Introduction
With the development of the social economy and
improvements in living standards, the incidence of cardiovascular
disease is increasing annually and is an important factor
endangering human health (1). The
heart develops compensatory cardiac hypertrophy in response to
stress, which is primarily characterised by an increase in
cardiomyocyte size, protein synthesis, foetal gene re-expression
and extracellular matrix (2).
Cardiac hypertrophy, as a compensatory mechanism, is beneficial to
maintain normal cardiac function in the early stages of stress.
However, sustained stress causes cardiac decompensation, which
eventually results in cardiac dilation, heart failure and sudden
death (3). Cardiac hypertrophy is
also an independent risk factor for increased cardiovascular
morbidity and mortality (4).
Numerous diseases cause cardiac hypertrophy,
including primary or secondary hypertension, valvular heart disease
and thyroid disease. The incidence of cardiovascular events, such
as myocardial ischaemia, ventricular arrhythmias, heart failure and
sudden death, increases with the development of left ventricular
(LV) hypertrophy (5–7). At the cellular and molecular level,
the cardiomyocyte hypertrophy process primarily includes three
aspects: Extracellular hypertrophic signal stimulation,
intracellular signal pathway transduction and nuclear gene
transcription activation (8,9). The
AKT signalling pathway plays a key role in the development of
cardiac hypertrophy. Under the stimulation of pressure overload and
neurohumoral factors, such as catecholamine hormone, angiotensin II
and endothelin, the AKT signalling pathway participates in the
pathological process of cardiac hypertrophy by activating specific
genes in the nucleus of cardiomyocytes (10,11).
However, the mechanism underlying cardiac hypertrophy is not fully
understood, and there are no effective prevention or treatment
methods. Therefore, it is of theoretical and clinical value to
further clarify the molecular mechanism underlying cardiac
hypertrophy, and to identify drug targets for the prevention and
treatment of cardiac hypertrophy.
Tripartite motif-containing protein (TRIM), also
known as N-terminal RING finger/B-box/coiled coil protein, is a
ring component protein of E3 ubiquitin ligase in general. TRIM14 is
a member of the TRIM family primarily distributed on the outer
membrane of the mitochondria. TRIM14 activates interferon
regulatory factor 3 and the NF-κB pathway via recruiting
NEMO for promoting ubiquitination reactions that recruited NEMO to
the MAVS signaling complex to participate in anti-RNA virus effects
(12). TRIM14 is also involved in
the DNA signalling pathway mediated by TRIM14 recruiting
ubiquitin-specific protease 14 (USP14) to cleave the ubiquitin
chains of cGAS at lysine (K) 414 and then inhibiting the
degradation of CGAs via autophagy (13). TRIM14 plays an important role in
tumours. For example, TRIM14 has been shown to be expressed at high
levels in gastric cancer and osteosarcoma, and it may be used as a
prognostic indicator for patients. TRIM14 is also related to
malignant pathological factors in gastric cancer, and it may be
involved in its occurrence and development (14,15).
However, TRIM14 inhibits the proliferation of cells in non-small
cell lung cancer by promoting the apoptosis of cancer cells and
inducing the production of interferon γ (16). TRIM14 also plays an important role
in innate immunity to pathogenic microorganisms; it inhibits
hepatitis C, Sindbis and influenza viruses (17,18).
However, whether TRIM14 is involved in cardiac hypertrophy is not
clear.
In the present study, the expression level of TRIM14
protein was first detected in mouse cardiac hypertrophy, and then
TRIM14 was overexpressed in vivo and in vitro to
determine its role in cardiomyocyte hypertrophy and cardiac
hypertrophy, and its mechanism was further explored.
Materials and methods
Generation of TRIM14-transgenic
(TRIM14-TG) mice
Cardiac-specific TG mice with TRIM14 overexpression
were established as previously described. Animals were provided by
Wuhan University Model Animal Center (19). First, the full-length TRIM14 mouse
cDNA was inserted into a vector (α-MHC-DN.JNK2; Addgene) with the
mouse α-MHC promoter. TG C57BL/6 mice expressing TRIM14 were
generated by microinjecting the α-MHC-TRIM14 vector into fertilized
mouse embryos, which were then implanted into pseudopregnant
females to obtain the desired TG mice. Mice were housed in a
specific pathogen-free environment with free access to food and
water. The Animal Care and Use Committee of Zhejiang Chinese
Medical University approved the present study (approval no.
IACUC-20200120-04; Hangzhou, China). All animal-related procedures
complied with the National Institutes of Health Guide for the Care
and Use of Laboratory Animals (19). Roughly 100 embryos and 8 female
pseudopregnant mice were used. The mice were raised in a specific
pathogen-free (SPF) environment with the conditions of temperature
20–25°C, humidity of 40–70% and a 12-h light/dark cycle.
Transverse aortic constriction (TAC)
surgery
8-10-week-old male C57BL/6J mice weighing 25 g were
subjected to sham or TAC surgery as previously described (19). Mice were anaesthetised via a 50
mg/kg intraperitoneal injection of sodium pentobarbital. With the
mouse in the supine position and in the presence of the toe-pinch
reflex, the skin over the middle chest was opened, the aortic arch
was exposed through the right side of the clavicle, and a 26-gauge
needle was used to ligate the aortic arch with 7–0 silk sutures.
The needle was removed quickly and the chest was closed. A
self-regulating heating pad maintained the mouse body temperature
at ~37°C during this process. Mice were placed at 37°C until
recovery. Sham-operated mice underwent the same surgery with the
exception of aortic constriction. The mice were divided into the
following 4 groups: 10 mice in the NTG Sham group, 10 mice in the
TRIM14-TG Sham group, 11 mice in the NTG TAC group and 11 mice in
the TRIM14-TG TAC group.
Echocardiography measurements
After 4 weeks of surgery, echocardiography
(Vevo2100; FUJIFILM VisualSonics) measurements were taken from mice
under anaesthesia with isoflurane inhalation; the induction and
maintenance concentrations of isoflurane were 3 and 1.5–2%,
respectively, as previously described (19). M-mode tracings from more than three
consecutive cardiac cycles were recorded at the papillary muscle
level of the LV short axis. LV end-diastolic/end-systolic
dimensions (LVEDd/LVESd) were measured, and LV fractional
shortening (FS) was calculated. FS was calculated as FS
(%)=(LVEDd-LVESd)/LVEDd ×100.
Histological analysis
Histological analysis was performed as previously
described (19). After the
echocardiography measurements were completed, mice were euthanized
by cervical dislocation after being anesthetized with 3% isoflurane
inhalation. The death of mice was confirmed by respiratory and
cardiac arrest, and pupil dilation. The body weight (BW) was
measured. Hearts were excised, washed with saline. The heart weight
(HW), lung weight (LW) and HW/tibia length (TL) were measured. The
ratios of HW/BW, LW/BW and HW/TL were calculated to evaluate the
degree of cardiac hypertrophy. Subsequently, hearts were fixed with
4% paraformaldehyde at room temperature for 24 h and embedded in
paraffin following standard histological procedures. Sections (5
µm) were taken at the papillary muscle level of the heart and
stained with H&E according to standard protocols to assess
tissue morphology, and picrosirius red (PSR) to assess collagen
deposition. Images of H&E staining and PSR staining from more
than five samples per group were captured using light microscopy
(Olympus Corp.), and cardiomyocyte cross-sectional area and cardiac
fibrosis were measured using image analysis software (Image-Pro
Plus 6.0; Media Cybernetics). The partially dissected heart tissue
of each mouse was fixed for pathological analysis, while the
remaining portion was snap-frozen in liquid nitrogen and stored at
−80°C for reverse transcription-quantitative PCR (RT-qPCR) and
western blotting.
Neonatal rat cardiomyocyte (NRCM)
culture, TRIM14 overexpression and phenylephrine (PE)
treatment
A total of 45 neonatal Sprague Dawley rats were used
for the cell experiment. Neonatal Sprague Dawley rats (age,
1–3-days), were euthanized by decapitation following anaesthesia
with 1% isoflurane inhalation and the heart was rapidly excised.
Subsequently, primary NRCMs were isolated from ventricles and
cultured at 37°C and 5% CO2 in high-glucose DMEM
(HyClone) containing 15% foetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), 1% penicillin/streptomycin and 0.1 mM
5-bromodeoxyuridine, as previously described (19). After 24 h, NRCMs were infected with
adenovirus at a multiplicity of infection of 50 and for 12 h at
37°C. NRCMs were placed in serum-free medium for starvation, 50
µmol/l PE (Abcam) was added, and the cells were cultured for
another 24 h and harvested (20).
The control group was treated with the same volume of PBS. To
overexpress TRIM14, the entire coding region of the rat TRIM14 gene
(NM_001399174.1) was inserted into a replication-defective
adenovirus vector (AdTRIM14) under the control of the
cytomegalovirus promoter. An adenovirus vector containing the gene
encoding green fluorescent protein was used as a control (AdGFP).
The pcDNA 3.1-TRIM14 plasmid was synthesized and subsequently
packaged into adenovirus by Shanghai Shenggong Corporation.
Immunofluorescence
Immunofluorescence staining was performed as
previously described (19). NRCMs
used for immunofluorescence were grown in dishes containing
coverslips. At the end of NRCM treatment, immunofluorescence was
performed as follows: NRCMs were fixed with 4% formaldehyde at room
temperature for 15 min, permeabilised with 0.1% Triton X-100 and
blocked in a 10% BSA solution at room temperature for 30 min. NRCMs
were incubated with primary antibodies against α-actinin (1:100
dilution; Abcam) at 4°C overnight, followed by incubation with
fluorescent secondary antibodies (1:200 dilution; Thermo Fisher
Scientific, Inc.) at room temperature for 1 h and staining with
DAPI. Details of all antibodies are provided in Table I. In total, ≥100 cells were
selected from five different fields of view in a single experiment
to calculate the average cell size. A fluorescence microscope
(Olympus Corp.) was used to collect the fluorescence staining
images of NRCMs. The cell surface areas of >100 NRCMs were
calculated using Image-Pro Plus 6.0. The experiment was repeated
twice and data from three biological replicates were used for
statistical analysis.
 | Table I.Antibodies used in the present
study. |
Table I.
Antibodies used in the present
study.
| Antibody | Manufacturer | Catalogue
number | Species raised
in | Dilution |
|---|
| TRIM14 | Santa Cruz
Biotechnology, Inc. | sc-79761 | Goat | 1:1,000 |
| p-AKT | CST | 4060 | Rabbit | 1:1,000 |
| AKT | CST | 4691 | Rabbit | 1:1,000 |
| p-GSK-3β | CST | 9322s | Rabbit | 1:1,000 |
| GSK-3β | CST | 12456s | Rabbit | 1:1,000 |
| p-mTOR | CST | 5536s | Rabbit | 1:1,000 |
| mTOR | CST | 2983s | Rabbit | 1:1,000 |
| p-p70 S6K | CST | 9208 | Rabbit | 1:1,000 |
| p70 S6K | CST | 2708 | Rabbit | 1:1,000 |
| GAPDH | Proteintech | HRP-60004 | Mouse | 1:10,000 |
RT-qPCR
RT-qPCR was performed as previously described
(19). Total RNA was extracted
from cultured cells or the left ventricles of mice using
TRIzol® reagent (Thermo Fisher Scientific, Inc.). cDNA
was synthesised using the Transcriptor First Strand cDNA Synthesis
Kit (Roche Diagnostics) according to manufacturer's instructions.
SYBR™ Green PCR Master Mix (Roche Diagnostics) was used for qPCR,
and the products were quantified using the LightCycler 480 System
(Roche Diagnostics). The thermal cycling conditions for qPCR are as
follows: denaturation at 95°C for 10 sec, annealing at 60°C for 10
sec, extension at 72°C for 10 sec, for a total of 40 cycles. GAPDH
was used as a reference gene. The primers used in RT-qPCR are
provided in Table II.
| Table II.Sequences of primers used for PCR
(5′-3′). |
Table II.
Sequences of primers used for PCR
(5′-3′).
| A, Mouse
primers |
|---|
|
|---|
| Gene name | Forward | Reverse primer |
|---|
| ANP |
TCGGAGCCTACGAAGATCCA |
TTCGGTACCGGAAGCTGTTG |
| BNP |
GAAGGACCAAGGCCTCACAA |
TTCAGTGCGTTACAGCCCAA |
| MYH7 |
CAACCTGTCCAAGTTCCGCA |
TACTCCTCATTCAGGCCCTTG |
| Collagen Iα |
TGCTAACGTGGTTCGTGACCGT |
ACATCTTGAGGTCGCGGCATGT |
| Collagen III |
ACGTAAGCACTGGTGGACAG |
CCGGCTGGAAAGAAGTCTGA |
| CTGF |
TGACCCCTGCGACCCACA |
TACACCGACCCACCGAAGACACAG |
| GAPDH |
ACTCCACTCACGGCAAATTC |
TCTCCATGGTGGTGAAGACA |
| TRIM14 |
GAGAATGGCGAGCGAGACTA |
TTGGCGTACGGGCGTATTT |
|
| B, Rat
primers |
|
| Gene
name | Forward | Reverse |
|
| ANP |
AAAGCAAACTGAGGGCTCTGCTCG |
TTCGGTACCGGAAGCTGTTGCA |
| MYH7 |
GTTTGCTGAAGGACACTCAAATCC |
TTCTTCTTCTGGTTGATGAGGCTGG |
| GAPDH |
TGTGAACGGATTTGGCCCTA |
GATGGTGATGGGTTTCCCGT |
| TRIM14 |
ATGAAAAATGGCGAGCGGGA |
TTGAGTCTGCAGAAACCTGCG |
Western blot analysis
Western blotting was performed as previously
described (19). Total protein was
extracted from cultured cardiomyocytes or left ventricle tissues
using RIPA lysis (Beyotime Institute of Biotechnology) buffer
containing PMSF, protease inhibitor and phosphatase inhibitor. The
BCA method was used to determine protein concentration. Equal
amounts of protein (20 µg) were separated by SDS-PAGE (10%) and
transferred to PVDF membranes (Millipore-Sigma). Membranes were
blocked with 5% skimmed milk at room temperature for 1 h and
incubated with primary antibodies (Table I) at 4°C overnight. After
incubation with secondary antibodies conjugated with horseradish
peroxidase (HRP) (1:10,000 dilution; Proteintech) at room
temperature for 1 h, protein bands were visualised using an ECL
imaging system (Bio-Rad Laboratories, Inc.). Protein expression
levels were quantified using ImageJ v1.48 (National Institutes of
Health). GAPDH was used as an internal reference.
Statistical analysis
Data are presented as the mean ± SD (n=3). Student's
t-test was used for comparisons between two groups. Comparisons
among more than two groups were carried out using one-way ANOVA
followed by Tukey's post-hoc test. All statistical analyses were
performed using SPSS (version 21.0; IBM Corp.). P<0.05 was
considered to indicate a statistically significant difference.
Results
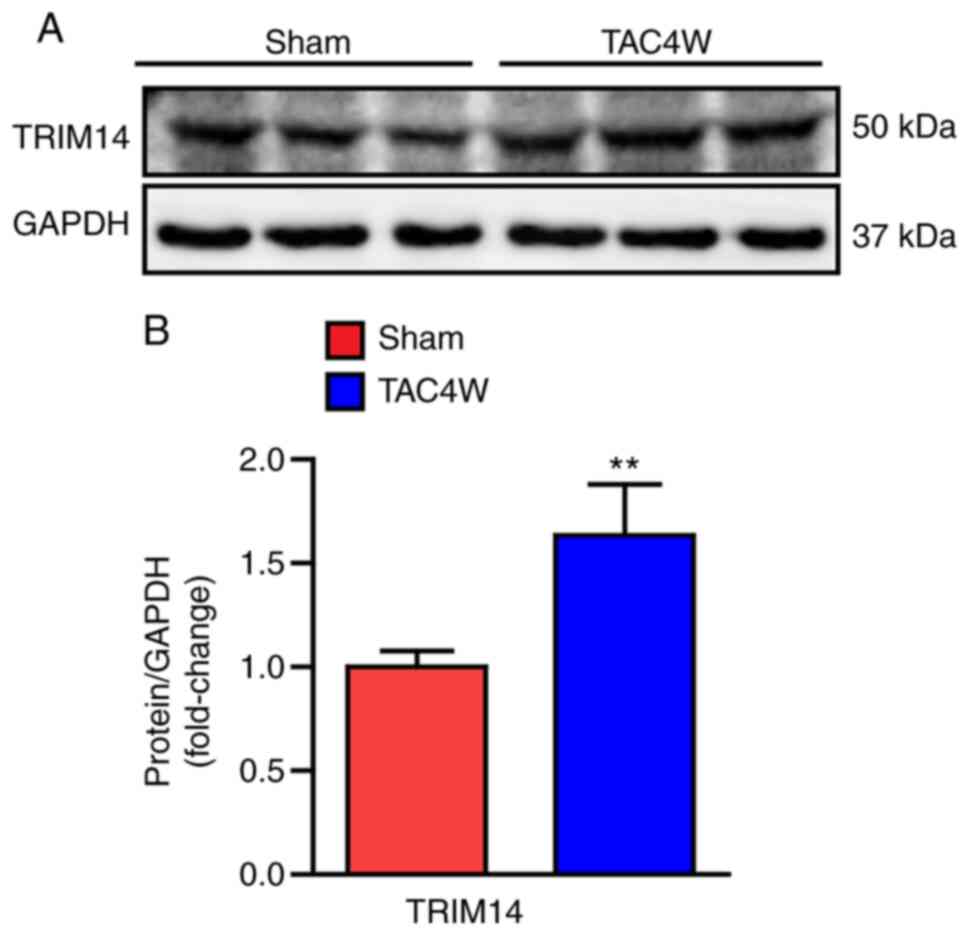
TRIM14 increases cardiac
hypertrophy
A model of cardiac hypertrophy was created in mice
that underwent TAC. The cardiac tissue of mice was collected and
the protein expression levels of TRIM14 were detected. The results
showed that the expression levels of TRIM14 were significantly
upregulated in the cardiac tissue of mice that underwent TAC
surgery compared with those in mice that received sham surgery
(Fig. 1A and B). These findings
suggested that TRIM14 was involved in the pathogenesis of cardiac
hypertrophy.
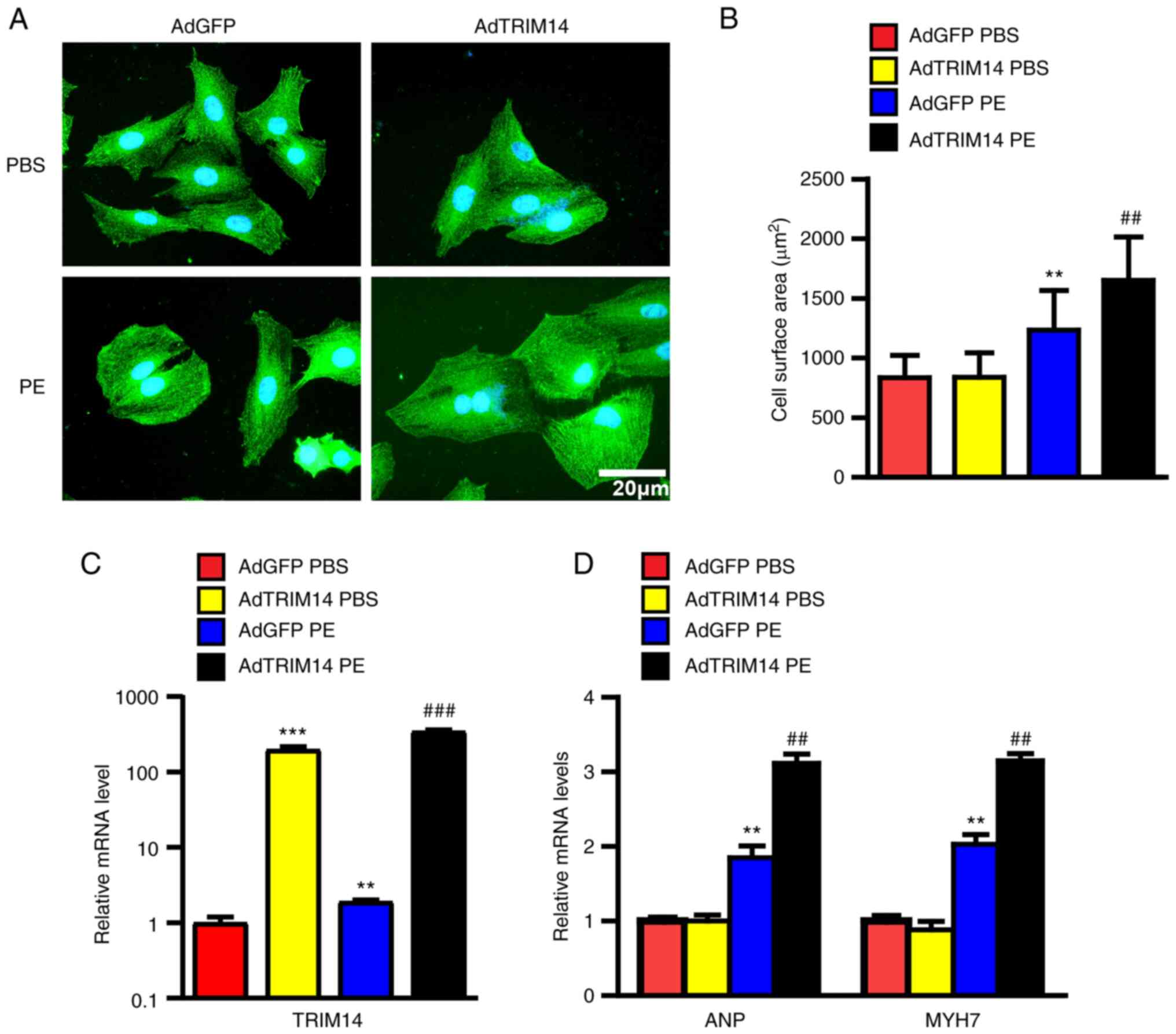
TRIM14 aggravates PE-induced
cardiomyocyte hypertrophy
To further assess the role of TRIM14 in cardiac
hypertrophy, primary cardiomyocytes were isolated from neonatal
rats. Cardiomyocyte hypertrophy was induced by PE stimulation
(Fig. 2A) and overexpression of
TRIM14 was mediated by adenovirus infection, which was confirmed by
a significant increase in mRNA levels (Fig. 2C). Following PE stimulation, the
surface area of cardiomyocytes in the AdTRIM14 group was
significantly higher than that in the AdGFP control group (Fig. 2B). The mRNA expression levels of
ANP and MYH7 in the after PE treatment AdTRIM14 group were also
significantly upregulated compared with those in the AdGFP group
(Fig. 2D). These results indicated
that TRIM14 promoted cardiomyocyte hypertrophy in vitro.
Cardiac-specific overexpression of
TRIM14 promotes dysfunction induced by pressure overload
To evaluate whether the overexpression of TRIM14 in
the heart aggravated cardiac hypertrophy, cardiac-specific
TRIM14-TG mice were created and bred as experimental animals, and
wild-type littermates [non-TG mice (NTG)] were used as controls.
Cardiac hypertrophy was induced by TAC surgery and echocardiography
was performed after 4 weeks. In total, ~10% of the mice that
underwent TAC surgery were excluded due to procedure failure and
other surgical complications, including acute heart failure and
wound infection caused by surgery. LVEDd, LVESd and FS were
measured. The results showed that LVEDd and LVESd were
significantly higher in the TRIM14-TG group than those in the NTG
group, and the FS percentage was lower than that in the NTG group
(Fig. 3A-C). After
echocardiography, the mice were sacrificed. The body weight (BW),
heart weight (HW) and lung weight (LW) of the sacrificed mice was
measured. The ratios of HW/BW, LW/BW and HW/tibia length (TL) were
calculated to evaluate the degree of cardiac hypertrophy. The
results showed that HW/BW, LW/BW and HW/TL in the TRIM14-TG group
were significantly higher than those in the NTG group (Fig. 3D-F). These results showed that the
degree of cardiac hypertrophy and deterioration of cardiac function
in TRIM14-TG mice after TAC was higher than that in NTG mice, which
indicated that TRIM14 had a positive regulatory effect on cardiac
hypertrophy and cardiac dysfunction caused by pressure load.
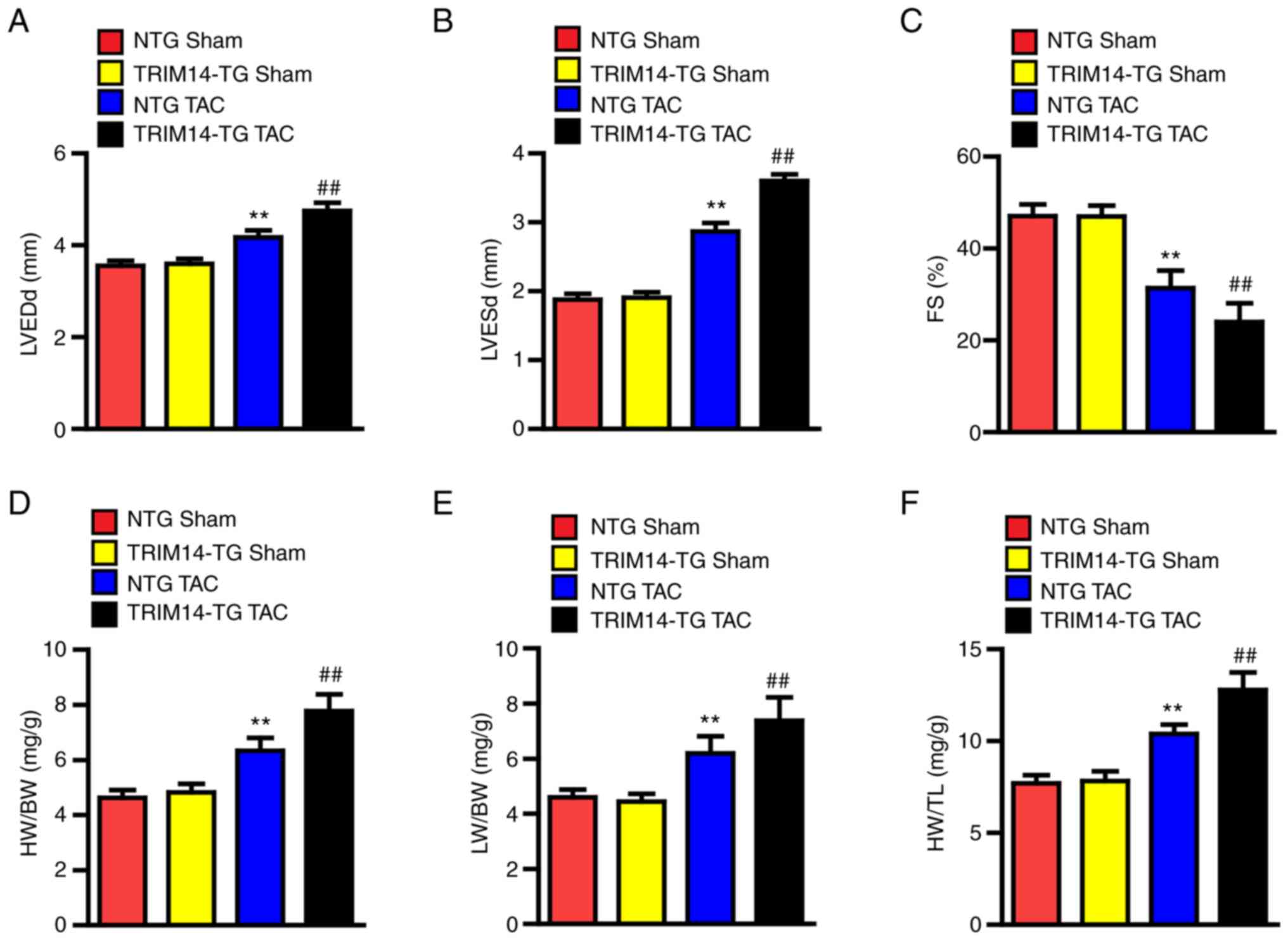 | Figure 3.TRIM14 overexpression exacerbates
cardiac hypertrophy and dysfunction caused by pressure overload. A
total of 4 weeks after sham or TAC surgery, the statistical results
of (A) LVEDd, (B) LVESd and (C) FS were obtained in 10 NTG mice and
10 TRIM14-TG mice. A total of 4 weeks after sham or TAC surgery,
the (D) HW/BW, (E) LW/BW and (F) HW/TL ratios of 10 NTG mice and 10
TRIM14-TG mice were analysed. **P<0.01 vs. NTG sham;
##P<0.01 vs. NTG TAC. TRIM14, tripartite
motif-containing 14; TAC, transverse aortic constriction; LVEDd,
left ventricular end diastolic diameter; LVESd, left ventricular
end systolic diameter; FS, fractional shortening; NTG,
non-transgenic; TG, transgenic; HW, heart weight; BW, body weight;
LW, lung weight; TL, tibia length. |
Cardiac-specific overexpression of
TRIM14 facilitates cardiomyocyte hypertrophy and fibrosis in
response to pressure overload
To further examine whether TRIM14 exacerbated
TAC-induced cardiac hypertrophy, histopathological and molecular
analyses were performed on mouse heart tissue. H&E and PSR
staining was used to observe cardiac tissue morphology,
cardiomyocyte cross-sectional area and the percentage of collagen
area. The results showed that the heart size, cardiomyocyte
cross-sectional area and percentage of collagen area (perivascular
and interstitial collagen; the statistical results of interstitial
fibrosis were not presented) of TRIM14-TG mice were significantly
increased after TAC compared with those in the NTG group (Fig. 4A-D). The mRNA levels of TRIM14 in
TRIM14-TG mice are significantly higher than those in NTG mice,
indicating the successful establishment of the TRIM14-TG mice
(Fig. 4E). The mRNA expression
levels of the hypertrophic markers ANP, BNP and MYH7, and those of
the fibrosis-related genes collagen I α, collagen III and CTGF,
were also detected in the left ventricle of mice. The mRNA
expression levels of ANP, BNP, MYH7, collagen I α, collagen III and
CTGF were significantly upregulated in TRIM14-TG mice after TAC
compared with those in NTG mice (Fig.
4F and G). These results suggested that TRIM14 contributed to
the development of cardiac hypertrophy and fibrosis.
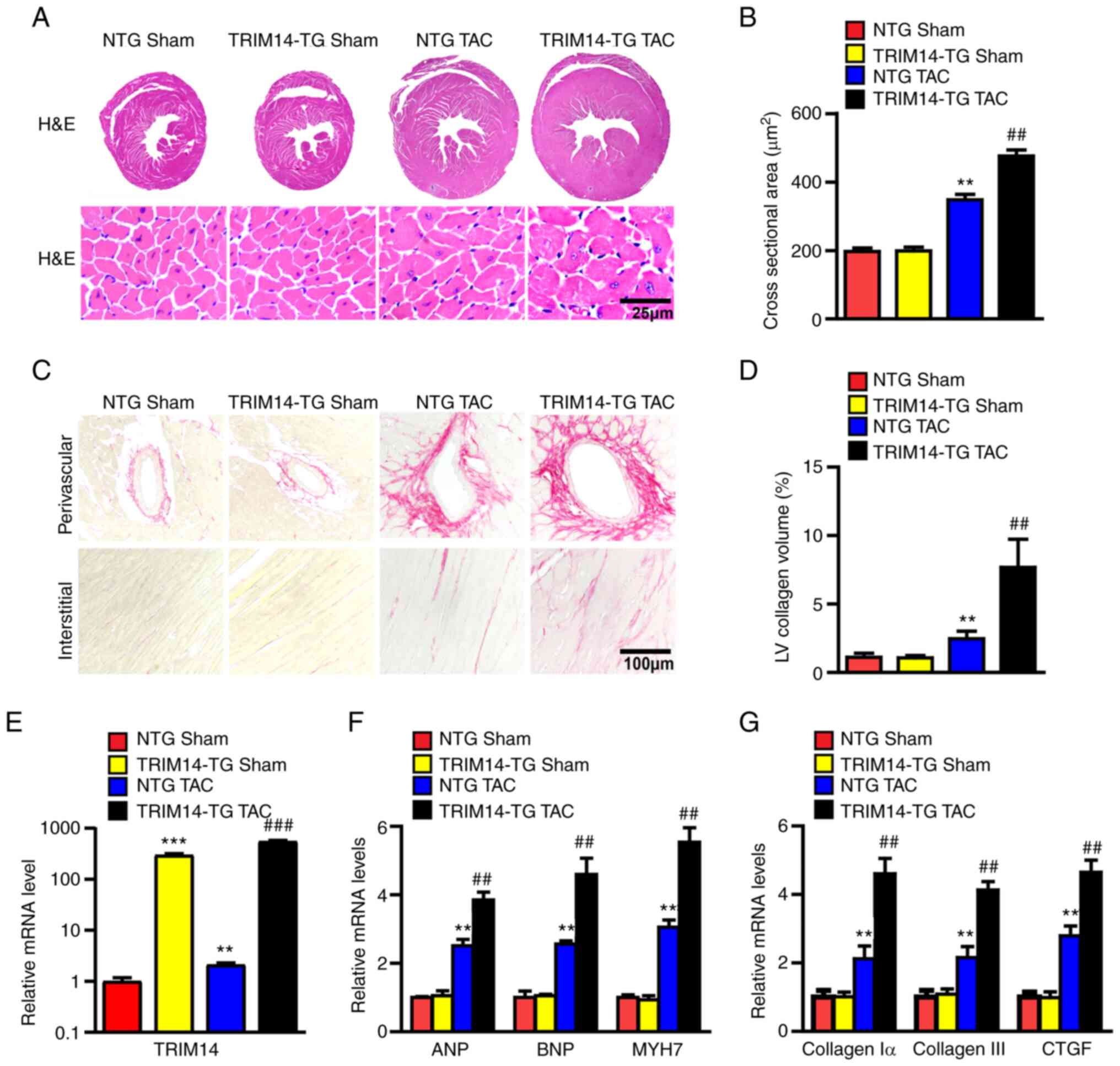 | Figure 4.Overexpression of TRIM14 promotes
cardiac hypertrophy and fibrosis. (A) Representative H&E
staining images of cardiac and cardiomyocyte cross-sectional area;
n=5. (B) Statistical results of cardiomyocyte cross-sectional area;
≥100 cells per group; scale bar, 25 µm; n=5. (C) Representative PSR
staining images of cardiac perivascular and interstitial collagen
fibres; scale bar, 100 µm; n=5. (D) Statistical results of cardiac
interstitial fibrosis; ≥40 fields per group. The relative mRNA
expression levels of (E) TRIM14, (F) ANP, BNP and MYH7, and (G)
collagen Iα, collagen III and CTGF were determined using reverse
transcription-quantitative PCR. n=6; **P<0.01, ***P<0.001 vs.
NTG sham; ##P<0.01, ###P<0.001 vs. NTG
TAC. TRIM14, tripartite motif-containing 14; PSR, picrosirius red;
NTG, non-transgenic; TAC, transverse aortic constriction; TG,
transgenic; LV, left ventricular. |
TRIM14 enhances activation of the AKT
signalling pathway under pressure overload
The results of the present study indicated that
TRIM14 promoted cardiac hypertrophy in vivo and in
vitro; however, the molecular mechanism underlying the effects
of TRIM14 in cardiac hypertrophy is not clear. Previous studies
have shown that activation of the AKT signalling pathway is closely
related to cardiac hypertrophy and that TRIM14 regulates the AKT
signalling pathway (15,21). Therefore, it was hypothesised that
TRIM14 may regulate cardiac hypertrophy via the AKT signalling
pathway. AKT signalling pathway-related proteins and their
phosphorylation levels were detected in cardiomyocytes and cardiac
tissues. Western blotting showed that the phosphorylation levels of
AKT, GSK-3β, mTOR and P70S6K in the AdTRIM14 group were
significantly elevated following PE treatment compared with those
in the AdGFP group (Fig. 5A and
B). The same trend was also observed in TRIM14-TG mice in
response to pressure overload (Fig. 5C
and D). These results suggested that TRIM14 overexpression
promoted cardiac hypertrophy by activating the AKT signalling
pathway.
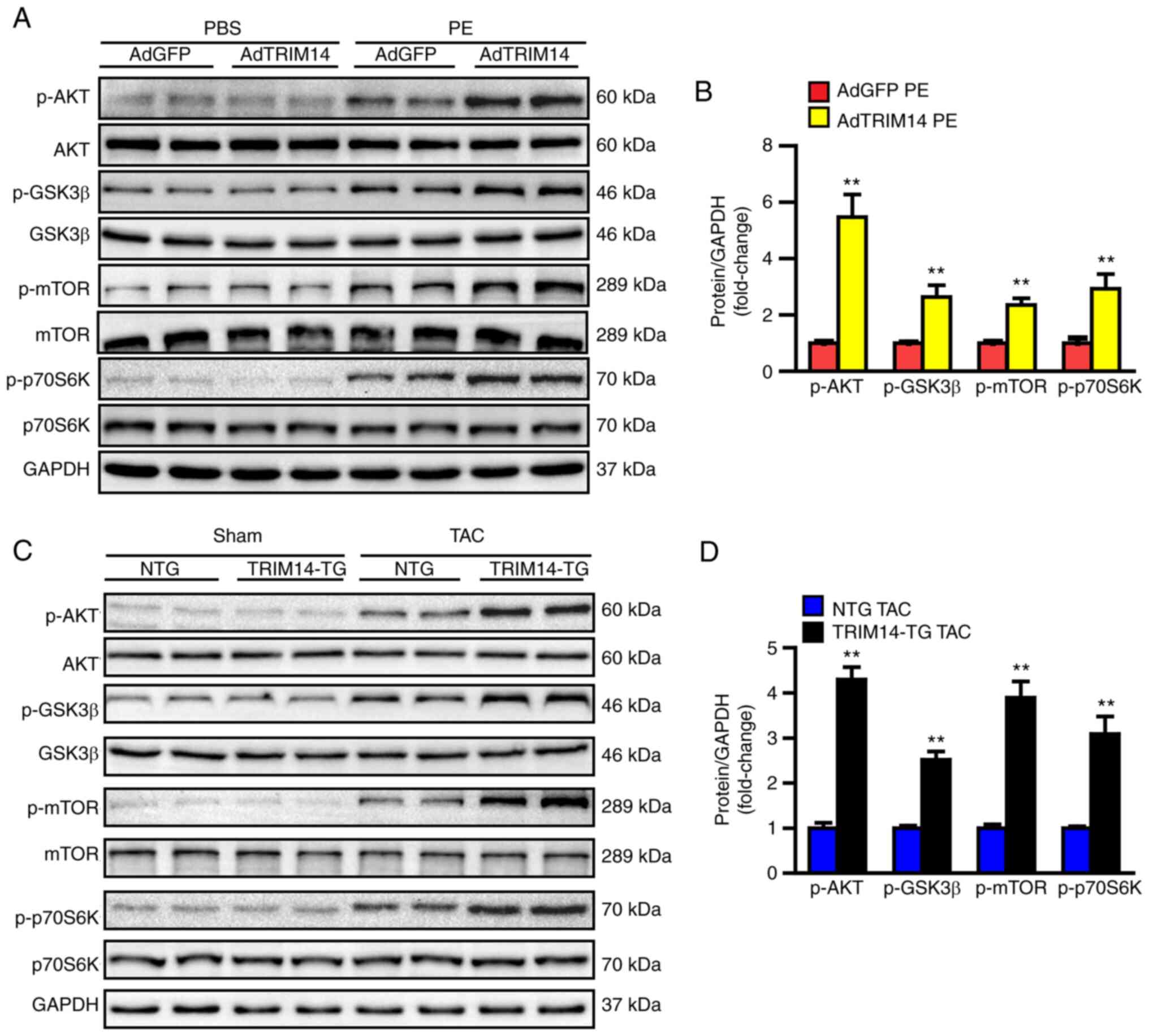 | Figure 5.TRIM14 enhances activation of the AKT
signalling pathway in response to hypertrophic stimulation. (A)
Western blotting of total and p-AKT, p-GSK-3β, p-mTOR and p-P70S6K
in NRCMs after infection with AdGFP or AdTRIM14 and treatment with
PBS or PE. (B) Semi-quantification of the protein expression levels
of p-AKT, p-GSK-3β, p-mTOR and p-P70S6K in NRCMs. **P<0.01 vs.
AdGFP PE; n=3. (C) Western blotting of total and p-AKT, p-GSK-3β,
p-mTOR and p-P70S6K in NTG and TRIM14-TG mice with TAC surgery. (D)
Semi-quantification of the protein expression levels of p-AKT,
p-GSK-3β, p-mTOR and p-P70S6K in mice. **P<0.01 vs. NTG TAC;
n=6. TRIM14, tripartite motif-containing 14; NRCMs, neonatal rat
cardiomyocytes; AdGFP, adenovirus GFP; AdTRIM14, adenovirus TRIM14;
PE, phenylephrine; p, phosphorylated; NTG, non-transgenic; TAC,
transverse aortic constriction; TG, transgenic. |
Discussion
The present study showed that TRIM14 played an
important role in cardiac hypertrophy and this effect may be
mediated by the AKT signalling pathway. To the best of our
knowledge, the current study is the first to propose the role of
TRIM14 in cardiac hypertrophy via activation of the AKT signalling
pathway. Findings revealed that the overexpression of TRIM14
increased cardiomyocyte surface area and hypertrophic markers in
NRCMs treated with PE, and it impaired cardiac function and
increased HW/BW, cardiomyocyte cross-sectional area, hypertrophic
markers, the percentage of collagen area and fibrosis-related genes
in mice following TAC surgery. In addition, the results revealed
that TRIM14 promoted cardiac hypertrophy by activating the AKT
signalling pathway in vivo and in vitro. The results
of the current study demonstrated that TRIM14 has a positive
regulatory effect on cardiac hypertrophy.
TRIM14 participates in multiple cell processes, such
as proliferation (22),
transformation (23), migration
(14), apoptosis (24), natural immune response (25) and protein degradation (17). Therefore, TRIM14 is associated with
a variety of pathological processes. TRIM14 is upregulated by
interferon I, and it is found in infection with human
immunodeficiency virus-1, hepatitis B virus, hepatitis C virus,
influenza A virus and other viruses, which suggests that TRIM14
participates in a wide range of natural immune regulatory responses
(17,18,26,27).
TRIM14 is also involved in cancer metastasis, invasion and cell
resistance (14,28). An increasing number of studies have
shown that tumour and immune regulatory factors, such as p53 and
IL6, have an important role in cardiac hypertrophy (29,30).
TRIM8 aggravates cardiac hypertrophy by enhancing the
TAK1-dependent signalling pathway, and TRIM32 suppresses cardiac
hypertrophy by inhibiting the AKT signalling pathway (31,32).
However, little attention has been given to TRIM14 in cardiac
hypertrophy. Molecules with similar structures generally have
similar functions, such as MYH6 and MYH7. Therefore, it was
hypothesised that TRIM14 may regulate cardiac hypertrophy. It was
observed that the protein expression levels of TRIM14 were
increased in mouse hypertrophic hearts, and that overexpression of
TRIM14 in NRCMs treated with PE increased cardiomyocyte surface
area and hypertrophic markers. HW/BW, LW/BW and HW/TL were measured
in TRIM14-TG mice following TAC surgery to test the proposed
hypothesis. It was demonstrated that the aforementioned ratios were
significantly increased, which suggested that TRIM14 promotes
pathological cardiac hypertrophy. Further experiments revealed that
the cardiomyocyte cross-sectional area, the percentage of collagen
area and the mRNA expression levels of the hypertrophic markers
ANP, BNP and MYH7, and of the fibrosis-related genes collagen I α,
collagen III and CTGF, were also significantly elevated in
TRIM14-TG mice. Under the stimulation of inappropriate body fluid
or pressure factors, such as increased cardiac volume load due to
aortic valve regurgitation and increased cardiac afterload due to
hypertension, TRIM14 may activate factors of cardiac hypertrophy
and fibrosis via its ubiquitination of these factors, which leads
to pathological changes in cardiac hypertrophy (11,22).
However, the mechanism underlying the effects of TRIM14 on
regulation of cardiac hypertrophy is not clear.
Numerous signalling pathways are involved in cardiac
hypertrophy, such as IGF-I/PI3K/AKT/PKB signalling pathway, mTOR
pathway and the CaN-NFAT (calcineurin-nuclear factor of activated T
cell) and MAPK (mitogen-activated protein kinase) pathways
(33–35). Yan et al (36) revealed that AKT1 was ubiquitinated
and simultaneously interacted with k63, which promoted AKT1
accumulation at the plasma membrane. AKT was activated via
phosphorylation of T308, which further activated the downstream
GSK-3β/mTOR/p70S6K signalling pathway to promote the synthesis of
hypertrophic factors and fibrin, which in turn led to cardiac
hypertrophy and dysfunction. A previous study reported that TRIM14
contributes to osteosarcoma cell proliferation by upregulating the
AKT signalling pathway, and inhibition of AKT activity reverses the
effect of TRIM14 on cell proliferation (15). Since the AKT signalling pathway
plays a key role in cardiac hypertrophy, it was hypothesized that
TRIM14 regulated cardiac hypertrophy via the AKT signalling
pathway. It was shown that the levels of phosphorylated (p)-p-AKT,
p-GSK-3β, p-mTOR and p-p70S6K were significantly increased in the
cardiac tissue of TRIM14-TG mice following TAC surgery and in the
cardiomyocytes infected with AdTRIM14 and treated with PE. These
findings suggested that TRIM14 may promote cardiac hypertrophy by
activating the AKT signalling pathway. However, the limitations of
the present study include the lack of verification as to whether
TRIM14 knockdown suppressed cardiac hypertrophy, and whether
inhibition of AKT reversed the pro-cardiac hypertrophy effect of
TRIM14. Therefore, further research is needed to investigate these
issues.
In the current study, the focus was primarily on the
effect of TRIM14 on the heart, and not on blood lipid levels;
therefore, serum samples were not collected. This is another
limitation of the present study and monitoring changes in blood
lipids will be taken under consideration in future studies.
In conclusion, the current study suggested that
TRIM14 may promote cardiac hypertrophy by activating the AKT
signalling pathway. Therefore, TRIM14 may be a novel target for
preventing cardiac hypertrophy.
Acknowledgements
Not applicable.
Funding
This work was supported by the General Project of Hubei
Provincial Health Commission of China (grant no. WJ2021M071).
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HH performed the experiments, analysed the data and
wrote the manuscript. YC and XF analysed the data. GX performed the
experiments. MY designed the study and performed most of the
experiments. HH and MY confirm the authenticity of all the raw
data. All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Animal experiments were approved by the Animal Care
and Use Committee of The Third Affiliated Hospital of Zhejiang
Chinese Medical University (approval no. IACUC-20200120-04;
Hangzhou, China). All animal-related procedures complied with the
National Institutes of Health Guide for the Care and Use of
Laboratory Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Roth GA, Mensah GA, Johnson CO, Addolorato
G, Ammirati E, Baddour LM, Barengo NC, Beaton AZ, Benjamin EJ,
Benziger CP, et al: Global burden of cardiovascular diseases and
risk factors, 1990–2019: Update from the GBD 2019 study. J Am Coll
Cardiol. 76:2982–3021. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carreño JE, Apablaza F, Ocaranza MP and
Jalil JE: Cardiac hypertrophy: Molecular and cellular events. Rev
Esp Cardiol. 59:473–486. 2006.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shimizu I and Minamino T: Physiological
and pathological cardiac hypertrophy. J Mol Cell Cardiol.
97:245–262. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ciarambino T, Menna G, Sansone G and
Giordano M: Cardiomyopathies: An Overview. Int J Mol Sci.
22:77222021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gallo S, Vitacolonna A, Bonzano A,
Comoglio P and Crepaldi T: ERK: A key player in the pathophysiology
of cardiac hypertrophy. Int J Mol Sci. 20:21642019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kawel-Boehm N, Kronmal R, Eng J, Folsom A,
Burke G, Carr JJ, Shea S, Lima JAC and Bluemke DA: Left ventricular
mass at MRI and long-term risk of cardiovascular events: The
multi-ethnic study of atherosclerosis (MESA). Radiology.
293:107–114. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Barbieri A, Bartolacelli Y, Bursi F,
Manicardi M and Boriani G: Remodeling classification system
considering left ventricular volume in patients with aortic valve
stenosis: Association with adverse cardiovascular outcomes.
Echocardiography. 36:639–650. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schirone L, Forte M, Palmerio S, Yee D,
Nocella C, Angelini F, Pagano F, Schiavon S, Bordin A, Carrizzo A,
et al: A review of the molecular mechanisms underlying the
development and progression of cardiac remodeling. Oxid Med Cell
Longev. 2017:39201952017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haque ZK and Wang DZ: How cardiomyocytes
sense pathophysiological stresses for cardiac remodeling. Cell Mol
Life Sci. 74:983–1000. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nakamura M and Sadoshima J: Mechanisms of
physiological and pathological cardiac hypertrophy. Nat Rev
Cardiol. 15:387–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang H, Wang XX, Zhou CY, Xiao X, Tian C,
Li HH, Yin CL and Wang HX: Tripartite motif 10 regulates cardiac
hypertrophy by targeting the PTEN/AKT pathway. J Cell Mol Med.
24:6233–6241. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu S, Chen J, Cai X, Wu J, Chen X, Wu YT,
Sun L and Chen ZJ: MAVS recruits multiple ubiquitin E3 ligases to
activate antiviral signaling cascades. Elife. 2:e007852013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen M, Meng Q, Qin Y, Liang P, Tan P, He
L, Zhou Y, Chen Y, Huang J, Wang RF and Cui J: TRIM14 inhibits cGAS
degradation mediated by selective autophagy receptor p62 to promote
innate immune responses. Mol Cell. 64:105–119. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang F, Ruan L, Yang J, Zhao Q and Wei W:
TRIM14 promotes the migration and invasion of gastric cancer by
regulating epithelialtomesenchymal transition via activation of AKT
signaling regulated by miR1955p. Oncol Rep. 40:3273–3284.
2018.PubMed/NCBI
|
|
15
|
Xu G, Guo Y, Xu D, Wang Y, Shen Y, Wang F,
Lv Y, Song F, Jiang D, Zhang Y, et al: TRIM14 regulates cell
proliferation and invasion in osteosarcoma via promotion of the AKT
signaling pathway. Sci Rep. 7:424112017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hai J, Zhu CQ, Wang T, Organ SL, Shepherd
FA and Tsao MS: TRIM14 is a putative tumor suppressor and regulator
of innate immune response in non-small cell lung cancer. Sci Rep.
7:396922017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang S, Chen Y, Li C, Wu Y, Guo L, Peng C,
Huang Y, Cheng G and Qin FX: TRIM14 inhibits hepatitis C virus
infection by SPRY domain-dependent targeted degradation of the
viral NS5A protein. Sci Rep. 6:323362016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu X, Wang J, Wang S, Wu F, Chen Z, Li C,
Cheng G and Qin FX: Inhibition of influenza A virus replication by
TRIM14 via its multifaceted protein-protein interaction with NP.
Front Microbiol. 10:3442019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu G, Liu Y, Huang H, Tang Y, Liu W, Mei
Y, Wan N, Liu X and Huang C: SH2B1 is critical for the regulation
of cardiac remodelling in response to pressure overload. Cardiovasc
Res. 107:203–215. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Z, Zhang XJ, Ji YX, Zhang P, Deng KQ,
Gong J, Ren S, Wang X, Chen I, Wang H, et al: The long noncoding
RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy.
Nat Med. 22:1131–1139. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nakamura Y, Kita S, Tanaka Y, Fukuda S,
Obata Y, Okita T, Kawachi Y, Tsugawa-Shimizu Y, Fujishima Y,
Nishizawa H, et al: A disintegrin and metalloproteinase 12 prevents
heart failure by regulating cardiac hypertrophy and fibrosis. Am J
Physiol Heart Circ Physiol. 318:H238–H251. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shen W, Jin Z, Tong X, Wang H, Zhuang L,
Lu X and Wu S: TRIM14 promotes cell proliferation and inhibits
apoptosis by suppressing PTEN in colorectal cancer. Cancer Manag
Res. 11:5725–5735. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Feng S, Cai X, Li Y, Jian X, Zhang L and
Li B: Tripartite motif-containing 14 (TRIM14) promotes
epithelial-mesenchymal transition via ZEB2 in glioblastoma cells. J
Exp Clin Cancer Res. 38:572019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu H, Sun B and Shen Q: TNF-α induces
apoptosis of human nucleus pulposus cells via activating the
TRIM14/NF-κB signalling pathway. Artif Cells Nanomed
Biotechnol. 47:3004–3012. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou Z, Jia X, Xue Q, Dou Z, Ma Y, Zhao Z,
Jiang Z, He B, Jin Q and Wang J: TRIM14 is a mitochondrial adaptor
that facilitates retinoic acid-inducible gene-I-like
receptor-mediated innate immune response. Proc Natl Acad Sci USA.
111:E245–E254. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nenasheva VV, Nikolaev AI, Martynenko AV,
Kaplanskaya IB, Bodemer W, Hunsmann G and Tarantul VZ: Differential
gene expression in HIV/SIV-associated and spontaneous lymphomas.
Int J Med Sci. 2:122–128. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tan G, Xu F, Song H, Yuan Y, Xiao Q, Ma F,
Qin FX and Cheng G: Identification of TRIM14 as a type I
IFN-stimulated gene controlling hepatitis B virus replication by
targeting HBx. Front Immunol. 9:18722018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Qiao CY, Qiao TY, Jin H, Liu LL, Zheng MD
and Wang ZL: LncRNA KCNQ1OT1 contributes to the cisplatin
resistance of tongue cancer through the KCNQ1OT1/miR-124-3p/TRIM14
axis. Eur Rev Med Pharmacol Sci. 24:200–212. 2020.PubMed/NCBI
|
|
29
|
Huang CY, Pai PY, Kuo CH, Ho TJ, Lin JY,
Lin DY, Tsai FJ, Padma VV, Kuo WW and Huang CY: p53-mediated miR-18
repression activates HSF2 for IGF-IIR-dependent myocyte hypertrophy
in hypertension-induced heart failure. Cell Death Dis. 8:e29902017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kumar S, Wang G, Zheng N, Cheng W, Ouyang
K, Lin H, Liao Y and Liu J: HIMF (hypoxia-induced mitogenic
factor)-IL (interleukin)-6 signaling mediates
cardiomyocyte-fibroblast crosstalk to promote cardiac hypertrophy
and fibrosis. Hypertension. 73:1058–1070. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen L, Huang J, Ji YX, Mei F, Wang PX,
Deng KQ, Jiang X, Ma G and Li H: Tripartite motif 8 contributes to
pathological cardiac hypertrophy through enhancing transforming
growth factor β-activated kinase 1-dependent signaling pathways.
Hypertension. 69:249–258. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen L, Huang J, Ji Y, Zhang X, Wang P,
Deng K, Jiang X, Ma G and Li H: Tripartite motif 32 prevents
pathological cardiac hypertrophy. Clin Sci (Lond). 130:813–828.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Al Asoom LI: Molecular mechanisms of
Nigella sativa- and Nigella sativa exercise-induced cardiac
hypertrophy in rats. Evid Based Complement Alternat Med.
2021:55530222021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen Y, Liu Z, Hu Z, Feng X and Zuo L:
Tripartite motif 27 promotes cardiac hypertrophy via PTEN/Akt/mTOR
signal pathways. Bioengineered. 13:8323–8333. 2022.PubMed/NCBI
|
|
35
|
Kumar S, Wang G, Liu W, Ding W, Dong M,
Zheng N, Ye H and Liu J: Hypoxia-induced mitogenic factor promotes
cardiac hypertrophy via calcium-dependent and hypoxia-inducible
factor-1α mechanisms. Hypertension. 72:331–342. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yan K, Ponnusamy M, Xin Y, Wang Q, Li P
and Wang K: The role of K63-linked polyubiquitination in cardiac
hypertrophy. J Cell Mol Med. 22:4558–4567. 2018. View Article : Google Scholar : PubMed/NCBI
|