Introduction
The Hox genes are a set of transcriptional
factor genes that play a crucial role in controlling the body plan
along the craniocaudal axis and specifying the segment identity of
tissues within the embryo. Hox genes are involved in early
developmental morphogenetic processes and remain expressed into
adulthood (1).
Hox genes are highly expressed within the
developing mesonephric duct, specifically Hoxa9, Hoxd9, Hoxa10,
Hoxd10, Hoxa11, Hoxa13, and Hoxd13 (1–3). The
Hox proteins family is highly conserved among species. In
vertebrates, there are 39 members organized into four tightly
clustered gene arrays (HOXA-HOXB-HOXC and
HOXD) (4).
In humans, the gene clusters are organized in a 3′
to 5′ orientation with paralog group 1 genes at the 3′ end of the
cluster (e.g., HOXA1, HOXB1, HOXD1) and higher number groups
located at the 5′ end. A cluster's temporal and spatial
collinearity is represented as a pattern of expression along the
anterior-posterior axis corresponding to the order of Hox gene in
its cluster within the 3′ to 5′ direction. Thus, during the
development, HOX genes located at the 3′ end, are firstly
expressed in the anterior regions and later on the 5′ genes
posteriorly (2–4). The role of mammalian HOX genes
in regulating segmental patterns of the hindbrain, skeleton axis,
and limb axis is extensively studied and well-known (5).
A total of 10 HOX genes have been linked to
human conditions (HOXA1, HOXA2, HOXA11, HOXA13, HOXB1, HOXB13,
HOXC13, HOXD4, HOXD10, and HOXD13) (4). Different patterns of inheritance and
variable penetrance and expressivity have been reported.
The most reported syndrome associated with
HOXA mutations is the Hand-foot-genital syndrome (HFGS;
OMIM# 140000). HFGS is a rare autosomal dominant condition
characterized by distal limb and genitourinary malformations and
caused by HOXA13 mutations (6–9).
Limb abnormalities include first-digit hypoplasia, comprising
short, proximally placed thumbs with hypoplastic thenar eminences
and short, medially deviated halluces and fusion or delayed
ossification of the wrist bones. These deformities appear to be
bilateral and symmetrical, with variable severity (10,11).
HOXA13 mutations are fully penetrant for limb defects with
variable expressivity and partially penetrant for genitourinary
tract anomalies with different fertility implications. Of note,
females with HFGS do not suffer systematically from infertility
(8–10,12).
Guttmacher (1993) reported a family with an HFG-like syndrome. The
particularities noted in this family include uniphalangeal second
toes with absent nails and postaxial polydactyly (13). Despite these distinct features, the
genetic investigation of the family reported by Guttmacher revealed
a missense mutation (p.Gln50Leu) in the HOXA13 gene,
indicating the phenotypic variability of the mutations (14).
The features of HFGS are found in another clinical
association with a broad spectrum of congenital malformations: the
VACTERL association (OMIM #192350). VACTERL is an acronym for
vertebral anomalies, anal atresia, cardiac defects,
tracheo-esophageal fistula, renal anomalies, and limb abnormalities
(15).
Thus, VACTERL association is typically defined by
the presence of at least three of its features (15). Although HFGS was associated only
with HOXA13 genetic variations, many non-Hox genes have been
linked to VACTERL association and other overlapping conditions
(15–17). Solomon et al have listed
more than 30 conditions with features in common with VACTERL and
their genetic etiology. From the HOX genes family, only
HOXA13 and HOXD13 genes are implicated in these
disorders (15). As an example,
clinical entities comprising cardiac defects such as Holt-Oram
syndrome, CHARGE syndrome, Andersen syndrome, and Alagille syndrome
are linked to TBX5, CHD7, KCNJ2, JAG2, and NOTCH2
genes respectively (15).
The molecular basis of VACTERL is poorly known.
Nevertheless, HOXD13 mutation has been associated with this
condition (18). The co-occurrence
of the VACTERL association and Mϋllerian duct defects has been
reported (19–22).
It should be noted that many of the overlapping
syndromes include specific features that tend to help a clinical
diagnosis of exclusion. Thus, a good clinical investigation is
crucial to rule out overlapping diagnoses.
Here, we report a female patient with a syndromic
clinical presentation including mainly arm agenesis, bicornuate
uterus, and bicuspid aortic valve (BAV).
Materials and methods
A peripheral blood sample was collected after
obtaining the written informed consent. Genomic DNA was extracted
by standard techniques.
Subsequently, whole-exome sequencing (WES) was
carried out using the Agilent Sure Select All Exon v4 kit (Agilent,
Santa Clara, CA, USA), and the sequencing was performed on an
Illumina HiSeq2000 sequencing apparatus (Illumina, SanDiego, CA,
USA). Raw fastQ files were aligned to the hg19 reference human
genome (University of California Santa Cruz, UCSC) using BWA
software. Variant calling workflow was performed according to the
GATK best practices. The output files were annotated using ANNOVAR
software. Variant annotation and prioritization process were
performed with the Variant Annotation and Filtering Tool (VarAFT),
version 2.17-2 (http://varaft.eu/).
To pinpoint putatively pathogenic and causal
variants we adopted the following filtering strategy: we first
excluded variants with a minor allele frequency (MAF) >1% in the
gnomAD database (http://gnomad.broadinstitute.org/). Then, we removed
non-coding and synonymous variants. Subsequently, the remaining
variants were filtered based on their in silico
pathogenicity prediction and clinical relevance.
Amino acids conservation was assessed by sequence
alignments using ClustalW (https://www.genome.jp/tools-bin/clustalw).
Results
Clinical and genetic findings
The patient is a female born to non-consanguineous
parents. Her clinical presentation comprises features of
VACTERL/HFGS syndrome including bicornuate uterus (double uterine
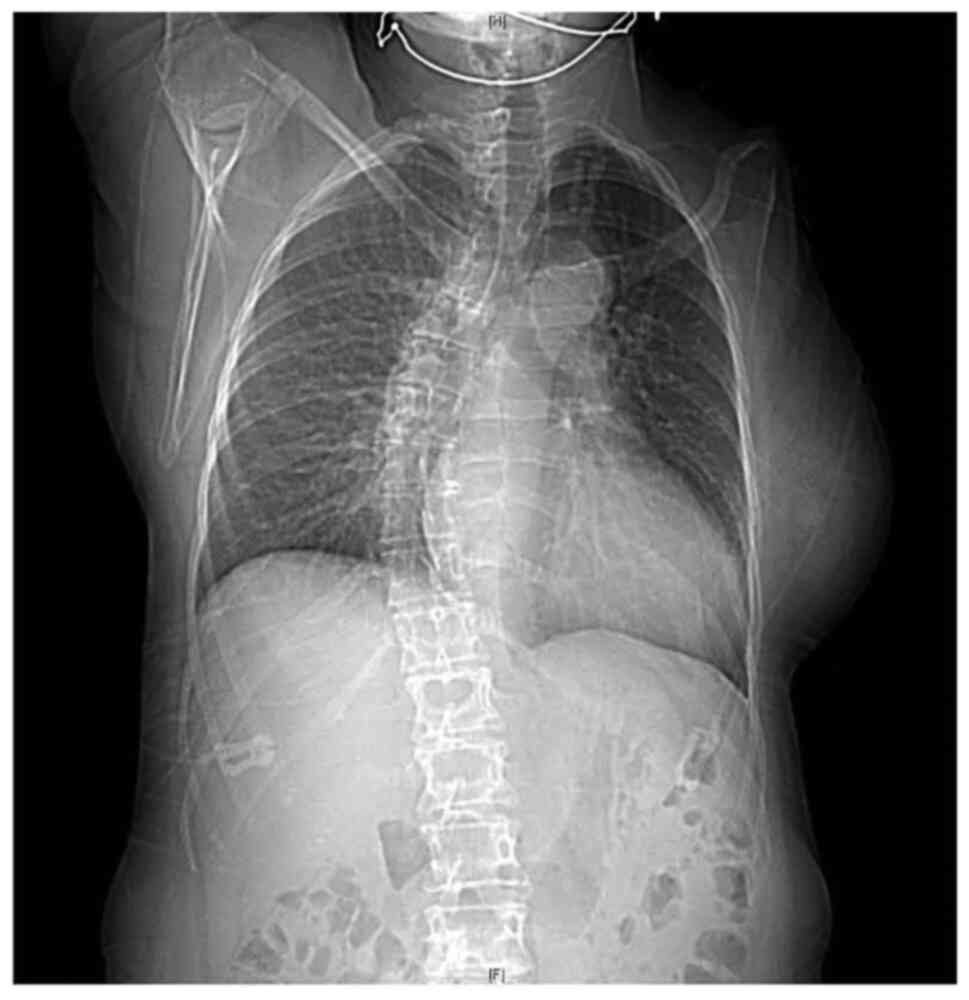
cavities, double cervix), left arm agenesis, scoliosis, and BAV
(Figs. 1 and 2). The patient was delivered at term with
an upper left arm agenesis. Any imperforate anus nor anal atresia
or tracheoesophageal fistula were noted in the patient's birth.
The patient had a history of normal intellectual
developmental milestones without over-susceptibility to childhood
diseases.
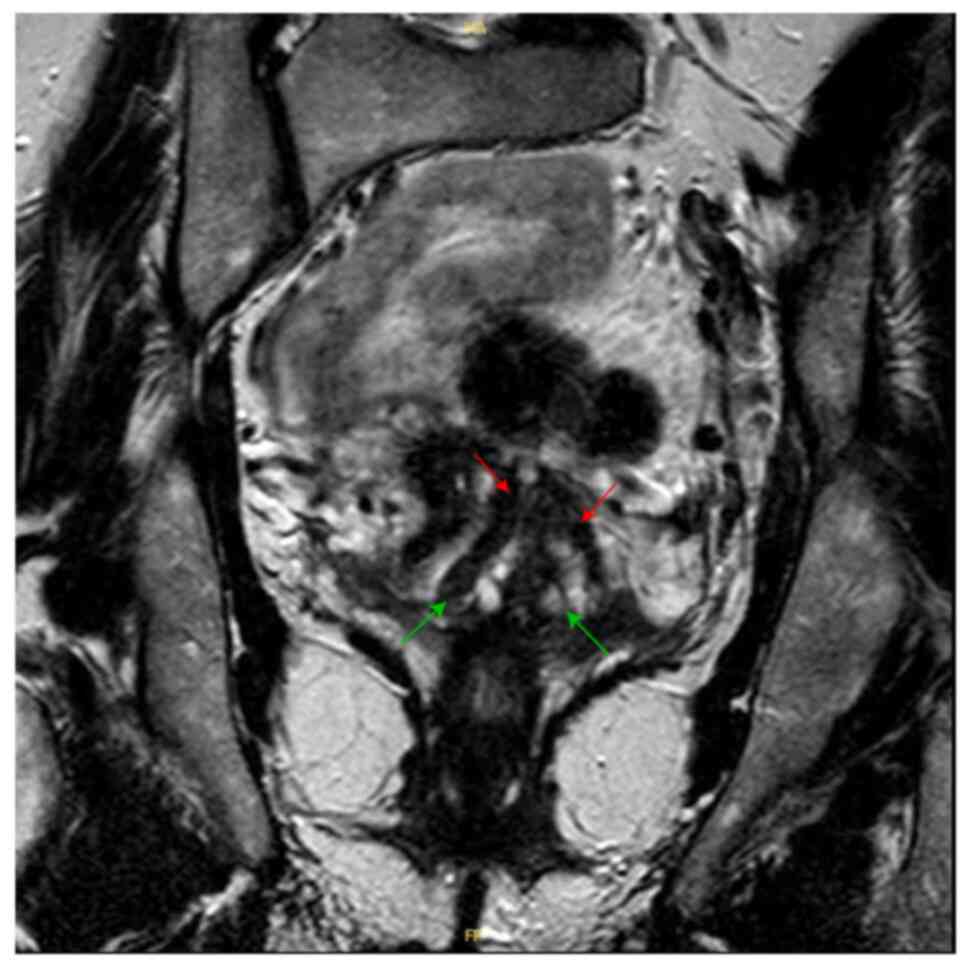
Pelvic MRI showed two uterine cavities and a double
cervix. On the T1-weighted sequence, there is no hematic retention
in the right or left side of the uterine horns. No anomalies in the
junction zones or the myometrium. Both appendages are of normal
size and regular outline (Fig. 1).
Of note, the patient did not have frequent pelvic pain or
miscarriage history due to her bicornuate uterus.
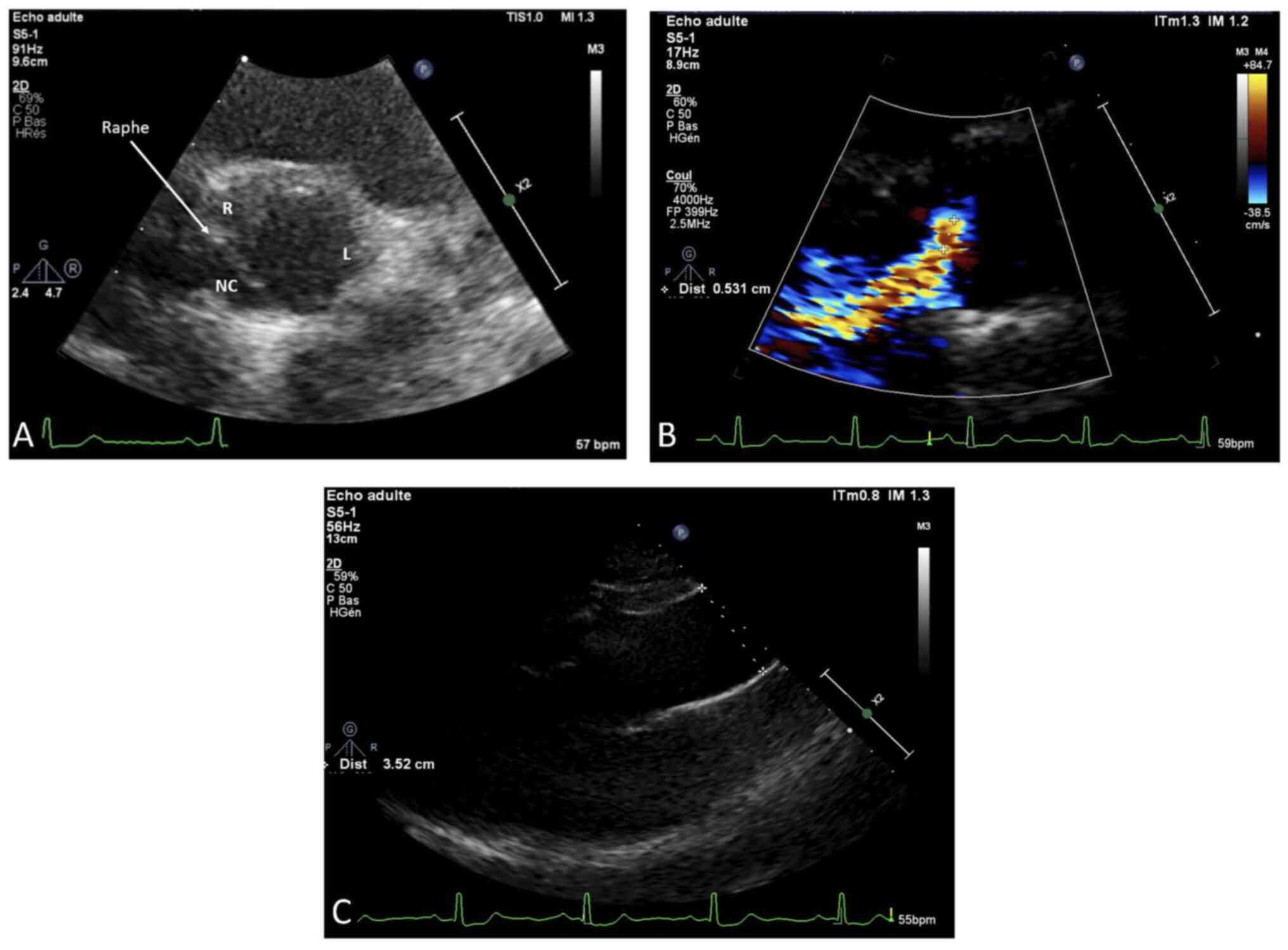
Her cardiac evaluation revealed a type 1
non-calcified BAV. Transthoracic echocardiography showed a normal
left ventricular ejection fraction (LVEF=70%) with a slight
diastolic LV dilation (Indexed end-diastole volume=108
ml/m2) and a raphe between the non-coronary and the
right coronary cusps corresponding to a type I, N-L, according to
the Sievers classification (Fig.
3A). A moderate aortic regurgitation was noted (ERO=20
mm2) due to a prolapse of the fused leaflet and a
restriction of the left coronary cusp (Fig. 3B). The aortic valve leaflets were
thin and un-calcified. There was neither BAV-related aortopathy nor
mitral regurgitation (Fig. 3C).
Systolic pulmonary arterial pressure was normal (25 mmHg).
The patient's parents were not examined but were
reportedly unaffected. The patient's mother did not accept to
participate in the genetic study by reason of iatrophobia.
Whole exome sequencing data analysis allowed us to
identify two missense variants in HOXA9 and HOXA13
genes.
The HOXA13: c.869 A>C: p.(Tyr290Ser)
variant is novel, located upstream of the homeodomain, and
predicted deleterious by SIFT, probably damaging by PolyPhen,
pathogenic by UMD-predictor and has a CADD_phred score=31.
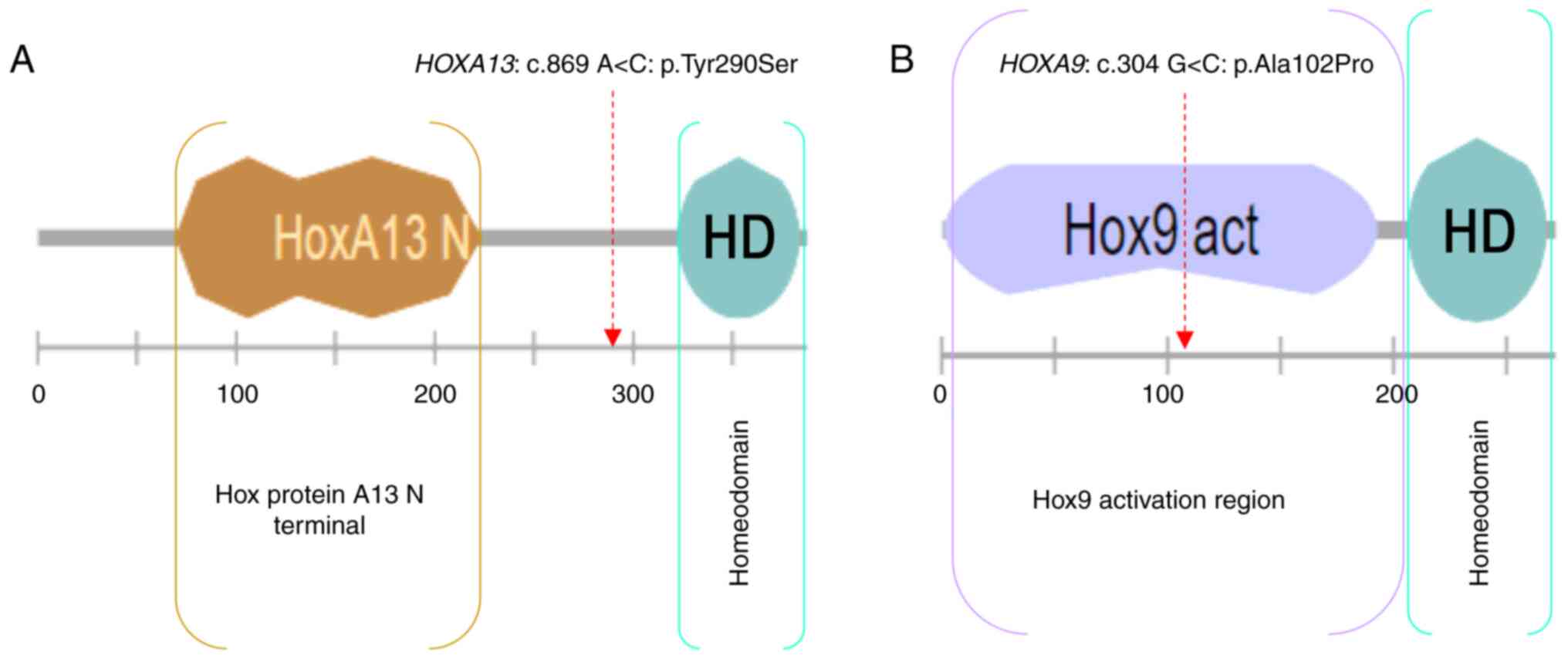
The HOXA9: c.304 G>C: p.(Ala102Pro)
variant is located in the HOX9 activation region and predicted
tolerated by SIFT, benign by PolyPhen, probably pathogenic by
UMD-predictor, and has a CADD_Phred score=22.8.
Both amino acids are conserved among species
(Fig. S1). Furthermore, the
variants were absent from our in-house database gathering 300
exomes.
In order to assess the impact of the variants on the
protein level, we used VarMap to retrieve protein structural
annotations (23). VarMap output
includes the predicted consequence of the variant and a mapping to
the residue in the closest 3D protein structure in the Protein Data
Bank. Both variants in HOXA9 and HOXA13 genes have a
high disease propensity value, 1.58 and 1.67 respectively (Fig. 4). The propensities quantify how
much more often a variant is observed in diseases than in the
natural variant data obtained from gnomAD. Values range from the
lowest (Ile->Val, propensity=0.25) to the highest (Cys->Arg,
propensity=3.27).
Sanger sequencing of the father's patient showed the
absence of both variants (Fig.
5).
As no familial history of limb or genitourinary
abnormalities was reported by the patient and the absence of the
variants in the father, the variants may have occurred de
novo.
Furthermore, no variants were found in genes related
to VACTERL and HFG syndromes with features in common with the
clinical presentation of our patient. The list of genes checked
includes JAG1, NOTCH1, NOTCH2, WNT7A, FBN2, LRP4, FOXF1, TBX3,
TBX5, HOXD13, MID1, RBM8A, SALL1 and ZIC3 genes.
Discussion
Here, we report a female patient with two missense
variants in HOXA9 and HOXA13 genes, which may explain
her clinical condition characterized by a complete arm agenesis,
bicornuate uterus, and BAV. According to the in silico
prediction, the novel HOXA13: p.(Tyr290Ser) is highly
pathogenic with a CADD score=31. The HOXA9: p.(Ala102Pro) is
predicted probably pathogenic by UMD-predictor with a high CADD
score as well (22.8). Moreover, HOXA9: p.(Ala102Pro) is
located in the activation domain of HOX9 protein (Fig. 6).
HOX genes are essential for reproductive
tract development and for adult function. During embryonic
development, HOXA genes play a key role in determining the
segmental identity of the female genital tract (5). HOXA9 and HOXA13 genes
are part of the A cluster on chromosome 7 and encode a DNA-binding
transcription factor that may regulate gene expression,
morphogenesis, and differentiation (5,24).
In adults, HOXA genes contribute to maintain
the developmental plasticity of reproductive tissues (5,25,26).
HOXA9 is highly expressed in areas destined to give rise to
the oviduct. HOXA10, HOXA11, and HOXA13 are expressed
in the developing uterus, the lower uterine segment and cervix, and
in the upper vagina, respectively (https://gtexportal.org/) (25).
Interestingly, HOXA genes expression persist
in the adult tissues which continue to undergo developmental
processes, such as hematopoiesis (bone marrow), menstrual cycle,
and pregnancy (reproductive tract) (5,25).
In this line, HOXA9 and HOXA13 genes have been
associated, respectively, with human myeloid leukemia and Mϋllerian
defects (8,27,28).
Indeed, the HOXA13 gene plays a crucial role in Mullerian
ducts fusion and ureter remodeling by the elimination of the common
caudal nephric duct. Hoxa13 mutant mice develop megaureters,
hydronephrosis, and malformations of the uterus (8).
Patients with HOXA13 mutations present
several digit anomalies including short thumbs, short middle
phalanges of the fingers, clinodactyly of the fifth fingers, and
the fusion of distal and middle phalanges of the toes (6,7,11,29).
The first HOXA13 mutation was identified in a large family
with HFGS reported originally by Stern et al 1970 (9,30).
The nonsense mutation identified in this family is located in the
HOXA13 homeodomain and leads to the truncation of 20 amino acids
which likely abolishes or reduces the protein's ability to bind to
DNA (9).
Thereafter, different mutations have been reported
in patients with HFGS including protein truncating variants and
missense variants (6,7,31,32).
Interestingly, missense mutations in HOXA13 have been
correlated to a severe limb phenotype (10,14).
The clinical presentation of the patient harboring the first
missense mutation in the human HOX protein, HOXA13:
p.(Asn51His), has been described as very severe. He had extremely
short thumbs, absent halluces, marked hypoplasia of all middle
phalanges, and granular hypospadias (10).
Both gain of function and dominant negative
mechanisms have been reported as the genetic basis of
HOXA13-related phenotypes. Indeed, missense mutations are
likely to perturb the DNA-binding properties of HOXA13, leading to
both loss and specific gain of function (11,14).
The association HOXA13/HOXD13 and
HOXA11 has been reported in patients with amegakaryocytic
thrombocytopenia and radio-ulnar synostosis. Furthermore, the
association HOXD13/HOXA13 has been as well reported as
linked to triphalangeal thumb brachyectrodactyly syndrome (29,33).
Moreover, patients double mutated in HOXD13 and
HOXA13 showed more severe digital abnormalities than
patients harboring monogenic mutations (29).
To our knowledge, congenital heart defects have not
been reported in syndromes linked to HOXA13 mutations.
However, a de novo 7 alanine deletion (Ala55-Ala61 del) in the
polyalanine tract of the HOXD13 gene has been identified in
a patient with VACTERL association (18). Her clinical presentation included:
anal atresia, tetralogy of Fallot, bilateral vesicoureteral reflux,
and fusion of the distal interphalangeal joints of 4th and 5th toes
(18). Of note, missense mutations
in the HOXA13 gene have been described as more pathogenic
than protein-truncating mutations or polyalanine expansion leading
to a more severe phenotype (11).
Overall, compared to the cases published previously,
our patient distinctively has a complete arm agenesis and BAV
making the association HOXA9/HOXA13 clinically
interesting.
According to the in silico prediction, the
literature, and ACMG classification, HOXA13: p.(Tyr290Ser)
is the most relevant variant that may explain the patient's
phenotype. Indeed, the HOXA13 variant meets the ACMG
pathogenic criteria: PS3 (Well-established in vitro or in
vivo functional studies supportive of a damaging effect on the
gene or gene product), PM6 (Assumed de novo but without
confirmation of paternity and maternity), PP2 (Missense variant in
a gene that has a low rate of benign missense variation and in
which missense variants are a common mechanism of disease) and PP4
(Patient's phenotype or family history is highly specific for a
disease with a single genetic etiology).
Although the novel HOXA13 variant is more
clinically relevant in the present case, the contribution of the
HOXA9/HOXA13 genes in BAV needs to be
investigated.
Our case study provides new insights into the likely
implication of the HOXA genes not only in limb and
genitourinary anomalies but also in cardiac defects found in
patients with HOXA morphopathies.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This research received a grant from the French Association
against Myopathies-AFM Telethon. This work was also supported by
the INSERM.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JFA and SZ conceived and designed the study. AT, GN,
JFA performed the clinical investigation of the patients and family
members. SZ and JFA confirm the authenticity of all the raw data.
HJ analyzed and interpreted the data, performed the molecular
investigation and wrote the original draft preparation. AT, JFA and
SZ critically reviewed and edited the manuscript. SZ supervised and
validated the study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All procedures performed in studies involving human
participants were in accordance with the ethical standards of the
institutional and/or national research committee and with the 1964
Helsinki declaration and its later amendments or comparable ethical
standards. Written informed consent was obtained from the patient
and family members included in this study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Krumlauf R: Hox genes, clusters and
collinearity. Int J Dev Biol. 62:659–663. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mallo M and Alonso CR: The regulation of
Hox gene expression during animal development. Development.
140:3951–3963. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Favier B and Dollé P: Developmental
functions of mammalian Hox genes. Mol Hum Reprod. 3:115–131. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Quinonez SC and Innis JW: Human HOX gene
disorders. Mol Genet Metab. 111:4–15. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Du H and Taylor HS: The role of Hox genes
in female reproductive tract development, adult function, and
fertility. Cold Spring Harb Perspect Med. 6:a0230022015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cao L, Chen C, Leng Y, Yan L, Wang S,
Zhang X and Luo Y: A missense mutation of HOXA13 underlies
hand-foot-genital syndrome in a Chinese family. J Genet.
96:647–652. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jorgensen EM, Ruman JI, Doherty L and
Taylor HS: A novel mutation of HOXA13 in a family with
hand-foot-genital syndrome and the role of polyalanine expansions
in the spectrum of Müllerian fusion anomalies. Fertil Steril.
94:1235–1238. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roux M, Bouchard M and Kmita M:
Multifaceted Hoxa13 function in urogenital development underlies
the hand-foot-genital syndrome. Hum Mol Genet. 28:1671–1681. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mortlock DP and Innis JW: Mutation of
HOXA13 in hand-foot-genital syndrome. Nat Genet. 15:179–180. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goodman FR, Bacchelli C, Brady AF, Brueton
LA, Fryns JP, Mortlock DP, Innis JW, Holmes LB, Donnenfeld AE,
Feingold M, et al: Novel HOXA13 mutations and the phenotypic
spectrum of hand-foot-genital syndrome. Am J Hum Genet. 67:197–202.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Imagawa E, Kayserili H, Nishimura G,
Nakashima M, Tsurusaki Y, Saitsu H, Ikegawa S, Matsumoto N and
Miyake N: Severe manifestations of hand-foot-genital syndrome
associated with a novel HOXA13 mutation. Am J Med Genet A.
164A:2398–2402. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wallis M, Tsurusaki Y, Burgess T, Borzi P,
Matsumoto N, Miyake N, True D and Patel C: Dual genetic diagnoses:
Atypical hand-foot-genital syndrome and developmental delay due to
de novo mutations in HOXA13 and NRXN1. Am J Med Genet A.
170:717–724. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guttmacher AE: Autosomal dominant preaxial
deficiency, postaxial polydactyly, and hypospadias. Am J Med Genet.
46:219–222. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Innis JW, Goodman FR, Bacchelli C,
Williams TM, Mortlock DP, Sateesh P, Scambler PJ, McKinnon W and
Guttmacher AE: A HOXA13 allele with a missense mutation in the
homeobox and a dinucleotide deletion in the promoter underlies
Guttmacher syndrome. Hum Mutat. 19:573–574. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Solomon BD, Bear KA, Kimonis V, de Klein
A, Scott DA, Shaw-Smith C, Tibboel D, Reutter H and Giampietro PF:
Clinical geneticists' views of VACTERL/VATER association. Am J Med
Genet A. 158A:3087–3100. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
van de Putte R, Dworschak GC, Brosens E,
Reutter HM, Marcelis CLM, Acuna-Hidalgo R, Kurtas NE, Steehouwer M,
Dunwoodie SL, Schmiedeke E, et al: A genetics-first approach
revealed monogenic disorders in patients with ARM and VACTERL
anomalies. Front Pediatr. 8:3102020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Solomon BD, Baker LA, Bear KA, Cunningham
BK, Giampietro PF, Hadigan C, Hadley DW, Harrison S, Levitt MA,
Niforatos N, et al: An approach to the identification of anomalies
and etiologies in neonates with identified or suspected VACTERL
(vertebral defects, anal atresia, tracheo-esophageal fistula with
esophageal atresia, cardiac defects, renal and limb anomalies)
association. J Pediatr. 164:451–457.e1. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Garcia-Barceló MM, Wong KK, Lui VC, Yuan
ZW, So MT, Ngan ES, Miao XP, Chung PH, Khong PL and Tam PK:
Identification of a HOXD13 mutation in a VACTERL patient. Am J Med
Genet A. 146A:3181–3185. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kang J, Mao M, Zhang Y, Ai FF and Zhu L:
Congenital anal atresia with rectovestibular fistula, scoliosis,
unilateral renal agenesis, and finger defect (VACTERL association)
in a patient with partial bicornuate uterus and distal vaginal
atresia: A case report. Medicine (Baltimore). 97:e128222018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Komura M, Kanamori Y, Sugiyama M, Tomonaga
T, Suzuki K, Hashizume K and Goishi K: A female infant who had both
complete VACTERL association and MURCS association: Report of a
case. Surg Today. 37:878–880. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Obeidat RA, Aleshawi AJ, Tashtush NA and
Alsarawi H: Unicornuate uterus with a rudimentary non-communicating
cavitary horn in association with VACTERL association: Case report.
BMC Womens Health. 19:712019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Balusamy SL, Kumar D and Subrahmanian SR:
MURCS and VACTERL association in a 27 year old female. J Obstet
Gynaecol. 29:762–763. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stephenson JD, Laskowski RA, Nightingale
A, Hurles ME and Thornton JM: VarMap: A web tool for mapping
genomic coordinates to protein sequence and structure and
retrieving protein structural annotations. Bioinformatics.
35:4854–4856. 2019.Available from:. https://academic.oup.com/bioinformatics/article/35/22/4854/5514476
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Goodman FR and Scambler PJ: Human HOX gene
mutations. Clin Genet. 59:1–11. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Taylor HS: The role of HOX genes in the
development and function of the female reproductive tract. Semin
Reprod Med. 18:81–89. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
He B, Ni ZL, Kong SB, Lu JH and Wang HB:
Homeobox genes for embryo implantation: From mouse to human. Animal
Model Exp Med. 1:14–22. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sitwala KV, Dandekar MN and Hess JL: HOX
proteins and leukemia. Int J Clin Exp Pathol. 1:461–474.
2008.PubMed/NCBI
|
|
28
|
Aryal S, Zhang Y, Wren S, Li C and Lu R:
Molecular regulators of HOXA9 in acute myeloid leukemia. FEBS J.
Nov 7–2021.(Epub ahead of print). PubMed/NCBI
|
|
29
|
Pérez-Cabrera A, Kofman-Alfaro S and
Zenteno JC: Mutational analysis of HOXD13 and HOXA13 genes in the
triphalangeal thumb-brachyectrodactyly syndrome. J Orthop Res.
20:899–901. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stern AM, Gall JC Jr, Perry BL, Stimson
CW, Weitkamp LR and Poznanski AK: The hand-food-uterus syndrome: A
new hereditary disorder characterized by hand and foot dysplasia,
dermatoglyphic abnormalities, and partial duplication of the female
genital tract. J Pediatr. 77:109–116. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Frisén L, Lagerstedt K, Tapper-Persson M,
Kockum I and Nordenskjöld A: A novel duplication in the HOXA13 gene
in a family with atypical hand-foot-genital syndrome. J Med Genet.
40:e492003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Innis JW, Mortlock D, Chen Z, Ludwig M,
Williams ME, Williams TM, Doyle CD, Shao Z, Glynn M, Mikulic D, et
al: Polyalanine expansion in HOXA13: Three new affected families
and the molecular consequences in a mouse model. Hum Mol Genet.
13:2841–2851. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Thompson AA and Nguyen LT: Amegakaryocytic
thrombocytopenia and radio-ulnar synostosis are associated with
HOXA11 mutation. Nat Genet. 26:397–398. 2000. View Article : Google Scholar : PubMed/NCBI
|