Introduction
Ocular angiogenesis is a complex pathophysiological
process, which is coordinated by a series of intricate and precise
molecular mechanisms. Pathological ocular angiogenesis, including
retinal, choroidal and corneal neovascularization (CoNV), is a
major cause of blindness globally (1). Amongst the most metabolically active
tissues in the body, the retina and choroid consume high levels of
oxygen and nutrients (2). The
principal function of the retinal vasculature is to metabolically
sustain the inner retina, while the outer retina is supplied by the
choroidal vasculature (3). The
innermost layer of all these ocular vessels is lined by endothelial
cells (ECs), which are metabolically active and simultaneously
maintain vascular homeostasis and systemic metabolism (4); they also play differential roles,
depending on their location. ECs differentiate into three distinct
subtypes, tip cells, stalk cells and phalanx cells, to permit their
adaptation to changes in supply and demand (5). Following induction by a large
variety of stimuli, including injury, infection and hypoxia,
disruptions in the functions of vascular ECs may lead to a range of
ocular vascular diseases due to abnormal angiogenesis, including
diabetic retinopathy (DR), retinopathy of prematurity (ROP) and
neovascular age-related macular degeneration (nAMD) (2). Anti-angiogenesis therapy, which
targets vascular endothelial growth factor (VEGF), has become the
primary therapy for the inhibition of pathological ocular
neovascularization. This therapy is effective for the majority of
patients. However, its use shows some limitations, which often
become prominent gradually. These include drug resistance, partly
due to the redundancy afforded by other angiogenic signals
(6), and non-compliance due to
the frequency of injections required. Thus, novel therapies are
still needed.
Metabolism is a key feature required for ECs to
survive, migrate, proliferate, and grow; thus, it is important for
the ocular vasculature (7).
Recently, metabolic pathways, including glucose metabolism, fatty
acid oxidation and amino acid metabolism, have been identified to
be crucial for angiogenesis in health and disease (4,8).
Glucose metabolism consists of two complementary pathways, glucose
anabolism and glucose catabolism, which are kept in balance.
Glucose catabolism is further divided into anaerobic glycolysis,
aerobic oxidation, and the pentose phosphate pathway (PPP). There
is increasing evidence to suggest that glucose metabolism controls
EC proliferation, migration and neovascularization (9,10).
EC activities primarily rely on glucose metabolism, particularly
glycolysis, as the source of energy (11,12). It is estimated that only 0.04% of
glucose is oxidized, while 98% of glucose is metabolized to lactate
in in rat coronary microvascular ECs (13). In human umbilical vein ECs,
compared with glutamine oxidation and fatty acid oxidation,
glycolytic flux is ~200-fold higher (12). There are three types of ECs: tip,
stalk, and phalanx cells. The phalanx cell, keeping a quiescent
state, maintains vascular integrity and inhibits inflammation. The
forkhead box O1 (FOXO1) protein induced-decrease in glycolysis can
keep ECs in a quiescent state and limit the overgrowth of vessels
(14). Concurrently, ECs store
glucose as glycogen in their intracellular reserves (15). When angiogenesis occurs, phalanx
cells activate and transform into tip/stalk cells, which migrate
and proliferate to form new blood vessels. This process is highly
dependent on glycolysis to provide energy. VEGF and FGF can enhance
glycolysis by increasing
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) and
hexokinase (HK)-2 to promote the activation of ECs (12,16). Hyperglycemia, a hallmark of
diabetes, inhibits glucose phosphorylation and contributes to
apoptosis of ocular microvascular ECs, causing diabetic retinopathy
through the suppression of HK2 expression (17,18). Therefore, the perturbation of EC
glycolytic metabolism results in EC dysfunction and vascular
pathologies (10). As 85% of the
energy required for ECs to function is obtained from glycolysis,
glycolysis may be a potential target for the management of
pathological retinal angiogenesis. Accordingly, the elucidation of
the underlying metabolic perturbations that occur is crucial for
the identification of EC metabolism-centric therapeutics (10).
Here, the processes involved in ocular angiogenesis
and the role of cell glucose metabolism are summarized. A deeper
understanding of the connection between glucose metabolism and
retinal angiogenesis may assist in the development of future
therapies and in prevention strategies.
Glucose metabolism
Glucose metabolism primarily consists of anaerobic
glycolysis and aerobic oxidation, and serves three important aims:
Energy generation, biosynthesis and metabolite production. Glucose
is first metabolized to generate pyruvate; pyruvate has two
different fates, depending on the availability of oxygen, namely
anaerobic glycolysis and aerobic oxidation, the latter coinciding
also with oxidative phosphorylation. The common factor between both
is that they use glucose for the production and sustenance of
sufficient supply of energy (19).
Glycolysis
Among the pathways of glucose metabolism present in
a cell, the most prevalent and important one is glycolysis
(Fig. 1). A glycolysis reaction,
literally the lysis of glucose, requires the conversion of glucose
into pyruvate and then into lactate as a waste product (20). The retina receives glucose and
oxygen from the afferent blood (via hemoglobin), where glucose
penetrates the cell membrane and enters cells by glucose
transporters, especially glucose transporter 1 (GLUT1) (4). The moment glucose enters the cells,
it is phosphorylated to glucose-6-phosphate (G6P) by HK, which is
the first rate-limiting step. G6P then converts to
fructose-6-phosphate (F6P). The conversion of F6P to
fructose-1,6-bisphosphate (F1,6P2) by 6-phosphofructo-1-kinase is
the second rate-limiting checkpoint of the glycolytic pathway.
Prior to this step, G6P under specific conditions can enter the PPP
or can be converted to glucose-1-phosphate, resulting in the
initiation of gluconeogenesis (10). Subsequently, F1,6P2 undergoes a
series of enzymatic reactions to produce phosphoenolpyruvate (PEP)
(12). Pyruvate kinase (PK), the
third rate-limiting enzyme, catalyzes the dephosphorylation of PEP
to produce pyruvate. In the absence of oxygen, pyruvate is reduced
to lactate, which is exported from the cell (20).
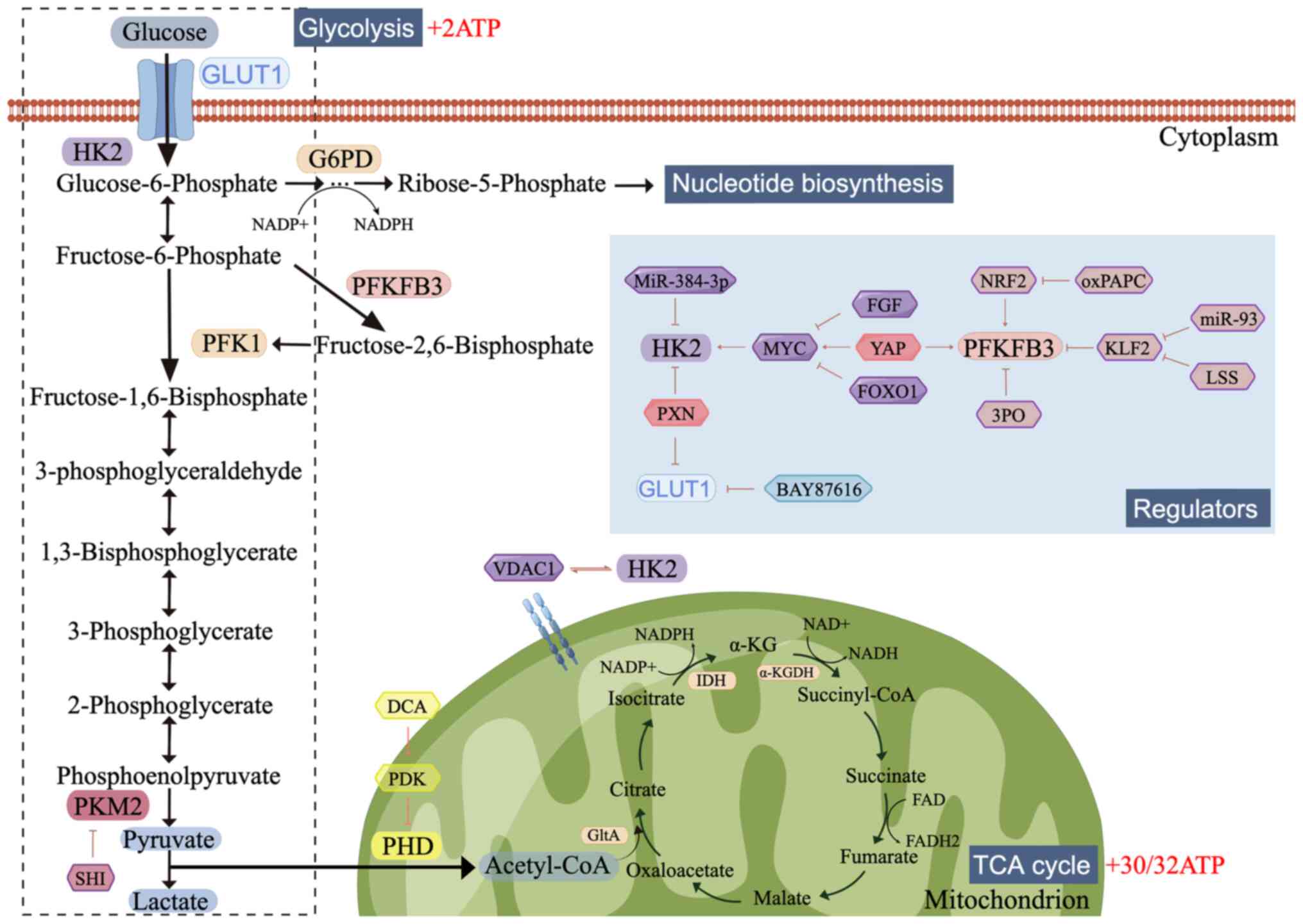 | Figure 1.Glucose metabolism and the regulators
involved. Glucose is first metabolized to generate pyruvate;
pyruvate is then reduced to lactate or enters the TCA cycle,
depending on the availability of oxygen. Several regulators,
including growth factors, transcription factors and miRNAs, mediate
glucose uptake and flux. The figure was created using Figdraw
(www.figdraw.com). TCA, tricarboxylic acid;
GLUT1, glucose transporter 1; HK2, hexokinase 2; G6PD, glucose
6-phosphate dehydrogenase; PFKFB3,
phosphofructokinase-2/fructose-2,6-bisphosphatase-3; PFK1,
phosphofructokinase-1; PKM2, Pyruvate kinase-2; PHD, prolyl
hydroxylase domain; Acetyl-CoA, acetyl coenzyme A; VDAC1, Voltage
Dependent Anion Channel 1; GltA, citrate synthase; IDH, isocitrate
dehydrogenase; α-KGDH, α-ketoglutarate dehydrogenase; SHI,
shikonin; PDK, pyruvate dehydrogenase kinase; DCA, Dichloroacetic
acid; PXN, Paxillin; FGF, fibroblast growth factor; YAP,
Yes-associated protein; FOXO1, Forkhead box O1; NRF2, Nuclear
factor E2-related factor; 3PO,
3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one; oxPAPC,
1-palmitoyl-2-arachidonoyl-sn-glycero-3phosphocholine; KLF2,
Krüppel-like factor 2; LSS, laminar shear stress. |
In cancer, the presence of a specific mode of
glycolysis occurs, which is termed aerobic glycolysis and
eponymously known as the ‘Warburg effect’. Glycolysis can occur in
the presence of oxygen to meet the increased energetic and
biosynthetic demands (20). The
Warburg effect is a hallmark of cancer and refers to a metabolic
shift in cancer cells. In addition, it has been demonstrated that
the mammalian retina also displays cancer-like metabolism (21).
Glucose oxidation
Under aerobic conditions, pyruvate can be delivered
into a mitochondrion where it is converted to acetyl coenzyme A
(acetyl-CoA), which enters the tricarboxylic acid (TCA) cycle,
ultimately resulting in the production of ATP via oxidation
reactions (Fig. 1). The TCA cycle
is an ubiquitous metabolic chain in all aerobic organisms. In the
first step, the pyruvate dehydrogenase complex catalyzes pyruvate
to form acetyl-CoA. Subsequently, acetyl-CoA is further condensed
with oxaloacetate to form citrate by citrate synthase. Citrate is
converted to isocitrate by isocitrate dehydrogenase, which is
further converted to α-ketoglutarate (α-KG) through oxidative
decarboxylation. The α-ketoglutarate dehydrogenase complex
catalyzes the conversion of succinyl-CoA from α-KG, and then
succinyl-CoA is converted to succinate. Finally, succinate, through
fumarate and malate, is reconverted into oxaloacetate.
Additionally, during the biochemical reactions involved in the TCA
cycle, NADH and FADH2 are generated as byproducts, which feed into
the process of oxidative phosphorylation for ATP generation.
Energy
Glucose metabolism plays a critical role in energy
generation, and it is the most efficient form of cellular energy
generation. The entire glycolytic reaction produces 2 single
molecules of ATP (22); however,
the complete oxidation of glucose can produce approximately 30/32
total ATP molecules (23).
Following the completion of the ocular vasculature development, the
majority of vascular ECs are quiescent. Under pathological
conditions, including hypoxia, infection and injury, the quiescent
ECs are potently activated to produce new vessels (24), and this process requires an
adequate supply of energy. Surprisingly, although glucose oxidation
produces significantly more ATP than glycolysis, the energy
consumed by ECs is primarily derived from glycolysis, accounting
for 85% of the energy supply, even when the oxygen supply is
sufficient (12). This phenomenon
may be explained by the observation that the mammalian retina
exhibits a cancer-like metabolism known as aerobic glycolysis
(25); thus, the dependence on
glycolysis is not surprising. At first glance, glycolysis is a
low-efficiency form of energy production; however, the rate of
production is faster (12).
Glycolysis is essential in allowing hypoxic tissues to restore
blood supply in a timely manner. In addition, when the ECs migrate
to avascular areas where the conditions are relatively hypoxic, a
shortage of oxygen supply makes glycolysis the only route of energy
production for ECs (26).
Additionally, the mitochondrial content in ECs is relatively
reduced (27) and glycolysis can
result in the avoidance of oxidative damage in ECs, by reducing the
production of reactive oxygen species (ROS) levels (28).
Carbon for nucleotide
biosynthesis
In addition to energy generation, glucose metabolism
has another significant function, being involved in biosynthesis
directly. For example, intermediates of glucose metabolism can act
as precursors for the de novo synthesis of nucleotides.
Glycolytic intermediates can enter the PPP, also referred to as the
phosphogluconate pathway and the hexose monophosphate shunt, to
provide carbons for nucleotide biosynthesis (29). In the PPP, G6P is reduced to
ribose-5-phosphate (R5P) by the rate-limiting enzymes glucose
6-phosphate dehydrogenase (G6PDH), 6-phosphogluconolactone (6PGL),
and 6-phosphogluconate dehydrogenase (6PGDH), concurrently
consuming NADP+ to generate NADPH, which is used for ROS scavenging
(30). R5P then serves as the
starting material in nucleotide biosynthesis. Thus, inhibition of
G6PDH compromises nucleotide biosynthesis and vessel sprouting
(31). In summary, glucose
metabolism is essential for nucleotide biosynthesis. The key
enzymes involved in these pathways also represent promising
anti-neovascularization therapeutic targets.
Epigenetics and glucose
metabolism
In addition to energy generation and biosynthesis,
glucose metabolism is required for epigenetic modifications and
gene regulation by providing metabolites that act as substrates or
co-factors for several important enzymatic reactions (32). Epigenetics refers to the
capability of the same genome to produce multiple distinct, yet
stable, phenotypes through chemical modifications of chromatin,
without alterations to the original DNA sequence (33,34). Epigenetics play crucial roles in
regulating gene expression and governing cellular phenotypes and
consist of histone modifications, DNA methylation and RNA-mediated
processes. In cancer, epigenetics and metabolism are highly
interconnected in an interdependent manner. Variations of the
expression of acetyl-CoA controlled by glycolysis greatly affect
the histone acetyltransferase-mediated histone acetylation
(35). Furthermore, high lactate
levels caused by glycolysis result in the generation of a local
acidic pH that promotes histone deacetylation (36). Promoter hypomethylation in turn
upregulates HK2 and facilitates glycolytic flux in glioblastoma and
hepatic carcinoma (37).
Collectively, glucose metabolism and epigenetics affects each other
to co-regulate a range of physiological and pathological activities
including angiogenesis.
Glucose metabolism in physiological ocular
angiogenesis
Physiological angiogenesis
The formation of the vasculature is primarily
mediated by two mechanisms: Vasculogenesis and angiogenesis
(3,38). Vasculogenesis refers to the
process of de novo vessel formation from undifferentiated
precursor cells during early embryonic development. Angiogenesis, a
complex process of new vessel formation from existing vessels, is
the predominant mode of retinal vessel growth (39). The mature retina consists of 10
layers from the inner limiting membrane to the retinal pigment
epithelium (RPE) and is supplied with dual blood supplies by both
the retinal and choroidal vasculatures that supply the inner and
outer layers, respectively (40).
During embryonic and early fetal development, oxygen and nutrients
are delivered to the retina by hyaloid vessels (41). In humans, hyaloid vasculature
formation, regression, and the majority of retinal vasculature
development occur before birth. In mice, the retinal vasculature
develops postnatally (3).
Hyaloid vasculature
The hyaloid vasculature arises from the central
hyaloid artery (HA), which enters from the optic fissure and
extends into the primitive vitreous at around 5 weeks gestation
(WG) (42). Subsequently, the HA
runs through the primitive vitreous to the posterior lens surface,
forming a dense capillary network, known as the tunica vasculosa
lentis (TVL), forming the vasculosa hyaloidea propria in a more
proximal position of the vitreous. The TVL expands further around
the anterior part of the lens forming the pupillary membrane and
eventually drains into the choroidal veins (43). The hyaloid vasculature is
characterized by the absence of veins; thus, all hyaloid vessels
are arteries and the venous drain is accomplished by the choroidal
vessels (43). The hyaloid
vessels exhibit clear evidence of regression at ~13-15 WG and
culminate in the involution of the entire hyaloid by 35–36 WG
(44).
Retinal vasculature
The retinal vasculature is an ocular circulatory
system that consists of the primary plexus, secondary plexus and
deeper plexus. The primary plexus of the retinal vasculature
emerges from an established capillary ring at the optic disc at ~15
WG and subsequently spreads across the inner surface of the retina
to the ora serrata nasally at ~36 WG, finally reaching the ora
serrata temporally at ~40 WG (38,39) (Fig.
2A). Consistent with the extension of the primary plexus, the
deeper plexus originates from the primary plexus veins at the optic
nerve head to the periphery in a manner of angiogenesis sprouting,
at ~25 WG (3,38). The development of a deeper plexus
continues after birth until the retinal vasculature is terminally
matured. Notably, during the entire development process, there is a
completely avascular region known as the fovea (38,45).
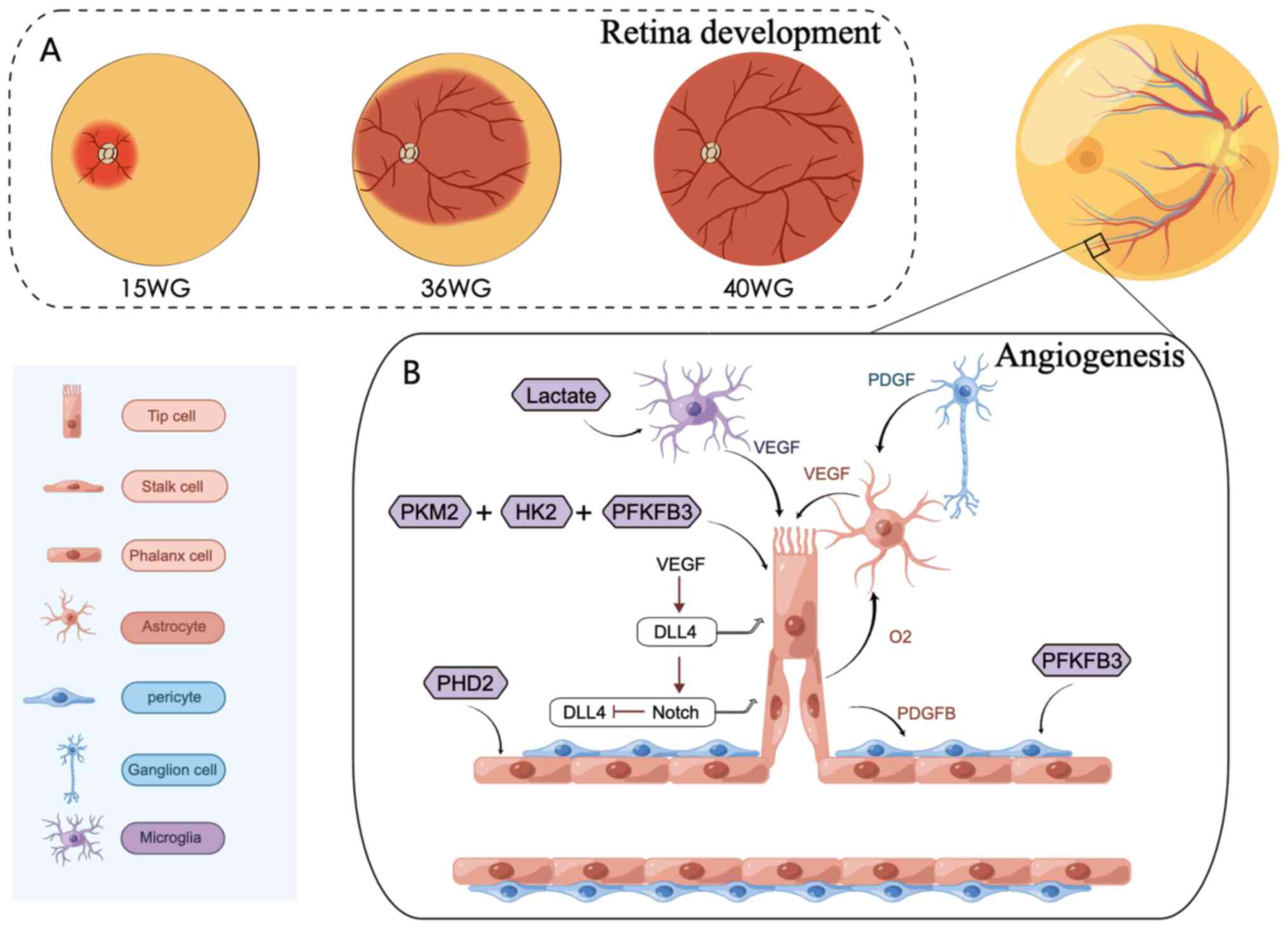 | Figure 2.The process of retinal angiogenesis.
(A) The primary plexus of the retinal vasculature emerges from an
established capillary ring at the optic disc at ~15 WG and
subsequently spreads across the inner surface of the retina to the
ora serrata nasally, at ~36 WG, finally reaching the ora serrata
temporally at ~40 WG. (B) Endothelial cells differentiate into
three subtypes: Tip cells, stalk cells and phalanx cells, depending
on their location, and interact with angiogenesis-related cells to
promote retinal vasculature development. This process is regulated
by metabolic enzymes. The figure was created using Figdraw
(www.figdraw.com). WG, week gestation; PKM2,
pyruvate kinase-2; HK2, hexokinase 2; PFKFB3,
phosphofructokinase-2/fructose-2,6-bisphosphatase-3; PHD, prolyl
hydroxylase domain; VEGF, vascular endothelial growth factor; PDGF,
platelet derived growth factor receptor; DLL4, Delta like 4. |
Retinal vasculature development is closely
associated with a multitude of cells, including ECs, astrocytes,
microglia and pericytes. Astrocytes develop from astrocyte
progenitor cells (APCs) and are present only within the
vascularized area; thus, the macular hole and the avascular region
of the peripheral retina lack astrocytes (3,38).
Under platelet-derived growth factor receptor (PDGF) stimulation,
which is secreted by retinal ganglion cells (Fig. 2B), APCs invade the retina through
the optic disc, then expand further across the nerve fiber layer
toward the peripheral margins of the retina forming a meshwork.
APCs invade prior to retinal vasculature development; however, APCs
do not differentiate or mature at this point. Along with the
transmigration of ECs into the retina, APCs are stimulated to
differentiate into mature astrocytes. Differentiated astrocytes,
located primarily at the leading edge of the invading ECs, and can
sense hypoxia signals and secrete VEGF, which mediates the
migration and proliferation of ECs. With the extension of vessels,
a sufficient oxygen supply in turn also contributes to astrocyte
maturation (38). In response,
astrocytes decrease VEGF synthesis and limit further growth of
retinal vessels by local feedback mechanisms (46).
ECs line the inner surface of vessels to support
tissue growth and repair. The growth of the retinal vasculature
depends on the sprout formation of ECs similar to plant
germination, which requires ECs to display different phenotypes
based on their location. ECs compete with each other for the
leading tip position (47)
(Fig. 2B). A specialized EC at
the distal end of each sprout extend long filopodial protrusions,
termed as the tip cell. Stalk cells form behind tip cells in
endothelial sprouts (47,48). Tip cells are nonproliferative and
can migrate, lead, and guide the vessel sprouts, while the
extension and perfusion of vessels mainly rely on stalk cells and
further vascular remodeling (48). The tip/stalk cell phenotype can be
regulated by the balance between a variety of angiogenic factors
and their downstream signaling pathways, such as VEGF/Notch
signaling (49). VEGF expression
is higher in the peripheral retina and this promotes the
transcription of the notch ligand Delta like 4 (DLL4) in tip cells,
concurrently inhibiting DLL4 transcription in adjacent cells
(50,51). As a result, the adjacent cells
finally differentiate into stalk cells. Tip cells tightly attach to
astrocytes and radially extend to the avascular area via various
long, dynamic actin-based filopodia along the astrocyte mesh
template under the stimulation of VEGF, guiding the direction of
growth (3). Following tip cells,
stalk cells extend fewer filopodia but proliferate to support
sprout elongation and form the vessel lumen. Adjacent sprouts fuse
to establish vessel loops (52).
The sprouting process iterates and creates a primitive plexus,
which is later remodeled into structured, hierarchical vascular
trees. There is a specialized population of ECs, the
cobblestone-like appearance of quiescent ECs, termed phalanx cells
(53). Under hypoxic conditions,
oxygen sensors become inactive, then phalanx cells can express
oxygen sensors to regulate vessel perfusion, mediated by prolyl
hydroxylase domain proteins (52). At the initiation of angiogenesis,
quiescent ECs are rapidly activated and switch to a proliferative
state, ultimately enhancing glycolysis (52). During sprouting, tip cells migrate
to avascular areas, where oxygen is absent and VEGF mRNA expression
is higher. In this condition, glycolysis provides adequate energy
for rapid vessel growth and mature. In turn, the new vessels can
provide adequate oxygen (12). In
response to adequate oxygen levels, the expression of VEGF
decreases, ECs stops migrating and switches from an activation
state (tip cells) into a quiescent state (phalanx cells). Thus, the
migration and proliferation of activated ECs relies on glycolysis.
However, with the establishment of vessels, ECs will again
transform into a quiescent state and reduce the glycolytic rate
(26).
Pericytes are specialized mural cells located at the
abluminal surface of capillary blood vessels (54) and together with ECs play a major
role in angiogenesis, participating in vessel formation,
remodeling, and stabilization (55). During angiogenesis, the
EC-specific ligand PDGFB can bind with high affinity to PDGF
receptor B (PDGFRb) secreted by pericytes, inducing the recruitment
and attachment of pericytes (Fig.
2B) (54). Studies have
revealed that the inactivation of PDGF-B/PDGFRb signaling results
in reduced retinal pericyte coverage, leading to endothelial
hyperplasia, abnormal vascular morphogenesis, and the formation of
microaneurysms (52,56,57). Compared to glucose oxidation,
glutamine oxidation and fatty acid oxidation, glycolysis also
provides up to 85% of the ATP required by pericyte (58). Under high glucose levels, Notch3
gene downregulation in pericytes reduces PDGFR levels, resulting in
disorders of endothelial-pericyte interactions, finally leading to
pericyte apoptosis (59). In
cancer, the treatment of pericytes with
3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO), an inhibitor
of PFKFB3, has been shown to reduce glycolysis, whereas adherence
to ECs is enhanced, which may be explained by the fact that 3PO
upregulates N-cadherin levels in pericytes (58). Overall, appropriate glycolysis may
facilitate the maintenance of pericytes in a quiescent state and
their adherence to ECs. However, whether this phenomenon exists in
the retina remains to be further confirmed.
Macrophage/microglia originate from retinal myeloid
cells, and possess high glycolytic characteristics similar to ECs
(11). Microglia are resident
macrophages in the central nervous system (60). Several unique stimulatory factors,
such as lactate, have been demonstrated to alter macrophage
metabolism (61) and induce
macrophage differentiation into an M2 phenotype, which in-turn
enhances retinal angiogenesis (11) (Fig.
2B). For example, proangiogenic cytokines, including VEGF,
released from macrophages, also facilitate EC glycolysis, resulting
in retinal neovascularization (RNV) (11). It also has been reported that the
deletion of macrophages by mannosylated clodronate liposomes
suppressed pathological angiogenesis and facilitated physiological
angiogenesis (62).
Choroidal vasculature
The development of the choroidal vasculature
precedes that of the retinal vasculature, and occurs via a
mechanisms termed hemo-vasculogenesis, which is distinguished from
vasculogenesis and angiogenesis (63). Hemo-vasculogenesis refers to the
blood vessels, blood cells having a common precursor, the
hemangioblast. The choroidal vasculature is divided into three
layers, namely the anterior choriocapillaris (CC), Sattler's layer
of intermediate vessels, and the outermost Haller's layer (63). The choroidal vasculature is
accountable for nourishing the outer retinal layers, particularly
photoreceptors and RPE. The CC initiates the formation of a single
layer, originating from islands of progenitors by
hemo-vasculogenesis during 6–8 WG (64). At 6–7 WG, erythroblasts can be
observed in the CC layer and distributed in the forming choroidal
stroma (63). By 8.5 WG, the
vascular lumens become apparent (64). At 11–12 WG, the deeper choroidal
vasculature can be observed in the posterior pole and the
equatorial choroid follows closely. Simultaneously, certain ECs
proliferate and sprout from the scleral side of CC, indicating that
the formation of intermediate vessels is mediated by angiogenesis,
apparently promoting the development of intermediate vessels and
the confluence of capillaries and large vessels (64). These developing vessels in the
central choroid are more mature, as pericyte-like cells are also
present in that position (65).
At 14–16 WG, cells surrounded by pericytes contact with ECs lining
the lumen via peg-in-socket-like contacts, a feature of the normal
adult microvasculature (65). By
21 WG in the posterior pole region, three layers of blood vessels
of CC have manifested and further expand and remodel after 22 WG
(63).
Key regulators of glucose metabolism
in physiological angiogenesis
In recent years, given the highly plastic phenotype
of ECs in retinal angiogenesis, the understanding of the regulators
controlling the glucose metabolism of ECs has substantially
increased, including the involvement of metabolic enzymes and
transcription factors. Targeting these regulators may hold
significant potential in maintaining vascular homeostasis. Crucial
elements regulating glucose metabolism are outlined below (Fig. 1):
HK2
HK2 is an isoform of the HK modulating the first
rate-limiting step in glycolysis, as described above. As previously
demonstrated, HK2 knockdown significantly reduced, while HK2
overexpression increased, glycolysis and retinal angiogenesis
(16). High glucose reduced HK2
levels and interactions with voltage-dependent anion channel, a
protein enriched in the outer mitochondrial membrane, resulting in
apoptosis of human umbilical vein ECs (HUVECs) by impairing
mitochondrial permeability (18).
Fibroblast growth factor (FGF) is an essential
growth factor that binds to the FGF receptor (FGFR) to activate
various signaling pathways (66).
FGFR1 and FGFR3 belong to the FGFR family and are expressed in mice
and humans. FGFR3 levels in human dermal lymphatic ECs were
previously demonstrated to be upregulated by FGFR1 knockdown;
however, FGFR3 knockdown did not affect FGFR1 levels (16). FGFR1 knockdown resulted in the
inhibition of HUVEC proliferation, decreased tip cell numbers, and
impaired retinal vascular branching and growth, whereas FGFR3
knockdown had no effect (16).
Further investigations indicated that FGF or FGFR1 knockdown
decreased HK2 expression (16).
Mechanistically, the link between FGF and HK2 involves MYC
regulation, which can coordinate cellular proliferation and
metabolism (16,67). FGF thus controls
glycolysis-mediated retinal angiogenesis through MYC-dependent
regulation of HK2 expression.
PFKFB3
PFKFB, a member of the bifunctional enzyme family,
can facilitate the synthesis of fructose-2,6-bisphosphate
(F2,6BP2), which is an allosteric activator of
phosphofructokinase-1 (68).
Among all the PFKFB isozymes, PFKFB3 has the highest kinase
activity (69). In a previous
study, the knockdown of PFKFB3 in vitro or the genetic
silencing of PFKFB3 in developing retina decreased F2,6BP2 levels,
reduced glycolytic flux, and decreased vessel sprouting and
vascular plexus expansion under physiological conditions in mice.
Conversely, PFKFB3 overexpression exerted the opposite effect
(12). Mechanistically, PFKFB3
inhibition induced these defects partly due to impaired EC
proliferation, but more prominently through the reduced
competitiveness of tip cells, and abrogated tge interactions
between PFKFB3 and actin (12).
1-Palmitoyl-2-arachidonoyl-sn-glycero-3phosphocholine (PAPC) is a
complex phospholipid enriched in cell membranes and is easily
susceptible to oxidation to oxPAPC, which can stimulate EC
sprouting and proliferation (70). Nuclear factor E2-related factor
(NRF2), a transcription factor, has been reported to be a mediator
of the oxPAPC response (71).
miR-93 is the most abundant miRNA in ECs; EC proliferation and
glycolysis were previously observed to be significantly induced
following miR-93 overexpression (72). The overexpression of oxPAPC led to
similar results as those for miR-93. Additionally, the effects of
miR-93 and oxPAPC overexpression on glycolysis and proliferation
could be reversed by the silencing of NRF2. Together, NRF2
regulates endothelial glycolysis and proliferation via miR-93 and
oxPAPC (71).
Shear stress, a force that blood flow exerts on the
ECs, is broadly categorized into disturbed or laminar shear stress
(LSS) (73,74). LSS plays a crucial role in the
activation of ECs from a quiescent state (73). In a previous study, when HUVECs
were exposed to LSS for 72 h, the glycolysis and mitochondrial
activity of the ECs was observed to be reduced. The silencing of
Krüppel-like factor 2 (KLF2), a transcription factor that
stabilizes the quiescent EC phenotype, reversed this alteration
(9). Subsequently, RNA sequencing
revealed that PFKFB3 was downregulated, following the
overexpression of KLF2. Thus, the simultaneous overexpression of
KLF2 and PFKFB3 revealed that the reduction of PFKFB3 was the cause
of KLF2-mediated inhibition of glycolysis (9). Overall, LSS inhibited EC glycolysis
contributing to the repression of PFKFB3 mediated by KLF2; however,
whether this occurs in vivo also remains to be established
(9). Taken together, multiple
means of inhibition of PFKFB3 reduce vessel formation under
physiological conditions.
PKM2
PKM2 is an isoform of PK, which is the final
catalytic step involved in glycolysis. As previously demonstrated,
PKM2 silencing inhibited glycolysis, generated fewer and shorter
sprouts with few filopodia in ECs in vitro, and diminished
radial vascular growth, although it had no effect on vascular
density in vivo (75).
High-resolution confocal microscopy revealed that PKM2 was enriched
at the VE-cadherin-mediated endothelial junctions and accumulated
at F-actin-rich filopodia and lamellipodia of tip cells (75). The lower number of junctions and
impaired endothelial barrier were detected in PKM2-deficient ECs.
In another study, ECs treated with shikonin (SHI), a
pharmacological inhibitor of PKM2, also exhibited a reduced
migratory ability; however, their proliferation levels remained
unaltered (75). Taken together,
whereas PKM2 is pivotal in retinal angiogenesis BY regulating EC
migration, the number of filopodia and VE-cadherin-mediated EC
junctions, it is not required for EC proliferation. However,
another study procured the opposite results, demonstrating that
PKM2 knockout suppressed EC proliferation via the upregulation of
p53, a transcription factor blocking cell cycle progression, a
process that is independent of the activity of PK (76). In cancer, PKM2 has been
demonstrated to interfere both with NF-κB/p65 and HIF-1α
activation, which ultimately triggers VEGF-A secretion and
subsequent blood vessel formation (77).
GLUT1
GLUT1 is one of the primary transporters of glucose
and is required for endothelial glycolysis. It has been revealed
that GLUT1 inhibition by BAY87616, a highly selective GLUT1
inhibitor, reduces glucose transport and glycolysis in human
retinal microvascular ECs and HUVEC. Of note, GLUT1 inhibition
attenuates EC proliferation and sprouting angiogenesis, but not
migration or viability (78).
Surprisingly, the number of tip cells was not significantly altered
under GLUT1 starvation, despite the fact that tip cells are highly
dependent on glycolysis. Overall, GLUT1 can regulate retinal
angiogenesis via glycolysis-mediated EC proliferation, but not via
migration (78). The Notch and
VEGF signaling pathways have been revealed to modulate the
expression of GLUT1 in ECs (12,78,79).
Other mediators
FOXO, a transcription factor, is a downstream
effector of the PIK3/AKT pathway that connects vascular growth with
metabolism (74,80). FOXO1 is a member of the FOXO
family and is highly enriched in ECs, according to the
immunofluorescence analysis data of a previous study (14). In that study, the loss of FOXO1 in
ECs resulted in abnormal retinal angiogenesis, and the ECs became
dense and hyperplastic, resulting in the inability of tip cells to
correctly sprout (14). The
overactivation of FOXO1 in ECs led to a hyper-pruned vasculature
network and a thinner lumen in retinal vessels (14). Thus, FOXO1 is a suppressor of EC
proliferation and retinal angiogenesis. As evidenced by a reduction
in glucose uptake, glycolytic flux and lactate production, FOXO1
activation was shown to lead to a robust reduction in glycolysis.
Moreover, transcriptional analysis revealed that MYC, a potent
driver of glycolysis, was downregulated under the same conditions.
Taken together, FOXO1 inhibited retinal angiogenesis via the
decreased glycolysis capacity mediated by MYC (14).
Glucose metabolism in pathological ocular
angiogenesis
Abnormal vessel growth, insufficient vessel growth,
or uncontrolled vessel growth, promotes ocular disease and poses a
threat to normal vision. As a consequence of dysregulated
angiogenesis, oxygen and nutrients are not correctly delivered,
leading to an imbalance in metabolic demand and supply and
disturbed neural retinal function (2). The pathological process of
angiogenesis is associated with several diseases, including ROP, DR
and age-related macular degeneration (AMD), amongst others.
Retinal neovascularization
ROP and DR
In humans, the majority of retinal vessels complete
development before birth, whereas normal retinal angiogenesis is
arrested in the preterm infant. Consequently, pathological
compensatory mechanisms are excessively triggered, and this is
hypothesized to finally result in the aberrant vascularization of
the retina, known as RNV (81).
This process is known as ROP. ROP has two postnatal phases: An
initial phase of vessel loss, followed by a second phase of vessel
over-proliferation (81,82). In the first stage, the relative
hyperoxic environment post-birth compared with that in utero
suppresses the expression of oxygen-regulated angiogenic growth
factors in astrocytes, Müller cells, pericytes and the RPE through
HIF-1α, leading to EC apoptosis, the cessation of retinal vessel
growth and the regression of existing vessels (2,83).
At this stage, only partial vascularization has a chance to be
observed in the developing retina. In the second stage, given the
increase in metabolic activity, dysplastic retinal vessels fail to
provide an adequate amount of oxygen and nutrients, resulting in
the pathological proliferation of vessels in response to VEGF
upregulation (81). HIF-1α is
also upregulated to initiate transcription of the genes as a
response to hypoxia, including VEGF (84). Ultimately, the retinal leakage and
detachment contribute to impaired vision.
DR, one of the most common complications of
diabetes, remains a leading cause of visual impairment and
blindness (85). Hyperglycemia
and other metabolic dysregulations lead to microvascular damage and
retinal function disorder (86).
At the onset of DR, the vessel wall is compromised due to the loss
of supporting pericytes and/or glial attachment leading to
capillary wall dilatation (microaneurysms), leakage (edema and hard
exudates), and rupture (hemorrhages) (86). As the severity of DR progresses,
capillary occlusion leads to retinal ischemia, which, in turn,
induces the upregulation of VEGF, driving pathological
neovascularization. Severe retinopathy may end with macular edema
and retinal detachment.
As described above, the neovascularization of ROP
and DR are both attributed to the severe imbalance in blood and
oxygen between demand and supply. The disruption of angiogenesis
reduces the oxygen supply, thus resulting in the upregulation of
pro-angiogenic factors, which can act directly on ECs to stimulate
excessive retinal angiogenesis. The mouse model of oxygen-induced
retinopathy (OIR) is a well-recognized model for RNV (87). As previously demonstrated, mice,
from postnatal day 7 (P7), were exposed to a hyperoxic environment
(where the concentration of oxygen was maintained at 75%) for 5
days (until P12), after which mice were subsequently maintained
under normal conditions (21% oxygen). RNV peaked at P17 (87). As described above, this process
involves initial vessel loss (P7-P12), neovascularization
(P12-P17), and neovascular regression (P17-P25) (87).
Key regulators of glucose metabolism
in RNV
miRNAs are small non-coding RNAs involved in almost
all biological processes, playing critical roles in cell
proliferation, growth, apoptosis and vascular neovascularization
(88). miR-384-3p has been
confirmed to inhibit the proliferation of human retinal
microvascular ECs (HRMECs) via the downregulation of HK2 and the
inhibition of RNV in DR (89).
Moreover, the genetic loss of GLUT1 results in a significant
reduction in glycolysis, EC proliferation and the branch point
density of retinal vessels in developing postnatal mice (78).
ECs treated with 3PO or
7,8-dihydroxy-3-(4-hydroxyphenyl)-chromen-4-one, small molecule
inhibitors of PFKFB3, present with dose-dependently reduced
glycolysis in ECs and the formation of RNV in mice with OIR
(69). However, 2-deoxyglucose
(2-DG), another glycolytic blocker, induced EC disintegration and
eventual death, and this may be attributed to the near-complete
inhibition of glycolysis achieved by 2-DG (69). In tumors, 3PO treatment had no
effect on tumor growth and cancer cell proliferation. However,
decreased glycolysis in pericytes, resulting in the higher coverage
of pericytes, which promoted tumor vessel normalization (58). It remains to be determined whether
3PO normalizes the previously formed pathological vessels in the
retina; however, it was previously demonstrated that in adult
healthy mice treated with 3PO for 15 days, there was no effect on
the healthy retinal vasculature system, and the perfusion of
retinal vessels (90).
Yes-associated protein (YAP) is a critical
downstream effector of the Hippo signaling pathway and functions as
a transcription cofactor that binds the TEA domain transcription
factor (TEAD) to initiate the expression of target genes (91,92). There is increasing evidence to
indicate that YAP is associated with both physiological and
pathological angiogenesis (93).
The YAP-TEAD1 complex has been demonstrated to activate the
transcription of PFKFB3 via binding to the PFKFB3 promoter, and it
has also been shown that blocking the YAP/PFKFB3 axis significantly
inhibits hypoxia-induced glycolysis by decreasing the secretion of
VEGFA and VEGFR1 (94). Based on
this observation, the role of the YAP/PFKFB3 axis in retinal
angiogenesis was further explored and it was found that the
inhibition of either YAP or PFKFB3 could restrict the biological
function of ECs and suppress RNV (94). Conversely, as demonstrated in
another study, the suppression of YAP transcription also reduced
glycolysis, damaged filopodia protrusion and contributed to
impaired retinal angiogenesis, and these effects were dependent on
the downregulation of MYC, another potent driver of glycolysis
(93).
Adenosine/adenosine receptor-mediated signaling has
been implicated in ischemic diseases and is regulated by hypoxia.
Ischemic proliferative retinopathy-induced hypoxia involves an
increase in adenosine and adenosine receptor levels (95). Peak adenosine levels are
temporally related to active vasculogenesis in the retina in the
model of OIR (96). The
expression of adenosine A2a receptor (Adora2a), an adenosine
receptor, was shown to increase markedly via a HIF-2α-dependent
mechanism in HRMECs and in mice with OIR. Activated Adora2a,
subsequently promoted glycolysis and retinal angiogenesis through
HIF-1α accumulation. The deletion of Adora2a reversed the increase
in the number and length of sprouts induced by the pharmacological
inhibition of Notch signaling (97). Collectively, as critical metabolic
regulators in ECs, miR-384-3p, HK2, PFKFB3, GLUT1, YAP and Adora2a
may be potential targets in the management of RNV.
Choroidal neovascularization (CNV)
AMD
AMD is also one of the leading causes of vision
loss. AMD can be classified into early and late stages. nAMD is the
advanced stage of AMD and is accompanied by pathological
angiogenesis; new blood vessels from pre-existing choroidal vessels
intrude through Brunch's membrane (BM) into the RPE or sub-retinal
space (98). This destructive
process is termed CNV and is referred to as subretinal
neovascularization (98). The
junction of the choroid and the retina is comprised of the RPE, BM,
and choroidal capillaries (63).
When certain triggers upregulate VEGF expression in RPE cells and
the BM is disrupted by proteases, choroidal vessel growth becomes
disorderly and extrudes into sub-retinal space, resulting in CNV
(98). Given that the choroidal
vasculature has not YET been adequately studied, the pathogenesis
of nAMD is not well defined and it is currently hypothesized to be
multifactorial (99).
Laser-induced CNV is the most efficient in vivo model
available to study the mechanisms of CNV. In mice with experimental
CNV, laser injury caused by the rupture of the BM has been shown to
result in cell damage and hypoxia, culminating in neovascular
lesions occurring on day 5 and reaching their peak on day 7
(100).
Key regulators of glucose metabolism
in CNV
The levels of lactic acid, the end-product of
glycolysis, can induce reprogramming in several cells and are
considered a marker of an underlying pathologies, including types
of cancer (101). Lactic acid
levels have been shown to be notably increased in the serum of mice
with laser-induced CNV on day 5 (102). Dichloroacetic acid (DCA) can
suppress pyruvate dehydrogenase kinase (PDK), which is a modulator
of lactate levels, by inactivating the pyruvate dehydrogenase
complex involved in the pyruvate conversion into acetyl-CoA. As
previously demonstrated, treatment with DCA significantly inhibited
CNV, with the optimal inhibition observed during the
late-angiogenic period (days 4–7) (103). Moreover, lactate has extra
functions in maintaining the M1/M2 macrophage balance in favor of
M2 macrophages by promoting the transformation M1 macrophages to M2
macrophages (103). Another
study further demonstrated that VEGFA mRNA and VEGFA protein
expression levels were only elevated in lactic acid-treated
macrophages. Thus, high VEGF levels enhanced neovascularization.
Finally, inhibiting lactate acid transport by the intravitreal
injection of α-cyanohydroxycinnamic acid, a monocarboxylate
transport blocker, downregulated VEGF levels and the subsequent CNV
(101). Vallée et al
(104) revealed that enhanced
WNT/β-catenin pathway activity stimulated the PI3K/Akt pathway and
HIF-α, which activated glycolytic enzymes (Glut, HK, PDK and
LDH-A). This process resulted in aerobic glycolysis, representing
the accumulation of lactate that initiates the expression of VEGF
to promote angiogenesis in nAMD. A previous study also demonstrated
that pyruvate, lactate levels and the lactate/pyruvate ratio were
increased in urine samples collected from patients with typical
AMD, indicating that glycolysis may be involved in the aggravation
of AMD (105).
In clinical samples, higher levels of the
intermediates of the TCA cycle were similarly detected in patients
with active nAMD. In a previous study, using ultrahigh-performance
liquid chromatography-tandem mass spectrometry, the detection of
energy metabolites in the aqueous humor of with AMD revealed that
citrate and isocitrate levels were significantly increased in the
AMD group, whereas succinate and α-ketoglutarate levels were
significantly decreased compared with the control group (106). Low α-ketoglutarate levels
contribute to the stabilization of HIF-1α and the secretion of
VEGF-A, which further promotes progression to CNV (107). Thus, the dysregulation of the
TCA cycle may also be a driving force in nAMD. However, the
underlying mechanisms require further investigation (106).
Another study also revealed that the deregulation
of glucose metabolism was associated with CNV (108). Following feeding with a
high-fructose/high-fat (HFHF) diet, rats began to exhibit fasting
hyperglycemia, glucose intolerance and insulin resistance,
indicative of an impaired glucose metabolism, which promoted CNV
compared to rats fed a standard diet. Simultaneously, using laser
photocoagulation to trigger CNV and feeding with an HFHF diet
induced the exacerbation of CNV. This result indicated that an HFHF
diet promotes a favorable environment for neovascular events.
Moreover, an HFHF diet increased the expression of glial fibrillary
acidic protein, an activator of glial cells. Sustained injury
induced by lasers in the BM also lead to the activation of glial
cells and an HFHF diet further promoted this activation to
propagate to the rest site of the retina (108).
3PO has been demonstrated to reduce the CNV lesion
volume in laser-injured mice by inhibiting PFKFB3 (69). SHI oral gavage has been confirmed
to alleviate the leakage, area and volume of mouse laser-induced
CNV lesions, and inhibit macrophage infiltration without any
evidence of ocular cytotoxicity through inhibiting PKM2 (109). Taken together, these findings
highlight a potential strategy for the management of CNV.
CoNV
The cornea is optically clear with the absence of
blood vessels, which is required for corneal transparency and the
maintenance of vision. The corneal epithelium combined with
protective factors forms a barrier that protects the cornea from
injuries and corneal infiltration by blood vessels. By contrast,
CoNV results from the invasion of blood vessels from the limbal
vascular plexus into the cornea in multiple pathological states,
including hypoxia, inflammation, infection, injury and degeneration
(110).
For example, inflammation-related injury disrupts
the homeostasis between proangiogenic cytokines and antiangiogenic
factors (111). Along with the
accumulation of proangiogenic stimuli, including VEGF, the vascular
ECs (VECs) surrounding the limbal vasculature begin to activate and
release proteolytic enzymes that degrade the BM of vessels and the
corneal extracellular matrix, ultimately permitting VECs to invade
the corneal stroma. The invaded VECs further proliferate, migrate,
sprout and grow, resulting in neovascularization in the cornea
(110). Metabolism plays a
critical role in the transportation of oxygen through the cornea.
High lactate levels were observed in the hypoxic cornea, resulting
in an increase in stromal lactate concentration (110).
Paxillin (PXN) is a vital component of focal
adhesion. The deletion of PXN reduces the levels of HK2 and GLUT1,
resulting in a decrease in lactate and ATP levels in HUVECs or the
VEGF-treated cornea, protecting against VEGF-induced EC invasion
and angiogenesis (112). In
addition, as previously demonstrated, rats with
streptozotocin-induced diabetes presented with a reduced degree of
alkali injury-induced CoNV compared with the control rats, although
the difference was not significant. This may be related to the
effect of high glucose on wound healing and the dissimilar to
circulatory high-glucose stimulation responses in CoNV development,
amongst different species (113).
To date, the relevance between CoNV and glucose
metabolism has not been intensively investigated. Considering the
strong link between glucose metabolism and ECs, however, the
regulators of glucose metabolism mentioned above may have potential
roles on CoNV, and further research is required in order to fully
elucidate this.
Conclusions and future perspectives
Over the past several decades, ocular vascular
development has been thoroughly studied at the molecular level;
however, studies at the metabolic level are lacking to a certain
extent. Glucose metabolism, particularly glycolysis, is a key
factor involved in ocular angiogenesis, both under physiological
and pathophysiological conditions. There are several factors,
including metabolic enzymes, transcription factors and epigenetics
involved in the regulation of glycolysis on angiogenesis.
Pre-pathological ocular angiogenesis can be suppressed from the
source, if the safe inhibition of EC glycolysis can be achieved
clinically. To date, the treatment of RNV by targeting VEGF
provides an effective therapy in clinical practice. However,
anti-VEGF therapy is only suitable for advanced ocular vascular
diseases. Therefore, the control of disease progression by directly
regulating endothelial metabolism is one of the most efficient
approaches during the early stages of ocular vascular diseases.
Thus, the process of ocular angiogenesis and the central
involvement of glucose metabolism were summarized in the present
review. Although an increasing number of studies have illustrated
the therapeutic potential of targeting glucose metabolism,
additional research is required to fully utilize this therapeutic
approach clinically.
Acknowledgement
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 82171081), the Shanghai Science and
Technology committee (grant no. 22ZR1478200), the Shanghai Pujiang
Program (grant no. 21PD068), the Shanghai Hospital Development
Center (grant nos. SHDC2020CR1043B and SHDC2020CR5014), the 234
Mountain Climbing Plan of Changhai Hospital (grant no. 2020YXK058),
the training program of the basic medical foundation of Changhai
Hospital (grant no. JC202117), and the Sailing program of the Naval
Medical University.
Availability of data and materials
Not applicable.
Authors' contributions
HS, WS and QL reviewed and edited the manuscript.
XG, HZ, WZ and RZ wrote part of the manuscript and prepared the
figures. All authors have read and approved the final manuscript.
Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Adamis AP, Aiello LP and D'Amato RA:
Angiogenesis and ophthalmic disease. Angiogenesis. 3:9–14. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sun Y and Smith LEH: Retinal vasculature
in development and diseases. Annu Rev Vis Sci. 4:101–122. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Selvam S, Kumar T and Fruttiger M: Retinal
vasculature development in health and disease. Prog Retin Eye Res.
63:1–19. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Theodorou K and Boon RA: Endothelial cell
metabolism in atherosclerosis. Front Cell Dev Biol. 6:822018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Geudens I and Gerhardt H: Coordinating
cell behaviour during blood vessel formation. Development.
138:4569–4583. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ebos JM and Kerbel RS: Antiangiogenic
therapy: Impact on invasion, disease progression, and metastasis.
Nat Rev Clin Oncol. 8:210–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li X and Carmeliet P: Targeting angiogenic
metabolism in disease. Science. 359:1335–1336. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Du W, Ren L, Hamblin MH and Fan Y:
Endothelial cell glucose metabolism and angiogenesis. Biomedicines.
9:1472021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Doddaballapur A, Michalik KM, Manavski Y,
Lucas T, Houtkooper RH, You X, Chen W, Zeiher AM, Potente M,
Dimmeler S and Boon RA: Laminar shear stress inhibits endothelial
cell metabolism via KLF2-mediated repression of PFKFB3.
Arterioscler Thromb Vasc Biol. 35:137–145. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eelen G, de Zeeuw P, Simons M and
Carmeliet P: Endothelial cell metabolism in normal and diseased
vasculature. Circ Res. 116:1231–1244. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu Z, Xu J, Ma Q, Zhang X, Yang Q, Wang
L, Cao Y, Xu Z, Tawfik A, Sun Y, et al: Glycolysis links reciprocal
activation of myeloid cells and endothelial cells in the retinal
angiogenic niche. Sci Transl Med. 12:eaay13712020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
De Bock K, Georgiadou M, Schoors S,
Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquière B,
Cauwenberghs S, Eelen G, et al: Role of PFKFB3-driven glycolysis in
vessel sprouting. Cell. 154:651–663. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Krützfeldt A: Metabolism of exogenous
substrates by coronary endothelial cells in culture. Journal of
Molecular and Cellular Cardiology. 22:1393–1404. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wilhelm K, Happel K, Eelen G, Schoors S,
Oellerich MF, Lim R, Zimmermann B, Aspalter IM, Franco CA, Boettger
T, et al: FOXO1 couples metabolic activity and growth state in the
vascular endothelium. Nature. 529:216–220. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vizan P, Sanchez-Tena S, Alcarraz-Vizan G,
Soler M, Messeguer R, Pujol MD, Lee WN and Cascante M:
Characterization of the metabolic changes underlying growth factor
angiogenic activation: Identification of new potential therapeutic
targets. Carcinogenesis. 30:946–952. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu P, Wilhelm K, Dubrac A, Tung JK, Alves
TC, Fang JS, Xie Y, Zhu J, Chen Z, De Smet F, et al: FGF-dependent
metabolic control of vascular development. Nature. 545:224–228.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eelen G, de Zeeuw P, Treps L, Harjes U,
Wong BW and Carmeliet P: Endothelial Cell Metabolism. Physiol Rev.
98:3–58. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang J, Guo Y, Ge W, Zhou X and Pan M:
High glucose induces apoptosis of HUVECs in a
mitochondria-dependent manner by suppressing hexokinase 2
expression. Exp Ther Med. 18:621–629. 2019.PubMed/NCBI
|
|
19
|
Bouche C, Serdy S, Kahn CR and Goldfine
AB: The cellular fate of glucose and its relevance in type 2
diabetes. Endocr Rev. 25:807–830. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gatenby RA and Gillies RJ: Why do cancers
have high aerobic glycolysis? Nat Rev Cancer. 4:891–899. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Agathocleous M, Love NK, Randlett O,
Harris JJ, Liu J, Murray AJ and Harris WA: Metabolic
differentiation in the embryonic retina. Nat Cell Biol. 14:859–864.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Romano AH and Conway T: Evolution of
carbohydrate metabolic pathways. Res Microbiol. 147:448–455. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fan T, Sun G, Sun X, Zhao L, Zhong R and
Peng Y: Tumor energy metabolism and potential of 3-Bromopyruvate as
an inhibitor of aerobic glycolysis: Implications in tumor
treatment. Cancers (Basel). 11:3172019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Carmeliet P and Jain RK: Molecular
mechanisms and clinical applications of angiogenesis. Nature.
473:298–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
DeBerardinis RJ and Cheng T: Q's next: The
diverse functions of glutamine in metabolism, cell biology and
cancer. Oncogene. 29:313–324. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li X, Kumar A and Carmeliet P: Metabolic
pathways fueling the endothelial cell drive. Annu Rev Physiol.
81:483–503. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Groschner LN, Waldeck-Weiermair M, Malli R
and Graier WF: Endothelial mitochondria-less respiration, more
integration. Pflugers Arch. 464:63–76. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
De Bock K, Georgiadou M and Carmeliet P:
Role of endothelial cell metabolism in vessel sprouting. Cell
Metab. 18:634–647. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wong BW, Marsch E, Treps L, Baes M and
Carmeliet P: Endothelial cell metabolism in health and disease:
impact of hypoxia. EMBO J. 36:2187–2203. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guan C, Cen HF, Cui X, Tian DY, Tadesse D
and Zhang YW: Proline improves switchgrass growth and development
by reduced lignin biosynthesis. Sci Rep. 9:201172019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Patra KC and Hay N: The pentose phosphate
pathway and cancer. Trends Biochem Sci. 39:347–354. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thakur C and Chen F: Connections between
metabolism and epigenetics in cancers. Semin Cancer Biol. 57:52–58.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hassell KN: Histone deacetylases and their
inhibitors in cancer epigenetics. Diseases. 7:572019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sharma U and Rando OJ: Metabolic inputs
into the epigenome. Cell Metab. 25:544–558. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Racey LA and Byvoet P: Histone
acetyltransferase in chromatin. Evidence for in vitro enzymatic
transfer of acetate from acetyl-coenzyme A to histones. Exp Cell
Res. 64:366–370. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McBrian MA, Behbahan IS, Ferrari R, Su T,
Huang TW, Li K, Hong CS, Christofk HR, Vogelauer M, Seligson DB and
Kurdistani SK: Histone acetylation regulates intracellular pH. Mol
Cell. 49:310–321. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Goel A, Mathupala SP and Pedersen PL:
Glucose metabolism in cancer. Evidence that demethylation events
play a role in activating type II hexokinase gene expression. J
Biol Chem. 278:15333–15340. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Provis J: Development of the primate
retinal vasculature. Prog Retin Eye Res. 20:799–821. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gariano R: Cellular mechanisms in retinal
vascular development. Prog Retin Eye Res. 22:295–306. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kolb H, Fernandez E and Nelson R:
Webvision: The Organization of the Retina and Visual System
[Internet]. University of Utah Health Sciences Center Copyright;
Salt Lake City, UT: 1995
|
|
41
|
Chase J: The evolution of retinal
vascularization in mammals. Ophthalmology. 89:1518–1525. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Baba T, McLeod DS, Edwards MM, Merges C,
Sen T, Sinha D and Lutty GA: VEGF 165 b in the developing
vasculatures of the fetal human eye. Dev Dyn. 241:595–607. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Saint-Geniez M and D'Amore PA: Development
and pathology of the hyaloid, choroidal and retinal vasculature.
Int J Dev Biol. 48:1045–1058. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhu M, Madigan MC, van Driel D, Maslim J,
Billson FA, Provis JM and Penfold PL: The human hyaloid system:
Cell death and vascular regression. Exp Eye Res. 70:767–776. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gariano RF and Gardner TW: Retinal
angiogenesis in development and disease. Nature. 438:960–966. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
West H, Richardson WD and Fruttiger M:
Stabilization of the retinal vascular network by reciprocal
feedback between blood vessels and astrocytes. Development.
132:1855–1862. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen W, Xia P, Wang H, Tu J, Liang X,
Zhang X and Li L: The endothelial tip-stalk cell selection and
shuffling during angiogenesis. J Cell Commun Signal. 13:291–301.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gerhardt H, Golding M, Fruttiger M,
Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C,
Alitalo K, Shima D and Betsholtz C: VEGF guides angiogenic
sprouting utilizing endothelial tip cell filopodia. J Cell Biol.
161:1163–1177. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Carmeliet P and Jain RK: Angiogenesis in
cancer and other diseases. Nature. 407:249–257. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Benedito R, Roca C, Sorensen I, Adams S,
Gossler A, Fruttiger M and Adams RH: The notch ligands Dll4 and
Jagged1 have opposing effects on angiogenesis. Cell. 137:1124–1135.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Suchting S, Freitas C, le Noble F,
Benedito R, Bréant C, Duarte A and Eichmann A: The Notch ligand
Delta-like 4 negatively regulates endothelial tip cell formation
and vessel branching. Proc Natl Acad Sci USA. 104:3225–3230. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Potente M, Gerhardt H and Carmeliet P:
Basic and therapeutic aspects of angiogenesis. Cell. 146:873–887.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fraisl P, Mazzone M, Schmidt T and
Carmeliet P: Regulation of angiogenesis by oxygen and metabolism.
Dev Cell. 16:167–179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Trost A, Lange S, Schroedl F, Bruckner D,
Motloch KA, Bogner B, Kaser-Eichberger A, Strohmaier C, Runge C,
Aigner L, et al: Brain and retinal pericytes: Origin, function and
role. Front Cell Neurosci. 10:202016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gerhardt H and Betsholtz C:
Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res.
314:15–23. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lindahl P, Johansson BR, Leveen P and
Betsholtz C: Pericyte loss and microaneurysm formation in
PDGF-B-deficient mice. Science. 277:242–245. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hellstrom M, Gerhardt H, Kalén M, Li X,
Eriksson U, Wolburg H and Betsholtz C: Lack of pericytes leads to
endothelial hyperplasia and abnormal vascular morphogenesis. J Cell
Biol. 153:543–553. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Cantelmo AR, Conradi LC, Brajic A, Goveia
J, Kalucka J, Pircher A, Chaturvedi P, Hol J, Thienpont B, Teuwen
LA, et al: Inhibition of the Glycolytic activator PFKFB3 in
endothelium induces tumor vessel normalization, impairs metastasis,
and improves chemotherapy. Cancer Cell. 30:968–985. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rangasamy S, Monickaraj F, Legendre C,
Cabrera AP, Llaci L, Bilagody C, McGuire P and Das A:
Transcriptomics analysis of pericytes from retinas of diabetic
animals reveals novel genes and molecular pathways relevant to
blood-retinal barrier alterations in diabetic retinopathy. Exp Eye
Res. 195:1080432020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhao J, Ha Y, Liou GI, Gonsalvez GB, Smith
SB and Bollinger KE: Sigma receptor ligand, (+)-pentazocine,
suppresses inflammatory responses of retinal microglia. Invest
Ophthalmol Vis Sci. 55:3375–3384. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Langston PK, Shibata M and Horng T:
Metabolism supports macrophage activation. Front Immunol. 8:612017.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhou Y, Yoshida S, Nakao S, Yoshimura T,
Kobayashi Y, Nakama T, Kubo Y, Miyawaki K, Yamaguchi M, Ishikawa K,
et al: M2 macrophages enhance pathological neovascularization in
the mouse model of oxygen-induced retinopathy. Invest Ophthalmol
Vis Sci. 56:4767–4777. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lutty GA, Hasegawa T, Baba T, Grebe R,
Bhutto I and McLeod DS: Development of the human choriocapillaris.
Eye (Lond). 24:408–415. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hasegawa T, McLeod DS, Bhutto IA, Prow T,
Merges CA, Grebe R and Lutty GA: The embryonic human
choriocapillaris develops by hemo-vasculogenesis. Dev Dyn.
236:2089–2100. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Baba T, Grebe R, Hasegawa T, Bhutto I,
Merges C, McLeod DS and Lutty GA: Maturation of the fetal human
choriocapillaris. Invest Ophthalmol Vis Sci. 50:3503–3511. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Vitale G, Cozzolino A, Malandrino P,
Minotta R, Puliani G, Saronni D, Faggiano A and Colao A: Role of
FGF system in neuroendocrine neoplasms: Potential therapeutic
applications. Front Endocrinol (Lausanne). 12:6656312021.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Stine ZE, Walton ZE, Altman BJ, Hsieh AL
and Dang CV: MYC, metabolism, and cancer. Cancer Discov.
5:1024–1039. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Van Schaftingen E, Lederer B, Bartrons R
and Hers HG: A kinetic study of pyrophosphate: Fructose-6-phosphate
phosphotransferase from potato tubers. Application to a microassay
of fructose 2,6-bisphosphate. Eur J Biochem. 129:191–195. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Schoors S, De Bock K, Cantelmo AR,
Georgiadou M, Ghesquière B, Cauwenberghs S, Kuchnio A, Wong BW,
Quaegebeur A, Goveia J, et al: Partial and transient reduction of
glycolysis by PFKFB3 blockade reduces pathological angiogenesis.
Cell Metab. 19:37–48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Lee S, Birukov KG, Romanoski CE,
Springstead JR, Lusis AJ and Berliner JA: Role of phospholipid
oxidation products in atherosclerosis. Circ Res. 111:778–799. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Jyrkkanen HK, Kansanen E, Inkala M, Kivelä
AM, Hurttila H, Heinonen SE, Goldsteins G, Jauhiainen S, Tiainen S,
Makkonen H, et al: Nrf2 regulates antioxidant gene expression
evoked by oxidized phospholipids in endothelial cells and murine
arteries in vivo. Circ Res. 103:e1–e9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kuosmanen SM, Kansanen E, Kaikkonen MU,
Sihvola V, Pulkkinen K, Jyrkkänen HK, Tuoresmäki P, Hartikainen J,
Hippeläinen M, Kokki H, et al: NRF2 regulates endothelial
glycolysis and proliferation with miR-93 and mediates the effects
of oxidized phospholipids on endothelial activation. Nucleic Acids
Res. 46:1124–1138. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Cecchi E, Giglioli C, Valente S, Lazzeri
C, Gensini GF, Abbate R and Mannini L: Role of hemodynamic shear
stress in cardiovascular disease. Atherosclerosis. 214:249–256.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Guo FX, Hu YW, Zheng L and Wang Q: Shear
stress in autophagy and its possible mechanisms in the process of
atherosclerosis. DNA Cell Biol. 36:335–346. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Gomez-Escudero J, Clemente C, Garcia-Weber
D, Acín-Pérez R, Millán J, Enríquez JA, Bentley K, Carmeliet P and
Arroyo AG: PKM2 regulates endothelial cell junction dynamics and
angiogenesis via ATP production. Sci Rep. 9:150222019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kim B, Jang C, Dharaneeswaran H, Li J,
Bhide M, Yang S, Li K and Arany Z: Endothelial pyruvate kinase M2
maintains vascular integrity. J Clin Invest. 128:4543–4556. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Azoitei N, Becher A, Steinestel K, Rouhi
A, Diepold K, Genze F, Simmet T and Seufferlein T: PKM2 promotes
tumor angiogenesis by regulating HIF-1α through NF-κB activation.
Mol Cancer. 15:32016. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Veys K, Fan Z, Ghobrial M, Bouché A,
García-Caballero M, Vriens K, Conchinha NV, Seuwen A, Schlegel F,
Gorski T, et al: Role of the GLUT1 glucose transporter in postnatal
cns angiogenesis and blood-brain barrier integrity. Circ Res.
127:466–482. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Yeh WL, Lin CJ and Fu WM: Enhancement of
glucose transporter expression of brain endothelial cells by
vascular endothelial growth factor derived from glioma exposed to
hypoxia. Mol Pharmacol. 73:170–177. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Hellström A, Smith LEH and Dammann O:
Retinopathy of prematurity. Lancet. 382:1445–1457. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Hartnett ME and Penn JS: Mechanisms and
management of retinopathy of prematurity. N Engl J Med.
367:2515–2526. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Pierce EA, Foley ED and Smith LE:
Regulation of vascular endothelial growth factor by oxygen in a
model of retinopathy of prematurity. Arch Ophthalmol.
114:1219–1228. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Hoppe G, Yoon S, Gopalan B, Savage AR,
Brown R, Case K, Vasanji A, Chan ER, Silver RB and Sears JE:
Comparative systems pharmacology of HIF stabilization in the
prevention of retinopathy of prematurity. Proc Natl Acad Sci USA.
113:E2516–E2525. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ogurtsova K, da Rocha Fernandes JD, Huang
Y, Linnenkamp U, Guariguata L, Cho NH, Cavan D, Shaw JE and
Makaroff LE: IDF diabetes atlas: Global estimates for the
prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract.
128:40–50. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Antonetti DA, Klein R and Gardner TW:
Diabetic retinopathy. N Engl J Med. 366:1227–1239. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Smith LE, Wesolowski E, McLellan A, Kostyk
SK, D'Amato R, Sullivan R and D'Amore PA: Oxygen-induced
retinopathy in the mouse. Invest Ophthalmol Vis Sci. 35:101–111.
1994.PubMed/NCBI
|
|
88
|
Bai Y, Bai X, Wang Z, Zhang X, Ruan C and
Miao J: MicroRNA-126 inhibits ischemia-induced retinal
neovascularization via regulating angiogenic growth factors. Exp
Mol Pathol. 91:471–477. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Xia F, Sun JJ, Jiang YQ and Li CF:
MicroRNA-384-3p inhibits retinal neovascularization through
targeting hexokinase 2 in mice with diabetic retinopathy. J Cell
Physiol. 234:721–730. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Schoors S, Cantelmo AR, Georgiadou M,
Stapor P, Wang X, Quaegebeur A, Cauwenberghs S, Wong BW, Bifari F,
Decimo I, et al: Incomplete and transitory decrease of glycolysis:
a new paradigm for anti-angiogenic therapy? Cell Cycle. 13:16–22.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu
J, Lin JD, Wang CY, Chinnaiyan AM, et al: TEAD mediates
YAP-dependent gene induction and growth control. Genes Dev.
22:1962–1971. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Pan D: The hippo signaling pathway in
development and cancer. Dev Cell. 19:491–505. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Kim J, Kim YH, Kim J, Park DY, Bae H, Lee
DH, Kim KH, Hong SP, Jang SP, Kubota Y, et al: YAP/TAZ regulates
sprouting angiogenesis and vascular barrier maturation. J Clin
Invest. 127:3441–3461. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Feng Y, Zou R, Zhang X, Shen M, Chen X,
Wang J, Niu W, Yuan Y and Yuan F: YAP promotes ocular
neovascularization by modifying PFKFB3-driven endothelial
glycolysis. Angiogenesis. 24:489–504. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Chen JF, Eltzschig HK and Fredholm BB:
Adenosine receptors as drug targets-what are the challenges? Nat
Rev Drug Discov. 12:265–286. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Lutty GA, Merges C and McLeod DS:
5′nucleotidase and adenosine during retinal vasculogenesis and
oxygen-induced retinopathy. Investigative Ophthalmol Visual Sci.
41:218–229. 2000.PubMed/NCBI
|
|
97
|
Liu Z, Yan S, Wang J, Xu Y, Wang Y, Zhang
S, Xu X, Yang Q, Zeng X, Zhou Y, Gu X, et al: Endothelial adenosine
A2a receptor-mediated glycolysis is essential for pathological
retinal angiogenesis. Nat Commun. 8:5842017. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Drake CJ and Fleming PA: Vasculogenesis in
the day 6.5 to 9.5 mouse embryo. Blood. 95:1671–1679. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
van Lookeren Campagne M, LeCouter J,
Yaspan BL and Ye W: Mechanisms of age-related macular degeneration
and therapeutic opportunities. J Pathol. 232:151–164. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Lambert V, Lecomte J, Hansen S, Blacher S,
Gonzalez ML, Struman I, Sounni NE, Rozet E, de Tullio P, Foidart
JM, et al: Laser-induced choroidal neovascularization model to
study age-related macular degeneration in mice. Nat Protoc.
8:2197–2211. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Draoui N and Feron O: Lactate shuttles at
a glance: From physiological paradigms to anti-cancer treatments.
Dis Model Mech. 4:727–732. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Song J, Lee K, Park SW, Chung H, Jung D,
Na YR, Quan H, Cho CS, Che JH, Kim JH, et al: Lactic acid
upregulates VEGF expression in macrophages and facilitates
choroidal neovascularization. Invest Ophthalmol Vis Sci.
59:3747–3754. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Lambert V, Hansen S, Schoumacher M,
Lecomte J, Leenders J, Hubert P, Herfs M, Blacher S, Carnet O, Yip
C, et al: Pyruvate dehydrogenase kinase/lactate axis: A therapeutic
target for neovascular age-related macular degeneration identified
by metabolomics. J Mol Med (Berl). 98:1737–1751. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Vallée A, Lecarpentier Y, Guillevin R and
Vallée JN: Aerobic glycolysis hypothesis through WNT/beta-catenin
pathway in exudative age-related macular degeneration. J Mol
Neurosci. 62:368–379. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Yokosako K, Mimura T, Funatsu H, Noma H,
Goto M, Kamei Y, Kondo A and Matsubara M: Glycolysis in patients
with age-related macular degeneration. Open Ophthalmol J. 8:39–47.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Han G, Wei P, He M and Teng H: Glucose
metabolic characterization of human aqueous humor in relation to
wet age-related macular degeneration. Invest Ophthalmol Vis Sci.
61:492020. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Joyal JS, Gantner ML and Smith LEH:
Retinal energy demands control vascular supply of the retina in
development and disease: The role of neuronal lipid and glucose
metabolism. Prog Retin Eye Res. 64:131–156. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Vidal E, Lalarme E, Maire MA, Febvret V,
Grégoire S, Gambert S, Acar N and Bretillon L: Early impairments in
the retina of rats fed with high fructose/high fat diet are
associated with glucose metabolism deregulation but not
dyslipidaemia. Sci Rep. 9:59972019. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Wang Y, Xie L, Zhu M, Guo Y, Tu Y, Zhon Y,
Zeng J, Zhu L, Du S, Wang Z, et al: Shikonin alleviates choroidal
neovascularization by inhibiting proangiogenic factor production
from infiltrating macrophages. Exp Eye Res. 213:1088232021.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Nicholas MP and Mysore N: Corneal
neovascularization. Exp Eye Res. 202:1083632021. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Clements JL and Dana R: Inflammatory
corneal neovascularization: Etiopathogenesis. Semin Ophthalmol.
26:235–245. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Yang W, Yang Y, Wan S, Xu Y, Li J, Zhang
L, Guo W, Zheng Y, Xiang Y and Xing Y: Exploring the mechanism of
the miRNA-145/paxillin axis in cell metabolism during
VEGF-A-induced corneal angiogenesis. Invest Ophthalmol Vis Sci.
62:252021. View Article : Google Scholar
|
|
113
|
Liu G, Chen L, Cai Q, Wu H, Chen Z, Zhang
X and Lu P: Streptozotocin induced diabetic mice exhibit reduced
experimental choroidal neovascularization but not corneal
neovascularization. Mol Med Rep. 18:4388–4398. 2018.PubMed/NCBI
|














