Introduction
Ferroptosis is an iron-dependent lipid reactive
oxygen species (ROS)-induced form of non-apoptotic cell death,
first proposed by Dixon et al in 2012 (1). This previous study revealed that the
Ras-selective lethal molecule erastin can affect glutathione (GSH)
peroxidase (GPX) activity, leading to redox imbalance, ROS
accumulation, membrane lipid peroxidation and ultimately to the
destruction of the integrity of the cell membrane (1). Ferroptosis induces morphological
changes that manifest as smaller mitochondria, increased
mitochondrial membrane density, decreased or lack of mitochondrial
cristae, and outer membrane breaks (2). The main characteristic of
ferroptosis is redox imbalance and the whole ferroptosis process,
which involves iron metabolism, lipid metabolism, oxidative stress
mechanisms, and the biosynthesis of nicotinamide adenine
dinucleotide phosphate, GSH and coenzyme Q10 (3), participates in the development and
progression of atherosclerosis (AS) (4). AS is characterized by the
accumulation of lipids in the arteries and the formation of
plaques. As the disease progresses, the blood vessels narrow, and
resulting in blood flow restriction or even blockage (5). In 1999, Ross (6) proposed that AS is a chronic and
progressive inflammatory disease, the mechanism of which involves
endothelial cell dysfunction and lipid accumulation, and that AS is
an important pathological cause of acute cardiovascular and
cerebrovascular diseases.
Epidemiological studies have reported that the
incidence rate of cardiovascular and cerebrovascular diseases
caused by AS has increased year on year, and that it has become the
primary cause of death in China and also worldwide (7). Notably, >75% of disabling and
fatal cardiovascular and cerebrovascular diseases in China are
caused by AS (8). Intracranial
atherosclerotic stenosis (ICAS) is an important cause of ischemic
stroke. A previous study on symptomatic intracranial arterial
stenosis and occlusion in China reported that the incidence of
intracranial AS in patients with ischemic stroke or transient
ischemic attack was 46.6% (9),
among which patients with ICAS had more severe symptoms, longer
hospital stays and higher stroke recurrence rates; moreover, the
recurrence rate increased with stenosis severity. Worldwide, ~2
billion individuals have carotid AS and the incidence of AS is
higher among male individuals than female individuals (7). AS is closely related to blood
pressure, blood sugar and blood lipid levels, smoking and obesity
among other risk factors, which over a period of 20–30 years can
gradually lead to vascular stenosis, causing a range of vascular
diseases.
AS is an inflammatory lesion caused by numerous risk
factors that damage vascular endothelial cells and is mainly
characterized by disordered lipid metabolism. It is well known that
lipid peroxidation, namely lipid oxidation degradation, generates
lipid peroxide radicals and hydrogen peroxide that serve crucial
roles in AS by causing inflammation and endothelial dysfunction,
whereas lipid peroxidation is also a core feature of ferroptosis
(10). Therefore, ferroptosis may
be a key factor in the occurrence and development of AS.
Ferroptosis is associated with various stages of AS development
through numerous physiological mechanisms, such as iron ion
metabolism, lipid metabolism and amino acid metabolism (11). Atherosclerotic stenosis is the
root cause of vascular-related diseases, such as coronary heart
disease, stroke and peripheral vascular disease. In particular, the
stability of neck AS plaques has been reported to be closely
related to the occurrence of ischemic stroke (12). According to global data (13,14), stroke currently remains the second
most common cause of mortality and disability, with ischemic stroke
accounting for 62.4% of all new stroke cases in 2019, thus placing
a heavy burden on society and the families of patients. A previous
meta-analysis on the subtypes of ischemic stroke demonstrated that
ischemic stroke due to large artery atheromatous sclerosis
accounted for 23% of cases and that the occurrence of cardiogenic
stroke was also closely related to major AS in the Asian population
(15). Therefore, a more precise
understanding of the causes and mechanisms of AS progression may be
beneficial for the treatment of patients.
Iron and ferroptosis
Regulation of iron homeostasis
Iron is the most abundant transition element in the
brain; it participates in the oxidation reaction, myelination,
neurotransmitter synthesis and metabolism, and it serves a key role
in cell respiration and energy generation (16). The body ingests iron through the
diet, and intestinal epithelial cells absorb iron and transport it
to the blood (17). In addition,
macrophages phagocytose aged red blood cells, which increases the
iron content in the blood (18),
which has been reported to be an independent predictor of vascular
damage (19). Extracellular
Fe3+ is transported into cells by transferrin receptor 1
(TfR1) and is converted into Fe2+, which participates in
the synthesis of heme in mitochondria (20) or is exported from cells by
ferroportin; iron is also stored in monocyte macrophages in the
form of ferritin, which helps to regulate iron storage and balance
in the body (Fig. 1). The
intracellular iron content is in a dynamic equilibrium and
reduction of the iron content in the body is protective (21). Nuclear receptor coactivator 4
(NCOA4) can recognize ferritin and promote its autophagic
degradation, leading to the release of free iron (22). Therefore, knocking out the NCOA4
gene can block ferritin deposition and avoid the accumulation of
free iron, thus protecting neurons from ferroptosis. Iron is
transported from the cytosol to the mitochondria by mitochondrial
ferritin (FTMT) and is used to synthesis iron porphyrin to reduce
free iron levels and protect mitochondria from oxidative stress.
Moreover, the upregulation of FTMT can increase iron consumption
and inhibit ferroptosis (23). It
has been reported that cyanidin-3-glucoside can inhibit the
occurrence of ferroptosis, affecting ischemia-reperfusion injury in
the myocardium by alleviating oxidative stress, reducing the free
iron content and downregulating TfR1 expression in cells and
tissues (24).
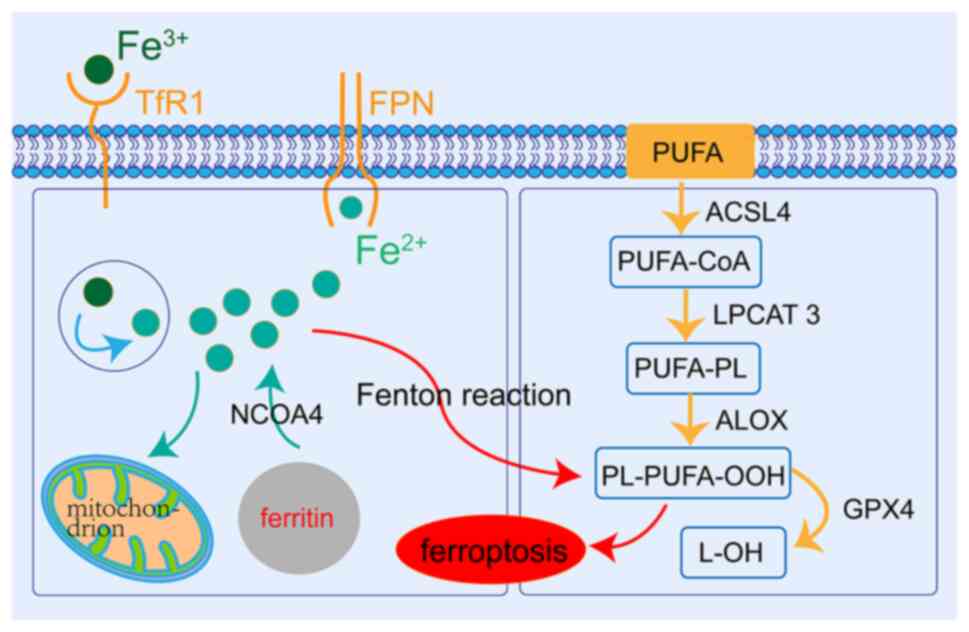 | Figure 1.Molecular mechanisms of ferroptosis.
Extracellular Fe3+ is transported to the cells byTfR1.
The Fe3+ is reduced to Fe2+ to play a role in
mitochondria or for storage in the form of ferritin, and the
remainder is exported from the cell through ferroportin to maintain
iron homeostasis. The reaction of PUFAs and excess reactive oxygen
species, which occurs on the cell membrane, is catalyzed by ACSL4
and LPCAT3, and mediated by ALOX, which leads to lipid
peroxidation. GPX4 can reduce ferroptosis by inhibiting lipid
peroxidation. PUFAs, n-3 polyunsaturated fatty acids; ACSL4,
acyl-CoA synthetase long chain family member 4; LPCAT3,
lysophosphatidylcholine acyltransferase 3; ALOX, arachidonate
lipoxygenase; PL, phospholipid; GPX4, glutathione peroxidase 4;
NCOA4, nuclear receptor coactivator 4; TfR1, transferrin receptor
1. |
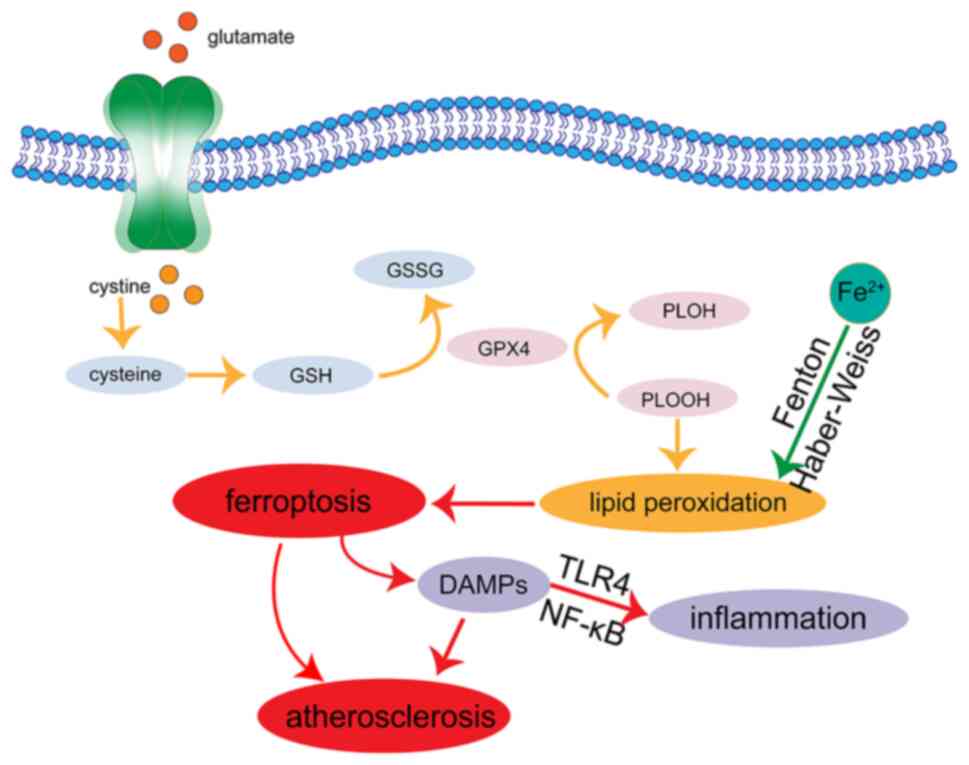
The imbalance of iron metabolism mediates the
occurrence of ferroptosis, and promotes the formation and
development of AS. Initially, a toxic iron reaction is associated
with an excess of stored iron (25), which can accelerate cerebral
tissue oxidation by increasing oxygen radical generation (26). This leads to super-oxidation
damage in the inner wall of the cerebral artery, further
aggravating the iron overload through the oxidized low-density
lipoprotein (ox-LDL)-mediated Toll-like receptor 4 (TLR4)/nuclear
factor κB (NF-κB) signaling pathway in macrophages (27). Moreover, the occurrence of
ferroptosis activates the TLR4/NF-κB signaling pathway and can
increase the expression of pro-inflammatory cytokine genes
(28). When intracellular free
iron levels increase, the intracellular labile iron pool also
increases (3). Excess divalent
iron may be involved in the Fenton reaction and other lipid
peroxidation processes that induce ROS overproduction and may be
involved in oxidative stress, with ROS promoting lipid peroxidation
and inducing ferroptosis (29).
ROS mainly include oxygen molecules, hydroxyl radicals, superoxide
anions and hydrogen peroxide radicals. Furthermore, iron is a
potent oxidant, which through the Haber-Weiss reaction catalyzed by
a large number of ROS, promotes the intracellular lipid
peroxidation reaction. The intracellular lipid peroxidation
reaction causes damage to proteins and nucleic acids, promotes
macrophage apoptosis and leads to the release of numerous
components of the cell contents; these substances can further
promote the infiltration of macrophages and enhance the lipid
peroxidation reaction (30).
LDL-C passes through damaged endothelial cells and enters
macrophages through oxidation, resulting in the formation of foam
cells. The aggregation of foam cells becomes the lipid core of
atherosclerotic plaques, which aggravates cell damage and lipid
peroxidation, increasing the production of ROS and lipid peroxide
products, such as MDA (31). In
addition, activated macrophages releases inflammatory factors, such
as TNF-α, IL-6 and IL-1β, increase the inflammatory response,
mediate the oxidation of lipoprotein, and further accelerate the
occurrence of AS.
Lipid metabolism pathway
Lipid peroxidation is the core process of
ferroptosis; specifically, the peroxidation of cell membrane
phospholipids by free radical-driven arachidonate lipoxygenases
(ALOXs) (32). Concurrently, iron
is associated with various stages of lipid peroxide generation,
including iron-catalyzed lipid oxidation and esterification, the
oxidation of polyunsaturated fatty acids and lipid ROS generation
via the Fenton reaction. Mitochondria are the main organelles where
iron utilization occurs in catabolic and anabolic pathways, and
they serve an important role in iron metabolism, as well as
material and energy metabolism (33). The Fenton reaction and other
peroxidation processes that incorporate iron (Fig. 2) can convert the mitochondrial
oxidation respiration product, hydrogen peroxide, into hydroxyl
radicals through the catalysis of ferrous ions. During the
oxidative phosphorylation of the electron transport chain, which
takes place on the inner mitochondrial membrane, electrons leak
from the complex and oxygen forms ROS through a series of redox
processes (34). Excessive ROS
generation can cause DNA damage, protein degeneration, lipid
peroxidation and can induce ferroptotic cell death. However, ROS
derived from mitochondria can also activate NLRP3 inflammatory
bodies and lead to the activation of the iron death signaling
pathway (35). Therefore, as a
mitochondrially derived antioxidant, the free radical scavenger
MitoQ can reduce ROS production by inhibiting mitochondrial
respiration and enhancing glycolytic function, thus protecting the
mitochondria and preventing GPX4-dependent ferroptosis (36). XJB-5-131 has been reported to have
a dual antioxidant effect, which can scavenge free radicals by
targeting mitochondria and inhibit DNA damage, providing protection
from ischemia-reperfusion-induced kidney injury and inflammation in
mice (37). Recently published
research has focused on the ROS produced by mitochondria, which may
promote the occurrence of ferroptosis by enhancing lipid
peroxidation, heme degradation and free iron overload, promoting
ferroptosis through HMOX-1 in the mitochondria (2). However, this process involves the
participation of multiple signaling pathways, and these factors
involved may have cross or offset effects, and the clear
participating mechanism requires further assessment in animal
experiments. Mitochondria-targeting antioxidants have been reported
to be effective in animal experiments (38); they can attenuate kidney injury
and promote tubular epithelial cells repair after
ischemia/reperfusion injury, and the overexpression of
mitochondrial ferritin has been shown to inhibit ferroptosis by
promoting the storage of mitochondrial iron.
GPX4 is a GPX that catalyzes the reduction of
hydrogen peroxide, organic hydroperoxide and lipid hydroperoxides,
thereby protecting cells from oxidative damage and ferroptosis
(39). As a ferroptosis inducer,
Ras-selective lethal small molecule 3 directly binds and inhibits
GPX4 activity, and can cause lipid ROS accumulation, mitochondrial
damage, disruption of ATP production, lipid peroxide accumulation
in cells and the promotion of ferroptosis (40). Acyl-CoA synthetase long chain
family member 4 (ACSL4) converts free arachidonic acid into
arachidonic arachidonoyl-CoA (41) and promotes unsaturated
phospholipid production, the main substrate for lipid peroxidation
(42). GPX4 can reduce lipid
peroxide, and following GPX4 inhibition (43), ACSL4 is considered to be required
for the occurrence of ferroptosis. Furthermore, microRNA-17-92 can
protect endothelial cells from ferroptosis in AS by mediating the
A20-ACSL4 axis (44). Doll et
al (45) reported that
knockdown of the ACLS4 gene significantly inhibited ferroptotic
cell death, thus suggesting that ACLS4 may participate in the
ferroptosis cascade via the lipid oxidation pathway. ACSL4 may also
aggravate neuronal damage through neuronal ferroptosis and promote
the release of pro-inflammatory cytokines from microglia (46). Baicalin has been reported to
inhibit ROS production, reduce ACSL4 expression, and mediate iron
uptake and autophagic degradation of ferritin to reduce
intracellular iron levels, which may participate in the prevention
of myocardial ischemia/reperfusion injury through anti-ferroptotic
mechanisms (47).
Ebselen (Ebs) is a small molecule organo-selenium
compound, which simulates GPX activity. As a lipid-soluble
compound, Ebs can easily enter the cell through the cell membrane
to exert antioxidant effects (48). Ebs can also inhibit the activity
of ALOX5 and ALOX15, and the synthesis of leukotriene; inhibition
of lipoxygenase and factors such as leukotriene are important for
preventing AS and inhibiting inflammation. It has been reported
that promoting the phosphorylation of AKT can increase the
expression of endothelial nitric oxide synthase in vascular
endothelial cells, increase nitric oxide release, and protect
against myocardial ischemia and reperfusion injury, while also
reducing oxidative stress and protecting the myocardial
mitochondria (49).
Amino acid metabolism pathway
Under normal physiological conditions (50), extracellular cystine is imported
into cells in exchange for intracellular glutamate via the
cystine/glutamate antiporter system xc−,
maintaining glutamate balance inside and outside the cell.
Intracellular cystine is converted into cysteine by cysteine
reductase, and GSH is generated by glutamate-cysteine ligase and
GSH synthetase. With antioxidants, GPX4 prevents ferroptosis by
reducing lipid peroxides to the alcohol form using GSH (Fig. 2). Furthermore, intracellular
cysteine deficiency caused by cysteine uptake disorder can lead to
the depletion of the antioxidant peptide, GSH, which is composed of
glutamate, cysteine and glycine, thus also leading to GPX4
inactivation and peroxide accumulation at lethal levels (Fig. 2). The depletion of GSH also leads
to glutamate-mediated iron and ROS generation, and triggers
oxidative cell death; specifically, ferroptosis through the amino
acid metabolic pathway (51). GSH
levels have been reported to be reduced in a mouse model of middle
cerebral artery occlusion where the infarct size was reduced
following intervention with the ferroptosis-related inhibitors,
liproxstatin-1 and ferrostatin-1 (46). Carvacrol has also been reported to
reduce the level of lipid peroxide in the ischemic brain tissue of
gerbils by increasing the expression of GPX4, inhibiting
ferroptosis, reducing cell death, and conserving the memory and
learning ability of gerbils following ischemia-reperfusion
(52).
It has been reported that the deposition of iron at
atherosclerotic plaques leads to the accumulation of ROS and the
death of macrophages; therefore, the loss of the antioxidant
capacity of macrophages directly leads to ferroptosis and plaque
formation (3). GSH, as a
tripeptide antioxidant and a cofactor of GPX4,is a key substrate
for GPX4, which can reduce the production of lipid peroxide. The
cell defense against lipid peroxidation is decreased due to the
depletion of GSH; however, cells do not reduce the amount of ROS
produced by Fenton reactions and other iron peroxidation reactions
in response to GSH depletion and therefore they are more sensitive
to ferroptosis (53). Erastin
functions as a ferroptosis inducer (54) that restrains the activity of the
xc− system, inhibiting cystine uptake, which
means that the cysteine in the cells cannot be used for GSH
synthesis. Intracellular reduced-GSH and oxidized GSH depletion
(55), the accumulation of
peroxidized phospholipids, the accumulation of lipid ROS and
protein or membrane damage can all trigger ferroptotic cell death.
GPX4 is a peroxide inhibitor protein discovered in 1982, which
belongs to the seleno-proteins, that produces water or alcohol and
protects cells by catalyzing certain reducing reactions of hydrogen
peroxide; therefore, a single dose of selenium delivered to the
brain can promote antioxidant GPX4 expression, protect neurons and
inhibit plaque growth in AS (56). Impaired GPX4 activity caused by
GPX4 deficiency or GSH depletion can lead to the inactivation of T
cells and promote ferroptosis; however, it can also promote the
differentiation of peripheral blood monocytes into B-cells and
natural killer cells (51).
Solute carrier family 7 member 11 (SLC7A11, also
known as xCT) is a key component of the cystine/glutamate
antiporter system xc−, which transports
extracellular cystine into cells in exchange for intracellular
glutamate, maintaining the glutamate balance inside and outside the
cell. The inhibition of cysteine-dependent GSH synthesis via
SLC7A11 leads to the inactivation of GPX4 and ultimately causes
ferroptosis in cells SLC7A11 has been reported as a well-validated
target for the prevention of ferroptotic cell death. Furthermore,
nuclear factor erythroid 2-related factor 2 (Nrf2) regulates the
occurrence of ferroptosis at the transcriptional level (57). Nrf2 regulates phagocytosis
following oxidative stress in macrophages by inhibiting cellular
iron uptake, reducing ROS production and upregulating SLC7A11
expression, as demonstrated by a high Nrf2 expression in
astrocytes, preventing neuronal cell death (58). Therefore, Nrf2 also functions as a
key regulator of lipid peroxidation and ferroptosis. Kaempferol has
been reported to protect cells from ferroptosis via activation of
the Nrf2/SLC7A11/GPX4 signaling pathway (59).
Nrf2 is a transcription factor, whose activation
promotes iron storage, reduces cellular iron uptake and limits ROS
production (55). Nrf2 is one of
the key regulators of the oxidative stress pathway (60), which negatively regulates
ferroptosis. Under normal physiological conditions, Kelch-like
ECH-associated protein 1 binds to Nrf2 via its C-terminal Kelch
domain and Nrf2 expression remains low through the ubiquitination
of the proteasome (61). When
oxidative stress occurs, Nrf2 dissociates from KEAP1 and undergoes
nuclear translocation, where it recognizes antioxidant response
sites and activates antioxidant genes, including heme oxygenase-1
(HO-1) and SLC7A11 (62). The
expression of SLC7A11, as a target of Nrf2, is upregulated when
Nrf2 is activated, protecting neuronal cells against ferroptosis,
and high Nrf2 expression in astrocytes protects against neuronal
cell death (63). HO-1 is a
stress-inducing enzyme encoded by the Hmox1 gene. The activation of
Nrf2 can initiate the downstream signal of HO-1, thus preventing
oxidative stress and scavenging free radicals (64). The knockout of Nrf2 can
significantly reduce the protein expression levels of SLC7A11 and
HO-1, and can promote the accumulation of lipid peroxides, trigger
ferroptosis through iron overload, excess ROS generation and lipid
peroxidation, and aggravate the progression of AS. Furthermore,
transglutaminase 2 can lead to neuronal death during stroke by
inducing GSH depletion, thereby promoting ROS accumulation and
ferroptosis (65).
Ferroptosis is involved in the formation and
progression of AS
Formation of AS
The pathogenesis of AS involves endothelial cell
dysfunction, lipid accumulation, foam cell formation, vascular
smooth muscle cell (SMC) migration and inflammatory factor
infiltration. The occurrence of AS begins with abnormal lipid
metabolism and a large amount of LDL being deposited on the intima
of the vascular wall of endothelial cells where it is oxidized to
ox-LDL during the process of oxidative stress (10). Free iron accelerates this
modification process through the action of hydroxyl free radicals.
Monocytes adhere to the vascular endothelium and are transferred to
the subendothelium through chemotaxis to transform into macrophages
(66). Macrophages serve a
pivotal role in AS progression, recognizing and destroying
endothelial cells. Macrophages phagocytize ox-LDL through protease
and oxygen free radicals secreted by scavenger receptors and are
then converted into foam cells, and excessive lipid deposition
forms lipid streaks that progress into lipid-containing plaques;
iron deposition is visible in the plaque formed by apolipoprotein
E-deficient mice and can be seen by staining analysis or imaging
(67). SMCs then migrate to the
inner membrane and proliferate, forming a fibrous cap of plaque,
which is the fibrous plaque phase of AS. Moreover, ox-LDL triggers
the immune response, damaged endothelial cells are activated and
express monocyte chemoattractant proteins (MCP)-1 and −8,
intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion
molecule-1 (VCAM-1), E-selectin, P-selectin and other inflammatory
factors (68). These factors
attract lymphocytes and monocytes to bind to endothelial cells and
infiltrate the arterial wall, which causes inflammation. For
macrophages, high mobility group protein B1 (HMGB1) is considered
necessary for the macrophage inflammatory response, as the
ferroptosis inducer erastin promotes the release of
damage-associated molecular patterns and HMGB1 (69). The plaque formation process
promotes the cooperation of iron and lipid accumulation in
macrophages, which lead to the development of AS (70). Moreover, AS plaques with
macrophage-derived foam cells as the main components are less
stable and more prone to rupture (71).
Immune responses are present throughout the
development of AS and inflammation relies on mediators, such as
immune cells and inflammatory factors, which are involved in AS
development through signaling pathways, such as TLR4, NF-κB and
JAK/STAT (72). As well as innate
immune cells, such as macrophages, adaptive immune cells
participate in the formation of AS by exerting pro-inflammatory or
anti-inflammatory effects through the secretion of various
cytokines or antibodies (73).
Th1 cells promote the development of AS by the secretion of IFN-γ,
TNF-α and IL-2. Th2 cells regulate the progression of AS
inflammation by the secretion of the anti-inflammatory factor IL-13
and the pro-inflammatory factor IL-4. IL-13 stimulates macrophage
polarization to the M2 isoform, releases IL-10 and TGF-β, and
functions against AS development through the activation of STAT3;
however, IL-4 increases CD36 expression, thereby enhancing
macrophage phagocytosis and promoting AS progression (74). Th17 accelerates the progression of
AS and Th17 cells produce IL-17, IL-22 and IL-23, recruit
neutrophils and promote inflammation at the site of infection
(75). Tregs inhibit the activity
of multiple immune cells, exerting anti-AS effects by secreting
TGF-β1 and IL-10 (76).
Effects of ferroptosis on AS
AS is mainly caused by vascular endothelial
dysfunction and plaques that form on blood vessel walls.
Atherosclerotic plaque progression is characterized by ox-LDL
accumulation within macrophages, macrophage death, necrotic core
formation, the rupture or shedding of unstable plaques, and entry
into the secondary lesion stage of AS. The majority of acute
ischemic events, such as acute coronary syndromes and stroke, are
attributed to vulnerable plaques (77). Typically, the core characteristics
of vulnerable plaques are active inflammation; in addition,
vulnerable plaques are more prone to plaque rupture, and they
include the following characteristics (78): i) A thin fibrous cap, large lipid
core and micro-calcifications; ii) intraplaque hemorrhage; iii)
fiber cap rupture or ulcer formation; and iv) numerous infiltrating
macrophages and neo-angiogenesis. The stability of atherosclerotic
plaques is closely related to the size of the lipid core, the
thickness of the fiber cap and the number of inflammatory cells
within the plaque (77).
Generally, macrophage death is considered to be a major factor in
necrotic core formation and plaque instability, and ferroptosis is
mainly involved in oxidative stress and the breakdown of
endothelial function (79).
Macrophages secrete matrix metalloproteases that degrade collagen
fibers in the plaque extracellular matrix, causing plaque rupture,
hemorrhage and thrombosis, the release of peroxides and nitrogen
radicals, and cause the death of surrounding cells (72). The lipid accumulation process
produces a large number of oxygen radicals involved in the
inflammatory reaction, promoting the formation of AS, while
releasing cytokines and proteases to degrade the collagen matrix of
the fiber cap, prompting the plaque to become brittle and rupture,
affecting its stability (80). A
previous study showed that GPX4 was highly expressed in the early
stage of plaque formation. With the progression of plaques, the
expression levels of NLRP3 and caspase-1 were increased, and in the
late stage, the expression of NLRP3 was significantly upregulated
and the expression of GPX4 was decreased. These vulnerable plaques
are closely related to a large number of acute ischemic events,
including acute coronary syndrome and ischemic stroke (77).
As well as the accumulation of LDL, numerous other
factors, such as high uric acid and high homocysteine levels are
considered independent risk factors for AS progression. High uric
acid levels cause vascular endothelial cell dysfunction by
inhibiting the protein expression of endothelial nitric oxide
synthase; furthermore, high uric acid levels stimulate monocytes to
produce IL-1, IL-6 and TNF-α, which is supported by homocysteine
(HCY) via JAK2/STAT3 signaling. Reducing HCY-enhanced STAT3
phosphorylation can significantly reduce HCY-induced microglial
activation, and IL-6 and TNF-α production (81). Moreover, endothelial cells
generate chemokines and adhesion molecules, promote the migration
and adhesion of SMCs, aggravate vascular stenosis and plaque
instability, promote vascular calcification, promote the
precipitation of uric acid crystallization, increase blood
viscosity, induce 5-HT release, increase platelet number and induce
internal platelet activation; thus promoting thrombosis. High HCY
levels damage endothelial cells, further exacerbating oxidative
injury and inflammatory processes, increasing fibrinogen
production, abnormal coagulation and platelet dysfunction (82). Platelets release arachidonic acid
to produce ROS, resulting in calcium and lipid deposits in the
endothelium, which reduces the elasticity of the arterial wall and
also causes oxidative stress by affecting cellular respiration,
resulting in LDL oxidation. In addition, vascular endothelial cells
are damaged, and the normal proportion of endothelin-1 and NO
secreted by them to maintain the vasomotor function is broken; the
impaired bioavailability of NO, and subsequent vasoconstriction and
proliferation of smooth muscle can lead to the progression of AS
(83).
Furthermore, gut dysbiosis exacerbates the
progression of AS by regulating intestinal structure, intestinal
barrier integrity, the inflammatory status and host metabolism
(84). It has been reported that
the inflammatory response caused by remote infection can aggravate
plaque development or cause plaque rupture, and that the metabolism
of cholesterol and lipids by intestinal microorganisms can affect
the development of atherosclerotic plaques (85). Notably, ferroptosis may also be
involved in this process. Chapkin et al observed that n-3
polyunsaturated fatty acids (PUFAs) and the short-chain fatty acid,
butyrate, induced apoptosis via a mitochondrially-targeted
antioxidant, mitoQ, which is a ubiquinone derivative, and
overexpression of GPX4. This mirrored the phenomenon that fatty
acid metabolism by the gut microbiota occurs by disrupting the
energy metabolism balance of mitochondria, eliciting abnormalities
in intracellular Ca2+ homeostasis systems and releasing
ROS, which induce cell ferroptosis (86). Similar reports, such as that by
Hayase and Jenq (87) reported
that the gut flora metabolite, trimethylamine N-oxide (TMAO),
enhanced M1 macrophage polarization through inflammasome formation
and the activation of NLRP3 in endothelial cells. Furthermore, TMAO
has been reported to cause the overexpression of TNF-α, IL-6 and
C-reactive proteins, directly inducing vascular inflammation and
endothelial dysfunction (88).
This process mainly promotes the occurrence and development of AS
through the NF-κB pathway. Concurrently, TMAO can increase platelet
hyperreactivity and promote arterial thrombosis through Toll
receptor signaling pathways (89). Therefore, it could be concluded
that AS risk factors break the intestinal ecological balance and
that gut microbial metabolites promote AS progression with the
involvement of ferroptosis and inflammation.
Following ischemic stroke, the NLRP3 inflammasome
has been reported to promote neuroinflammation, and to trigger the
apoptosis of glial cells and neurons. NLRP3 activates caspase-1,
triggers the release of inflammatory mediators, such as the
cytokines IL-18 and IL-1, exacerbates oxidative stress and
endoplasmic reticulum stress, and promotes brain edema and
atherosclerotic processes (90).
Ferroptosis is involved in AS
progression
The progression of AS is often inevitable and is
generally considered to occur due to the accumulation of oxidative
lipids in the intima, activation of the inflammatory process and
the presence of two types of macrophages in its plaque (91). In the early stages, M2 macrophages
are the main type of infiltrated cells, the plaque is relatively
stable and the anti-inflammatory M2 macrophages produce IL-10 and
IL-13, which inactivate Th1 lymphocytes, reduce inflammation and
promote tissue damage repair (92). As the disease progresses,
long-term iron overload causes the number of M1 macrophages to
increase and dominate, further promoting the development of AS; M1
macrophages have a potent phagocytic activity and secrete
proinflammatory factors, such as TNF-α (93) (Fig.
3). Blood vessels that are constantly infiltrated by
inflammatory factors for a long time are prone to the aggregation
and oxidation of LDL, and the deposition of atherosclerotic
plaques, promoting AS and the ferroptosis of macrophages in the
plaque (94).
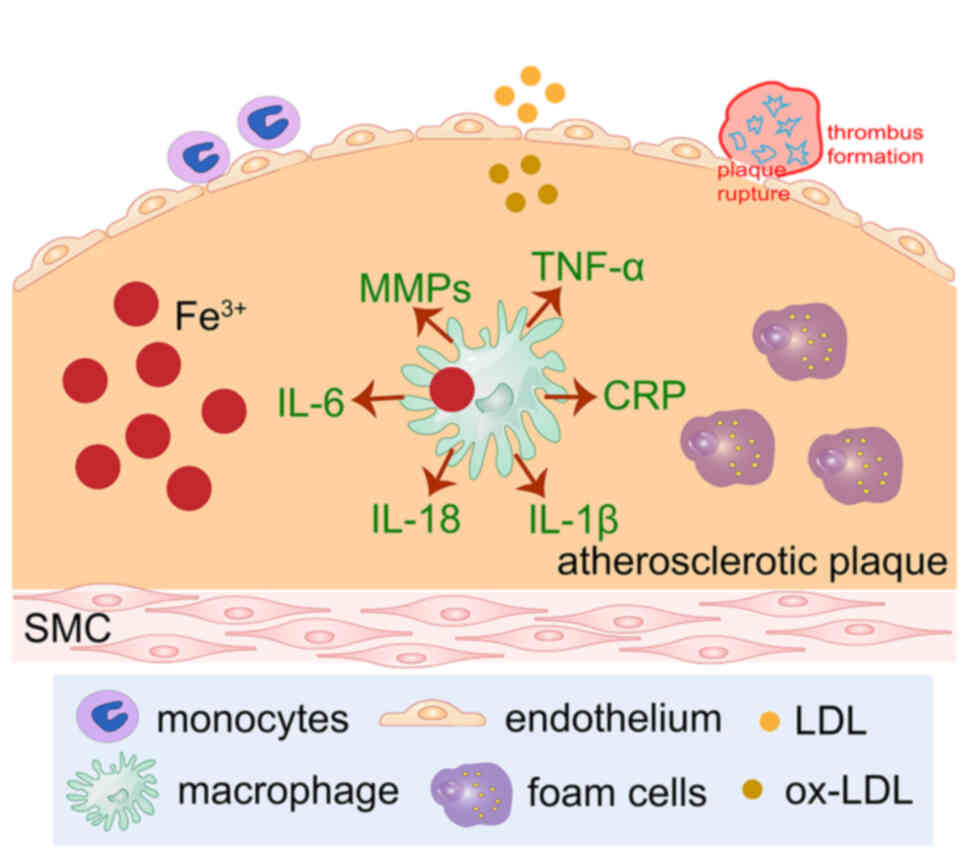 | Figure 3.Ferroptosis is triggered by excessive
lipid peroxidation. ox-LDL induces iron accumulation. Free iron has
strong endothelial cytotoxicity and, at the same time, lipid-laden
macrophages accumulate in the arterial subendothelial space, which
promotes the inflammatory response of the arterial wall. Iron
deposition in plaques causes ox-LDL to activate the TLR4/NF-κB
signaling pathway and promotes foam cell formation. Ferric iron
retention in atherosclerotic plaques promotes oxidative stress,
lipid peroxidation and increases the instability of plaque. The
occurrence of ferroptosis is involved in numerous processes of
atherosclerosis. Lipid peroxidation, plaque hemorrhage and iron
deposition are important characteristics of advanced
atherosclerotic plaques. LDL, low-density lipoprotein; ox-LDL,
oxidized LDL; MMPs, matrix metalloproteases; CRP, C-reactive
protein; SMC, smooth muscle cell. |
In the early stage, AS appears in the form of
hemosiderin deposition. In the highly oxidative environment of
atherosclerotic lesions, red blood cells rapidly dissolve and
release hemoglobin, which is easily oxidized and releases heme. The
pro-oxidant and pro-inflammatory effects of heme affect the
functions of endothelial cells and macrophages, and promote the
oxidation of LDL. Therefore, cholesterol levels are closely related
to iron deposition, and activation of the cellular heme
oxygenase-1/ferritin system can slow down the progression of this
process. Notably, iIncreased iron deposition has been reported to
occur in the cerebral cortex of rabbits fed a high-cholesterol diet
(95). In MRI
T2-weighted images, intra-plaque hemorrhage and iron
deposition have been observed (96). Therefore, macrophage ferroptosis
may be considered to be involved in the progression of AS (97) and could serve as a potential
target for intervention with AS progression. Vinchi et al
(98) reported that AS was
profoundly aggravated in iron-loaded mice. It has also been
reported that iron can exacerbate AS by lipid profile alterations,
vascular permeabilization, sustained endothelial activation,
elevation of pro-atherogenic inflammatory mediators and reduced
nitric oxide availability (99).
Treatment with iron chelators in mice has been reported to restore
vascular endothelial function, reduce the levels of IL-6, TNF-α and
MCP-1, and suppress AS (100).
Iron overload leads to endothelial cell dysfunction through
pro-oxidative and proinflammatory effects, directly causing AS
progression, whereas the occurrence of ferroptosis affects AS
progression through increased ROS production and cytokine secretion
(101). Consistent with this, it
has been confirmed in human coronary artery endothelial cells in
vitro that Tanshinone IIA can inhibit ferroptosis-induced
endothelial lipid peroxidation and dysfunction by activating the
Nrf2 pathway, thus ultimately alleviating AS (102). Furthermore, it has also been
reported that ferroptosis is involved in the progression of AS
mainly through oxidative stress and the exacerbated breakdown of
endothelial function (67).
In a previous study, iron content was directly
quantified by nuclear magnetic resonance spectroscopy and it was
reported that the level of redox active iron was increased in
carotid atherosclerotic lesions compared with in normal healthy
human endothelium, and it was further increased in late AS,
inducing oxidative stress and inflammatory responses (103). Notably, Bai et al
(104) assessed
ApoE−/− mice with high-fat diet-induced AS and evaluated
the expression of ferroptosis-related factors. This previous study
reported significant reductions in the mRNA and protein expression
levels of SLC7A11, a decrease in endothelial cell and angiogenic
markers, such as CD31, and mitochondrial damage in endothelial
cells. Moreover, the opposite results were reported in the same
model treated with an iron chelator; the expression levels of the
pro-angiogenic factor VEGF, and the adhesion molecules ICAM-1 and
VCAM-1 were decreased, and the lesion area in AS was reduced, which
indicated that ferroptosis may be involved in the progression of AS
(104).
Processes, such as lipid oxidation, inflammatory
reactions and iron accumulation, can occur during the pathogenesis
of AS. Currently, the applied targets of ferroptosis inhibitors
focus on the imbalance of lipid peroxidation and ferrous ions. Iron
chelators, antioxidants and free radical scavengers reduce the
ferroptosis of endothelial cells by reducing the lipid peroxidation
of the plaques in the AS. In vitro, vascular endothelial
cells treated with ox-LDL have ferroptotic properties and the use
of ferroptotic inhibitors can interfere with this process (Table I).
 | Table I.Ferroptosis-related factors and
possible targets. |
Table I.
Ferroptosis-related factors and
possible targets.
| First author
(year) | Ferroptotic
factor | Regulatory
mechanism | Possible
intervention target to inhibit ferroptosis | (Refs.) |
|---|
| Lu et al
(2020) | TFR1 | lncRNA PVT1
regulates miR-214-mediated TFR1 expression | Silencing of lncRNA
PVT1 and miR-214 overexpression markedly decrease PVT1 levels to
suppress ferroptosis in vivo | (125) |
| Li et al
(2021) | NCOA4 | Degradation of
ferritin leads to free iron release | Knockout of NCOA4
notably abrogates ferritinophagy and thus inhibits ferroptosis | (126) |
| Chen et al
(2021) | ACSL4 | Enhancing lipid
peroxidation | ROSI inhibits ACSL4
and blocks the lipid peroxidation process | (127) |
| Lu et al
(2018) | ROS | Nrf2/NADPH/ROS
pathway | Artesunate
suppresses oxidative toxicity and inflammatory by activating Nrf2
and downregulating ROS | (128) |
| Liu et al
(2020) | GPX4 |
xc−/GSH/GPX4 | Sulforaphane
alleviates the cytotoxicity of erastin by promoting the expression
of genes related to GSH synthesis | (129) |
| Dong et al
(2020) | Nrf2 |
Nrf2/SLC7A11/HO-1 | Nrf2 alleviates
OGD/R-induced ferroptosis by upregulating SLC7A11 and HO-1 | (64) |
| Wang et al
(2020) | HMOX1 | Nrf2/HO-1 | The upregulation of
Nrf2 iron-related target gene HMOX-1 exerts antioxidant and
anti-inflammatory effects | (2) |
| Ratan (2020) | HIF-1α | HIF-1α/HO-1 | Adaptaquin
selectively inhibits HIF prolyl hydroxylases and stabilizes HIF-1
to protect neurons from ferroptosis | (57) |
Ferroptosis accelerates the progression of
AS, leading to ischemic stroke
Ferroptosis accelerates AS progression mainly
through vascular endothelial disorder and the lipid peroxidation of
vascular endothelial cells, endothelial function impairment,
platelet adhesion aggregation and eventually, thrombosis (105). The causal association between
iron and AS is as follows. Firstly, iron overload leads to the
activation of lipoxygenase (106), the upregulation of ACSL4 and
ALOX, and catalyzes the lipid peroxidation of phospholipids
containing PUFAs in the lipid bilayer of the cell membrane, which
are degraded by oxidation, resulting in damage to the cell
membrane. Secondly, the interaction between iron and oxygen
radicals leads to lipid peroxidation and neuronal death, promotes
thrombosis by accelerating the progression of AS and intravascular
platelet activation, and eventually, ischemic stroke (98). It has previously been reported
that ALOX15 knockout can reduce iron deposition in the cerebral
cortex, the level of ROS and the level of 4-hydroxynonenol, which
is the final product of lipid peroxidation, and reduces nerve
injury through the spermidine/spermine N1-acetyltransferase
1/ALOX15 axis (107).
During the process of an ischemic stroke, iron
accumulation can exacerbate neuronal injury in patients or in
animal models; iron accumulation is associated with AS and the
occurrence of ferroptosis accelerates plaque formation (3). However, this process can be
prevented by iron chelation therapy (108). A previous study reported a
reduction in iron accumulation and attenuated neuronal degeneration
in mice that had been treated with Fer1 (109). Clinical trials have reported
that reducing the systemic iron content within 1–3 days of ischemic
stroke may provide benefits for patients with acute ischemic stroke
(110). Hypoxia-inducible factor
(HIF)-1 prolyl hydroxylases (PHDs) may be a target of iron
chelators to inhibit ferroptosis. In the brains of hypobaric
hypoxic rats pre-treated with deferoxamine, hypoxia inactivated
PHDs, causing the accumulation of HIF-1α and the level of HIF-1α
protein to be significantly upregulated (111). Cells were more able to tolerate
the hypoxic environment and hypoxic cells could recover faster,
eventually reducing the volume of cerebral infarction. Moreover,
HIF-1 may downregulate ACSL4 expression by binding the ACSL4
promoter to inhibit its transcription and alleviate ischemic brain
injury (112).
Neuronal death and secondary inflammation due to
cerebral ischemia are directly related to poor functional outcomes,
with inflammation occurring throughout the entire phase of
ischemia-reperfusion injury and ferroptosis interlinking with
inflammation. Following cerebral ischemia, the release of
inflammatory cytokines and neurotoxic mediators can be induced
through multiple signaling pathways, such as NF-κB and STAT3,
leading to neuronal damage and death (113). NF-κB activation following
ischemia, and the expression of TNF-α, IL-1 and IL-6, is
upregulated in cells to promote the inflammatory response (11). The high expression of IL-6
promotes the continuous phosphorylation of signal transducers and
STAT3, and the transcription factor NF-κB enters the nucleus from
the cytoplasm, regulating the expression of inflammatory cytokines,
causing endothelial cell dysfunction and macrophage polarization,
and promoting inflammation (114). IL-6/STAT3 is an important
pathway for mediating intracellular inflammatory signaling, which
can mediate the production of the proinflammatory cytokines, TNF-α
and IL-1, and the anti-inflammatory cytokines, IL-4 and IL-10.
Furthermore, the NF-κB/IL-6/STAT3 signaling pathway participates in
the regulation of ferroptosis through the inflammatory response
after cerebral ischemia and increases the expression of hepcidin
(115). Notably, red wine
polyphenol extract has been shown to efficiently suppress the
inflammation of intestinal epithelial cells by inhibiting JAK/STAT
and promoting Nrf2 pathways (116). Therefore, it may be hypothesized
that enhancing the activity of Nrf2 and inhibiting the JAK/STAT
signaling pathway, may reduce inflammation and monocyte
differentiation to macrophages, and regulate SLC7A11 to inhibit
ferroptosis. Artesunate is an antimalarial drug, and research has
indicated that it also has antitumor and anti-inflammatory effects;
it can inactivate the generation of pro-inflammatory mediators in
microglia by affecting the NF-κB, p38/MAPK and Nrf2/ARE-dependent
signaling pathways, thus inhibiting activation of the immune
response following cerebral ischemia (117).
The presence of ferroptosis will continuously damage
neural tissue for days to weeks; following ferroptosis,
danger-associated molecular patterns (DAMPs) trigger neutrophil
recruitment, neutrophil infiltration, proinflammatory cytokine
expression (118), leukocyte
death, and changes the immune status of the body (119). DAMPs associated with ferroptosis
include HMGB1 and IL-33 (120).
HMGB1 is released by ferroptotic cells and acts as an adjuvant to
activate the recognition receptor of the NF-κB pathway (20); moreover, it triggers an
inflammatory response in peripheral macrophages and exacerbates the
poor prognosis of ischemic stroke, including cerebral edema and the
risk of ischemia-reperfusion (121,122). Furthermore, it has been reported
that ferroptosis is the main cause of neuronal death after ischemic
stroke. Abnormal tau phosphorylation aggregation leads to neuronal
winding, which is involved in the mechanism of ischemic and
hemorrhagic stroke. Therefore, inhibiting tau protein expression
can inhibit the excitatory cytotoxicity of cells, promote iron
outflow to prevent the occurrence of ferroptosis and reduce damage
to nerve cells (123). Moreover,
the main role of tau protein in neurons is to promote the formation
of neuronal microtubule structures and play a key role in axonal
transport and cognitive function, but as mice grow older, tau has
double effects (124); the tau
protein is hyperphosphorylated and amyloid protein is formed,
causing nerve fiber damage and nerve fiber degeneration, thus
aggravating the neurotoxic iron accumulation.
Conclusion and future perspectives
The mechanisms of AS are complex and involve
multiple processes in numerous cell types. Crucially, with the
participation of foam cells and macrophages, ferroptosis drives the
progression of AS through oxidative stress and inflammatory
responses. The present review summarized that ferroptosis is
involved in the entire period of atherogenesis and progression
through numerous signaling pathways, including lipid pattern,
atherosclerotic plaque, fiber plaque and plaque rupture, while
interlinking with inflammation to exacerbate the poor prognosis of
AS-related diseases. During the development of AS, lipids are
deposited under the vascular endothelium forming fatty streak
plaques. With the progression of the disease, vascular endothelial
cells are damaged, ferroptosis and inflammation participate in the
atherosclerotic plaque stage, smooth muscle cells gradually
migrate, and the formation of fibrous caps on the surface of the
plate indicates the progression of the fibrous plaque stage. The
presence of ferroptosis and inflammation further damages
endothelial cells, rupture the plaque and forms a series of
ischemic events. Undoubtedly, advances in the study of
ferroptosis-associated mechanisms will change the traditional
concept of AS, and may improve the ability to manage AS risk and
address the inevitable risks that remain following current
interventions. In conclusion, ferroptosis serves a crucial role in
the pathogenesis of AS, ischemic stroke and coronary heart disease,
and with the exploration of clinical feasibility, the targeting of
ferroptosis may provide novel insights into the treatment of
vascular-related diseases.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Dalian High-level Talents
Innovation Support Program (grant no. 2018RQ54).
Availability of data and materials
Not applicable.
Authors' contributions
JL, LX contributed to the conception and design of
this study. JL prepared the tables and figures, and wrote the
manuscript. YXZ, XQC revised the manuscript critically and and
added relevant relevant literature. HTC was responsible for
revising the manuscript and given final approval of the version to
be published. All authors read and approved the final version of
the manuscript. Data authentication is not applicable..
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang H, Liu C, Zhao Y and Gao G:
Mitochondria regulation in ferroptosis. Eur J Cell Biol.
99:1510582020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qiu Y, Cao Y, Cao W, Jia Y and Lu N: The
application of ferroptosis in diseases. Pharmacol Res.
159:1049192020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lin L, Zhang MX, Zhang L, Zhang D, Li C
and Li YL: Autophagy, pyroptosis, and ferroptosis: New regulatory
mechanisms for atherosclerosis. Front Cell Dev Biol. 9:8099552022.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen X, Li X, Xu X, Li L, Liang N, Zhang
L, Lv J, Wu YC and Yin H: Ferroptosis and cardiovascular disease:
Role of free radical-induced lipid peroxidation. Free Radic Res.
55:405–415. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ross R: Atherosclerosis-an inflammatory
disease. N Engl J Med. 340:115–126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Barquera S, Pedroza-Tobias A, Medina C,
Hernández-Barrera L, Bibbins-Domingo K, Lozano R and Moran AE:
Global overview of the epidemiology of atherosclerotic
cardiovascular disease. Arch Med Res. 46:328–338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Herrington W, Lacey B, Sherliker P,
Armitage J and Lewington S: Epidemiology of atherosclerosis and the
potential to reduce the global Burden of atherothrombotic disease.
Circ Res. 118:535–546. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Y, Zhao X, Liu L, Soo YO, Pu Y, Pan
Y, Wang Y, Zou X, Leung TW, Cai Y, et al: Prevalence and outcomes
of symptomatic intracranial large artery stenoses and occlusions in
China: The Chinese Intracranial Atherosclerosis (CICAS) study.
Stroke. 45:663–669. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Falk E: Pathogenesis of atherosclerosis. J
Am Coll Cardiol. 47 (Suppl 8):C7–C12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Y, Zhao Y, Ye T, Yang L, Shen Y and
Li H: Ferroptosis signaling and regulators in atherosclerosis.
Front Cell Dev Biol. 9:8094572021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jiang C, Zhang J, Zhu J, Wang X, Wen Z,
Zhao X and Yuan C; CARE-II Investigators, : Association between
coexisting intracranial artery and extracranial carotid artery
atherosclerotic diseases and ipsilateral cerebral infarction: A
Chinese atherosclerosis risk evaluation (CARE-II) study. Stroke
Vasc Neurol. 6:595–602. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
GBD 2019 Stroke Collaborators, . Global,
regional, and national burden of stroke and its risk factors,
1990–2019: A systematic analysis for the Global Burden of disease
study 2019. Lancet Neurol. 20:795–820. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Saini V, Guada L and Yavagal DR: Global
epidemiology of stroke and access to acute ischemic stroke
interventions. Neurology. 97 (Suppl 2):S6–S16. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ornello R, Degan D, Tiseo C, Di Carmine C,
Perciballi L, Pistoia F, Carolei A and Sacco S: Distribution and
temporal trends from 1993 to 2015 of ischemic stroke subtypes: A
systematic review and meta-analysis. Stroke. 49:814–819. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gong L, Tian X, Zhou J, Dong Q, Tan Y, Lu
Y, Wu J, Zhao Y and Liu X: Iron dyshomeostasis induces binding of
APP to BACE1 for amyloid pathology, and impairs APP/Fpn1 complex in
microglia: Implication in pathogenesis of cerebral microbleeds.
Cell Transplant. 28:1009–1017. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang C: Essential functions of
iron-requiring proteins in DNA replication, repair and cell cycle
control. Protein Cell. 5:750–760. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pisano G, Lombardi R and Fracanzani AL:
Vascular damage in patients with nonalcoholic fatty liver disease:
Possible role of iron and ferritin. Int J Mol Sci. 17:6752016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Valenti L, Dongiovanni P, Motta BM,
Swinkels DW, Bonara P, Rametta R, Burdick L, Frugoni C, Fracanzani
AL and Fargion S: Serum hepcidin and macrophage iron correlate with
MCP-1 release and vascular damage in patients with metabolic
syndrome alterations. Arterioscler Thromb Vasc Biol. 31:683–690.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu J, Kuang F, Kroemer G, Klionsky DJ,
Kang R and Tang D: Autophagy-dependent ferroptosis: Machinery and
regulation. Cell Chem Biol. 27:420–435. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ma J, Qian C, Bao Y, Liu MY, Ma HM, Shen
MQ, Li W, Wang JJ, Bao YX, Liu Y, et al: Apolipoprotein E
deficiency induces a progressive increase in tissue iron contents
with age in mice. Redox Biol. 40:1018652021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yuan H, Pratte J and Giardina C:
Ferroptosis and its potential as a therapeutic target. Biochem
Pharmacol. 186:1144862021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fuhrmann DC, Mondorf A, Beifuß J, Jung M
and Brune B: Hypoxia inhibits ferritinophagy, increases
mitochondrial ferritin, and protects from ferroptosis. Redox Biol.
36:1016702020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shan X, Lv ZY, Yin MJ, Chen J, Wang J and
Wu QN: The protective effect of cyanidin-3-glucoside on myocardial
ischemia-reperfusion injury through ferroptosis. Oxid Med Cell
Longev. 2021:88801412021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Weinberg ED: The hazards of iron loading.
Metallomics. 2:732–740. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang F, Bao Q, Wang Z, Ma M, Shen J, Ye F
and Xie X: Sex-specific genetically predicted iron status in
relation to 12 vascular diseases: A mendelian randomization study
in the UK Biobank. Biomed Res Int. 2020:62460412020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ouyang S, You J, Zhi C, Li P, Lin X, Tan
X, Ma W, Li L and Xie W: Ferroptosis: The potential value target in
atherosclerosis. Cell Death Dis. 12:7822021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xiao Z, Kong B, Fang J, Qin T, Dai C,
Shuai W and Huang H: Ferrostatin-1 alleviates
lipopolysaccharide-induced cardiac dysfunction. Bioengineered.
12:9367–9376. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Naito Y, Masuyama T and Ishihara M: Iron
and cardiovascular diseases. J Cardiol. 77:160–165. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kajarabille N and Latunde-Dada GO:
Programmed cell-death by ferroptosis: Antioxidants as mitigators.
Int J Mol Sci. 20:49682019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ellulu MS, Patimah I, Khaza'ai H, Rahmat
A, Abed Y and Ali F: Atherosclerotic cardiovascular disease: A
review of initiators and protective factors. Inflammopharmacology.
24:1–10. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Su LJ, Zhang JH, Gomez H, Murugan R, Hong
X, Xu D, Jiang F and Peng ZY: Reactive oxygen species-induced lipid
peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med
Cell Longev. 2019:50808432019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gan B: Mitochondrial regulation of
ferroptosis. J Cell Biol. 220:e2021050432021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu J, Kang R and Tang D: Signaling
pathways and defense mechanisms of ferroptosis. FEBS J. Jun
6–2021.(Epub ahead of print). View Article : Google Scholar
|
|
35
|
Mishra SR, Mahapatra KK, Behera BP, Patra
S, Bhol CS, Panigrahi DP, Praharaj PP, Singh A, Patil S, Dhiman R
and Bhutia SK: Mitochondrial dysfunction as a driver of NLRP3
inflammasome activation and its modulation through mitophagy for
potential therapeutics. Int J Biochem Cell Biol. 136:1060132021.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jelinek A, Heyder L, Daude M, Plessner M,
Krippner S, Grosse R, Diederich WE and Culmsee C: Mitochondrial
rescue prevents glutathione peroxidase-dependent ferroptosis. Free
Radic Biol Med. 117:45–57. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhao Z, Wu J, Xu H, Zhou C, Han B, Zhu H,
Hu Z, Ma Z, Ming Z, Yao Y, et al: XJB-5-131 inhibited ferroptosis
in tubular epithelial cells after ischemia-reperfusion injury. Cell
Death Dis. 11:6292020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Friedmann Angeli JP, Schneider M, Proneth
B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch
A, Eggenhofer E, et al: Inactivation of the ferroptosis regulator
Gpx4 triggers acute renal failure in mice. Nat Cell Biol.
16:1180–1191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ursini F and Maiorino M: Lipid
peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic
Biol Med. 152:175–185. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li N, Jiang W, Wang W, Xiong R, Wu X and
Geng Q: Ferroptosis and its emerging roles in cardiovascular
diseases. Pharmacol Res. 166:1054662021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tuo QZ, Liu Y, Xiang Z, Yan HF, Zou T, Shu
Y, Ding XL, Zou JJ, Xu S, Tang F, et al: Thrombin induces
ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion.
Signal Transduct Target Ther. 7:592022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Oh BM, Lee SJ, Park GL, Hwang YS, Lim J,
Park ES, Lee KH, Kim BY, Kwon YT, Cho HJ and Lee HG: Erastin
inhibits septic shock and inflammatory gene expression via
suppression of the NF-kappaB pathway. J Clin Med. 8:22102019.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chu B, Kon N, Chen D, Li T, Liu T, Jiang
L, Song S, Tavana O and Gu W: ALOX12 is required for p53-mediated
tumour suppression through a distinct ferroptosis pathway. Nat Cell
Biol. 21:579–591. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xiao FJ, Zhang D, Wu Y, Jia QH, Zhang L,
Li YX, Yang YF, Wang H, Wu CT and Wang LS: miRNA-17-92 protects
endothelial cells from erastin-induced ferroptosis through
targeting the A20-ACSL4 axis. Biochem Biophys Res Commun.
515:448–454. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Doll S, Proneth B, Tyurina YY, Panzilius
E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A,
et al: ACSL4 dictates ferroptosis sensitivity by shaping cellular
lipid composition. Nat Chem Biol. 13:91–98. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cui Y, Zhang Y, Zhao X, Shao L, Liu G, Sun
C, Xu R and Zhang Z: ACSL4 exacerbates ischemic stroke by promoting
ferroptosis-induced brain injury and neuroinflammation. Brain Behav
Immun. 93:312–321. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fan Z, Cai L, Wang S, Wang J and Chen B:
Baicalin prevents myocardial ischemia/reperfusion injury through
inhibiting ACSL4 mediated ferroptosis. Front Pharmacol.
12:6289882021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Noguchi N: Ebselen, a useful tool for
understanding cellular redox biology and a promising drug candidate
for use in human diseases. Arch Biochem Biophys. 595:109–112. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chu D and Zhang Z: Trichosanthis
pericarpium aqueous extract protects H9c2 cardiomyocytes from
Hypoxia/Reoxygenation injury by regulating PI3K/Akt/NO pathway.
Molecules. 23:24092018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen X, Yu C, Kang R, Kroemer G and Tang
D: Cellular degradation systems in ferroptosis. Cell Death Differ.
28:1135–1148. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Guan X, Li X, Yang X, Yan J, Shi P, Ba L,
Cao Y and Wang P: The neuroprotective effects of carvacrol on
ischemia/reperfusioninduced hippocampal neuronal impairment by
ferroptosis mitigation. Life Sci. 235:1167952019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhang Y, Lu X, Tai B, Li W and Li T:
Ferroptosis and its multifaceted roles in cerebral stroke. Front
Cell Neurosci. 15:6153722021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wei X, Yi X, Zhu XH and Jiang DS:
Posttranslational modifications in ferroptosis. Oxid Med Cell
Longev. 2020:88320432020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W and
Wang J: Molecular mechanisms of ferroptosis and its role in cancer
therapy. J Cell Mol Med. 23:4900–4912. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Alim I, Caulfield JT, Chen Y, Swarup V,
Geschwind DH, Ivanova E, Seravalli J, Ai Y, Sansing LH, Ste Marie
EJ, et al: Selenium drives a transcriptional adaptive program to
block ferroptosis and treat stroke. Cell. 177:1262–1279. e252019.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ratan RR: The chemical biology of
ferroptosis in the central nervous system. Cell Chem Biol.
27:479–498. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Song X and Long D: Nrf2 and Ferroptosis: A
new research direction for neurodegenerative diseases. Front
Neurosci. 14:2672020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yuan Y, Zhai Y, Chen J, Xu X and Wang H:
Kaempferol ameliorates oxygen-glucose
deprivation/reoxygenation-induced neuronal ferroptosis by
activating Nrf2/SLC7A11/GPX4 axis. Biomolecules. 11:9232021.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Anandhan A, Dodson M, Schmidlin CJ, Liu P
and Zhang DD: Breakdown of an ironclad defense system: the critical
role of NRF2 in mediating ferroptosis. Cell Chem Biol. 27:436–447.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M,
Buchfelder M and Savaskan N: Nrf2-Keap1 pathway promotes cell
proliferation and diminishes ferroptosis. Oncogenesis. 6:e3712017.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ren JX, Li C, Yan XL, Qu Y, Yang Y and Guo
ZN: Crosstalk between oxidative stress and ferroptosis/oxytosis in
ischemic stroke: Possible targets and molecular mechanisms. Oxid
Med Cell Longev. 2021:66433822021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Dodson M, Castro-Portuguez R and Zhang DD:
NRF2 plays a critical role in mitigating lipid peroxidation and
ferroptosis. Redox Biol. 23:1011072019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Dong H, Qiang Z, Chai D, Peng J, Xia Y, Hu
R and Jiang H: Nrf2 inhibits ferroptosis and protects against acute
lung injury due to intestinal ischemia reperfusion via regulating
SLC7A11 and HO-1. Aging (Albany NY). 12:12943–12959. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Colak G and Johnson GV: Complete
transglutaminase 2 ablation results in reduced stroke volumes and
astrocytes that exhibit increased survival in response to ischemia.
Neurobiol Dis. 45:1042–1050. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yang Y, Wang Y, Guo L, Gao W, Tang TL and
Yan M: Interaction between macrophages and ferroptosis. Cell Death
Dis. 13:3552022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Marques VB, Leal MAS, Mageski JGA, Fidelis
HG, Nogueira BV, Vasquez EC, Meyrelles SDS, Simões MR and Dos
Santos L: Chronic iron overload intensifies atherosclerosis in
apolipoprotein E deficient mice: Role of oxidative stress and
endothelial dysfunction. Life Sci. 233:1167022019. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Bosseboeuf E and Raimondi C: Signalling,
metabolic pathways and iron homeostasis in endothelial cells in
health, atherosclerosis and Alzheimer's disease. Cells. 9:20552020.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wen Q, Liu J, Kang R, Zhou B and Tang D:
The release and activity of HMGB1 in ferroptosis. Biochem Biophys
Res Commun. 510:278–283. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Xiao L, Luo G, Guo X, Jiang C, Zeng H,
Zhou F, Li Y, Yu J and Yao P: Macrophage iron retention aggravates
atherosclerosis: Evidence for the role of autocrine formation of
hepcidin in plaque macrophages. Biochim Biophys Acta Mol Cell Biol
Lipids. 1865:1585312020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Luo Y, Duan H, Qian Y, Feng L, Wu Z, Wang
F, Feng J, Yang D, Qin Z and Yan X: Macrophagic CD146 promotes foam
cell formation and retention during atherosclerosis. Cell Res.
27:352–372. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhu Y, Xian X, Wang Z, Bi Y, Chen Q, Han
X, Tang D and Chen R: Research progress on the relationship between
atherosclerosis and inflammation. Biomolecules. 8:802018.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Gao Z, Xu X, Li Y, Sun K, Yang M, Zhang Q,
Wang S, Lin Y, Lou L, Wu A, et al: Mechanistic Insight into PPARү
and Tregs in Atherosclerotic Immune Inflammation. Front Pharmacol.
12:7500782021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Gistera A and Hansson GK: The immunology
of atherosclerosis. Nat Rev Nephrol. 13:368–380. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Lee GR: The balance of Th17 versus treg
cells in autoimmunity. Int J Mol Sci. 19:7302018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Meng X, Yang J, Dong M, Zhang K, Tu E, Gao
Q, Chen W, Zhang C and Zhang Y: Regulatory T cells in
cardiovascular diseases. Nat Rev Cardiol. 13:167–179. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Libby P: The changing landscape of
atherosclerosis. Nature. 592:524–533. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Bentzon JF, Otsuka F, Virmani R and Falk
E: Mechanisms of plaque formation and rupture. Circ Res.
114:1852–1866. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Kobayashi M, Suhara T, Baba Y, Kawasaki
NK, Higa JK and Matsui T: Pathological roles of iron in
cardiovascular disease. Curr Drug Targets. 19:1068–1076. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Martinet W, Coornaert I, Puylaert P and De
Meyer GRY: Macrophage death as a pharmacological target in
atherosclerosis. Front Pharmacol. 10:3062019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Chen S, Dong Z, Cheng M, Zhao Y, Wang M,
Sai N, Wang X, Liu H, Huang G and Zhang X: Homocysteine exaggerates
microglia activation and neuroinflammation through microglia
localized STAT3 overactivation following ischemic stroke. J
Neuroinflammation. 14:1872017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zhang T, Jiang Y, Zhang S, Tie T, Cheng Y,
Su X, Man Z, Hou J, Sun L, Tian M, et al: The association between
homocysteine and ischemic stroke subtypes in Chinese: A
meta-analysis. Medicine (Baltimore). 99:e194672020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Kumar A, Palfrey HA, Pathak R, Kadowitz
PJ, Gettys TW and Murthy SN: The metabolism and significance of
homocysteine in nutrition and health. Nutr Metab (Lond). 14:782017.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Zhou W, Cheng Y, Zhu P, Nasser MI, Zhang X
and Zhao M: Implication of gut microbiota in cardiovascular
diseases. Oxid Med Cell Longev. 2020:53940962020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Jonsson AL and Backhed F: Role of gut
microbiota in atherosclerosis. Nat Rev Cardiol. 14:79–87. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Chapkin RS, Navarro SL, Hullar MAJ and
Lampe JW: Diet and gut microbes act coordinately to enhance
programmed cell death and reduce colorectal cancer risk. Dig Dis
Sci. 65:840–851. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Hayase E and Jenq RR: Too much TMAO and
GVHD. Blood. 136:383–385. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Janeiro MH, Ramirez MJ, Milagro FI,
Martinez JA and Solas M: Implication of trimethylamine N-Oxide
(TMAO) in disease: Potential biomarker or new therapeutic target.
Nutrients. 10:13982018. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Lassiger-Herfurth A, Pontarollo G, Grill A
and Reinhardt C: The gut microbiota in cardiovascular disease and
arterial thrombosis. Microorganisms. 7:6912019. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Tuttolomondo A, Puleo MG, Velardo MC,
Corpora F, Daidone M and Pinto A: Molecular biology of
atherosclerotic ischemic strokes. Int J Mol Sci. 21:93722020.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Cornelissen A, Guo L, Sakamoto A, Virmani
R and Finn AV: New insights into the role of iron in inflammation
and atherosclerosis. EBioMedicine. 47:598–606. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Tabas I and Bornfeldt KE: Macrophage
phenotype and function in different stages of atherosclerosis. Circ
Res. 118:653–667. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chen X, Kang R, Kroemer G and Tang D:
Ferroptosis in infection, inflammation, and immunity. J Exp Med.
218:e202105182021. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wolf D and Ley K: Immunity and
inflammation in atherosclerosis. Circ Res. 124:315–327. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Jeney V, Balla G and Balla J: Red blood
cell, hemoglobin and heme in the progression of atherosclerosis.
Front Physiol. 5:3792014. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Raman SV, Winner MW III, Tran T,
Velayutham M, Simonetti OP, Baker PB, Olesik J, McCarthy B,
Ferketich AK and Zweier JL: In vivo atherosclerotic plaque
characterization using magnetic susceptibility distinguishes
symptom-producing plaques. JACC Cardiovasc Imaging. 1:49–57. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Hu H, Chen Y, Jing L, Zhai C and Shen L:
The link between ferroptosis and cardiovascular diseases: A novel
target for treatment. Front Cardiovasc Med. 8:7109632021.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Vinchi F, Porto G, Simmelbauer A, Altamura
S, Passos ST, Garbowski M, Silva AMN, Spaich S, Seide SE, Sparla R,
et al: Atherosclerosis is aggravated by iron overload and
ameliorated by dietary and pharmacological iron restriction. Eur
Heart J. 41:2681–2695. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Neven E, De Schutter TM, Behets GJ, Gupta
A and D'Haese PC: Iron and vascular calcification. Is there a link?
Nephrol Dial Transplant. 26:1137–1145. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Kempf T and Wollert KC: Iron and
atherosclerosis: Too much of a good thing can be bad. Eur Heart J.
41:2696–2698. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Le Y, Zhang Z, Wang C and Lu D:
Ferroptotic cell death: New regulatory mechanisms for metabolic
diseases. Endocr Metab Immune Disord Drug Targets. 21:785–800.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
He L, Liu YY, Wang K, Li C, Zhang W, Li
ZZ, Huang XZ and Xiong Y: Tanshinone IIA protects human coronary
artery endothelial cells from ferroptosis by activating the NRF2
pathway. Biochem Biophys Res Commun. 575:1–7. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Stadler N, Lindner RA and Davies MJ:
Direct detection and quantification of transition metal ions in
human atherosclerotic plaques: Evidence for the presence of
elevated levels of iron and copper. Arterioscler Thromb Vasc Biol.
24:949–954. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Bai T, Li M, Liu Y, Qiao Z and Wang Z:
Inhibition of ferroptosis alleviates atherosclerosis through
attenuating lipid peroxidation and endothelial dysfunction in mouse
aortic endothelial cell. Free Radic Biol Med. 160:92–102. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Chen Z, Yan Y, Qi C, Liu J, Li L and Wang
J: The role of ferroptosis in cardiovascular disease and its
therapeutic significance. Front Cardiovasc Med. 8:7332292021.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Huang F, Yang R, Xiao Z, Xie Y, Lin X, Zhu
P, Zhou P, Lu J and Zheng S: Targeting ferroptosis to treat
cardiovascular diseases: A new continent to be explored. Front Cell
Dev Biol. 9:7379712021. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Zhao J, Wu Y, Liang S and Piao X:
Activation of SSAT1/ALOX15 axis aggravates cerebral
ischemia/reperfusion injury via triggering neuronal ferroptosis.
Neuroscience. 485:78–90. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Selim M: Treatment with the iron chelator,
deferoxamine mesylate, alters serum markers of oxidative stress in
stroke patients. Transl Stroke Res. 1:35–39. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Xie BS, Wang YQ, Lin Y, Mao Q, Feng JF,
Gao GY and Jiang JY: Inhibition of ferroptosis attenuates tissue
damage and improves long-term outcomes after traumatic brain injury
in mice. CNS Neurosci Ther. 25:465–475. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Millan M, DeGregorio-Rocasolano N, Perez
de la Ossa N, Reverté S, Costa J, Giner P, Silva Y, Sobrino T,
Rodríguez-Yáñez M, Nombela F, et al: Targeting pro-oxidant iron
with deferoxamine as a treatment for ischemic stroke: Safety and
optimal dose selection in a randomized clinical trial. Antioxidants
(Basel). 10:12702021. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Li W, Xiang Z, Xing Y, Li S and Shi S:
Mitochondria bridge HIF signaling and ferroptosis blockage in acute
kidney injury. Cell Death Dis. 13:3082022. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Bai YT, Xiao FJ, Wang H, Ge RL and Wang
LS: Hypoxia protects H9c2 cells against Ferroptosis through
SENP1-mediated protein DeSUMOylation. Int J Med Sci. 18:1618–1627.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Liu H, Wu X, Luo J, Wang X, Guo H, Feng D,
Zhao L, Bai H, Song M, Liu X, et al: Pterostilbene attenuates
astrocytic inflammation and neuronal oxidative injury after
ischemia-reperfusion by inhibiting NF-kB phosphorylation. Front
Immunol. 10:24082019. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Yang L, Wang H, Yang X, Wu Q, An P, Jin X,
Liu W, Huang X, Li Y, Yan S, et al: Auranofin mitigates systemic
iron overload and induces ferroptosis via distinct mechanisms.
Signal Transduct Target Ther. 5:1382020. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Erta M, Quintana A and Hidalgo J:
Interleukin-6, a major cytokine in the central nervous system. Int
J Biol Sci. 8:1254–1266. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Nunes C, Teixeira N, Serra D, Freitas V,
Almeida L and Laranjinha J: Red wine polyphenol extract efficiently
protects intestinal epithelial cells from inflammation via opposite
modulation of JAK/STAT and Nrf2 pathways. Toxicol Res (Camb).
5:53–65. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Zuo S, Li Q, Liu X, Feng H and Chen Y: The
potential therapeutic effects of artesunate on stroke and other
central nervous system diseases. Biomed Res Int. 2016:14890502016.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Khoshnam SE, Winlow W, Farzaneh M, Farbood
Y and Moghaddam HF: Pathogenic mechanisms following ischemic
stroke. Neurol Sci. 38:1167–1186. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Iadecola C, Buckwalter MS and Anrather J:
Immune responses to stroke: Mechanisms, modulation, and therapeutic
potential. J Clin Invest. 130:2777–2788. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Zhang Y, Xin L, Xiang M, Shang C, Wang Y,
Wang Y, Cui X and Lu Y: The molecular mechanisms of ferroptosis and
its role in cardiovascular disease. Biomed Pharmacother.
145:1124232022. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Yu Y, Yan Y, Niu F, Wang Y, Chen X, Su G,
Liu Y, Zhao X, Qian L, Liu P and Xiong Y: Ferroptosis: A cell death
connecting oxidative stress, inflammation and cardiovascular
diseases. Cell Death Discov. 7:1932021. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Yan HF, Tuo QZ, Yin QZ and Lei P: The
pathological role of ferroptosis in ischemia/reperfusion-related
injury. Zool Res. 41:220–230. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Derry PJ, Hegde ML, Jackson GR, Kayed R,
Tour JM, Tsai AL and Kent TA: Revisiting the intersection of
amyloid, pathologically modified tau and iron in Alzheimer's
disease from a ferroptosis perspective. Prog Neurobiol.
184:1017162020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Tuo QZ, Lei P, Jackman KA, Li XL, Xiong H,
Li XL, Liuyang ZY, Roisman L, Zhang ST, Ayton S, et al:
Tau-mediated iron export prevents ferroptotic damage after ischemic
stroke. Mol Psychiatry. 22:1520–1530. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Lu J, Xu F and Lu H: LncRNA PVT1 regulates
ferroptosis through miR-214-mediated TFR1 and p53. Life Sci.
260:1183052020. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Li C, Sun G, Chen B, Xu L, Ye Y, He J, Bao
Z, Zhao P, Miao Z, Zhao L, et al: Nuclear receptor coactivator
4-mediated ferritinophagy contributes to cerebral ischemia-induced
ferroptosis in ischemic stroke. Pharmacol Res. 174:1059332021.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Chen J, Yang L, Geng L, He J, Chen L, Sun
Q, Zhao J and Wang X: Inhibition of Acyl-CoA synthetase long-chain
family member 4 facilitates neurological recovery after stroke by
regulation ferroptosis. Front Cell Neurosci. 15:6323542021.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Lu H, Wang B, Cui N and Zhang Y:
Artesunate suppresses oxidative and inflammatory processes by
activating Nrf2 and ROSdependent p38 MAPK and protects against
cerebral ischemia-reperfusion injury. Mol Med Rep. 17:6639–6646.
2018.PubMed/NCBI
|
|
129
|
Liu Z, Lv X, Song E and Song Y: Fostered
Nrf2 expression antagonizes iron overload and glutathione depletion
to promote resistance of neuron-like cells to ferroptosis. Toxicol
Appl Pharmacol. 407:1152412020. View Article : Google Scholar : PubMed/NCBI
|