Introduction
Congenital cataract is the leading cause of
blindness or visual impairment in children. The incidence of
congenital cataract among newborns is estimated at 5–15
cases/10,000 live births worldwide (1). Of congenital cataract cases, 8–25%
are hereditary and the predominant mode of inheritance is autosomal
dominant. However, autosomal recessive or X-linked patterns of
inheritance have also emerged (2).
At least 34 genes have been associated with the onset of congenital
cataract, including lens-, cytoskeletal structure- and
membrane-related genes and transcription factors (3–6).
Advances and integration of several methods of genetic analysis
have accelerated the study of hereditary cataract. Therefore,
genetic defects associated with particular phenotypes have been
identified. A significant number of pathogenic genes and mutations
have been successfully characterized in congenital cataract through
the application of linkage analysis, DNA probe microarray and gene
sequencing (7,8).
In the lens, >90% of the water-soluble
cytoplasmic proteins are composed of crystallines, which are
divided into α-, β- and γ-crystallines (9). β-crystalline accounts for ~35% of
total lens proteins. Its highly conserved structure and unique
spatial arrangement are the basis of lens transparency (10). It has been reported that the normal
expression of β-crystalline is of great significance for the
formation of lens and provides the potential of normal vision
(11). Gap junction proteins are
the most common components of gap junction. These proteins mainly
mediate the transcellular transport of nutrients, metabolites, ions
and second messengers and serve a significant role in maintaining
intracellular metabolic balance and homeostasis (12). A previous study revealed that
connexin (Cx)43, Cx46 and Cx50, encoded by GJAJ, GJA3 and
GJA8, respectively, were expressed in the lens (6).
The current study aimed to identify mutations in
three Chinese families with congenital cataract via using whole
exome sequencing (WES). The results of the present study could
further expand the pathogenic genetic spectrum of cataract, thus
providing the foundation for unraveling its complex molecular basis
and pathogenesis. Additionally, the data could be considered as a
significant reference for the development of gene-targeted drugs
and personalized therapeutic approaches for patients with
congenital cataract.
Materials and methods
Clinical evaluation and collection of
familial blood samples
The current study was approved by the Ethics
Committee of the Jinling Hospital, Nanjing University School of
Medicine and all research subjects signed informed consent. All
methods were performed according to the relevant guidelines and
regulations. All investigators adhered to the principles expressed
in the Declaration of Helsinki. Samples from three patients
diagnosed with hereditary congenital cataract were collected from
the Department of Ophthalmology of Jinling Hospital. Pedigree
investigation revealed three affected Chinese families. Family A
consisted of 11 affected and 27 unaffected members, family B of two
affected and six unaffected members and family C of four and eight
affected and unaffected members, respectively. All family members
with congenital cataract underwent ophthalmic examination and
general physical checkup, including assessment of visual function
and slit-lamp examination. In addition, the detailed family history
was recorded, while information regarding disease onset and
symptoms was also collected. Finally, the pedigree map was plotted
according to the examination results.
Collection and DNA extraction of
peripheral blood
Firstly, peripheral venous blood (4 ml per person)
was collected from the probands of each genetic family, their
parents or children and individual family members. A total of 11
peripheral blood samples were collected from three cataract
families, including four patients from A, four patients from B and
three patients from C family. Meanwhile, peripheral blood samples
from 100 normal people were collected as controls. Subsequently,
total DNA was extracted from peripheral blood using the TIANamp
blood DNA kit (Tiangen Biotech Co., Ltd.). The DNA samples were
analyzed for protein and RNA contamination, as well as for
degradation using agarose gel electrophoresis. Agarose gel solution
(1.5%) was prepared and 1 µl 6XLoading Buffer and 5 µl PCR product
were absorbed and mixed for sample loading and electrophoresis
performed at 110 V at constant pressure for 30 min. The gel was
imaged using Gel-Red (Biotium) was observed. The concentration of
the DNA samples was measured using the Qubit 3.0 fluorometer. Only
samples with a concentration of >0.6 µg were selected for the
follow-up experiments.
WES and variant analysis
The proband's DNA was analyzed by high-throughput
WES (Beijing Zhiyin Oriental Translational Medicine Research Center
Co., Ltd.). WES was performed using the Nimblegen whole exon
capture chip and DNA was sequenced on an Illumina HiSeq series
sequencer (Illumina, Inc.). The sequencing coverage of the target
sequence was not <99%. Finally, data analysis was performed.
Following screening, the data were aligned to the reference
sequence and the variants were detected using the BWA, SAM and
Pindel tools (13–15). To screen for suspected mutations,
the variation data were compared in the dbSNP database (http://www.ncbi.nlm.nih.gov/), 1000 Genome (http://browser.1000genomes.org/index.html), ExAC
(http://exac.broadinstitute.org/), OMIM
(https://www.ncbi.nlm.nih.gov/omim/),
HapMap (https://www.ncbi.nlm.nih.gov/variation/news/NCBI_retiring_HapMap)
and Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/). Variants in 25, 15 and 17
known disease-causing genes were detected in family A, family B and
family C, respectively. To screen for suspected pathogenic genes
and mutations, the following screening strategies were applied: i)
Single nucleotide polymorphism/insertion and deletions (InDel) loci
and the reported congenital cataract-related pathogenic genes were
listed and loci with a mutation frequency of >0.01 in Genome
Aggregation Database, ESP6500 and 1000 Genome databases were
removed. ii) Suspicious variants were filtered according to the
family inheritance pattern. In terms of recessive genes, when only
one heterozygous mutation was detected, this mutation was excluded.
Non-synonymous mutations, such as nonsense, missense, frameshift
and splicing mutations were retained. iii) According to the
American College of Medical Genetics and Genomics score, pathogenic
variants, likely pathogenic variants and variants of uncertain
significance were retained. The pathogenicity of mutations was
predicted using SIFT (http://sift-dna.org), Polymorphism Phenotyping v2
(http://genetics.bwh.harvard.edu/pph2/) and Mutation
Taster (http://www.mutationtaster.org/) tools. All suspected
pathogenic mutations were retained. iv) To verify whether the
reported pathogenic inheritance pattern of the suspected gene was
consistent with the inheritance pattern of the family, the data
were analyzed using OMIM (https://www.omim.org/) or PubMed (https://pubmed.ncbi.nlm.nih.gov/) databases.
Following multiple comparisons, a mutation was considered as a
candidate pathogenic mutation of the family And the data were
summarized for verification.
Sanger sequencing and bioinformatics
analysis
Mutations with high pathogenic possibility were
screened from the exon sequencing results and the corresponding
exon sequences were then located in NCBI (https://www.ncbi.nlm.nih.gov/). PCR primer sequences
were designed according to the candidate mutation sites and
amplification of the target-genes was performed using the DNA
obtained from the family members as a template. To verify whether
there were mutations in the candidate sites, the qualified
amplification products of the first generation were sequenced and
the sequencing results were analyzed using Chromas 2 software
(Technelysium Pty Ltd). Subsequently, the Chromas software was also
used to assess whether the mutations in this family was pathogenic
and conformed to the law of genotype co-segregation. Amino acid
conservation analysis was performed using DNAMAN 9.0 software
(LynnonBiosoft), while amino acid hydrophobicity analysis was
performed via searching for protein structure models in the UniProt
database (https://www.uniprot.org/) combined
with Protscale (https://web.expasy.org/protscale/) database.
Results
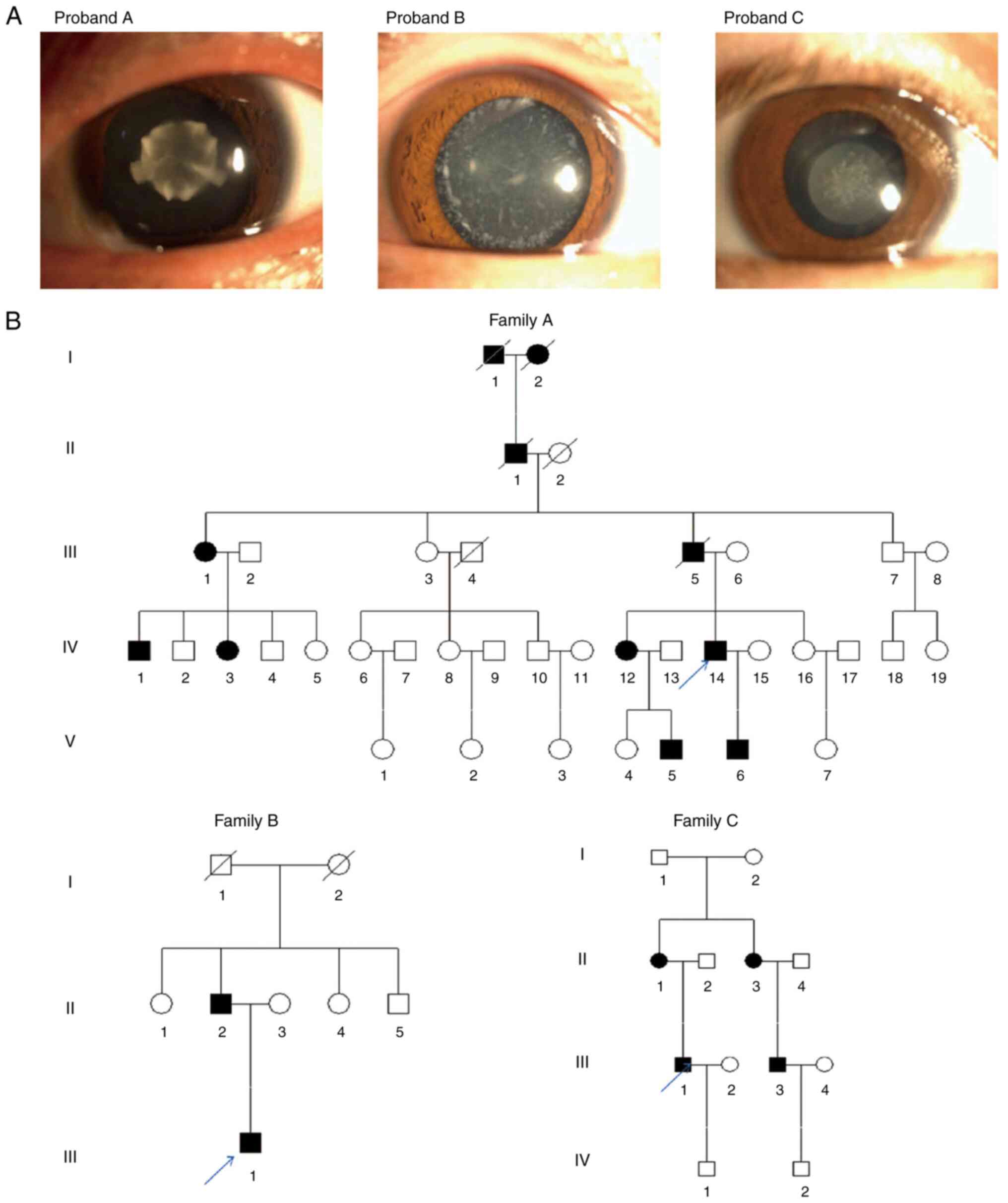
Clinical evaluation and pedigree
investigation
The male proband of family A had attended Jinling
Hospital due to poor vision since childhood and he was diagnosed
with bilateral congenital cataract. His sister (IV-12) and her son
(V-5) suffered from poor vision since childhood. However, they did
not undergo surgery. The proband's son (V-6) underwent
ophthalmological surgery due to poor vision. The proband of family
B, an 18-year-old male, presented with unexpected and progressive
vision loss in both eyes, and more severe in the left eye, one year
ago. Eye examination revealed white cloudy crystals in both eyes
and the proband was eventually diagnosed with bilateral congenital
cataract. His father (II-2) had been previously diagnosed with
bilateral congenital cataract, while his mother (II-3) had normal
vision. The proband of family C, a male subject, had been also
diagnosed with irrational progressive vision loss at the age of
three due to bilateral congenital cataract. His mother (II-1) and
sister also suffered from congenital cataract, his father (II-2)
had normal vision, while his cousin (III-3) had undergone surgery
for poor vision. The proband's son (IV-1), 8 years old, also had
normal vision. All probands denied any existence of family history
of hypertension and diabetes mellitus. Furthermore, slit-lamp
examination showed that the proband of family A suffered from
nuclear cataract opacity, while 11 affected members were recorded
in the family. Additionally, the proband of family B was diagnosed
with white punctate opacity, with two affected members in the
family. Finally, the proband of family C also suffered from nuclear
cataract, with four affected members in the family. There were both
male and female patients with cataract in each family. Therefore, a
sex-linked pattern of inheritance was excluded and the inheritance
pattern was autosomal dominant (Fig.
1).
Mutational bioinformatics
analysis
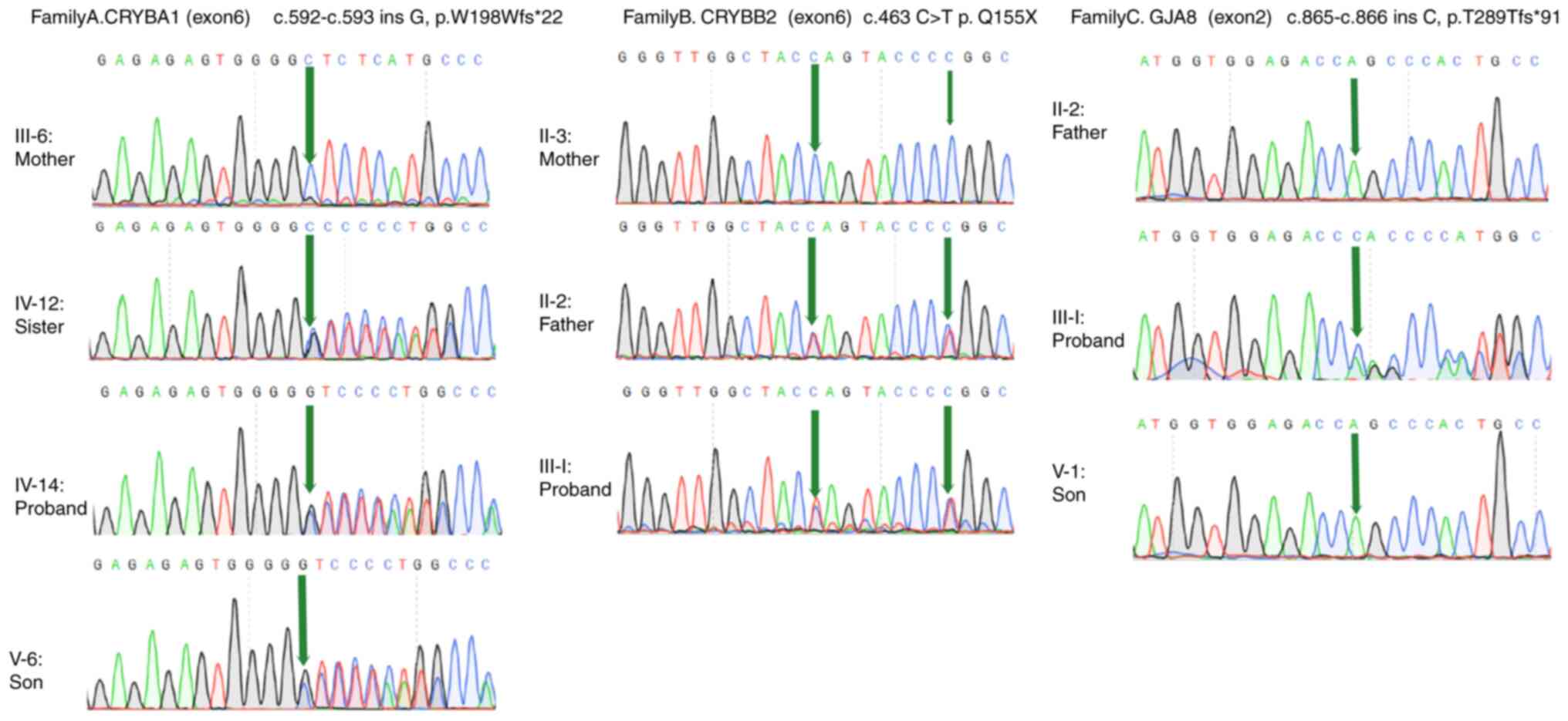
In the proband of family A (IV-14), a mutation in
exon 6 of CRYBA1/A3, c.592-593insG, was identified,
resulting in a shift in the amino acid coding sequence
(p.W198Wfs*22). In the proband of family B (III-1), a mutation in
exon 6 of CRYBB2 (c.463C > T) was found, resulting in a
premature stop codon (p.Q155X). Another mutation (c.471C > T) in
exon 6 of CRYBB2 was also detected in the proband and other
members of family B. However, the above mutation did not alter the
amino acid coding sequence. In the proband of family C (III-5), a
mutation in exon 2 of GJA8 (c.865-866insC) was detected,
resulting in a shift of amino acid coding sequence (p. T289Tfs*91).
In the corresponding family, the same mutation was found in all
affected, but not in unaffected, members. However, the three
mutations were not detected in 100 healthy individuals (Fig. 2). Among all three mutations, two
were frameshift mutations and one was nonsense mutation.
Hypothetically, the above two mutations could lead to abnormal
changes in the amino acid sequence of the protein or produce a
truncated protein with a high probability of being structurally
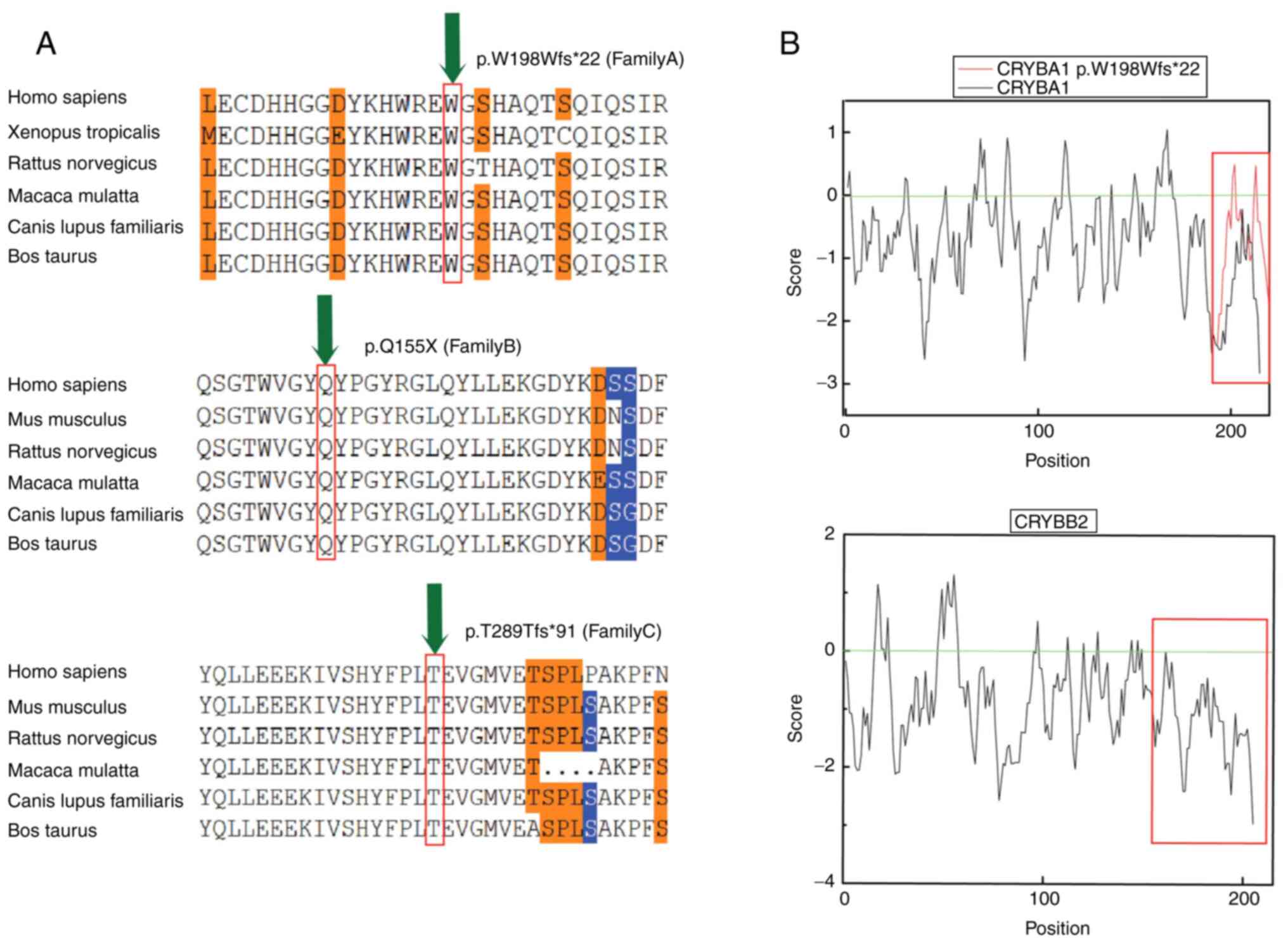
deleterious. Furthermore, sequence conservation analysis was
performed (Fig. 3A). Since
crystallines occupy a large proportion of the lens and their
solubility can affect the their transparency, hydrophobicity
analysis of the two crystallines was performed. Therefore, analysis
of the amino acids encoded by CRYBA1/A3 and CRYBB2
demonstrated that the hydrophobicity of the amino acid was
increased when the tryptophan codon 198 was mutated. In addition,
the solubility of the mutant crystallin beta a1/a3 (cryba1/a3)
protein was decreased compared with that of the native cryba1/a3
protein. Hydrophobicity analysis also demonstrated that after
glutamine, located at position 155 of crybb2, the deleted amino
acids were all hydrophilic, with a mean value of −1.28. This effect
resulted in reduced solubility of the mutated protein compared with
the native one (Fig. 3B).
Discussion
The members of the β-crystalline family Are the most
abundant water-soluble cytoplasmic proteins in human lens. This
family consists of two groups with seven members. Cryba1-a4 are
acidic proteins, while crybb1-b3 are basic proteins and are encoded
by CRYBA1/A3, CRYBA2, CRYBA4 and CRYBB1-B3,
respectively. Cryba1 and cryba3 are encoded by CRYBAl/A3
(16,17). All β-crystallines encompass two
domains, each consisting of two highly conserved ‘Greek key’ motifs
(17,18). The physiological expression of
β-crystalline is associated with normal eye development and normal
vision.
CRYBA1/A3 is located on 17q11.2 and consists
of six exons. Exons 3, 4, 5 and 6 mainly encode the ‘Greek key’
motifs, the linker polypeptide and the carboxy terminus (17,19).
In family A, an unreported mutation in exon 6 of CRYBA1/A3
(c.592-c.593insG) was detected. The above mutation caused a shift
in the codon sequence. Therefore, compared with the native protein,
22 amino acids after tryptophan at position 198 changed, while the
amino acid sequence was increased by five amino acids. Previous
studies revealed several splice site mutations and a small number
of missense mutations in CRYBA1, resulting in congenital
Y-suture cataract (19,20). In the present study, an insertion
mutation in CRYBA1 was identified for the first time in a
Chinese family with congenital cataract and a nuclear cataract
phenotype. This insertion mutation caused a frameshift in the
protein coding sequence, thus leading to an altered protein
structure, eventually resulting in congenital cataract.
CRYBB2 is located on 22q11.23 and consists of
six exons. The complete crybb2 is composed of 205 amino acids
(21). In the present study, in
family B, a nonsense mutation in exon 6 of CRYBB2 (c.463C
> T) was identified. The fourth ‘Greek key’ motif of crybb2
protein is encoded by amino acids 149–191, while glutamine at
position 155 is located at the beginning of this motif. Mutant
protein was 51 amino acids shorter compared with the native one,
since the fourth ‘Greek key’ motif and the C terminal domain were
lost. This mutation has been previously reported in a family with
congenital cerulean cataract, in a four-generation Swiss family
with autosomal dominant Coppock-like cataract, in a five-generation
Indian family with sutural cataract with punctate and cerulean
opacities and in a four-generation Chilean family segregating
autosomal dominant cataract with variable location, morphology,
color and density of opacities among affected family members
(16,22–24).
Different families with the above mutation could show a different
phenotype. In the present study, the phenotype of the family was
cataract with white punctate opacities.
Gap junction channels allow the selective passage of
ions and other molecules to promote the formation of electrical and
biochemical coupling between cells, thus maintaining normal lens
fiber cell physiology and tissue function. The above process is
also essential for regulating the microcirculation system of the
lens to preserve their stability (12,13,25).
A previous study demonstrated that Cx50, encoded by GJA8,
was expressed in the epithelial and fiber cells of the lens
(26). The p.T289Tfs*91 frameshift
mutation in GJA8 has not been previously reported. This
mutation resulted in changes in the topological domain at the end
of the protein. Additionally, the mutated protein was 53 amino
acids shorter compared with the native protein. Cx50, a link
protein, consists of four transmembrane domains (T1-T4), two
extracellular loops (EL1, EL2), an intercellular loop (IL), the N
terminal domain and the cytoplasmatic C terminal domain. Pathogenic
loci in patients with GJA8-related cataract are continuously
being identified. A previous study in a family with lamellar
cataract, identified several mutations in GJA8, that could
affect the T2 transmembrane domain of Cx50 (27). Additionally, the missense variant
V64G was detected in the developmentally conserved EL1 (28). Other studies also demonstrate that
the glu48lys mutation is associated with the development of banding
zonular nuclear cataract, while the autosomal dominant lamellar
cataract is associated with two mutations in GJA8, namely
P88S and P88Q (29,30). The insertion mutation in
GJA8 at codon 203, producing a truncated protein and the
missense mutation c.217T > C, are both associated with autosomal
recessive cataract (31,32). High throughput sequencing of
samples derived from the members of a family with congenital
nuclear cataract detected a novel variant (c.166A > C) at
position 166 of the coding region of Cx50 (33). In the present study, a mutation
(c.865-c.866insC) in exon 2 of GJA8 was detected in family
C, resulting in a shift in the amino acid coding sequence
(p.T289Tfs*91). This mutation was associated with nuclear cataract.
These findings further supported the significant role of
GJA8 in maintaining the normal function of the lens and its
association with congenital cataract. Mutations at different
positions of the gene may exhibit different effects on clinical
signs.
The above phenomena, from the increased risk of
age-related and congenital cataract to the development of band
nuclear cataract, lamellar powder cataract, congenital aphakia and
corneal sclerosis, indicate that fully understanding the
association between different mutation sites and phenotype is of
great importance (34,35). Additionally, further investigation
of the association between CRYBA1, CRYBB2 and GJA8,
three significant candidate genes and different cataract phenotypes
is urgently needed.
In the current study, three mutations associated
with congenital cataract were identified in three Chinese families
using WES technology and Sanger sequencing. More specifically, a
frameshift mutation in exon 6 of CRYBA1 (c.592-593insG), a
nonsense mutation in exon 6 of CRYBB2 (c.471C > T) and a
frameshift mutation in exon 2 of GJA8 (c.865-866 ins C) were
detected. Biological analysis revealed that all three mutations
were associated with congenital cataract in all three families.
Among the above mutations, two, one in CRYBA1 and one in
GJA8, were reported for the first time and were involved in
the development of congenital cataract. This finding further
expanded the pathogenic gene spectrum of cataract and lay the
foundation for unraveling the complex molecular basis and
pathogenesis of congenital cataract. However, the association
between the mechanism underlying the development of cataract and
genotype/phenotype should be further investigated. Each mutation
site was identified by only one family cohort. Due to the genetic
causes of the disease, there are more gene mutations involved, in
the sense that it is less likely that the mutation sites in the
collected families will be the same. To date, we have not come
across any other family with the same disease-causing locus of
CRYBA1 (p.W198Wfs*22) and CRYBB2 (p.Q155X). The authors will
continue to collect related cases in future studies. If multiple
families with the same gene mutation site are verified, its
association with different phenotypes of congenital cataract can be
further discussed, providing a theoretical basis for further study
of its molecular basis and further expand the genotype-phenotype
map with congenital cataract.
Acknowledgements
Part of this study was completed with the assistance
of the sequencing company (Beijing Zhiyin Oriental Translational
Medicine Research Center Co., Ltd.) which the authors thank.
Funding
The present study was supported by Medical Innovation Project of
Logistics Service (grant no. 18JS005), Open Subject of Jiangsu
Population Society (grant nos. JSPA2019017 and JSPA2019020) and
Jiangsu Funding Program for Excellent Postdoctoral Talent.
Availability of data and materials
The data generated in the present study may be found
in the SRA under accession number PRJNA944388, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA944388.
Authors' contributions
CQ and YH are responsible for designing the present
study, data analysis and drafting the manuscript. CJ and XZ were
responsible for collecting data, sorting literature, checking the
correctness of language and correcting errors. PZ and WL
participated in designing the present study and collecting samples.
HZ participated in the statistical analysis of the data. PZ and XX
confirm the authenticity of all the raw data. CX and XX
participated in designing the present study and critical
discussion. All the authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
the Jinling Hospital, Nanjing University School of Medicine and all
research subjects signed an informed consent. All methods were
performed in accordance with relevant guidelines and regulations.
All investigators adhered to the principles expressed in the
Declaration of Helsinki.
Patient consent for publication
The participants consented to the use of their blood
samples for the purpose of scientific research. The patients
consented to the images being taken for the purpose of research and
also consented to their publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sun W, Xiao X, Li S, Guo X and Zhang Q:
Exome sequencing of 18 Chinese families with congenital cataracts:
A new sight of the NHS gene. PLoS One. 9:e1004552014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li J, Chen X, Yan Y and Yao K: Molecular
genetics of congenital cataracts. Exp Eye Res. 191:1078722020.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiao X, Khan SY, Irum B, Khan AO, Wang Q,
Kabir F, Khan AA, Husnain T, Akram J, Riazuddin S, et al:
Correction: missense mutations in CRYAB are liable for recessive
congenital cataracts. PLoS One. 12:e01714032017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kumar M, Agarwal T, Kaur P, Kumar M,
Khokhar S and Dada R: Molecular and structural analysis of genetic
variations in congenital cataract. Mol Vision. 19:2436–2450.
2013.PubMed/NCBI
|
|
5
|
Khan I, Chandani S and Balasubramanian D:
Structural study of the G57W mutant of human gamma-S-crystallin,
associated with congenital cataract. Mol Vis. 22:771–782.
2016.PubMed/NCBI
|
|
6
|
Yue B, Haddad BG, Khan U, Chen H, Atalla
M, Zhang Z, Zuckerman DM, Reichow SL and Bai D: Connexin 46 and
connexin 50 gap junction channel properties are shaped by
structural and dynamic features of their N-terminal domains. J
Physiol. 599:3313–3335. 2021. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Valiunas V, Brink PR and White TW: Lens
connexin channels have differential permeability to the second
messenger cAMP. Invest Ophthalmol Vis Sci. 60:3821–3829. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brink PR, Valiunas V and White TW: Lens
connexin channels show differential permeability to signaling
molecules. Int J Mol Sci. 21:69432020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Berry V, Ionides A, Pontikos N, Georgiou
M, Yu J, Ocaka LA, Moore AT, Quinlan RA and Michaelides M: The
genetic landscape of crystallins in congenital cataract. Orphanet J
Rare Dis. 15:3332020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mothobi ME, Guo S, Liu Y, Chen Q, Yussuf
AS, Zhu X and Fang Z: Mutation analysis of congenital cataract in a
Basotho family identified a new missense allele in CRYBB2. Mol Vis.
15:1470–1475. 2009.PubMed/NCBI
|
|
11
|
Pauli S, Söker T, Klopp N, Illig T, Engel
W and Graw J: Mutation analysis in a German family identified a new
cataract-causing allele in the CRYBB2 gene. Mol Vis. 13:962–967.
2007.PubMed/NCBI
|
|
12
|
Micheal S, Niewold ITG, Siddiqui SN, Zafar
SN, Khan MI and Bergen AAB: Delineation of novel autosomal
recessive mutation in GJA3 and autosomal dominant mutations in GJA8
in Pakistani congenital cataract families. Genes (Basel).
9:1122018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li H and Homer N: A survey of sequence
alignment algorithms for next-generation sequencing. Brief
Bioinform. 11:473–483. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ghoneim DH, Myers JR, Tuttle E and
Paciorkowski AR: Comparison of insertion/deletion calling
algorithms on human next-generation sequencing data. BMC Res Notes.
7:8642014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bateman JB, von-Bischhoffshaunsen FR,
Richter L, Flodman P, Burch D and Spence MA: Gene conversion
mutation in crystallin, beta-B2 (CRYBB2) in a Chilean family with
autosomal dominant cataract. Ophthalmology. 114:425–432. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hegde S, Kesterson RA and Srivastava OP:
CRYβA3/A1-crystallin knockout develops nuclear cataract and causes
impaired lysosomal cargo clearance and calpain activation. PLoS
One. 11:e01490272016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hejtmancik JF: Congenital cataracts and
their molecular genetics. Semin Cell Dev Biol. 19:134–149. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang Z, Li Q, Ma Z, Guo Y, Zhu S and Ma X:
A G→T splice site mutation of CRYBA1/A3 associated with autosomal
dominant suture cataracts in a Chinese family. Mol Vis.
17:2065–2071. 2011.PubMed/NCBI
|
|
20
|
Ni SH, Zhang JM and Zhao J: A novel
missense mutation of CRYBA1 in a northern Chinese family with
inherited coronary cataract with blue punctate opacities. Eur J
Ophthalmol. 32:193–199. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Weisschuh N, Aisenbrey S, Wissinger B and
Riess A: Identification of a novel CRYBB2 missense mutation causing
congenital autosomal dominant cataract. Mol Vis. 18:174–180.
2012.PubMed/NCBI
|
|
22
|
Litt M, Carrero-Valenzuela R, LaMorticella
DM, Schultz DW, Mitchell TN, Kramer P and Maumenee IH: Autosomal
dominant cerulean cataract is associated with a chain termination
mutation in the human β-crystallin gene CRYBB2. Hum Mol Genet.
6:665–668. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gill D, Klose R, Munier FL, McFadden M,
Priston M, Billingsley G, Ducrey N, Schorderet DF and Héon E:
Genetic heterogeneity of the Coppock-like cataract: A mutation in
CRYBB2 on chromosome 22q11.2. Invest Ophthalmol Vis Sci.
41:159–165. 2000.PubMed/NCBI
|
|
24
|
Vanita, Sarhadi V, Reis A, Jung M, Singh
D, Sperling K, Singh JR and Bürger J: A unique form of autosomal
dominant cataract explained by gene conversion between
beta-crystallin B2 and its pseudogene. J Med Genet. 38:392–396.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Valiunas V and White TW: Connexin43 and
connexin50 channels exhibit different permeability to the second
messenger inositol triphosphate. Sci Rep. 10:87442020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xin L and Bai D: Functional roles of the
amino terminal domain in determining biophysical properties of Cx50
gap junction channels. Front Physiol. 4:3732013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Meşe G, Richard G and White TW: Gap
junctions: Basic structure and function. J Invest Dermatol.
127:2516–2524. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zheng JQ, Ma ZW and Sun HM: A heterozygous
transversion of connexin 50 in a family with congenital nuclear
cataract in the northeast of China. Zhonghua Yi Xue Yi Chuan Xue Za
Zhi. 22:76–78. 2005.PubMed/NCBI
|
|
29
|
Berry V, Mackay D, Khaliq S, Francis PJ,
Hameed A, Anwar K, Mehdi SQ, Newbold RJ, Ionides A, Shiels A, et
al: Connexin 50 mutation in a family with congenital ‘zonular
nuclear’ pulverulent cataract of Pakistani origin. Hum Genet.
105:168–170. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Berry V, Ionides A, Pontikos N, Moghul I,
Moore AT, Quinlan RA and Michaelides M: Whole exome sequencing
reveals novel and recurrent disease-causing variants in lens
specific gap junctional protein encoding genes causing congenital
cataract. Genes. 11:5122020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li L, Fan DB, Zhao YT, Li Y, Yang ZB and
Zheng GY: GJA8 missense mutation disrupts hemichannels and induces
cell apoptosis in human lens epithelial cells. Sci Rep.
9:191572019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ponnam SP, Ramesha K, Tejwani S,
Ramamurthy B and Kannabiran C: Mutation of the gap junction protein
alpha 8 (GJA8) gene causes autosomal recessive cataract. J Med
Genet. 44:e852007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hadrami M, Bonnet C, Veten F, Zeitz C,
Condroyer C, Wang P, Biya M, Sidi Ahmed MA, Zhang Q, Cheikh S, et
al: A novel missense mutation of GJA8 causes congenital cataract in
a large Mauritanian family. Eur J Ophthalmol. 29:621–628. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ceroni F, Aguilera-Garcia D, Chassaing N,
Bax DA, Blanco-Kelly F, Ramos P, Tarilonte M, Villaverde C, da
Silva LRJ, Ballesta-Martínez MJ, et al: New GJA8 variants and
phenotypes highlight its critical role in a broad spectrum of eye
anomalies. Hum Genet. 138:1027–1042. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Devi RR and Vijayalakshmi P: Novel
mutations in GJA8 associated with autosomal dominant congenital
cataract and microcornea. Mol Vis. 12:190–195. 2006.PubMed/NCBI
|