Introduction
Opioids, such as morphine, are important drugs for
the treatment of clinical pain. The main limitation associated with
the long-term use of morphine is the occurrence of physiological
tolerance and dependence (1).
Morphine tolerance may develop through the use of multiple
administration routes, the use of different doses and various
durations (2). Morphine tolerance
leads to poor analgesic effects, constipation, drowsiness, skin
itching, and even to morphine addiction and other adverse reactions
(3). Therefore, how to
effectively reduce the occurrence of morphine tolerance provides
the main focus of the present study.
The main mechanisms underlying morphine tolerance
are opioid receptor desensitization and endocytosis (4), alteration of the glutamate receptor
(5) and glial activation
(6). In addition, previous
studies have shown that the release of inflammatory factors has an
important role in morphine-induced analgesic tolerance (7,8).
Activated microglial cells are able to produce numerous
pro-inflammatory cytokines, including interleukin (IL)-6, tumor
necrosis factor-α (TNF)-α and IL-1β, which contribute towards the
development of morphine tolerance (9). Intrathecal injection of microglial
inhibitors has previously been shown to significantly reduce
tolerance to chronic opioids (10). Therefore, effectively inhibiting
the activity of microglia is of great importance in terms of
alleviating morphine tolerance.
A previous study demonstrated that angiotensin (Ang)
II receptor type 1 (AT1R) is expressed in microglial cells
(11). The angiotensin-converting
enzyme (ACE)/Ang II/AT1R axis can lead to activation of the renin
angiotensin system (RAS) thus activating vasoconstriction,
inflammation, fibrosis, cell growth and migration (12). Notably, renin cleaves Ang I from
angiotensinogen, which is further cleaved to Ang II by
Ang-converting enzyme, and Ang II ultimately produces two
receptors: AT1R and AT2R (13).
Previous studies have shown that the brain RAS mediates microglial
polarization. For example, Ang II can activate NADPH oxidase
complexes of microglial cells through the AT1R, which leads to an
enhancement of neuroinflammatory responses (14,15). In concordance with this finding,
AT1R activation has been shown to intensify microglial inflammatory
responses, oxidative stress and dopaminergic degeneration in the
mitochondrial permeability transition pore model of Parkinson's
disease, and AT1R blockers were shown to inhibit these responses
(16–18). Therefore, it was hypothesized that
regulation of the AT1R may exert an influence on the development of
morphine tolerance.
Candesartan is an AT1R antagonist, the
pharmacological action of which is to antagonize Ang II-induced
vasoconstriction by binding to vascular smooth muscle AT1R, thereby
reducing peripheral vascular resistance (19). A recent study reported that
candesartan is able to regulate neuroinflammatory responses through
inhibiting the release of pro-inflammatory cytokines and
stimulating anti-inflammatory cytokines in lipopolysaccharide
(LPS)- and interferon-γ-stimulated BV2 cells (20). Candesartan has also been shown to
reduce microglial activation in young and aged animals (21).
Therefore, it was hypothesized that candesartan may
reduce the inflammatory responses and microglial activation that
are induced by morphine tolerance. The present study sought to
address these hypotheses.
Materials and methods
Cell culture
BV2 microglia cells were obtained from the BeNa
Culture Collection; Beijing Beina Chunglian Institute of
Biotechnology and were cultured in Dulbecco's modified Eagle's
medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml
penicillin and 100 µg/ml streptomycin (all from Gibco; Thermo
Fisher Scientific, Inc.) at 37°C in an atmosphere containing 5%
CO2/95% air. BV2 cells were treated with morphine
(Northeast Pharmaceutical Group Co., Ltd.) at concentrations of 50,
100 and 200 µM for 24 h at 37°C, and 200 µM was ultimately selected
as the optimal concentration of morphine (22). For further experiments, 1×105
cells were plated in a 6-well plate overnight and the next morning
BV2 cells were treated with candesartan (1 or 5 µmol/l) or morphine
(200 µM) alone for 24 h, or were co-treated with candesartan and
morphine, at 37°C for 24 h. The cells were divided into the
control, 1 µmol/l candesartan, 5 µmol/l candesartan, 200 µM
morphine, 200 µM morphine + 1 µmol/l candesartan and 200 µM
morphine + 5 µmol/l candesartan treatment groups. Untreated cells
were regarded as the control group. In an alternative set of
experiments, BV2 cells were treated with morphine (200 µM) the
following morning after plating with or without candesartan (1 and
5 µmol/l; MedChemExpress) for 24 h at 37°C (23). The cells were divided into the
control, 200 µM morphine, 200 µM morphine + 1 µmol/l candesartan
and 200 µM morphine + 5 µmol/l candesartan treatment groups.
Untreated cells were regarded as the control group. For the
mechanistic studies, the BV2 cells were pre-treated with the
peroxisome proliferator-activated receptor-γ (PPARγ) antagonist,
GW9662 (10 mM; MedChemExpress) or with the AMPK inhibitor, compound
C (1 mM; MedChemExpress) for 30 min at 37°C. The BV2 cells were
divided into the control, 200 µM morphine, 200 µM morphine + 5
µmol/l candesartan, 200 µM morphine + 5 µmol/l candesartan +
GW9662, and 200 µM morphine + 5 µmol/l candesartan + compound C
treatment groups. Untreated cells were regarded as the control
group.
Reverse transcription-quantitative PCR
(RT-qPCR) analysis
Total RNA was extracted from BV2 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) combined with treatment with DNase (Promega Corporation).
cDNA was synthesized from 1 µg total RNA in a 20-µl reaction volume
using the ImProm-II™ Reverse Transcription System (Promega
Corporation). All procedures were performed according to the
manufacturer's instructions. qPCR was performed using a LightCycler
480 SYBR-Green 1 Master mix (cat. no. 04707516001; Roche
Diagnostics). The qPCR thermocycling reaction conditions were as
follows: 95°C for 10 min, followed by 35 cycles of denaturation at
95°C for 1 min, annealing at 64°C for 1 min and elongation at 72°C
for 1 min, and a final extension step at 72°C for 7 min, before
maintaining the reaction mixture at 4°C. RNA expression was
quantitatively analyzed using the 2-ΔΔCq method (24). The primer sequences were as
follows: AT1R, forward 5′-GCCGTCGCTCAGGTTATTCT-3′ and reverse
5′-CAGGAACTTTGCCCCTTTGC-3′; ionized calcium-binding adaptor
molecule 1 (IBA-1), forward 5′-TGAGGAGATTTCAACAGAAGCTGA-3′ and
reverse 5′-CCTCAGACGCTGGTTGTCTT-3′; GAPDH, forward
5′-CCCTTAAGAGGGATGCTGCC-3′ and reverse
5′-ACTGTGCCGTTGAATTTGCC-3′.
Western blot analysis
The BV2 cells were lysed with RIPA buffer (Thermo
Fisher Scientific, Inc.), and the protein concentration was
detected using a BCA kit (Beyotime Institute of Biotechnology).
Proteins (20 µg) were separated by SDS-PAGE on 10% gels and were
then transferred to PVDF membranes (Merck KGaA). Subsequently, the
membranes were blocked with 5% non-fat milk for 1.5 h at 37°C.
Primary antibodies were then incubated with the membranes at 4°C
overnight. The next day, after washing, the PVDF membranes were
incubated with Goat Anti-Mouse IgG H&L (HRP)-conjugated
secondary antibody (1:5,000 dilution; cat. no. ab7063; Abcam) or
Goat Anti-rabbit IgG H&L (HRP)-conjugated secondary antibody
(1:2,000 dilution; cat. no. ab6721; Abcam). The proteins were
visualized using an ECL detection solution (Merck KGaA) and were
analyzed with ImageJ software (version 1.46; National Institutes of
Health). The primary antibodies used were as follows: Anti-AT1R
(cat no. ab124505; 1:2,000), anti-TNF-α (cat no. ab255275;
1:2,000), anti-IL-1β (cat no. ab254360; 1:2,000), anti-IL-6 (cat
no. ab259341; 1:2,000), anti-PPARγ (cat no. ab178860; 1:2,000),
anti-phosphorylated (p)-AMP-activated protein kinase (AMPK; cat no.
ab133448; 1:2,000), anti-AMPK (cat no. ab32047; 1:2,000),
anti-IBA-1 (cat no. ab178846; 1:2,000) and anti-GAPDH (cat no.
ab8245; 1:5,000) (all from Abcam). The intensities of the protein
bands were normalized against those of GAPDH.
ELISA
Briefly, BV2 cells were seeded into 96-well plates
(5×103 cells/well) and were stimulated with the relevant treatment
the next day. After centrifuging at 2,000 × g for 5 min at 4°C, the
cell supernatants were collected. The levels of TNF-α (cat no.
H052-1), IL1-β (cat no. H001) and IL-6 (cat no. H007-1-1) in the
supernatant were assessed using ELISA kits (Nanjing Jiancheng
Bioengineering Institute) according to the manufacturer's
instructions.
Immunofluorescence (IF) staining
The cells were fixed with 4% formaldehyde for 30 min
at 4°C and then permeabilized with 0.1% Triton X-100 in PBS for 15
min at 4°C. After blocking with 10% FBS for 30 min at 37°C, the
cells were incubated with anti-IBA (cat no. ab178846; 1:200) and
anti-AT1R primary antibodies (cat no. ab124505; 1:200) (both from
Abcam) at 4°C overnight. Subsequently, the cells were incubated
with goat anti-rabbit IgG H&L Alexa Fluor® 488
antibody (cat no. ab150077; 1:1,000; Abcam) for 30 min at 37°C. The
nuclei were stained with DAPI after IF staining for 15 min at room
temperature. Images were visualized under a fluorescence microscope
(Eclipse 80i; Nikon Corporation).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 8 software (GraphPad Software, Inc.). Data are presented as
the mean ± standard deviation and all experiments were repeated
three times. One-way analysis of variance analysis followed by
Tukey's post-hoc test of variance was used to compare the
differences among multiple groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
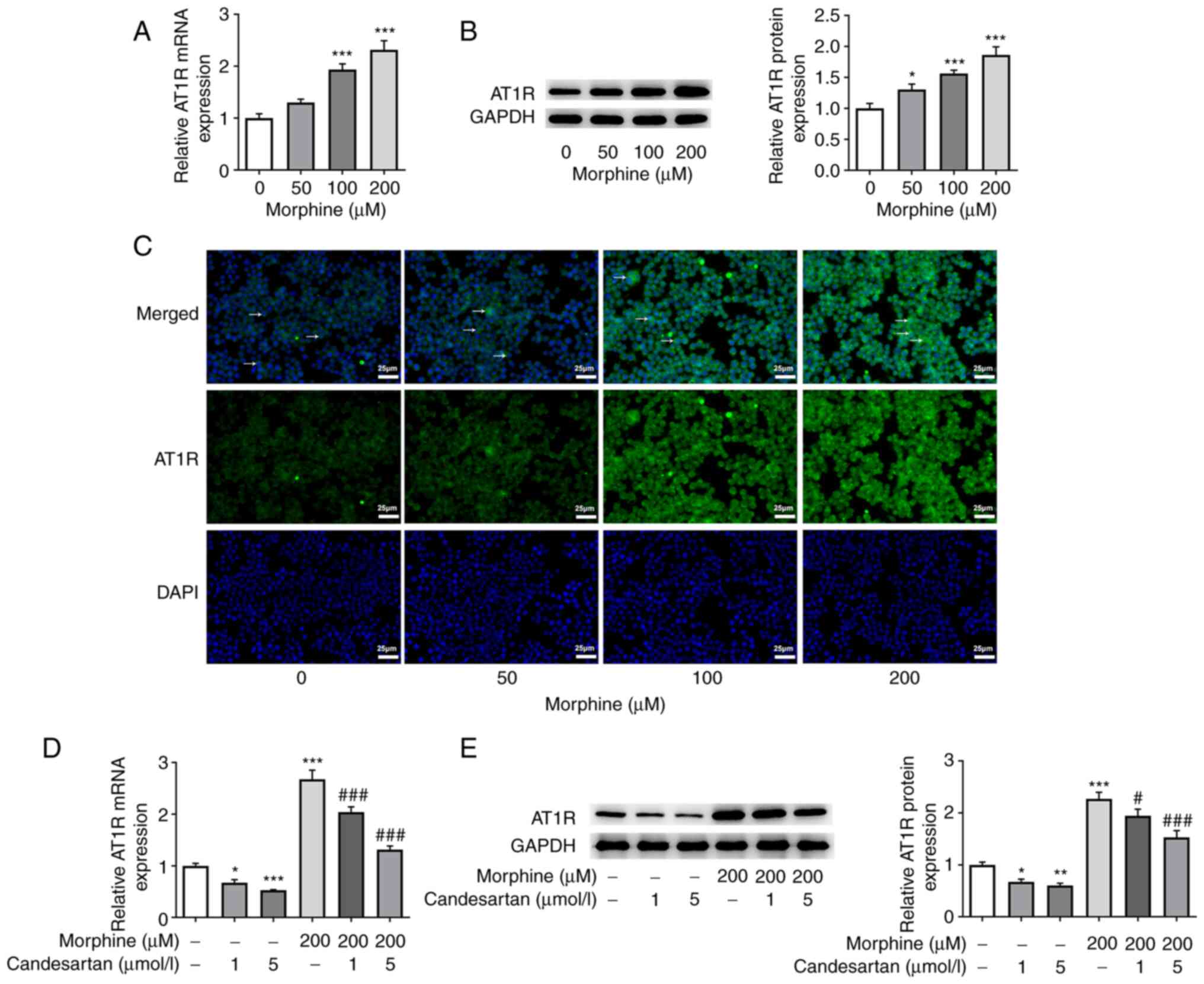
Candesartan inhibits the expression of
AT1R in morphine-induced BV2 cells
After BV2 cells were treated with morphine at
different doses (50, 100 and 200 µM), the expression levels of AT1R
were detected by RT-qPCR, western blotting and IF staining. The
results revealed that AT1R expression levels were increased in the
morphine treatment groups compared with those in the control group
(Fig. 1A-C). The expression of
AT1R was most significantly increased in the 200 µM morphine
treatment group; therefore, this concentration of morphine was
chosen for subsequent experiments. Subsequently, the effects of
different concentrations of candesartan on the expression levels of
AT1R in morphine-induced cells were investigated. These experiments
revealed that the expression levels of AT1R in BV2 cells were
significantly decreased following treatment with 1 and 5 µmol/l
candesartan compared with those in the control group. Candesartan
also caused a significant decrease in AT1R expression levels in
co-treated BV2 cells compared with those in the 200 µM morphine
treatment group (Fig. 1D and
E).
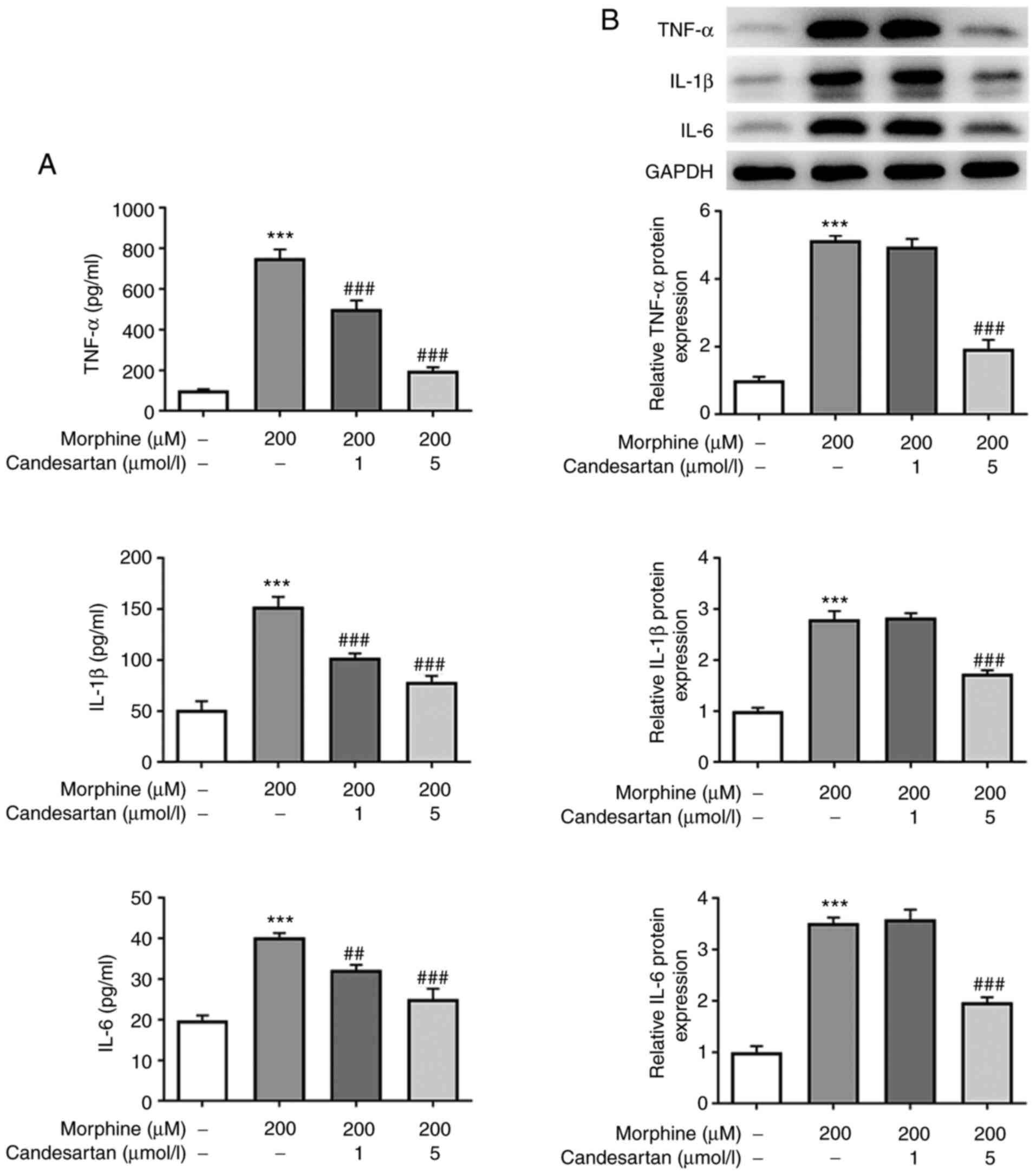
Candesartan dose-dependently inhibits
the inflammatory response in morphine-induced BV2 cells
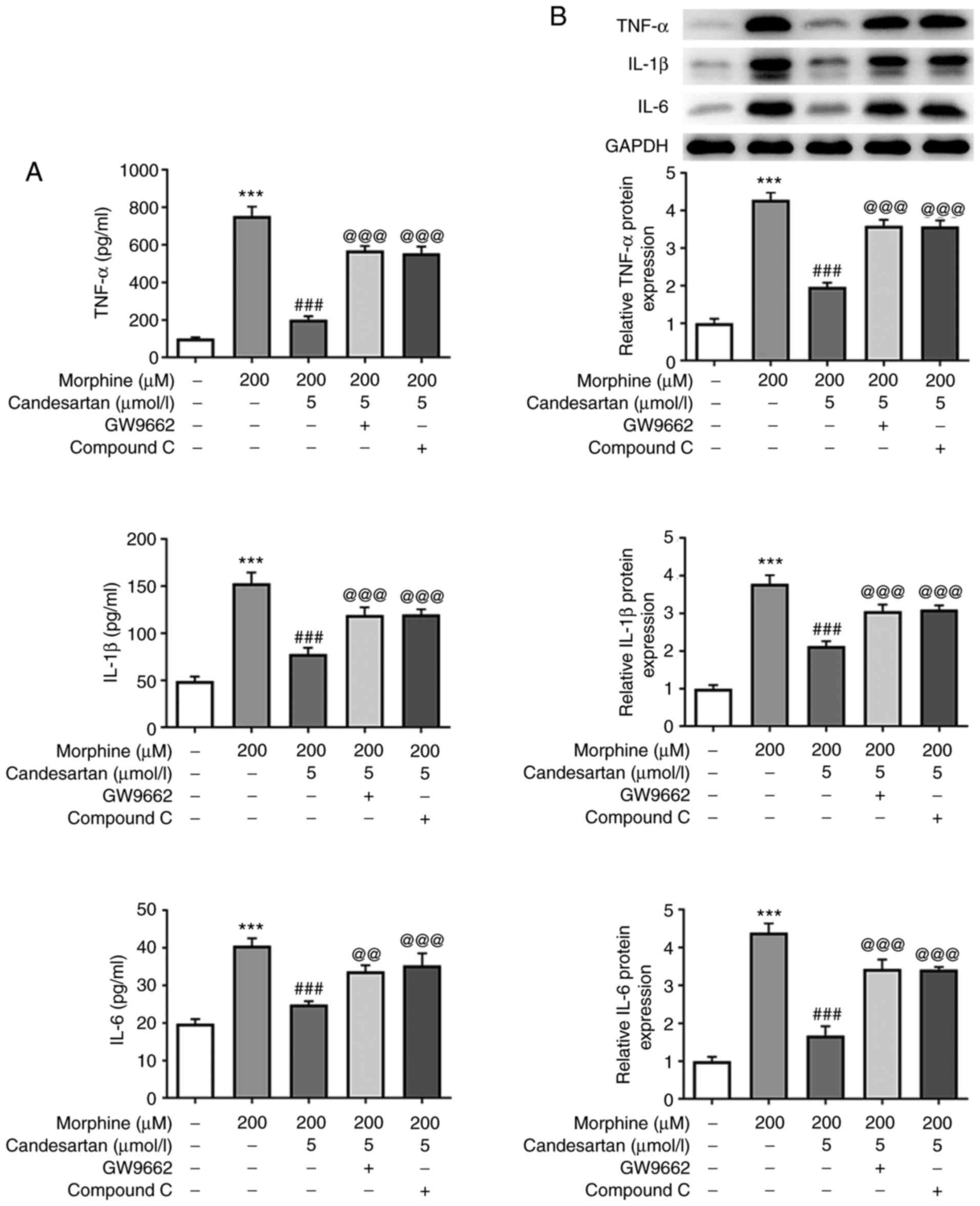
ELISA and western blot analysis were then used to
detect the intracellular levels of the inflammatory factors. The
results obtained revealed that the levels of inflammatory cytokines
were significantly increased in the 200 µM morphine group compared
with those in the control group. However, further administration of
candesartan reversed the morphine-induced increases in the levels
of inflammatory factors (Fig. 2A and
B).
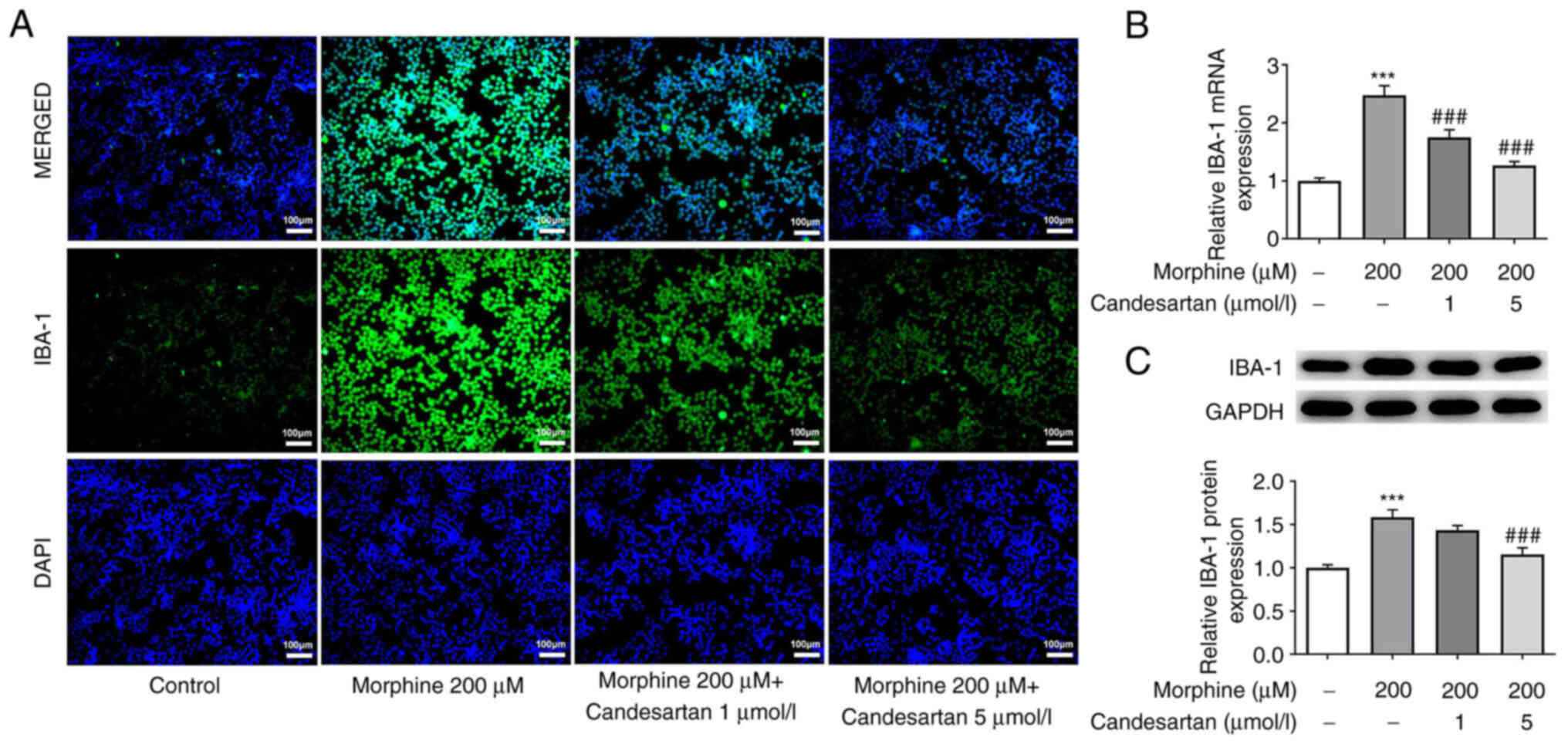
Candesartan dose-dependently inhibits
cell activation in morphine-induced BV2 cells
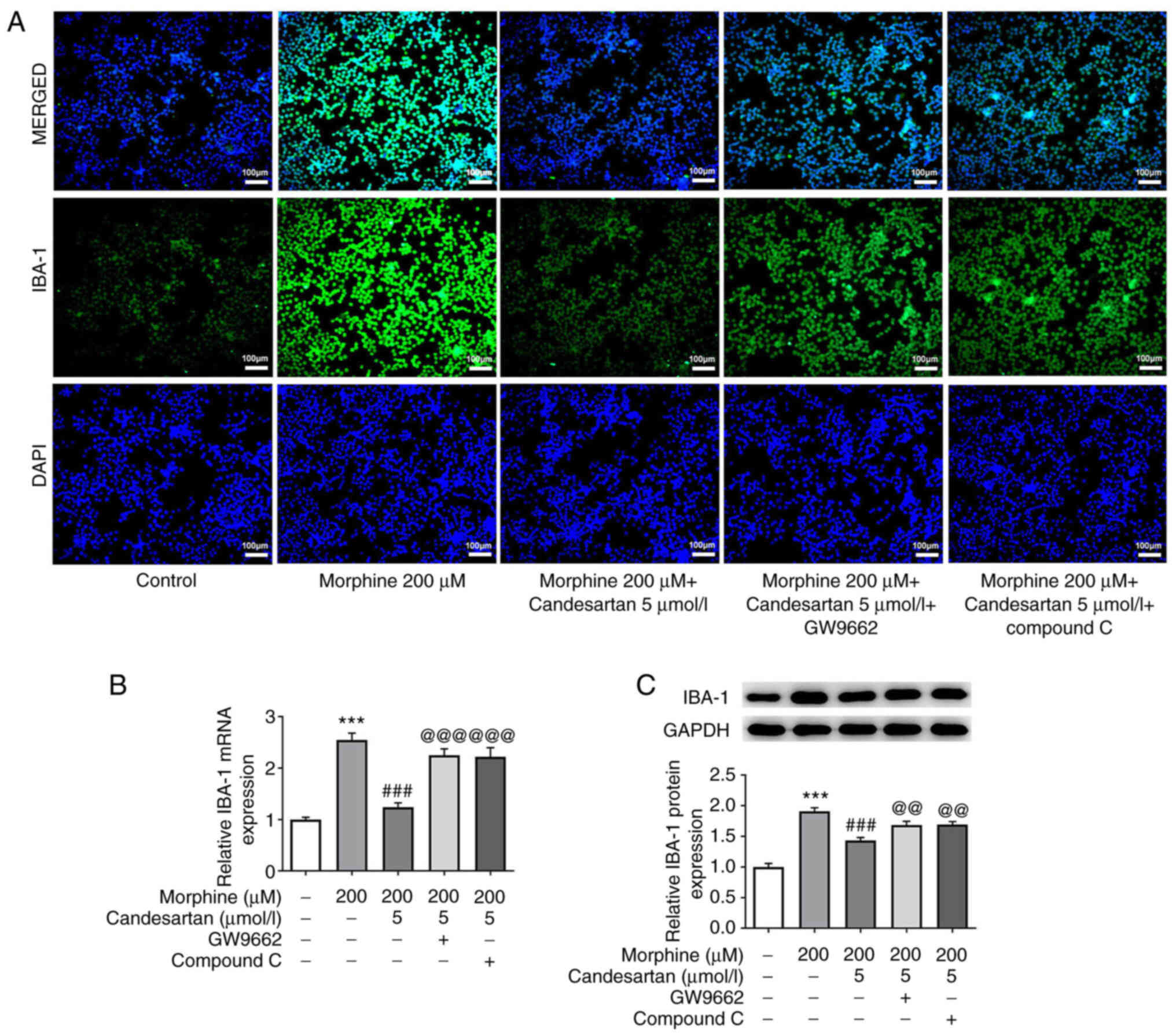
Subsequently, IF analysis was used to detect the
expression levels of IBA-1, a marker of microglial activation.
IBA-1 expression in the 200 µM morphine treatment group was
markedly increased compared with that in the control. After further
administration of candesartan, the expression levels of IBA-1 were
markedly decreased (Fig. 3A).
RT-qPCR and western blot analysis were subsequently used to verify
the expression levels of IBA-1. These experiments revealed that the
expression trend of IBA-1 was consistent with that revealed in the
IF staining experiments (Fig. 3B and
C).
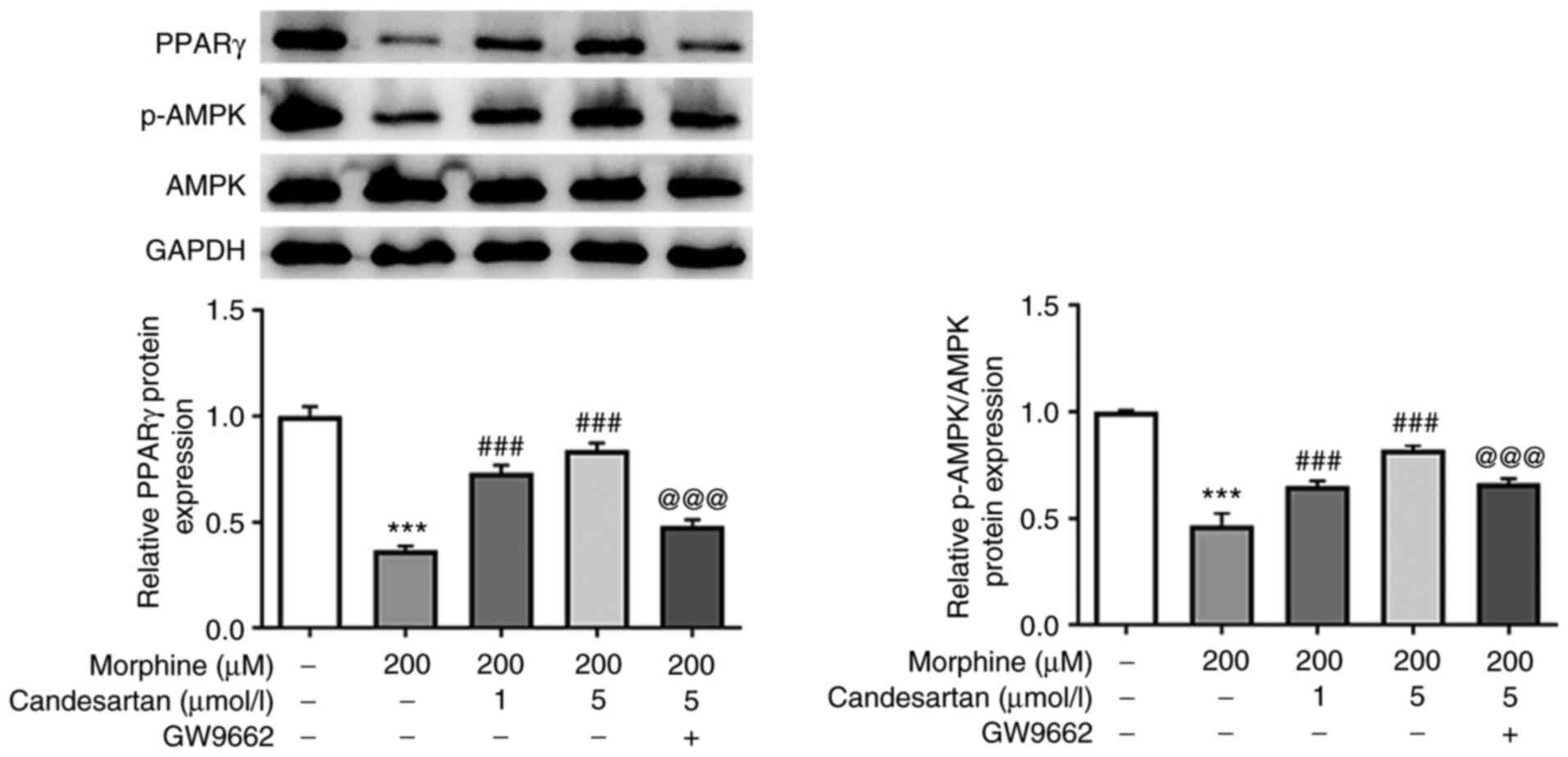
Candesartan activates the expression
of PPARγ and AMPK in morphine-induced BV2 cells
Mechanistic studies were subsequently performed to
investigate whether candesartan could exert effects on the
expression levels of PPARγ and AMPK in morphine-induced BV2 cells.
The expression levels of PPARγ and p-AMPK in the PPARγ/AMPK
signaling pathway were significantly decreased following treatment
with morphine compared with those in the control group. Following
candesartan treatment (1 or 5 µmol/l) in morphine-treated cells,
PPARγ and p-AMPK expression levels were dose-dependently elevated.
Conversely, after further treatment with the PPARγ antagonist
GW9662, the trends in the altered expression levels of PPARγ and
p-AMPK were reversed compared with those in the 200 µM morphine + 5
µmol/l candesartan group (Fig.
4). Therefore, it was possible to conclude that candesartan
could activate expression of the PPARγ and AMPK proteins in the
PPARγ/AMPK signaling pathway in morphine-induced BV2 cells.
Candesartan attenuates inflammatory
factors in morphine-induced BV2 cells via the PPARγ/AMPK signaling
pathway
To further explore the mechanism, inhibitors of the
PPARγ/AMPK signaling pathway were added to the cells; namely, the
PPARγ antagonist GW9662 and the AMPK inhibitor compound C.
According to the dose-dependent effect of candesartan on
morphine-induced BV2 cells, candesartan at a concentration of 5
µmol/l was selected for cell treatment in the following
experiments. The levels of TNF-α, IL-1β and IL-6 in the 200 µM
morphine + 5 µmol/l candesartan + GW9662, and 200 µM morphine + 5
µmol/l candesartan + compound C groups were significantly increased
compared with those in the 200 µM morphine + 5 µmol/l candesartan
treatment group (Fig. 5A and B).
These findings indicated that inhibition of PPARγ or AMPK reversed
the suppressive effects of candesartan on the inflammatory response
in morphine-induced BV2 cells.
Candesartan attenuates microglial
activation in morphine-induced BV2 cells via PPARγ/AMPK
signaling
IBA-1 expression in the 200 µM morphine + 5 µmol/l
candesartan + GW9662, and 200 µM morphine + 5 µmol/l candesartan +
compound C groups were markedly increased compared with that in the
200 µM morphine + 5 µmol/l candesartan group (Fig. 6A). Subsequently, RT-qPCR and
western blot analysis were performed, and the results obtained
revealed the same trends as identified in the IF staining analysis
(Fig. 6B and C). These findings
suggested that inhibition of PPARγ or AMPK reversed the suppressive
effects of candesartan on microglial activation in morphine-induced
BV2 cells.
Discussion
Opioids are the first choice of drug for the
clinical treatment of severe cancer pain and perioperative pain
(25). Morphine, as one of the
most widely used opioid drugs for reducing pain, has a short
half-life and a variety of dosage forms (26). However, the major limitation of
long-term use of morphine is its gradual loss of efficacy, which is
known as morphine drug tolerance (27). To make further medical advances,
resolving the issue of morphine tolerance is essential.
Activation of microglia has been reported to have an
important role in morphine tolerance. Morphine is able to activate
microglia by acting on morphine receptors or Toll-like receptor-4
(TLR-4) on the surface of microglia (28). Activated microglia release a large
number of cytokines and inflammatory factors, including IL-1β, IL-6
and TNF-α, which act on their corresponding receptors, resulting in
increased excitability of pain-associated neurons and pain.
Subsequently, the analgesic effect of morphine is weakened and the
process of tolerance is accelerated (7). Therefore, morphine tolerance may be
effectively circumvented through inhibiting microglial activation
and the inflammatory response. In the present study, it was shown
that the expression of inflammatory factors in BV2 microglia cells
was increased and microglia were activated following morphine
induction.
Microglia express µ-opioid receptors (29), as well as TLR-4 (30) and P2X receptors (31). It has been shown that blocking
TLR-4 can lead to a reduction in morphine tolerance (32,33). Furthermore, activation of the
NLRP3 inflammasome, which is dependent on TLR-4/P2X7 receptors in
the spinal cord, can lead to promotion of the development of
morphine tolerance (34). These
findings suggested that the regulation of microglial receptors may
have an influence on the development of morphine tolerance. A
previous study reported that AT1R is expressed in microglia
(35). Inhibition of AT1R has
been shown to alleviate neuroinflammation in various neurological
diseases, and AT1R antagonists may inhibit microglia-mediated
neuroinflammatory responses and microglial activation in
neurological diseases (36,37). In addition, it has been reported
that morphine can regulate the activation of AT1R and vitamin D
receptor to induce T-cell apoptosis (38). Therefore, it was reasonable to
hypothesize that regulation of AT1R may also affect morphine
tolerance. In the present study, it was revealed that the
expression levels of AT1R in morphine-induced BV2 cells were
significantly increased with an increase in the dose of morphine
used for induction. However, following treatment with the AT1R
antagonist candesartan, the expression levels of AT1R were
decreased. Moreover, candesartan was also shown to dose-dependently
inhibit morphine-induced BV2 cell inflammation and activation. A
previous study demonstrated that candesartan is able to reduce the
release of inflammation cytokines in the brain in Alzheimer's
disease (23). Candesartan has
also been shown to improve stroke-induced neuronal injury by
transferring microglia to the M2 phenotype (20). In addition, candesartan has been
reported to inhibit LPS-induced neuroinflammation in rats, and to
also inhibit the LPS-induced BV2 cell inflammatory response
(39). Notably, candesartan is a
common drug used in the clinical treatment of elderly hypertension,
which is known to have a good safety profile. The use of the AT1R
blocker candesartan could reduce the cost of drug development,
which is beneficial for the clinical treatment of morphine
tolerance.
Candesartan is an AT1R blocker, and it has been
shown that interference with AT1R can reduce atherosclerotic damage
in diabetic mice via activating PPARγ (40). Activation of PPARγ has also been
reported to improve morphine analgesia and morphine tolerance
(41,42). Moreover, PPARγ can activate AMPK
signaling (43,44). Notably, metformin, an AMPK
activator, has been reported to reverse the increase in AT1R
detected in response to a high-fructose diet (45). AT1R antagonists can regulate the
proliferation and migration of vascular smooth muscle cells through
AMPK/mTOR (46), and blocking
AT1R can effectively inhibit activation of the AMPK/p38
MAPK/MCPIP1/ER pathway in macrophages (47). Activation of AMPK signaling, in
itself, may also reduce morphine tolerance (48). Therefore, effective activation of
AMPK may have an important role in reducing morphine tolerance.
Consequently, the mechanism through which candesartan could
regulate morphine-induced microglial activation and the
inflammatory response was further explored in the present study by
treating cells with the PPARγ antagonist GW9662 and the AMPK
inhibitor compound C. These experiments revealed that GW9662 and
compound C were able to significantly reverse the inhibitory
effects of candesartan on morphine-induced microglial inflammation
and activation, suggesting that candesartan may activate PPARγ/AMPK
signaling via inhibiting AT1R, thereby reducing morphine
tolerance.
The present study has some limitations. Firstly,
some indicators were not detected. Notably, OX-42 and IBA-1 are
both important markers of microglia; however, the present study
only detected the expression of IBA-1; therefore, the role of OX-42
will be further verified in future experiments. Furthermore, the
levels of inflammatory cytokines TNF-α, IL-1β and IL-6 were
detected; however, the levels of NF-κB, IFN or IL-1 were not
detected, which we aim to further verify in future experiments. In
addition, morphine significantly decreased AT1R at the
transcriptional level, which indicates that morphine may inactivate
the transcription factor for AT1R; the candidates for transcription
factors of ATR1 will be further discussed in future
experiments.
In conclusion, to the best of our knowledge, the
present study was the first to explore the effects of the AT1R
blocker candesartan on the inflammation and activation of microglia
induced by morphine, in order to determine the association between
AT1R, microglial activation and morphine tolerance, and the
underlying mechanism was explored. The present study demonstrated
that candesartan reduced the morphine-induced inflammatory response
and cellular activation of BV2 cells via the PPARγ/AMPK signaling
pathway, suggesting that candesartan may improve morphine
tolerance. Consequently, the present study provided a theoretical
basis for the use of candesartan in the treatment of morphine
tolerance.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WZ and ZZ contributed to the conception and design
of the present study, analyzed and interpreted the data, and
critically revised the manuscript for important intellectual
content. FS, JY and SS contributed to designing the study, analyzed
the data, and drafted and revised the manuscript. WZ and ZZ confirm
the authenticity of all the raw data. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhang TJ, Qiu Y and Hua Z: The emerging
perspective of morphine tolerance: MicroRNAs. Pain Res Manag.
2019:94329652019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kornetsky C and Bain G: Morphine:
Single-dose tolerance. Science. 162:1011–1012. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schaefer CP, Tome ME and Davis TP: The
opioid epidemic: A central role for the blood brain barrier in
opioid analgesia and abuse. Fluids Barriers CNS. 14:322017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Martini L and Whistler JL: The role of mu
opioid receptor desensitization and endocytosis in morphine
tolerance and dependence. Curr Opin Neurobiol. 17:556–564. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang M, Luo L, Zhang Y, Wang W, Dong J,
Du W, Jiang W and Xu T: Metabotropic glutamate receptor 5
signalling induced NMDA receptor subunits alterations during the
development of morphine-induced antinociceptive tolerance in mouse
cortex. Biomed Pharmacother. 110:717–726. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jokinen V, Sidorova Y, Viisanen H,
Suleymanova I, Tiilikainen H, Li Z, Lilius TO, Mätlik K, Anttila
JE, Airavaara M, et al: Differential spinal and supraspinal
activation of Glia in a rat model of morphine tolerance.
Neuroscience. 375:10–24. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eidson LN and Murphy AZ: Inflammatory
mediators of opioid tolerance: Implications for dependency and
addiction. Peptides. 115:51–58. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin CP and Lu DH: Role of
neuroinflammation in opioid tolerance: Translational evidence from
human-to-rodent studies. Adv Exp Med Biol. 1099:125–139. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Berrios I, Castro C and Kuffler DP:
Morphine: Axon regeneration, neuroprotection, neurotoxicity,
tolerance, and neuropathic pain. P R Health Sci J. 27:119–128.
2008.PubMed/NCBI
|
|
10
|
Hutchinson MR, Coats BD, Lewis SS, Zhang
Y, Sprunger DB, Rezvani N, Baker EM, Jekich BM, Wieseler JL,
Somogyi AA, et al: Proinflammatory cytokines oppose opioid-induced
acute and chronic analgesia. Brain Behav Immun. 22:1178–1189. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miyoshi M, Miyano K, Moriyama N, Taniguchi
M and Watanabe T: Angiotensin type 1 receptor antagonist inhibits
lipopolysaccharide-induced stimulation of rat microglial cells by
suppressing nuclear factor kappaB and activator protein-1
activation. Eur J Neurosci. 27:343–351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chappell MC: Biochemical evaluation of the
renin-angiotensin system: The good, bad, and absolute? Am J Physiol
Heart Circ Physiol. 310:H137–H152. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ji J, Tao P and He L: Kangxianling
decoction prevents renal fibrosis in rats with 5/6 nephrectomy and
inhibits Ang II-induced ECM production in glomerular mesangial
cells. J Pharmacol Sci. 139:367–372. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Labandeira-Garcia JL, Rodriguez-Perez AI,
Garrido-Gil P, Rodriguez-Pallares J, Lanciego JL and Guerra MJ:
Brain renin-angiotensin system and microglial polarization:
Implications for aging and neurodegeneration. Front Aging Neurosci.
9:1292017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rodriguez-Perez AI, Borrajo A,
Rodriguez-Pallares J, Guerra MJ and Labandeira-Garcia JL:
Interaction between NADPH-oxidase and Rho-kinase in angiotensin
II-induced microglial activation. Glia. 63:466–482. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Joglar B, Rodriguez-Pallares J,
Rodriguez-Perez AI, Rey P, Guerra MJ and Labandeira-Garcia JL: The
inflammatory response in the MPTP model of Parkinson's disease is
mediated by brain angiotensin: Relevance to progression of the
disease. J Neurochem. 109:656–669. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rey P, Lopez-Real A, Sanchez-Iglesias S,
Muñoz A, Soto-Otero R and Labandeira-Garcia JL: Angiotensin
type-1-receptor antagonists reduce 6-hydroxydopamine toxicity for
dopaminergic neurons. Neurobiol Aging. 28:555–567. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rodriguez-Pallares J, Rey P, Parga JA,
Muñoz A, Guerra MJ and Labandeira-Garcia JL: Brain angiotensin
enhances dopaminergic cell death via microglial activation and
NADPH-derived ROS. Neurobiol Dis. 31:58–73. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bulsara KG and Makaryus AN: Candesartan.
In: StatPearls. Treasure Island (FL): StatPearls Publishing;
2022
|
|
20
|
Qie S, Ran Y, Lu X, Su W, Li W, Xi J, Gong
W and Liu Z: Candesartan modulates microglia activation and
polarization via NF-κB signaling pathway. Int J Immunopathol
Pharmacol. 34:20587384209749002020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Timaru-Kast R, Gotthardt P, Luh C, Huang
C, Hummel R, Schäfer MKE and Thal SC: Angiotensin II receptor 1
blockage limits brain damage and improves functional outcome after
brain injury in aged animals despite age-dependent reduction in AT1
Expression. Front Aging Neurosci. 11:632019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang L, Yin C, Xu X, Liu T, Wang B, Abdul
M, Zhou Y, Cao J and Lu C: Pellino1 contributes to morphine
tolerance by microglia activation via MAPK signaling in the spinal
cord of mice. Cell Mol Neurobiol. 40:1117–1131. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Torika N, Asraf K, Apte RN and
Fleisher-Berkovich S: Candesartan ameliorates brain inflammation
associated with Alzheimer's disease. CNS Neurosci Ther. 24:231–242.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stein C: New concepts in opioid analgesia.
Expert Opin Investig Drugs. 27:765–775. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sverrisdottir E, Lund TM, Olesen AE,
Drewes AM, Christrup LL and Kreilgaard M: A review of morphine and
morphine-6-glucuronide's pharmacokinetic-pharmacodynamic
relationships in experimental and clinical pain. Eur J Pharm Sci.
74:45–62. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mercadante S, Arcuri E and Santoni A:
Opioid-induced tolerance and hyperalgesia. CNS Drugs. 33:943–955.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Selfridge BR, Wang X, Zhang Y, Yin H,
Grace PM, Watkins LR, Jacobson AE and Rice KC: Structure-activity
relationships of (+)-naltrexone-inspired toll-like receptor 4
(TLR4) antagonists. J Med Chem. 58:5038–5052. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hickman SE, el Khoury J, Greenberg S,
Schieren I and Silverstein SC: P2Z adenosine triphosphate receptor
activity in cultured human monocyte-derived macrophages. Blood.
84:2452–2456. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lim KH and Staudt LM: Toll-like receptor
signaling. Cold Spring Harb Perspect Biol. 5:a0112472013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bernier LP: Purinergic regulation of
inflammasome activation after central nervous system injury. J Gen
Physiol. 140:571–575. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Eidson LN and Murphy AZ: Blockade of
Toll-like receptor 4 attenuates morphine tolerance and facilitates
the pain relieving properties of morphine. J Neurosci.
33:15952–15963. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang H, Huang M, Wang W, Zhang Y, Ma X,
Luo L, Xu X, Xu L, Shi H, Xu Y, et al: Microglial TLR4-induced TAK1
phosphorylation and NLRP3 activation mediates neuroinflammation and
contributes to chronic morphine-induced antinociceptive tolerance.
Pharmacol Res. 165:1054822021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang H, Zhang Y, Ma X, Wang W, Xu X, Huang
M, Xu L, Shi H, Yuan T, Jiang W, et al: Spinal TLR4/P2X7
receptor-dependent NLRP3 inflammasome activation contributes to the
development of tolerance to morphine-induced antinociception. J
Inflamm Res. 13:571–582. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Biancardi VC, Stranahan AM, Krause EG, de
Kloet AD and Stern JE: Cross talk between AT1 receptors and
Toll-like receptor 4 in microglia contributes to angiotensin
II-derived ROS production in the hypothalamic paraventricular
nucleus. Am J Physiol Heart Circ Physiol. 310:H404–H415. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sun H, Wu H, Yu X, Zhang G, Zhang R, Zhan
S, Wang H, Bu N, Ma X and Li Y: Angiotensin II and its receptor in
activated microglia enhanced neuronal loss and cognitive impairment
following pilocarpine-induced status epilepticus. Mol Cell
Neurosci. 65:58–67. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rodriguez-Perez AI, Garrido-Gil P, Pedrosa
MA, Garcia-Garrote M, Valenzuela R, Navarro G, Franco R and
Labandeira-Garcia JL: Angiotensin type 2 receptors: Role in aging
and neuroinflammation in the substantia nigra. Brain Behav Immun.
87:256–271. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chandel N, Sharma B, Salhan D, Husain M,
Malhotra A, Buch S and Singhal PC: Vitamin D receptor activation
and downregulation of renin-angiotensin system attenuate
morphine-induced T cell apoptosis. Am J Physiol Cell Physiol.
303:C607–C615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bhat SA, Goel R, Shukla R and Hanif K:
Angiotensin receptor blockade modulates NFκB and STAT3 signaling
and inhibits glial activation and neuroinflammation better than
angiotensin-converting enzyme inhibition. Mol Neurobiol.
53:6950–6967. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tiyerili V, Becher UM, Aksoy A, Lütjohann
D, Wassmann S, Nickenig G and Mueller CF: AT1-receptor-deficiency
induced atheroprotection in diabetic mice is partially mediated via
PPARγ. Cardiovasc Diabetol. 12:302013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
de Guglielmo G, Kallupi M, Scuppa G,
Stopponi S, Demopulos G, Gaitanaris G and Ciccocioppo R: Analgesic
tolerance to morphine is regulated by PPARγ. Br J Pharmacol.
171:5407–5416. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ghavimi H, Hassanzadeh K, Maleki-Dizaji N,
Azarfardian A, Ghasami S, Zolali E and Charkhpour M: Pioglitazone
prevents morphine antinociception tolerance and withdrawal symptoms
in rats. Naunyn Schmiedebergs Arch Pharmacol. 387:811–821. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xu X, Wang Y, Wei Z, Wei W, Zhao P, Tong
B, Xia Y and Dai Y: Madecassic acid, the contributor to the
anti-colitis effect of madecassoside, enhances the shift of Th17
toward Treg cells via the PPARγ/AMPK/ACC1 pathway. Cell Death Dis.
8:e27232017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Huang H, Wang ZJ, Zhang HB, Liang JX, Cao
WD, Wu Q, He CP and Chen C: The function of PPARγ/AMPK/SIRT-1
pathway in inflammatory response of human articular chondrocytes
stimulated by advanced glycation end products. Biol Pharm Bull.
42:1303–1309. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chao YM, Wu KLH, Tsai PC, Tain YL, Leu S,
Lee WC and Chan JYH: Anomalous AMPK-regulated angiotensin AT1R
expression and SIRT1-mediated mitochondrial biogenesis at RVLM in
hypertension programming of offspring to maternal high fructose
exposure. J Biomed Sci. 27:682020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhao Y, Shang F, Shi W, Zhang J, Zhang J,
Liu X, Li B, Hu X and Wang L: Angiotensin II receptor type 1
antagonists modulate vascular smooth muscle cell proliferation and
migration via AMPK/mTOR. Cardiology. 143:1–10. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shu S, Zhang Y, Li W, Wang L, Wu Y, Yuan Z
and Zhou J: The role of monocyte chemotactic protein-induced
protein 1 (MCPIP1) in angiotensin II-induced macrophage apoptosis
and vulnerable plaque formation. Biochem Biophys Res Commun.
515:378–385. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang Y, Tao GJ, Hu L, Qu J, Han Y, Zhang
G, Qian Y, Jiang CY and Liu WT: Lidocaine alleviates morphine
tolerance via AMPK-SOCS3-dependent neuroinflammation suppression in
the spinal cord. J Neuroinflammation. 14:2112017. View Article : Google Scholar : PubMed/NCBI
|