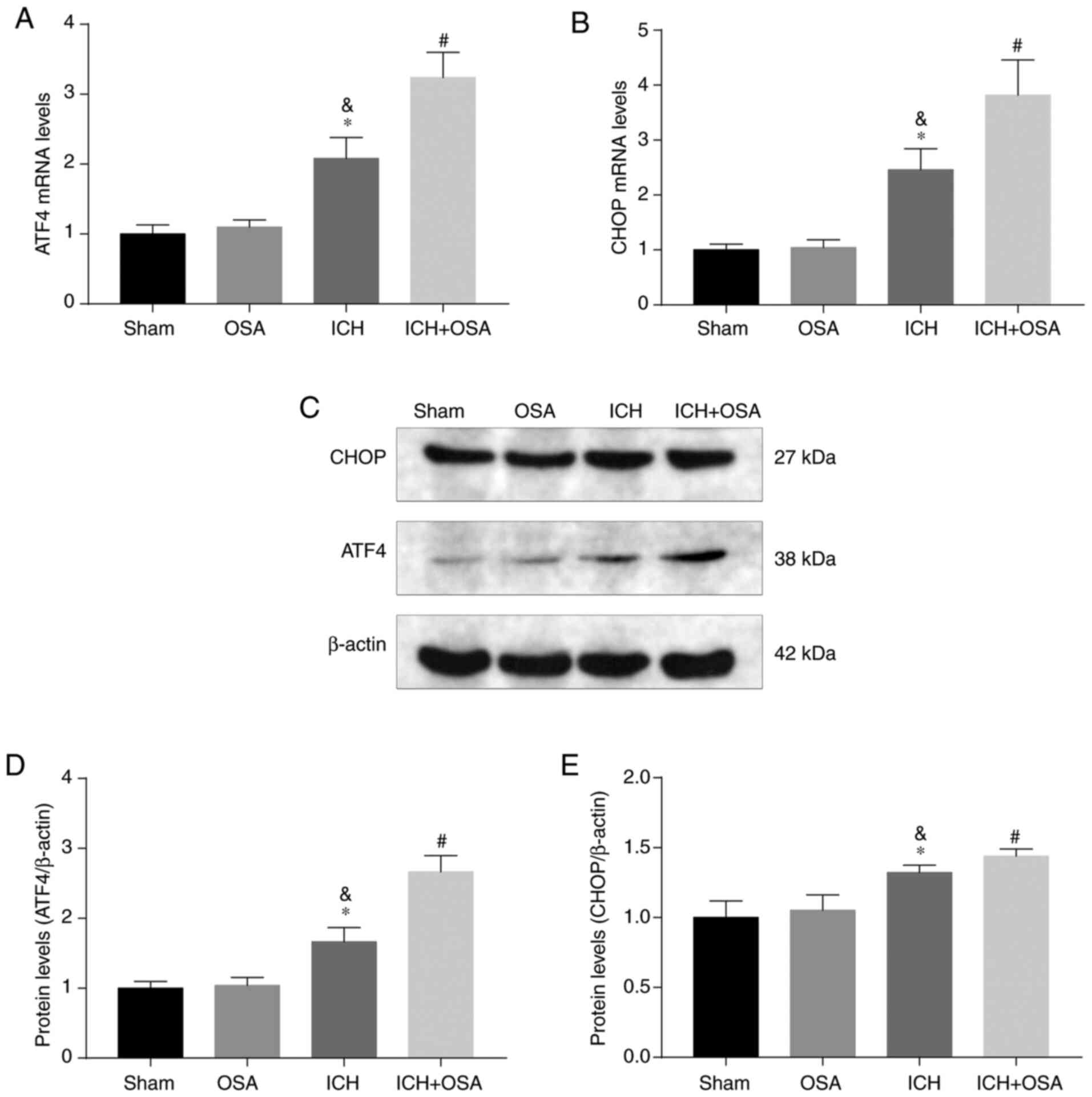
OSA aggravates EBI via the ATF4/CHOP
signaling pathway following ICH
Deng et al (39) reported that the ATF4/CHOP signaling
pathway is a key mechanism by which brain injury is ameliorated in
ICH rats. Therefore, the present study investigated whether OSA
aggravated apoptosis and neuroinflammation by regulating the
ATF4/CHOP signaling pathway. First, protein and gene expression
levels of ATF4 and CHOP were assessed following ICH and OSA by
western blotting and RT-qPCR. The results showed that mRNA
expression levels of ATF4 and CHOP increased following ICH and
further increased following OSA (Fig.
5A and B). Similar results were demonstrated by western blot
analysis of protein expression levels (Fig. 5C-E). Hence, it was hypothesized that
OSA aggravated EBI by inhibiting the ATF4/CHOP signaling pathway
following ICH.
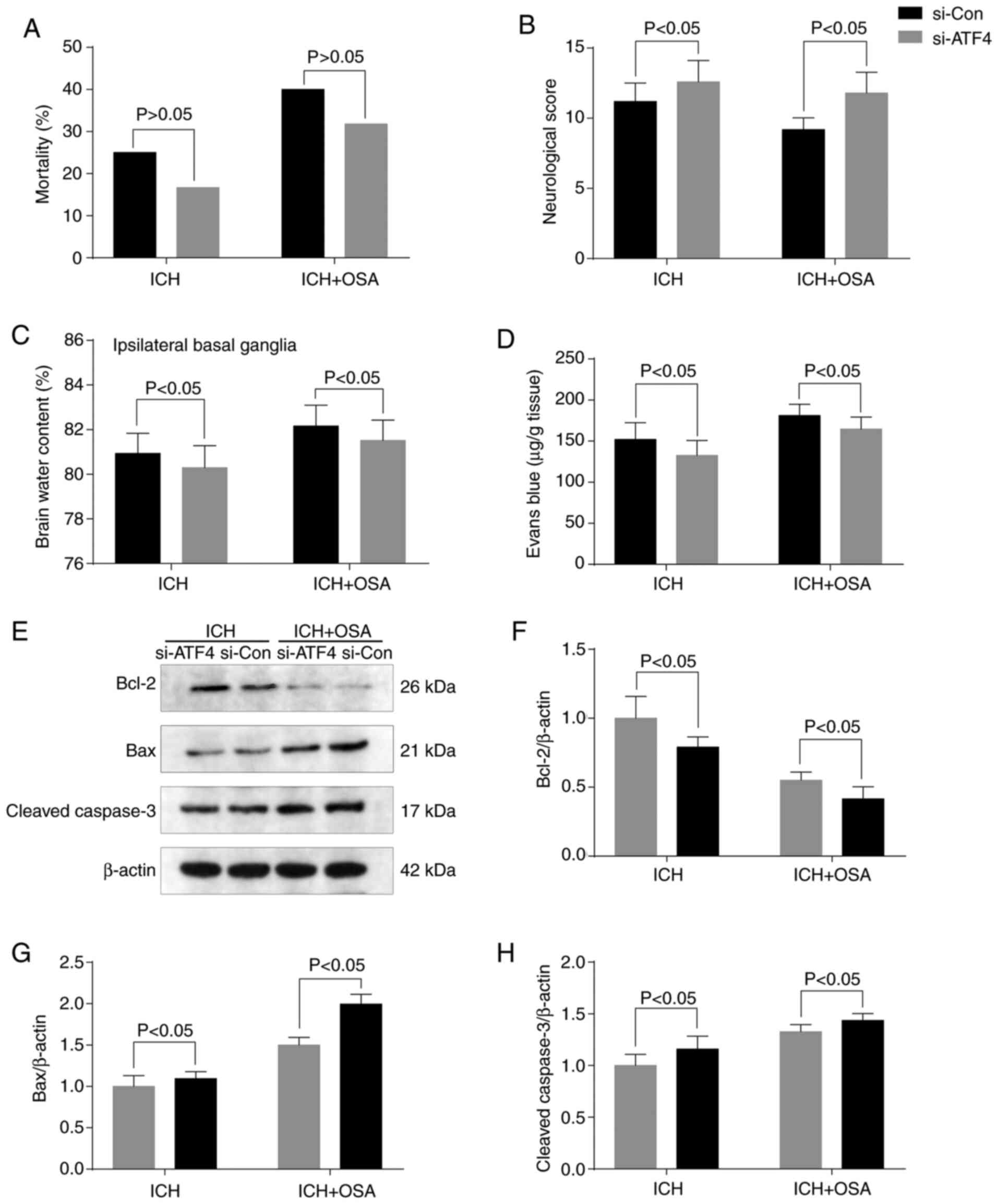
Knockdown of ATF4 reverses
OSA-aggravated EBI following ICH
To investigate the role of ATF4 in EBI, transfection
with targeted siRNA (si-ATF4) was performed to downregulate
expression of ATF4. There was no significant difference in
mortality rate following ATF4 knockdown (Fig. 6A). RT-qPCR was used to confirm that
transfection was successful and ATF4 expression was efficiently
silenced (Fig. S1). Following ATF4
knockdown, neurological score, ipsilateral basal ganglia brain
edema and BBB permeability were improved (Fig. 6B-D), indicating a reversal of
OSA-induced EBI. The levels of apoptosis-associated proteins
(Cleaved-Caspase-3, Bcl-2 and Bax) were assessed by western
blotting (Fig. 6E). The expression
levels of Cleaved-Caspase-3 and Bax were decreased and those of
Bcl-2 increased significantly following ATF4 knockdown (Fig. 6F-H).
Discussion
The present study evaluated the mechanisms by which
OSA aggravates EBI in a mouse ICH model. The present study
demonstrated that OSA aggravates EBI, neurological dysfunction,
brain damage, neuroinflammation, apoptosis and neuronal death
following ICH. The potential mechanism may be associated with the
ATF4/CHOP signaling pathway.
OSA is characterized by repetitive narrowing or
collapsing of the upper airways, resulting in recurrent hypoxia
during sleep (40). Interruption of
breathing by OSA results in IH, leading to decreased blood oxygen
saturation and impaired sleep quality; prolonged hypoxia induces
inflammatory responses, which affect the function of the vascular
endothelium (41). OSA not only
seriously affects the quality of life but also induces or
aggravates various comorbidities, including hypertension (40), coronary heart disease (42), ICH (25) and stroke (43). The potential molecular mechanisms of
OSA-induced disease are complicated. OSA induces oxidative stress
and activation of the inflammatory system, resulting in the
production of reactive oxygen species (ROS); ROS may be a potential
therapeutic target for ameliorating hypoxia-induced cell death and
tissue injury (44). Furthermore,
an increasing number of studies have demonstrated an association
between OSA and cognitive impairment and CNS disease (19–22,24–26).
A previous study reported that OSA induces cognitive
impairment via neuronal death in the hippocampus (45). Lim and Pack (46) reported that OSA alters BBB
permeability by changing the expression levels of influx and efflux
transporters, causing an acute leak via the tight junctions of the
BBB. Halder and Milner (47)
demonstrated that chronic hypoxia triggers BBB disruption and
subsequent neurological dysfunction. In the present study, brain
water content increased and BBB permeability was enhanced following
ICH and further aggravated by subsequent OSA. Zolotoff et al
(48) found that IH and sustained
hypoxia can induce BBB disruption and increase BBB permeability via
production of ROS, Nrf2 and HIF-1α. ICH increases brain water
content via BBB disruption, cerebral vasospasm and vascular
endothelial dysfunction (13,35).
Hence, it was hypothesized that OSA combined with ICH increases
brain water content and brain damage and that BBB permeability and
vascular endothelial dysfunction serve an important role. The
underlying molecular mechanisms are unclear and need further
study.
Neuroinflammation is the primary pathophysiological
change following cerebral hemorrhage; excessive neuroinflammatory
response causes hippocampal neuronal apoptosis and EBI (27). Molecular markers of
neuroinflammation include pro-inflammatory cytokines IL-1β, IL-6,
TNF-α and NF-κB. Numerous studies have demonstrated that OSA
induces cognitive deficits or brain damage, partly via oxidative
stress and neuroinflammation, and that brain damage and neuronal
apoptosis are alleviated if neuroinflammation is prevented
(49–51). Gong et al (49) reported that OSA aggravates neuronal
and microglial damage; damaged microglia release large amounts of
proinflammatory cytokines, and an excessive inflammatory response
leads to neurotoxicity and neuronal apoptosis by activating
BNIP3-dependent mitophagy via the JNK/ERK signaling pathway.
Additionally, OSA-induced nerve damage and stimulation of the
interaction between toll-like receptor 2 and its aptamer MyD88 are
associated with microglial aggregation, NF-κB activation, oxidative
stress and elevated levels of inflammatory cytokines in mice
(50). In the present study,
expression levels of pro-inflammatory cytokines, including IL-1β,
IL-6, TNF-α and NF-κB, were increased significantly following ICH
combined with OSA, thus aggravating EBI. The molecular mechanism
may be associated with the ATF4/CHOP signaling pathway.
ATF4 serves different roles in different tissues,
organs, and cells. In mammals, ATF4 is involved in a variety of
physiological activities, such as eye development, bone formation,
metabolism, energy balance, stress reaction, learning, memory and
CNS disease (52,53). Overexpressed ATF4 enters the nucleus
and activates the transcription factor CHOP, which is involved in
endoplasmic reticulum (ER) stress-induced neuronal apoptosis
(54). ATF4 mRNA is expressed in
all tissues, but ATF4 protein is only expressed at low levels under
normal physiological conditions; the translation of ATF4 is
upregulated by phosphorylation of the α subunit of eukaryotic
initiation factor 2 (eIF2), which is triggered by ER and oxidative
stress and amino acid deficiency (55). Baleriola et al (56) reported that the expression of ATF4
protein is increased in neuronal axons, as well as near amyloid
plaques and in bead axons in the Alzheimer's disease brain.
Inhibition of local translation and retrograde transport or
knockdown of axonal ATF4 mRNA abolishes amyloid β-induced ATF4
transcriptional activity and cell loss (57). In a CIH rat model, a
H2/O2 mixture inhibits ER stress-induced
apoptosis via the PERK/eIF2α/ATF4 signaling pathway and notably
improves cardiac dysfunction and myocardial fibrosis (58). Furthermore, overexpression of ATF4
upregulates transcription of factors such as CHOP; this upregulates
expression the apoptotic regulator mitochondrial apoptosis protein
Bcl-2, which interacts with apoptotic regulators, activates the
pro-apoptotic protein Bax and suppresses expression levels of
anti-apoptotic gene Bcl-2 family proteins, thus promoting oxidative
stress-induced nerve cell death (59). In the present study, expression
levels of ATF4 and CHOP were increased significantly following ICH
and further aggravated following OSA; this was associated with
increased neuronal death and EBI. The present data showed that
neuronal death and EBI following ICH/OSA were partially prevented
following ATF4 knockdown. To the best of our knowledge, however,
there have been no reports concerning the regulation of the
downstream molecular pathway of CHOP following ICH/OSA and the
exact mechanism requires further investigation.
In conclusion, OSA aggravated neurological
dysfunction, brain edema, neuroinflammation and neuronal injury
following ICH by promoting ER stress-associated apoptosis and
neuroinflammation in hippocampal neurons. The mechanisms underlying
these effects involved activation of ATF4/CHOP-mediated ER stress
and apoptosis. Additionally, further investigations into the
effects of OSA treatment combined with ICH in the clinic are
required.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WF wrote the manuscript. WF, WJ, XF and YW performed
the experiments and prepared the figures. WF and YW confirm the
authenticity of all the raw data. XC and YW designed the study and
revised the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Wuxi Clinical College of Anhui Medical University
(approval no. YXLL-2020-112; Wuxi, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
van Asch CJ, Luitse MJ, Rinkel GJ, van der
Tweel I, Algra A and Klijn CJ: Incidence, case fatality, and
functional outcome of intracerebral haemorrhage over time,
according to age, sex, and ethnic origin: A systematic review and
meta-analysis. Lancet Neurol. 9:167–176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang Y, Zhang X, Wei Q, Leng S, Li C, Han
B, Bai Y, Zhang H and Yao H: Activation of sigma-1 receptor
enhanced pericyte survival via the interplay between apoptosis and
autophagy: Implications for blood-brain barrier integrity in
stroke. Transl Stroke Res. 11:267–287. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang Z, Cho S, Rehni AK, Quero HN, Dave
KR and Zhao W: Automated assessment of hematoma volume of rodents
subjected to experimental intracerebral hemorrhagic stroke by bayes
segmentation approach. Transl Stroke Res. 11:789–798. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gross BA, Jankowitz BT and Friedlander RM:
Cerebral intraparenchymal hemorrhage: A review. JAMA.
321:1295–1303. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu X, Luo J, Liu H, Cui W, Guo K, Zhao L,
Bai H, Guo W, Guo H, Feng D and Qu Y: Recombinant adiponectin
peptide ameliorates brain injury following intracerebral hemorrhage
by suppressing astrocyte-derived inflammation via the inhibition of
Drp1-mediated mitochondrial fission. Transl Stroke Res. 11:924–939.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hanley DF, Thompson RE, Rosenblum M,
Yenokyan G, Lane K, McBee N, Mayo SW, Bistran-Hall AJ, Gandhi D,
Mould WA, et al: Efficacy and safety of minimally invasive surgery
with thrombolysis in intracerebral haemorrhage evacuation (MISTIE
III): A randomised, controlled, open-label, blinded endpoint phase
3 trial. Lancet. 393:1021–1032. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen J, Zhu J, He J, Wang Y, Chen L, Zhang
C, Zhou J and Yang L: Ultra-early microsurgical treatment within 24
h of SAH improves prognosis of poor-grade aneurysm combined with
intracerebral hematoma. Oncol Lett. 11:3173–3178. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen J, Wang Y, Wu J, Yang J, Li M and
Chen Q: The potential value of targeting ferroptosis in early brain
injury after acute CNS disease. Front Mol Neurosci. 13:1102020.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Adeoye O and Broderick JP: Advances in the
management of intracerebral hemorrhage. Nat Rev Neurol. 6:593–601.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mendelow AD, Gregson BA, Rowan EN, Murray
GD, Gholkar A and Mitchell PM; STICH II Investigators, : Early
surgery versus initial conservative treatment in patients with
spontaneous supratentorial lobar intracerebral haematomas (STICH
II): A randomised trial. Lancet. 382:397–408. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou Y, Wang Y, Wang J, Anne Stetler R and
Yang QW: Inflammation in intracerebral hemorrhage: From mechanisms
to clinical translation. Prog Neurobiol. 115:25–44. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xue M and Yong VW: Neuroinflammation in
intracerebral haemorrhage: Immunotherapies with potential for
translation. Lancet Neurol. 19:1023–1032. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen JH, Yang LK, Chen L, Wang YH, Wu Y,
Jiang BJ, Zhu J and Li PP: Atorvastatin ameliorates early brain
injury after subarachnoid hemorrhage via inhibition of AQP4
expression in rabbits. Int J Mol Med. 37:1059–1066. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bao WD, Zhou XT, Zhou LT, Wang F, Yin X,
Lu Y, Zhu LQ and Liu D: Targeting miR-124/Ferroportin signaling
ameliorated neuronal cell death through inhibiting apoptosis and
ferroptosis in aged intracerebral hemorrhage murine model. Aging
Cell. 19:e132352020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gautam J, Xu L, Nirwane A, Nguyen B and
Yao Y: Loss of mural cell-derived laminin aggravates hemorrhagic
brain injury. J Neuroinflammation. 17:1032020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Karuppagounder SS, Alim I, Khim SJ,
Bourassa MW, Sleiman SF, John R, Thinnes CC, Yeh TL, Demetriades M,
Neitemeier S, et al: Therapeutic targeting of oxygen-sensing prolyl
hydroxylases abrogates ATF4-dependent neuronal death and improves
outcomes after brain hemorrhage in several rodent models. Sci
Transl Med. 8:328ra3292016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen J, Xuan Y, Chen Y, Wu T, Chen L, Guan
H, Yang S, He J, Shi D and Wang Y: Netrin-1 alleviates subarachnoid
haemorrhage-induced brain injury via the PPARγ/NF-KB signalling
pathway. J Cell Mol Med. 23:2256–2262. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou Y, Tao T, Liu G, Gao X, Gao Y, Zhuang
Z, Lu Y, Wang H, Li W, Wu L, et al: TRAF3 mediates neuronal
apoptosis in early brain injury following subarachnoid hemorrhage
via targeting TAK1-dependent MAPKs and NF-κB pathways. Cell Death
Dis. 12:102021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chernyshev OY, Bir SC, Maiti TK, Patra DP,
Sun H, Guthikonda B, Kelley RE, Cuellar H, Minagar A and Nanda A:
The relationship between obstructive sleep apnea and ruptured
intracranial aneurysms. J Clin Sleep Med. 15:1839–1848. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mason RH, Ruegg G, Perkins J, Hardinge M,
Amann-Vesti B, Senn O, Stradling JR and Kohler M: Obstructive sleep
apnea in patients with abdominal aortic aneurysms: Highly prevalent
and associated with aneurysm expansion. Am J Respir Crit Care Med.
183:668–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zaremba S, Albus L, Schuss P, Vatter H,
Klockgether T and Güresir E: Increased risk for subarachnoid
hemorrhage in patients with sleep apnea. J Neurol. 266:1351–1357.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Geer JH, Falcone GJ, Vanent KN, Leasure
AC, Woo D, Molano JR, Sansing LH, Langefeld CD, Pisani MA, Yaggi HK
and Sheth KN: Obstructive sleep apnea as a risk factor for
intracerebral hemorrhage. Stroke. 52:1835–1838. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bir SC, Nanda A, Cuellar H, Sun H,
Guthikonda B, Liendo C, Minagar A and Chernyshev OY: Coexistence of
obstructive sleep apnea worsens the overall outcome of intracranial
aneurysm: A pioneer study. J Neurosurg. 128:735–746. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pontes-Neto OM, Fernandes RM, Sander HH,
da Silva LA, Mariano DC, Nobre F, Simão G, de Araujo DB, dos Santos
AC and Leite JP: Obstructive sleep apnea is frequent in patients
with hypertensive intracerebral hemorrhage and is related to
perihematoma edema. Cerebrovasc Dis. 29:36–42. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Orrù G, Storari M, Scano A, Piras V, Taibi
R and Viscuso D: Obstructive sleep apnea, oxidative stress,
inflammation and endothelial dysfunction-an overview of predictive
laboratory biomarkers. Eur Rev Med Pharmacol Sci. 24:6939–6948.
2020.PubMed/NCBI
|
|
26
|
Hung MW, Tipoe GL, Poon AM, Reiter RJ and
Fung ML: Protective effect of melatonin against hippocampal injury
of rats with intermittent hypoxia. J Pineal Res. 44:214–221. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen J, Zhang C, Yan T, Yang L, Wang Y,
Shi Z, Li M and Chen Q: Atorvastatin ameliorates early brain injury
after subarachnoid hemorrhage via inhibition of pyroptosis and
neuroinflammation. J Cell Physiol. Mar 31–2021.(Epub ahead of
print). View Article : Google Scholar
|
|
28
|
Wu X, Luo J, Liu H, Cui W, Guo W, Zhao L,
Guo H, Bai H, Guo K, Feng D and Qu Y: Recombinant adiponectin
peptide promotes neuronal survival after intracerebral haemorrhage
by suppressing mitochondrial and ATF4-CHOP apoptosis pathways in
diabetic mice via Smad3 signalling inhibition. Cell Prolif.
53:e127592020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu Z, Wang Z, Yu L, Ding Y, Xu Y, Xu N, Li
R, Tang J, Chen G and Zhang JH: GCN2 reduces inflammation by
p-eIF2α/ATF4 pathway after intracerebral hemorrhage in mice. Exp
Neurol. 313:16–25. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
National Research Council Institute for
Laboratory Animal Research, . Guide for the Care and Use of
Laboratory Animals National Academies Press. National Academy of
Sciences; Washington, DC: 1996
|
|
31
|
Deng S, Sherchan P, Jin P, Huang L, Travis
Z, Zhang JH, Gong Y and Tang J: Recombinant CCL17 enhances hematoma
resolution and activation of CCR4/ERK/Nrf2/CD163 signaling pathway
after intracerebral hemorrhage in mice. Neurotherapeutics.
17:1940–1953. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li L, Ren F, Qi C, Xu L, Fang Y, Liang M,
Feng J, Chen B, Ning W and Cao J: Intermittent hypoxia promotes
melanoma lung metastasis via oxidative stress and inflammation
responses in a mouse model of obstructive sleep apnea. Respir Res.
19:282018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen JH, Wu T, Xia WY, Shi ZH, Zhang CL,
Chen L, Chen QX and Wang YH: An early neuroprotective effect of
atorvastatin against subarachnoid hemorrhage. Neural Regen Res.
15:1947–1954. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li G, Dong Y, Liu D, Zou Z, Hao G, Gao X,
Pan P and Liang G: NEK7 coordinates rapid neuroinflammation after
subarachnoid hemorrhage in mice. Front Neurol. 11:5512020.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen JH, Wu T, Yang LK, Chen L, Zhu J, Li
PP, Hu X and Wang YH: Protective effects of atorvastatin on
cerebral vessel autoregulation in an experimental rabbit model of
subarachnoid hemorrhage. Mol Med Rep. 17:1651–1659. 2018.PubMed/NCBI
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nguyen JQN, Resnick CM, Chang YH, Hansen
RM, Fulton AB, Moskowitz A, Calabrese CE and Dagi LR: Impact of
obstructive sleep apnea on optic nerve function in patients with
craniosynostosis and recurrent intracranial hypertension. Am J
Ophthalmol. 207:356–362. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wei C, Guo S, Liu W, Jin F, Wei B, Fan H,
Su H, Liu J, Zhang N, Fang D, et al: Resolvin D1 ameliorates
inflammation-mediated blood-brain barrier disruption after
subarachnoid hemorrhage in rats by modulating A20 and NLRP3
inflammasome. Front Pharmacol. 11:6107342020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Deng S, Liu S, Jin P, Feng S, Tian M, Wei
P, Zhu H, Tan J, Zhao F and Gong Y: Albumin reduces oxidative
stress and neuronal apoptosis via the ERK/Nrf2/HO-1 pathway after
intracerebral hemorrhage in rats. Oxid Med Cell Longev.
2021:88913732021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Prabhakar NR, Peng YJ and Nanduri J:
Hypoxia-inducible factors and obstructive sleep apnea. J Clin
Invest. 130:5042–5051. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tan J, Xing H, Sha S, Li J, Miao Y and
Zhang Q: Analysis of circulating microvesicles levels and effects
of associated factors in elderly patients with obstructive sleep
apnea. Front Aging Neurosci. 13:6092822021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Strausz S, Ruotsalainen S, Ollila HM,
Karjalainen J, Kiiskinen T, Reeve M, Kurki M, Mars N, Havulinna AS,
Luonsi E, et al: Genetic analysis of obstructive sleep apnoea
discovers a strong association with cardiometabolic health. Eur
Respir J. 57:20030912021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li J, McEvoy RD, Zheng D, Loffler KA, Wang
X, Redline S, Woodman RJ and Anderson CS: Self-reported snoring
patterns predict stroke events in high-risk patients with osa: Post
hoc analyses of the save study. Chest. 158:2146–2154. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yu LM, Zhang WH, Han XX, Li YY, Lu Y, Pan
J, Mao JQ, Zhu LY, Deng JJ, Huang W and Liu YH: Hypoxia-induced ROS
contribute to myoblast pyroptosis during obstructive sleep apnea
via the NF-κB/HIF-1α signaling pathway. Oxid Med Cell Longev.
2019:45963682019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhu Y, Fenik P, Zhan G, Mazza E, Kelz M,
Aston-Jones G and Veasey SC: Selective loss of catecholaminergic
wake active neurons in a murine sleep apnea model. J Neurosci.
27:10060–10071. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lim DC and Pack AI: Obstructive sleep
apnea and cognitive impairment: Addressing the blood-brain barrier.
Sleep Med Rev. 18:35–48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Halder SK and Milner R: Mild hypoxia
triggers transient blood-brain barrier disruption: A fundamental
protective role for microglia. Acta Neuropathol Commun. 8:1752020.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zolotoff C, Voirin AC, Puech C, Roche F
and Perek N: Intermittent hypoxia and its impact on Nrf2/HIF-1α
expression and ABC transporters: An in vitro human blood-brain
barrier model study. Cell Physiol Biochem. 54:1231–1248. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gong LJ, Wang XY, Gu WY and Wu X:
Pinocembrin ameliorates intermittent hypoxia-induced
neuroinflammation through BNIP3-dependent mitophagy in a murine
model of sleep apnea. J Neuroinflammation. 17:3372020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Deng Y, Liu K, Pan Y, Ren J, Shang J, Chen
L and Liu H: TLR2 antagonism attenuates the hippocampal neuronal
damage in a murine model of sleep apnea via inhibiting
neuroinflammation and oxidative stress. Sleep Breath. 24:1613–1621.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Johnson SM, Randhawa KS, Epstein JJ,
Gustafson E, Hocker AD, Huxtable AG, Baker TL and Watters JJ:
Gestational intermittent hypoxia increases susceptibility to
neuroinflammation and alters respiratory motor control in neonatal
rats. Respir Physiol Neurobiol. 256:128–142. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang C and Guo F: Effects of activating
transcription factor 4 deficiency on carbohydrate and lipid
metabolism in mammals. IUBMB Life. 64:226–230. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Aimé P, Karuppagounder SS, Rao A, Chen Y,
Burke RE, Ratan RR and Greene LA: The drug adaptaquin blocks
ATF4/CHOP-dependent pro-death Trib3 induction and protects in
cellular and mouse models of Parkinson's disease. Neurobiol Dis.
136:1047252020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Galehdar Z, Swan P, Fuerth B, Callaghan
SM, Park DS and Cregan SP: Neuronal apoptosis induced by
endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated
induction of the Bcl-2 homology 3-only member PUMA. J Neurosci.
30:16938–16948. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Harding HP, Zhang Y, Zeng H, Novoa I, Lu
PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al: An
integrated stress response regulates amino acid metabolism and
resistance to oxidative stress. Mol Cell. 11:619–633. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Baleriola J, Walker CA, Jean YY, Crary JF,
Troy CM, Nagy PL and Hengst U: Axonally synthesized ATF4 transmits
a neurodegenerative signal across brain regions. Cell.
158:1159–1172. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Liu Y: Hydrogen peroxide induces nucleus
pulposus cell apoptosis by ATF4/CHOP signaling pathway. Exp Ther
Med. 20:3244–3252. 2020.PubMed/NCBI
|
|
58
|
Zhao YS, An JR, Yang S, Guan P, Yu FY, Li
W, Li JR, Guo Y, Sun ZM and Ji ES: Hydrogen and oxygen mixture to
improve cardiac dysfunction and myocardial pathological changes
induced by intermittent hypoxia in rats. Oxid Med Cell Longev.
2019:74152122019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lange PS, Chavez JC, Pinto JT, Coppola G,
Sun CW, Townes TM, Geschwind DH and Ratan RR: ATF4 is an oxidative
stress-inducible, prodeath transcription factor in neurons in vitro
and in vivo. J Exp Med. 205:1227–1242. 2008. View Article : Google Scholar : PubMed/NCBI
|