Introduction
Lung cancer is one of the most diagnosed types of
cancer, the most common in China and the leading cause of
cancer-related mortality worldwide, with an estimated 2 million new
cases and 1.76 million deaths every year (1,2).
Treatment options for small cell lung cancer (SCLC) are limited
compared with other types of lung cancer and provide only a
transient benefit for most patients (3). SCLC accounts for >15% of all lung
cancers (4) and most cases are
associated with smoking (5). SCLC
is characterized by rapid disease progression and early widespread
metastasis. Thus, 80–85% of patients have extensive-stage small
cell lung cancer (ES-SCLC) at the time of initial diagnosis
(6). For several years, the
standard treatment for patients with ES-SCLC has been
platinum-based combination chemotherapy, which has shown a survival
benefit; however, despite favorable initial treatment effects,
median survival has rarely exceeded 1 year (5). Most patients with ES-SCLC eventually
die due to cancer recurrence, with only 10–20% of patients
surviving longer than 2 years. Therefore, an in-depth search for
drug targets is needed to develop effective combination therapies
with minimal toxicity (5,7,8).
It is well known that the incidence of central
nervous system tumors has a place in the statistics table of new
cancers in China in 2022 (2).
Glioblastoma (GBM) is the most common malignant primary brain
tumor. GBM is classified by the World Health Organization as grade
IV and the median survival of patients is only 8–18 months
(8). Currently, treatment
strategies for glioma include surgery, radiotherapy, chemotherapy,
immunotherapy and targeted therapy (9,10).
However, chemotherapeutic drugs have limited therapeutic effects
when used alone (11). Therefore,
combining chemotherapeutic drugs with other carriers can overcome
drug resistance and reduce their toxic side effects (10). For example, drug delivery systems
such as polymeric micelles, liposomes and nanoparticles can all
synergistically facilitate the passage of chemotherapy drugs across
the blood-brain barrier to target the desired sites (10,12,13).
The main obstacles to effective chemotherapy for glioma are drug
resistance and toxicity. Accordingly, there is an urgent need for
new drugs and drug delivery systems to alleviate these two
challenges for clinical application.
The pioneering drug paclitaxel (PTX), extracted from
Taxus brevifolia, is a chemical drug derived from natural
plants approved by the U.S. Food and Drug Administration (14). There is considerable evidence to
suggest that PTX is one of the most successful and widely used
natural antitumor drugs in various tumors, including glioma
(15–17) and lung cancer (18,19).
Indeed, PTX selectively targets microtubules and causes cell cycle
arrest at the G2/M phase, inducing cytotoxic effects in
a dose- and time-dependent manner. Recently, development of several
innovative drug delivery formulations, such as nanoemulsions,
nanosuspensions, nanoparticles, liposomes and polymeric micelles,
have been used to enhance the targeted cell delivery of PTX
(20–23). However, the biological
characteristics of PTX, such as low bioavailability, poor water
solubility and high incidence of toxicity (24), may affect its antitumor efficacy to
a certain extent and cause allergies, gastrointestinal reactions,
neurotoxicity and other adverse reactions. However, the
aforementioned defects can be partially overcome with the help of
nano-drug delivery systems (25).
Nanoparticles have been the topic of emerging
research interest for their unique properties, including nanometer
size, biocompatibility, large surface-to-volume ratio and easy
surface modification (26).
Traditional antitumor chemotherapy drugs, such as single and
non-targeted drugs, are slowly being discontinued. For example,
resistance to PTX monotherapy can occur in the treatment of
prostate cancer, so docetaxel in combination with prednisone is now
the recommended treatment for this disease (27). Since combination therapy has the
synergistic effect of multiple drugs, which may act through
different pathways, it can improve efficacy, reduce drug dosage and
toxicity, and gradually replace the current cancer treatment
strategy (28). Pulvirent et
al (29) prepared a hybrid
system including the iron oxide magnetic nanoparticles and a
metal-organic framework subclass constituted by trivalent
transition metals and bi- or tri-carboxylic ligands (MNPs@MIL) with a particle size of ≤50 nm,
which retains the magnetic properties of the iron core and has the
loading capacity of porous containing iron ions and MILs. Moreover,
the same study showed that MNPs@MIL
could carry, load and release a higher quantity of drugs. In the
treatment of GBM with temozolomide, the MIL-modified MNPs were more
endocytosed than the naked MNPs at all concentrations due to the
capability of in situ PTX-loaded MNPs@MIL targeting to the nucleus and
cytoplasm to produce antitumor effects, suggesting that the hybrid
system is able to overcome the blood-brain barrier and target brain
tumors. Recently, iron-based iron oxide nanoparticles (IONP) have
been used in clinical practice (30). Drug-encapsulating nanoparticles can
enter the blood system and reach specific tumor sites through their
enhanced permeability and retention effect, which is defined as the
process of extravasation of large molecules from leaky tumor
vasculature and accumulation in the tumor tissue, continuously
releasing the drug and eventually producing a significant
inhibitory effect on tumor growth (31). It was shown that the PTX-loaded
IONPs (IONP@PTX) synthesized by the
present research group possess lower toxicity than PTX monotherapy,
suggesting that IONP can reduce the toxicity of drugs and improve
biosafety (32).
Autophagy is an evolutionarily conserved process for
cellular degradation, yet it is frequently viewed in cancer biology
as a double-edged sword with the twin functions of tumor
development and tumor suppression (33). Autophagy-related protein (ATG),
including Beclin 1 and mTOR, and autophagic pathways (i.e., the
autophagy-lysosome pathway, ubiquitin-like protein system and mTOR
signaling pathway) are involved in the pathological processes of
cancer development (33,34). As one of the most crucial protein
complexes in the creation of the autophagic pathway, Beclin1 is the
first mammalian discovered tumor-associated ATG protein and it also
plays a vital role in the production of autophagosomes as well as
their expansion and maturation (35). mTOR1, a key autophagy regulator
connected to endosomes and lysosome membranes, interacts with its
effectors through phosphorylation and plays a role in the
development of lysosomes as well as the suppression of the
autophagic process (36,37). Sequestosome1 (p62) is a
multifunctional adapter protein that regulates the accumulation of
protein aggregates and autophagic clearance (38). Histone deacetylase 6 (HDAC6) is a
member of class IIb HDAC family and it deacetylates microtubule
proteins, resulting in microtubule depolymerization and
disconnection of the autophagosomal lysosomal fusion pathway
(39–41), thereby inhibiting a member of the
ATG8 family, LC3, which is a marker of the autophagic process.
Moreover, the state of cellular redox homeostasis has a significant
impact on autophagy (42). A
growing body of evidence suggests that excessive autophagy and
lysosome activation may cause lipid peroxidation (LPD), promoting
ferroptosis (43). Accordingly, it
has also been recommended to induce autophagy-mediated cellular
ferroptosis to kill cancer cells (43).
Ferroptosis is a nonapoptotic programmed cell death
process that can eradicate tumors via reactive oxygen species (ROS)
accumulation and iron-dependent pathways (44). In the acidic environment of tumors,
Fe2+ and Fe3+ ions released from the
iron-based nanoparticles participate in the Fenton reaction, which
generates ROS and triggers LPD, eventually leading to excessive
accumulation of iron ions and inducing tumor cell death by
ferroptosis (45). In addition,
ferroptosis can be triggered by inhibiting two types of
small-molecule substances, namely system Xc-mediated cystine uptake
and glutathione peroxidase 4 (GPX4) (46). On the other hand, depleting
L-cysteine (Cys) can also sensitize cells to ferroptosis through
direct or indirect cysteine dioxygenase type 1-mediated metabolism
of Cys, thereby reducing glutathione (GSH) expression (46). In a previous study, the nuclear
factor erythroid 2-related factor 2 (Nrf2) was demonstrated to be
involved in ferroptosis regulation and the treatment of
neurodegenerative diseases (47).
PTX is one of the most frequently prescribed
medications in Japanese clinical practice to treat recurrent SCLC
and the efficacy of nanoparticle albumin-bound (nab)-PTX
monotherapy might be moderate for heavily treated, relapsed SCLC
patients (19). Several studies
reported the use of PTX combined with carboplatin, gemcitabine or
albumin-bound PTX nanoparticles for the treatment of metastatic or
recurrent SCLC, indicating that PTX as second-line chemotherapy is
a good choice for the treatment of patients with SCLC (48–50).
After nano-microsized modification, PTX is an ideal drug for the
treatment of SCLC due to its improved therapeutic efficiency and
diminished side effects. Based on nanoformulations, PTX can improve
drug solubility, targeted activity, attenuate side effects after
excisional surgery and effectively inhibit the growth of GBM cells
(51,52). Accumulating evidence suggests that
PTX is a broad-spectrum antitumor drug and has been used against
human cancers, including glioma (25,53–55).
For instance, it has recently been reported that an in situ
targeted nanoparticle-hydrogel hybrid system modified with PTX
could enhance the therapeutic effect of chemo-immunotherapy on
residual infiltrative glioma (56). Nonetheless, to the best of our
knowledge, there are only a few reports on the effects of IONP@PTX on NCI-H446 and M059K cells.
Therefore, elucidating the possible mechanisms underlying the
treatment of IONP@PTX in vitro
is of profound importance for the development of universal
application targeted therapies against SCLC and GBM, which may
provide new therapeutic ideas for clinical applications,
particularly for non-invasive therapeutic approaches. On this
basis, the present study focused on the possible synergistic effect
of IONP@PTX on SCLC H446 and GBM
M059K cells and further explored its potential mechanisms.
Materials and methods
Preparation of IONP@PTX
High-temperature pyrolysis was used to create oleic
acid-coated IONP. Briefly, 0.7 g Fe(acac)3 (Shanghai
Aladdin Chemical Reagent Co., Ltd.), 3.1 ml oleic acid (Shanghai
Aladdin Chemical Reagent Co., Ltd), and 0.9 ml oleylamine (Shanghai
Aladdin Chemical Reagent Co., Ltd.) were dissolved in 20 ml benzyl
ether (Shanghai Aladdin Chemical Reagent Co., Ltd.), preheated at
220°C for 1 h and subsequently heated to 280–300°C for 30 h under
the protection of nitrogen. After cooling to room temperature, the
mixture was transferred into 30 ml anhydrous ethanol to collect the
oleic acid-coated IONP through magnetic separation and then
dissolved in 10 ml chloroform. Subsequently, 150 mg
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino
(polyethylene glycol)-2000] (DSPE-PEG 2000) (Shanghai Advanced
Vehicle Technology, Co., Ltd.) and 5.5 mg PTX (Shanghai Aladdin
Chemical Reagent Co., Ltd) were weighed and put into an
eggplant-shaped container with 5 ml trichloromethane. Then, 11 mg
oleic acid-coated IONP and 11 ml distilled water were added and
mixed ultrasonically to make an emulsion, as previously described
(32). Thereafter, the suspension
was concentrated through gentle solvent evaporation using a
rotavapor for 50 min in a 70°C water bath until the product was
clear and transparent without visible bubbles, as previously
described (32). Finally, the
mixed emulsion was transferred to an ultrafiltration tube (0.5 ml,
3 kDa) to separate and purify the IONP@PTX, as previously described (32).
Drug loading capacity
Briefly, 1 mg/ml IONP@PTX solution was aspirated and diluted
25 times to measure the peak area by high performance liquid
chromatography at 227 nm, as descried in our previous study
(32). The peak area, which was
293522, was substituted into the standard curve y=26736 × + 1154.9,
R2=0.9987, to obtain the concentration and then to
calculate the loading capacity of PTX/mg (iron concentration)
solution using the following formula: Loading
capacity=concentration × dilution × volume.
Cell culture
Human SCLC NCI-H446 cells and human GBM M059K brain
malignant cells were obtained from Jiangsu KGI Biotechnology Co.,
Ltd. NCI-H446 cells were cultured in 90% RPMI-1640 (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml
streptomycin (P/S). Similarly, M059K cells were cultured in 90%
DMEM/F12 (Gibco; Thermo Fisher Scientific, Inc.) supplemented with
10% FBS and P/S (Beijing Solarbio Science & Technology Co.,
Ltd.). Both cell lines were placed in an incubator at 37°C with 5%
CO2 and saturated humidity (Thermo Fisher Scientific,
Inc.).
Cell viability
Following digestion with 0.25% EDTA-free trypsin
(cat. no. C0205; Shanghai Biyuntian Biotechnology Co., Ltd.), the
NCI-H446 and M059K cells were separately counted and adjusted to a
cell suspension of 5×104 cells/ml. Subsequently, 100 µl
cell suspension was transferred to each well of a 96-well cell
culture plate and cultured for 24 h. After discarding the old
culture medium, 100 µl NCI-H446 cells (or M059K cells) were treated
with IONP (0.7 µg/ml), PTX (0.3 µg/ml), IONP (0.7 µg/ml) + PTX (0.3
µg/ml), and IONP@PTX (1.0 µg/ml) at
37°C for 24 h. Simultaneously, untreated cells were set as a
negative control. Thereafter, 10 µl Cell Counting Kit-8 solution
(cat. no. KGA317; Nanjing KeyGen Biotech Co., Ltd.) was added to
each well and incubated for another 2 h. After shaking gently for
10 min, the optical density (OD) of each well was detected at 450
nm on a microplate reader (BioTek ELx800; BioTek) and cell growth
inhibitory rate was calculated using the formula: Inhibitory rate
(%)=[(OD of control group-OD of experimental group)/(OD of control
group-OD of blank group)] ×100. As shown in a previous study
(57), the calculation formula of
drug interaction was calculated using the following formula:
combination index=Ea+b/(Ea +
Eb-Ea × Eb), where a and b are any
two treatments, Ea+b is the inhibitory rate of the
combined a and b treatments, and Ea and Eb
are separately the inhibitory rates of a and b. A combination index
between 0.85 and 1.15 shows an additive effect and its value
>1.15 shows synergistic interactions.
Detection of iron ion
Total iron ion concentration was quantified with an
Iron Colorimetric Assay Kit (cat. no. E1042; Applygen Technologies,
Inc.). Under the premise of the above grouping, the autophagy
promoter rapamycin (50 nM; MedChemExpress) was separately added to
each group, namely IONP (0.7 µg/ml) + rapamycin (50 nM), PTX (0.3
µg/ml) + rapamycin (50 nM), IONP (0.7 µg/ml) + PTX (0.3 µg/ml) +
rapamycin (50 nM), IONP@PTX (1.0
µg/ml) + rapamycin (50 nM), and untreated cells (negative control).
Following the aforementioned treatment of NCI-H446 and M059K cells,
both cells were separately washed twice with cold PBS (cat. no.
C0221A; Shanghai Biyuntian Biotechnology Co., Ltd.) and lysed with
200 µl lysis buffer per well on a shaker for 2 h. In 100 µl cell
lysate, 100 µl PBS and 4.5% potassium permanganate mixture were
added, mixed gently, incubated at 60°C for 1 h and then cooled to
room temperature. Subsequently, 30 µl Iron ion detection agent was
added, mixed gently and incubated at room temperature for another
30 min. Finally, all liquid was placed into a 96-well plate and
absorbance was measured at 550 nm on a microplate reader (BioTek
ELx800; BioTek).
Detection of intracellular ROS
Diacetyldichlorofluorescein (DCFH-DA) fluorescent
probe (cat. no. KGT010-1; Jiangsu Kaiji Biotechnology Co., Ltd.)
was used to detect the level of intracellular ROS. In brief,
NCI-H446 and M059K cells were separately prepared for a cell
suspension at a density of 5×105 cells/ml, plated into a
6-well plate for 24 h and then treated as aforementioned.
Meanwhile, untreated cells were set as a negative control. The
cells were digested with 0.25% EDTA-free trypsin, collected and
washed with PBS three times and centrifuged at 110 × g for 5 min at
24°C. Thereafter, 1×106 cells/ml were suspended in
DCFH-DA (1:1,000) at a final concentration of 10 µmol/l, routinely
cultured at 37°C for 20 min and mixed at 3–5 min intervals.
Finally, the stained cells were washed with serum-free RPMI-1640
three times to fully remove the free DCFH-DA and then intracellular
ROS was detected (excitation wavelength, 488 nm; Emission
wavelength, 530 nm) using flow cytometry (CytoFLEX; Beckman
Coulter, Inc.).
Detection of lipid peroxidation
Intracellular LPD was detected using the
C11-BODIPYTM (cat. no. D3861; Thermo Fisher Scientific, Inc.)
fluorescent probe. Briefly, the pretreated NCI-H446 and M059K cells
were separately washed three times with PBS at 110 × g for 5 min at
24°C, collected, and adjusted to 1×106 cells/ml. The
cells were suspended in the 1:1,000 diluted C11-BODIPYTM at a final
concentration of 10 µmol/l, routinely cultured at 37°C for 20 min,
and mixed at 3–5 min intervals. After washing the stained cells
three times with serum-free RPMI-1640 to fully remove the free
C11-BODIPYTM, the intracellular LPD was finally detected using flow
cytometry (CytoFLEX; Beckman Coulter, Inc.).
Western blotting assay
The pretreated NCI-H446 cells and M059K cells were
separately collected to extract the protein from the cell lysate
following 15 min of centrifugation at 21,912 × g at 4°C, and the
protein concentration was determined using a BCA protein content
detection kit (cat. no. KGA902; Nanjing KeyGen Biotech Co., Ltd.).
First, identical amounts of protein (20 µg/lane) were separated on
10% gels using SDS-PAGE, transferred to PVDF membranes and blocked
with fresh 5% nonfat dry milk at room temperature for 2 h. The
membrane was incubated with diluted primary antibodies, including
rabbit anti-P62 (1:1,000; cat. no. ab91526; Abcam), Nrf2 (1:5,000;
cat. no. ab62352; Abcam), GPX4 (1:2,000; cat. no. ab123066; Abcam),
mammalian target of rapamycin complex 1 (mTORC1) (1:2,000; cat. no.
ab40768; Abcam), LC3II/I (1:2,000; cat. no. ab192890; Abcam),
Beclin1 (1:1,000; cat. no. ab62557; Abcam), HDAC6 (1:10,000; cat.
no. ab133493; Abcam), and GAPDH (1:5,000; cat. no. KGAA002; Nanjing
KeyGen Biotech Co., Ltd.). After washing with Tris-buffered
saline-Tween (TBST) three times, the membrane was subsequently
incubated with the Goat Anti-Rabbit IgG/HRP antibody (1:10,000;
cat. no. KGAA002; Nanjing KeyGen Biotech Co., Ltd.) for 1 h at room
temperature. Following washing with TBST again, the blots were
developed using a chemiluminescence detection system (ECL Luminata
Crescendo; cat. no. WBLUR0500; MilliporeSigma).
Statistical analysis
GraphPad Prism 8.0.1 software (GraphPad Software;
Dotmatics) was used for statistical analysis. All data in this
study are based on at least three replicated experiments. All data
are expressed as the mean ± standard deviation and analyzed using
either one-way ANOVA or two-way ANOVA followed by Tukey's post hoc
test among groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Characterization of IONP@PTX
As reported in our previous study (32), the synthesized IONP@PTX was uniformly distributed, with a
core particle size of 10 nm, a hydrated particle size of
36.18±11.76 nm, a zeta potential of-29±7.65 mV and a drug loading
value of 273.5 µg/mg (iron concentration) solution.
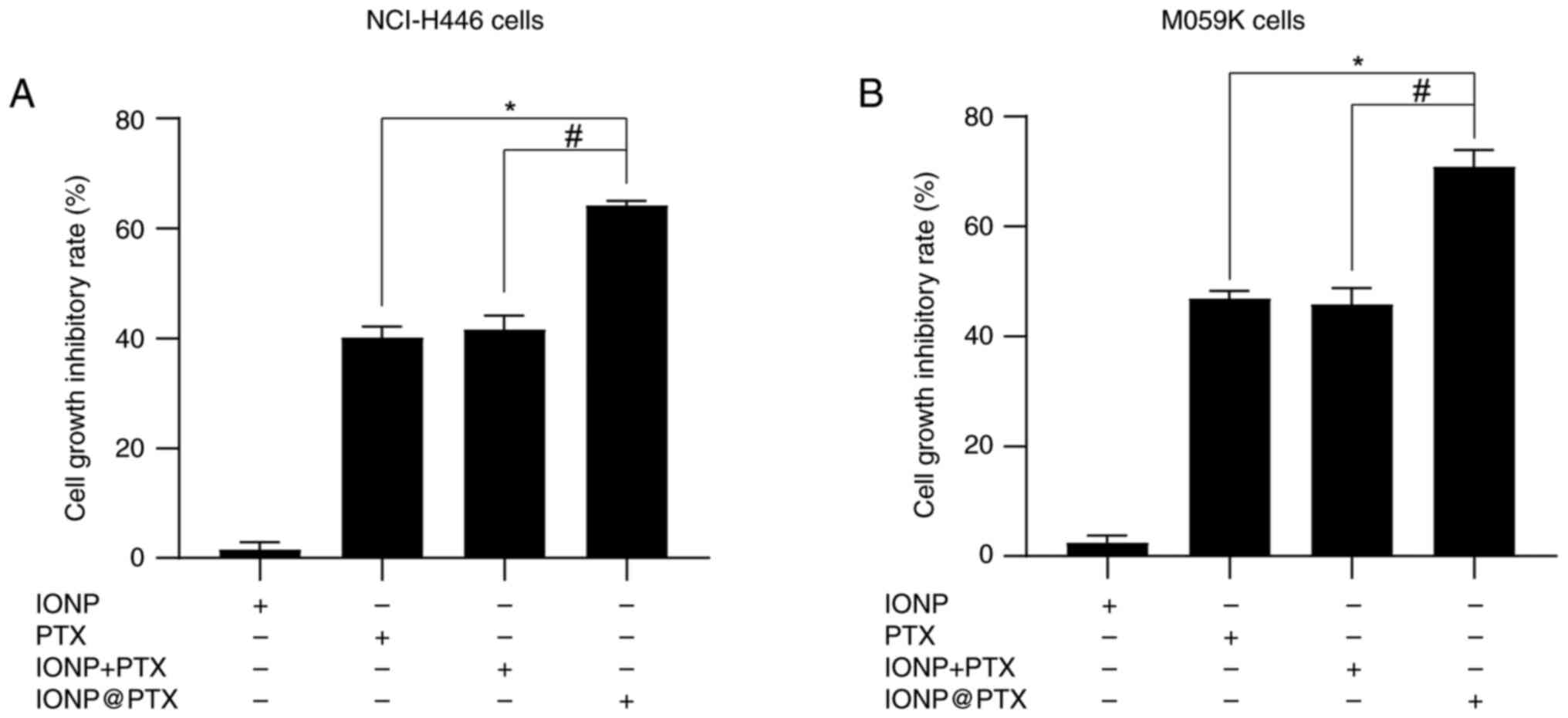
Effects of IONP@PTX on the cell growth inhibitory
rates
After 24 h of treatment with IONP, PTX, IONP + PTX,
and IONP@PTX, the cell growth
inhibitory rates, both for NCI-H446 and M059K cells, were
significantly higher in the IONP@PTX
group, compared with those in the PTX monotherapy group or the IONP
+ PTX group (P<0.001); however, there was no significant
difference between the PTX monotherapy group and the IONP + PTX
group (P>0.05; Fig. 1A and
B).
According to the cell inhibition rate calculation,
the cell growth inhibitory rate of IONP + PTX was 41.70±0.99% in
NCI-H446 cells and 45.88±2.23% in M059K cells, and the combination
index was 0.953 in NCI-H446 cells and 1.03 in M059K cells, which is
between 0.85 and 1.15, therefore showing an additive effect.
However, the cell growth inhibitory rate of IONP@PTX was 64.24±0.53% in NCI-H446 cells
and 70.90±2.68% in M059K cells, and the combination index was 1.46
in NCI-H446 cells and 1.59 in M059K cells, which is >1.15,
therefore showing synergistic interactions. These results in H446
and MO59K cells indicate that IONP@PTX significantly exerts a synergistic
effect on cell viability, compared with either PTX monotherapy or
IONP + PTX (Fig. 1; Table I).
 | Table I.Q value of drug interaction in
NCI-H446 cells and M059K cells. |
Table I.
Q value of drug interaction in
NCI-H446 cells and M059K cells.
| Group | Cell growth
inhibitory rate, % | Q value |
|---|
| NCI-H446 cells |
|
|
|
IONP | 1.66±0.51 | - |
|
PTX | 40.24±0.79 | - |
| IONP +
PTX | 41.70±0.99 | 0.953 |
|
IONP@PTX | 64.24±0.53 | 1.46 |
| M059K cells |
|
|
|
IONP | 2.49±1.72 | - |
|
PTX | 46.93±1.34 | - |
| IONP +
PTX | 45.88±2.23 | 1.03 |
|
IONP@PTX | 70.90±2.68 | 1.59 |
Effects of IONP@PTX on the induction of ferroptosis in
NCI-H446 and M059K cells
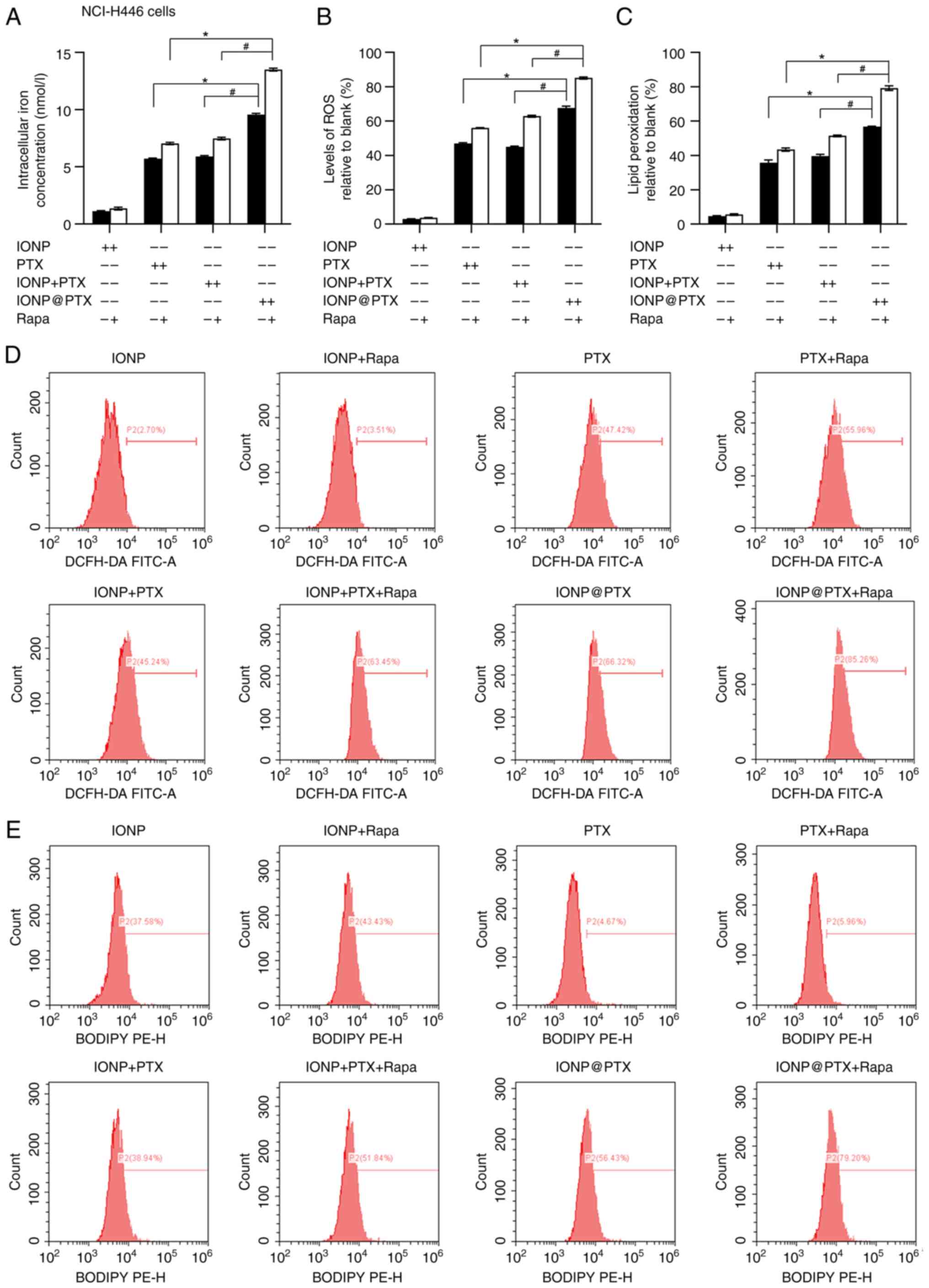
After 24 h of co-incubation of NCI-H446 cells with
IONP, PTX, IONP + PTX, and IONP@PTX,
the total iron ion content, ROS and LPD levels measured using
DCFH-DA and C11-BODIPYTM, respectively, were
significantly higher in the IONP@PTX
group than those in the other groups (all P<0.05; Fig. 2A-E). However, the levels of
ferroptosis-related protein Nrf2 in the IONP@PTX group were slightly lower than those
in the other groups, while the levels of GPX4 were slightly lower
in the IONP@PTX group than those in
the IONP + PTX group but there were no significant differences
among them (P>0.05; Fig. 3A, G and
H).
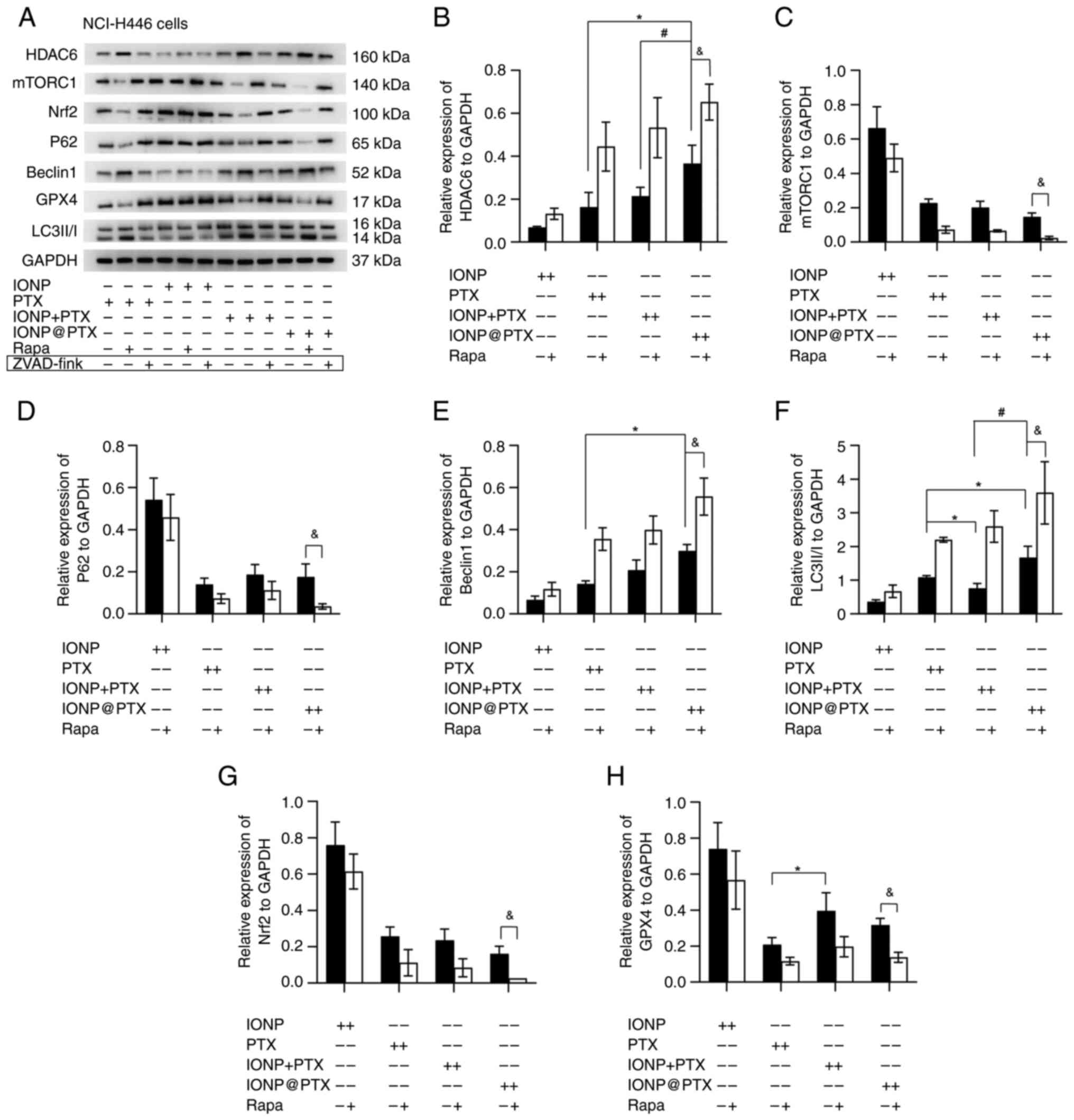 | Figure 3.Effects of IONP@PTX on the expression of
ferroptosis-related proteins and autophagy-related proteins in
NCI-H446 cells. (A) Representative western blot analysis of
ferroptosis-related proteins and autophagy-related proteins in
NCI-H446 cells following treatment with different drugs for 24 h.
(B-F) Relative expression of autophagy-related proteins HDAC6,
mTORC1, p62, Beclin 1 and LC3-II/I in NCI-H446 cells following
treatment with or without 50 nmol/l rapamycin for 24 h.*P<0.05
vs. PTX group. #P<0.05 vs. IONP + PTX group;
&P<0.05 vs. IONP@PTX monotherapy (without rapamycin)
group. (G) Relative expression of ferroptosis-related protein Nrf2
in NCI-H446 cells following treatment with or without 50 nmol/l
rapamycin for 24 h. (H) Relative expression of ferroptosis-related
protein GPX4 in NCI-H446 cells following treatment with or without
50 nmol/l rapamycin for 24 h. *P<0.05 vs. PTX group (without
rapamycin). &P<0.05 vs. IONP@PTX monotherapy (without rapamycin).
Data were obtained from three independent repeated experiments. In
the western blot analysis, the IONP group was used as the control
group due to the large loading of IONP samples, which had lower
toxicity on NCI-H446 cells. The results of the circled strips are
not reflected in this article. IONP, iron oxide nanoparticles; PTX,
paclitaxel; Nrf2, nuclear factor erythrocyte 2 related factor 2;
GPX4, glutathione peroxidase 4; p62, sequestosome1; Rapa,
rapamycin. |
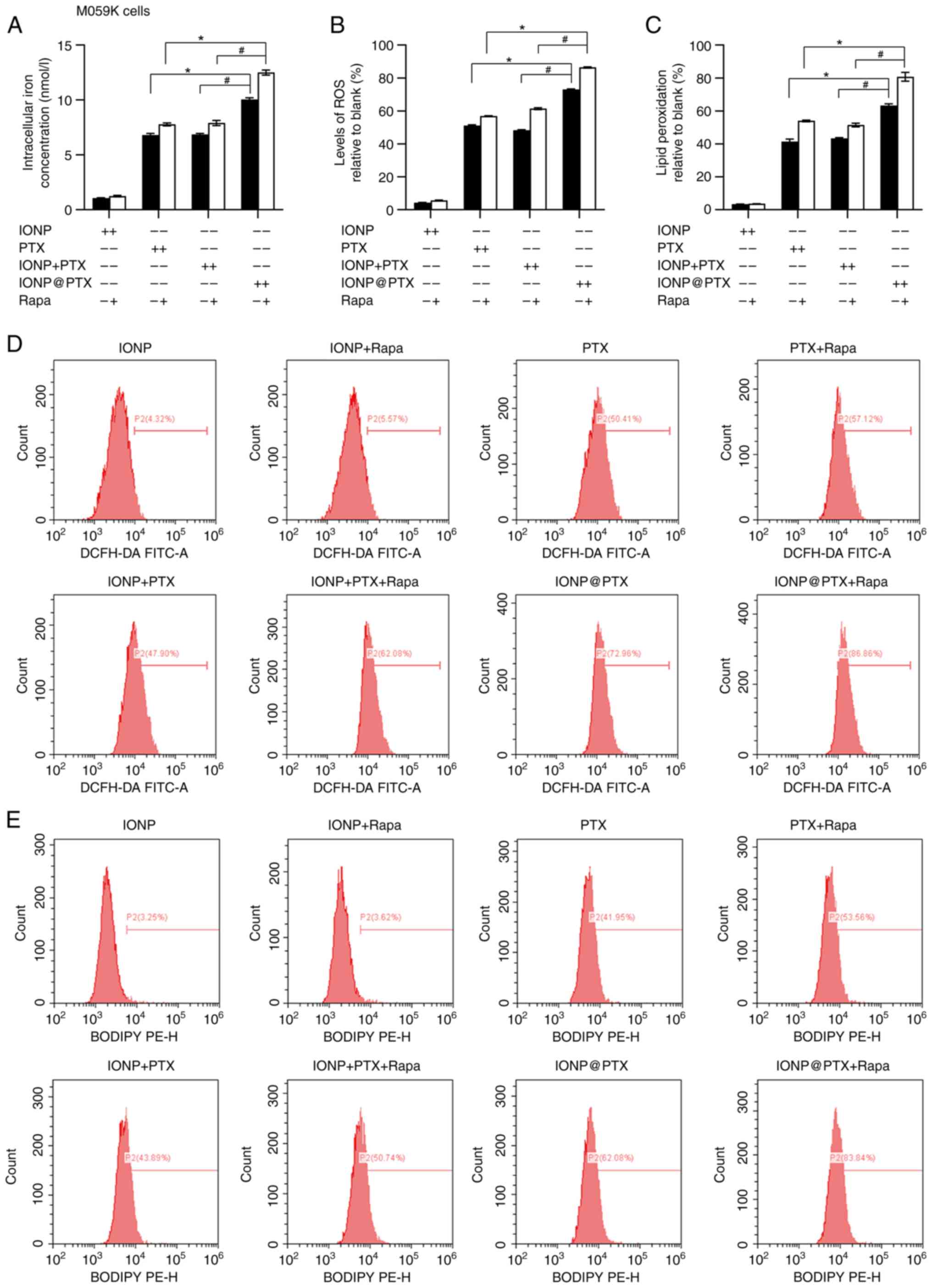
Similar results were also obtained in M059K cells in
terms of total iron ion content, ROS and LPD levels (P<0.05;
Fig. 4A-E). In addition, a
significantly lower expression of ferroptosis-related protein GPX4
was measured in the IONP@PTX group
compared with that in the IONP + PTX group (P<0.05). In
addition, the protein expression of Nrf2 was slightly lower but did
not reach a statistical significance (P>0.05; Fig. 5A, G and H). These results were
consistent in both cell lines following incubation with or without
rapamycin (P<0.05; Figs. 3A, G and
H, and 5A, G and H).
Collectively, IONP@PTX significantly
increases the levels of ROS, ferric ions and LPD in both cell
lines. Moreover, the synergistic effect of IONP@PTX can be enhanced by addition of
rapamycin, indicating that the autophagy promoter rapamycin
enhances the IONP@PTX-induced
cellular ferroptosis.
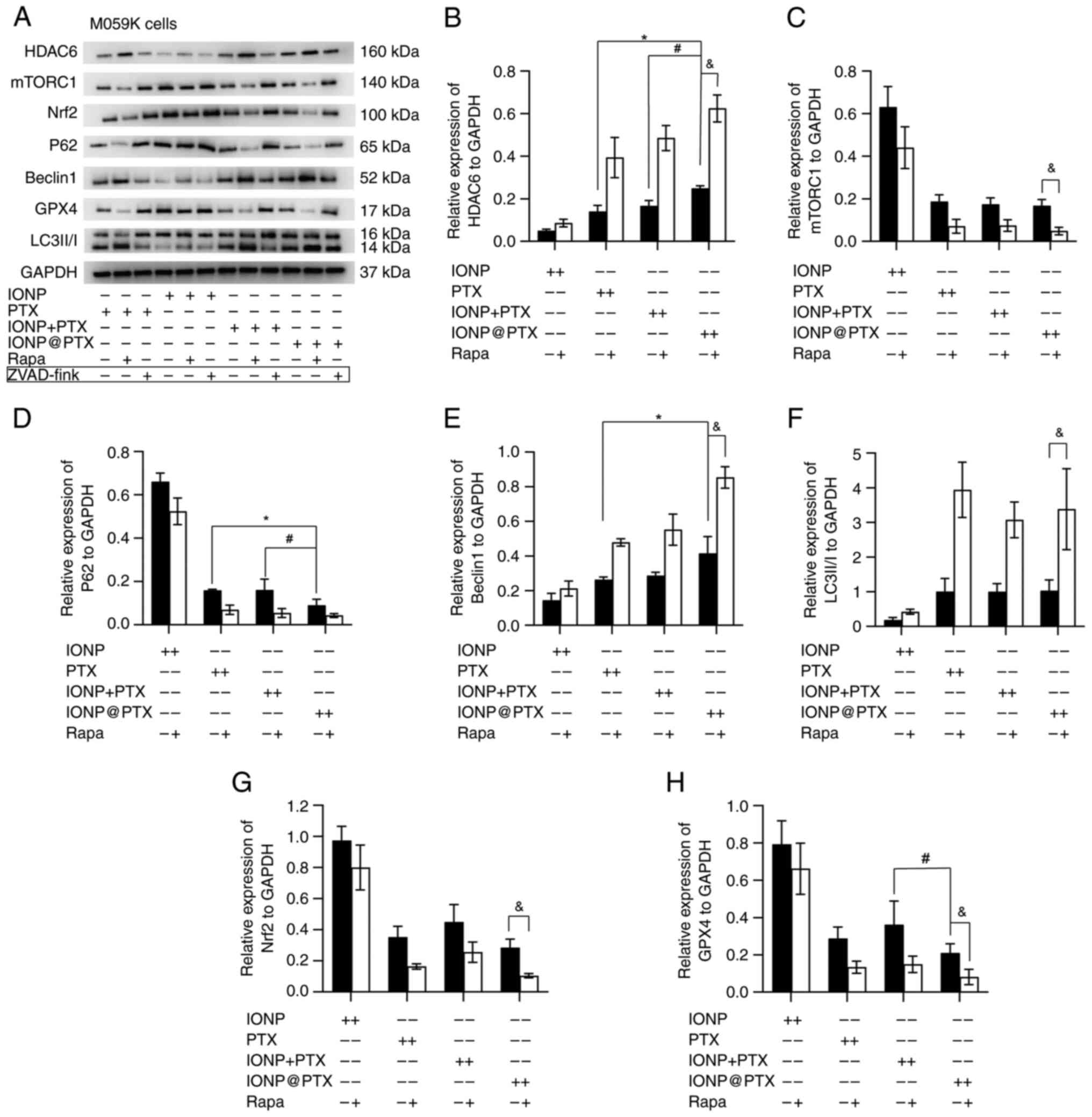 | Figure 5.Effects of IONP@PTX on the expression of
ferroptosis-related proteins and autophagy-related proteins in
M059K cells. (A) Representative western blot analysis of
ferroptosis-related proteins and autophagy-related proteins in
M059K cells following treatment with different drugs for 24 h.
(B-F) Relative expression of autophagy-related proteins HDAC6,
mTORC1, p62, Beclin1 and LC3-II/I in the M059K cells following
treatment with or without 50 nmol/l rapamycin for 24 h. *P<0.05
vs. PTX group. #P<0.05, vs. IONP + PTX group.
&P<0.05, vs. IONP@PTX (without rapamycin) group. (G)
Relative expression of ferroptosis-related protein Nrf2 in M059K
cells following treatment with or without 50 nmol/l rapamycin for
24 h. (H) The relative expression of ferroptosis-related protein
GPX4 in M059K cells following treatment with or without 50 nmol/l
rapamycin for 24 h. #P<0.05 vs. IONP + PTX group.
&P<0.05 vs. IONP@PTX (no rapamycin) group. Data were
obtained from three independent repeated experiments. In the
western blot analysis, the IONP group was used as the control group
due to the large loading of IONP samples, which had lower toxicity
in M059K cells. The results of the circled strips are not reflected
in this article. IONP, iron oxide nanoparticles; PTX, paclitaxel;
Nrf2, nuclear factor erythrocyte 2 related factor 2; GPX4,
glutathione peroxidase 4; HDAC6, histone deacetylase 6; mTORC1,
mammalian target of rapamycin complex 1; LC3, light chain 3; p62,
sequestosome1; Rapa, rapamycin. |
Effects of IONP@PTX on the induction of autophagy in
NCI-H446 and M059K cells
Compared with PTX, IONP@PTX significantly increased the
expression of autophagy-related proteins Beclin 1, LC3-II/I and
HDAC6 (P<0.05) in NCI-H446 cells; however, there were no
significant differences in the p62 and mTORC1 expression. Notably,
compared with IONP + PTX, IONP@PTX
significantly increased the expression of autophagy-associated
proteins LC3 and HDAC6 in NCI-H446 cells (P<0.05; Fig. 3A-F).
Compared with PTX, IONP@PTX significantly increased the
expression of autophagy-related proteins Beclin 1, HDAC6 and
significantly decreased the expression of p62 protein (P<0.05)
in M059K cells; however, there were no significant differences in
the LC3-II/I and mTORC1 expression (P>0.05). Notably, compared
with the IONP + PTX, IONP@PTX
significantly increased the expression of autophagy-associated
HDAC6 and significantly decreased the expression of p62 protein in
M059K cells (P<0.05; Fig.
5A-F).
Furthermore, additional rapamycin enhanced the
IONP@PTX-induced the
upregulation of Beclin1, LC3 and HDAC6, as well as the
downregulation of mTORC1 in NCI-H446 and M059K cells (P<0.05).
Moreover, rapamycin enhanced the IONP@PTX-induced downregulation of
p62 protein in NCI-H446 cells (P<0.05; Figs. 3A-F and 5A-F). These results indicate that the
autophagic signaling pathway may be involved in the IONP@PTX-induced ferroptosis of H446
and M059K cells.
Discussion
It has been demonstrated in our previous study that
IONP is effective for the induction of ferroptosis in GBM U251
cells (58) and IONP can
effectively synergize with chemotherapeutic drugs against tumors
via ferroptosis (59). It can
therefore be inferred that IONP is expected to be a carrier for
effectively transporting PTX and synergizing PTX against cancer
cells.
However, it is not clear whether the observed effect
was produced by bundling IONP with PTX to form a compound drug or
by only simply adding nanoparticles with PTX. Unlike the
nanoparticles used to induce ferroptosis investigated in our
previous studies, the IONP@PTX used
in the present study is a composite drug formed by bundling
nanoparticles with the antitumor drug PTX. Considering that brain
metastasis of lung cancer is one of the most common distant
metastatic sites (60), the
present authors chose two tumor cell lines (NCI-H446 and M059K)
that differ from the ones used in our previous study (U251 and
HMC3) to investigate the possible antitumor effect. The
experimental results have demonstrated that PTX has a certain
inhibitory effect on viability in NCI-H446 and M059K cells, whereas
IONP monotherapy does not. Notably, the combination index of the
IONP + PTX was between 0.85–1.15 and that of IONP@PTX was >1.15 in NCI-H446 and M059K
cells, implying that IONP@PTX
produces a synergistic effect in different types of intracranial
tumors, such as primary and secondary brain tumors, and the
synergistic effect of IONP@PTX is not
a coincidence. Accordingly, IONP@PTX
could be used in different kinds of brain tumors, laying the
foundation for subsequent application-based studies.
Ferroptosis is usually accompanied by massive iron
accumulation and iron-dependent LPD (61). The GPX family was shown to be
capable of breaking down aberrant endogenous peroxides, among which
GPX4 is the only enzyme that can reduce lipid membrane peroxides
and its inactivation is central in ferroptosis (62). In turn, GSH is a key cofactor for
the selenoprotein GPX4. When intracellular GSH levels fall, GPX4
activity decreases and intracellular ROS and lipid peroxide
accumulation increases, eventually leading to cellular ferroptosis
(46). Nrf2 is a basic leucine
zipper transcription factor that participates in antioxidant
reduction processes, lipid metabolism control and iron metabolism
(47). The present study found
that IONP@PTX substantially raised
total iron ion concentration, ROS, and LPD levels in NCI-H446 and
M059K cells when compared with PTX or PTX + IONP. This finding was
similar to that of our previous investigation on GBM U251 cells
(32). Using malignant GBM M059K
cells in vitro, the present study confirmed that IONP@PTX significantly decreased the
expression of ferroptosis-related GPX4 protein compared with the
IONP + PTX, but the expression levels of Nrf2 and GPX4 were not
significantly reduced in other groups. It can be inferred that the
IONP@PTX-induced ferroptosis
does not occur through the normal Nrf2 and GPX4 routes, most likely
due to the cells' heightened sensitivity to ferroptosis caused by
GSH depletion. We hypothesized that IONP@PTX significantly increased total iron
ion concentration and abnormal labile iron pools, thus making cells
more vulnerable to ferroptosis (63–66).
Although IONP@PTX did not reduce Nrf2
and GPX4 levels, it caused a significant increase in ROS and LPD
accumulation in both cell lines. The main reason for this is that
the increased intracellular GSH demand is insufficient to support
the effective elimination of phospholipid hydroperoxides,
eventually resulting in ferroptosis (67). Notably, the IONP@PTX + rapamycin can release more iron
ions, causing LPD and ferroptosis in NCI-H446 and M059K cells.
These findings imply that IONP@PTX-induced ferroptosis may be
associated with autophagy.
Recently, the relationship between autophagy and
cancer has attracted considerable attention (68). Autophagy is a defense mechanism
that maintains cellular homeostasis and promotes cell survival by
eliminating damaged organelles or abnormal proteins; it is also a
lysosome-mediated degradation pathway (69) that affects all stages of tumor
initiation and progression (68,70,71).
Microtubule-associated proteins LC3, mTOR, Beclin 1 and p62 are
central autophagy-related genes involved in the regulation of the
autophagic process (72). Among
them, Beclin 1 and LC3 mainly regulate autophagosome formation
(73), while p62 is an effector of
selective autophagy as well as a substrate of nuclear autophagy
(73,74). A recent study has documented that
rapamycin, a mTORC1 receptor-specific inhibitor, can regulate the
mTOR pathway to activate autophagy (71). It is known that at the core of the
autophagy process is the conversion from LC3-I to LC3-II, and the
expression of LC3-II has been shown to be a marker of autophagic
activity, therefore the expression of the LC3-II/I ratio can be
used to evaluate the level of autophagy (75). The present results showed that
IONP@PTX increased the level of
LC3-II/I and decreased that of mTORC1 in NCI-H446 cells. Moreover,
additional rapamycin enhanced the IONP@PTX-induced upregulation of LC3
and downregulation of mTORC1 in both cell lines. IONP@PTX did not significantly increase the
expression level of LC3 but it showed a significant increase in the
conversion from LC3-I to LC3-II in M095K cells. In our previous
study, it was documented that, compared with PTX monotherapy,
IONP@PTX significantly increased the
expression of LC3 protein in glioma U251 cells (32). Accumulating evidence has indicated
that PTX plays a vital role in antagonizing the production of HDAC6
protein (76–78). The detection of autophagy-related
proteins in the present study revealed that only the expression of
autophagy-related proteins HDAC6 and Beclin1 jointly increased in
H446 and M059K cells, without marked changes in other indicators.
Notably, additional rapamycin enhanced IONP@PTX-induced upregulation of
Beclin1 and HDAC6 in both cell lines. These results suggest that
IONP@PTX induces a synergistic effect
on cellular ferroptosis, most likely, through the autophagy
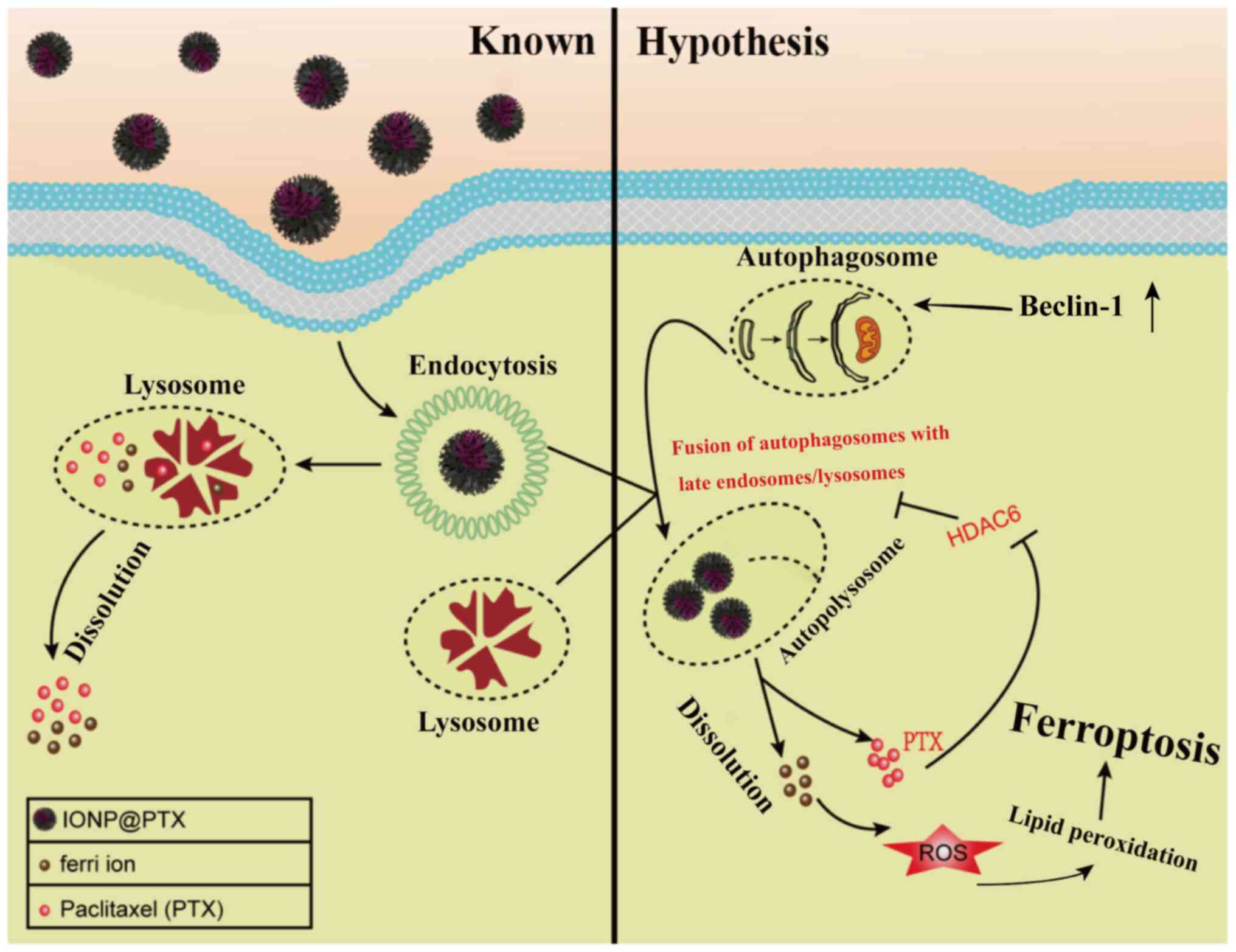
pathway. Based on the above findings, it is hypothesized for the
role of IONP@PTX, that is, IONP@PTX synergistically induces cellular
ferroptosis by affecting the Beclin1-HDAC6 autophagic pathway.
Specifically, IONP@PTX enhances
autophagy and increases autophagosome formation by upregulating
Beclin1 after endocytosis and induces ferroptosis by promoting the
recruitment of HDAC6 to facilitate the fusion of autophagosomes
with late endosomes/lysosomes. In turn, HDAC6 acetylates
microtubulin to destabilize it and inhibits autophagosome-lysosome
fusion, thereby creating a feedback inhibition loop between
autophagosome-lysosome fusion and HDAC6, and inhibiting IONP@PTX degradation. Once the balance is
disturbed, the autophagic degradation results in a continuous
degradation of IONP@PTX, subsequently
releasing PTX and iron ions. The PTX released from the IONP@PTX complex following endocytosis
antagonizes this feedback inhibition loop of HDAC6, thus breaking
this balance and promoting autophagosome-lysosome fusion. On the
other hand, the released PTX and iron ions synergistically promoted
ferroptosis, ultimately producing a significant and enhanced
synergistic induction of ferroptosis compared with PTX monotherapy
or IONP + PTX (Fig. 6). Further
studies are required to test the validity of this hypothesis. In
the following study, tumor models will be created using NCI-H446
(or M059K) cells in nude mice to further investigate whether
IONP@PTX can synergistically induce
ferroptosis through the autophagic pathway in vivo.
Subsequently, Beclin1 and HDAC6 knocked down or overexpressing nude
mice models will be created to verify the effects and key roles of
these two genes in the synergistic effect of IONP@PTX in inducing ferroptosis through
autophagy. In addition, the effect of IONP@PTX on the capacity of migrate, invade
and apoptosis should be further studied in NCI-H446 and M059K cell
lines.
In conclusion, IONP bundled with PTX exerts a
synergistic effect on the viability of NCI-H446 and M059K cell
lines by inducing cellular ferroptosis, which may be associated
with autophagy.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (grant no. 32060228), the Guangxi Natural
Science Foundation Project (grant nos. 2023GXNSFAA026385,
2022JJA140211, 2017GXNSFAA198112 and 2019GXNSFAA245077), Research
and Innovation Base for Basic and Clinical Application of Nerve
Injury and Repair (grant no. ZY21195042), Guangxi Key Laboratory of
Big Data Intelligent Cloud Management for Neurological Diseases
(grant no. ZTJ2020005) and Guilin Scientific Research and
Technology Development Project (grant no. 20190219-2).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QN was responsible for conceptualization, subject
design, experimental framework construction and in vitro
experiments, and was a major contributor in writing the manuscript.
WC performed in vitro experiments, western blotting, data
analysis and management, and draft revision. TZ assisted with the
experimental design, in vitro experiments, data analysis and
literature review. SY was responsible for data analysis,
statistical analysis, design research, literature search and
collation. ZR helped with the experimental approach, and the
concept and design of the study. PZ was responsible for assisting
in the design of experimental methods and the production of
manuscript images. JW was responsible for methodology, project
funding acquisition, manuscript framework design, experimental
supervision and critical revision of important intellectual
content. QN and WC confirm the authenticity of all the raw data.
All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Thai AA, Solomon BJ, Sequist LV, Gainor JF
and Heist RS: Lung cancer. Lancet. 398:535–554. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xia C, Dong X, Li H, Cao M, Sun D, He S,
Yang F, Yan X, Zhang S, Li N and Chen W: Cancer statistics in China
and United States, 2022: Profiles, trends, and determinants. Chin
Med J (Engl). 135:584–590. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Caeser R, Egger JV, Chavan S, Socci ND,
Jones CB, Kombak FE, Asher M, Roehrl MH, Shah NS, Allaj V, et al:
Genomic and transcriptomic analysis of a library of small cell lung
cancer patient-derived xenografts. Nat Commun. 13:21442022.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yuan M, Zhao Y, Arkenau HT, Lao T, Chu L
and Xu Q: Signal pathways and precision therapy of small-cell lung
cancer. Signal Transduct Target Ther. 7:1872022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mak DWS, Li S and Minchom A: Challenging
the recalcitrant disease-developing molecularly driven treatments
for small cell lung cancer. Eur J Cancer. 119:132–150. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
George J, Lim JS, Jang SJ, Cun Y, Ozretić
L, Kong G, Leenders F, Lu X, Fernández-Cuesta L, Bosco G, et al:
Comprehensive genomic profiles of small cell lung cancer. Nature.
524:47–53. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Low JT, Ostrom QT, Cioffi G, Neff C, Waite
KA, Kruchko C and Barnholtz-Sloan JS: Primary brain and other
central nervous system tumors in the United States (2014–2018): A
summary of the CBTRUS statistical report for clinicians. Neurooncol
Pract. 9:165–182. 2022.PubMed/NCBI
|
|
9
|
Lapointe S, Perry A and Butowski NA:
Primary brain tumours in adults. Lancet. 392:432–446. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou YS, Wang W, Chen N, Wang LC and Huang
JB: Research progress of anti-glioma chemotherapeutic drugs
(review). Oncol Rep. 47:1012022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tan AC, Ashley DM, López GY, Malinzak M,
Friedman HS and Khasraw M: Management of glioblastoma: State of the
art and future directions. CA Cancer J Clin. 70:299–312. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang Y, Zhai M, Chen Z, Han X, Yu F, Li
Z, Xie X, Han C, Yu L, Yang Y and Mei X: Dual-modified liposome
codelivery of doxorubicin and vincristine improve targeting and
therapeutic efficacy of glioma. Drug Deliv. 24:1045–1055. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
d'Angelo M, Castelli V, Benedetti E,
Antonosante A, Catanesi M, Dominguez-Benot R, Pitari G, Ippoliti R
and Cimini A: Theranostic nanomedicine for malignant gliomas. Front
Bioeng Biotechnol. 7:3252019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu L and Chen L: Progress in research on
paclitaxel and tumor immunotherapy. Cell Mol Biol Lett. 24:402019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zeng W, Kwan Law BY, Wai Wong VK, Bik Chan
DS, Fai Mok SW, Ying Gao JJ, Yan Ho RK, Liang X, Li JH, Lee MT, et
al: HM30181A, a potent P-glycoprotein inhibitor, potentiates the
absorption and in vivo antitumor efficacy of paclitaxel in an
orthotopic brain tumor model. Cancer Biol Med. 17:9862020.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang YH, Mao JW and Tan XL: Research
progress on the source, production, and anti-cancer mechanisms of
paclitaxel. Chin J Nat Med. 18:890–897. 2020.PubMed/NCBI
|
|
17
|
Gonzalez-Angulo AM and Hortobagyi GN:
Optimal schedule of paclitaxel: Weekly is better. J Clin Oncol.
26:1585–1587. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Joos G, Schallier D, Pinson P, Sterckx M
and Van Meerbeeck JP: Paclitaxel (PTX) as second line treatment in
patients (pts) with small cell lung cancer (SCLC) refractory to
carboplatin-etoposide: A multicenter phase II study. J Clin Oncol.
22 (14 Suppl):S72112004. View Article : Google Scholar
|
|
19
|
Nakao M, Fujita K, Suzuki Y, Arakawa S,
Sakai Y, Sato H and Muramatsu H: Nab-paclitaxel monotherapy for
relapsed small cell lung cancer: Retrospective analysis and review.
Anticancer Res. 40:1579–1585. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ahmed Khalil A, Rauf A, Alhumaydhi FA,
Aljohani ASM, Javed MS, Khan MA, Khan IA, El-Esawi MA, Bawazeer S,
Bouyahya A, et al: Recent developments and anticancer therapeutics
of paclitaxel: An update. Curr Pharm Des. 28:3363–3373. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Silva P, Nascimento A, Martinho O, Reis R
and Bousbaa H: Targeting BUB3 in combination with paclitaxel
inhibits proliferation of glioblastoma cells by enhancing cellular
senescence. Sci Lett. 1:12022.
|
|
22
|
Erthal LCS, Shi Y, Sweeney KJ, Gobbo OL
and Ruiz-Hernandez E: Nanocomposite formulation for a sustained
release of free drug and drug-loaded responsive nanoparticles: An
approach for a local therapy of glioblastoma multiforme. Sci Rep.
13:50942023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu HC, Feng Y, Song XY, Song CY, Chen JL,
Wang YC, He XL, Liang RC, Li JH and Tan H: Implantable polyurethane
scaffolds loading with PEG-paclitaxel conjugates for the treatment
of glioblastoma multiforme. Chin J Polym Sci. 40:491–503. 2022.
View Article : Google Scholar
|
|
24
|
Jibodh RA, Lagas JS, Nuijen B, Beijnen JH
and Schellens JH: Taxanes: Old drugs, new oral formulations. Eur J
Pharmacol. 717:40–46. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sharifi-Rad J, Quispe C, Patra JK, Singh
YD, Panda MK, Das G, Adetunji CO, Michael OS, Sytar O, Polito L, et
al: Paclitaxel: Application in modern oncology and
nanomedicine-based cancer therapy. Oxid Med Cell Longev.
2021:36877002021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mali P and Sherje AP: Cellulose
nanocrystals: Fundamentals and biomedical applications. Carbohydr
Polym. 275:1186682022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kahn B, Collazo J and Kyprianou N:
Androgen receptor as a driver of therapeutic resistance in advanced
prostate cancer. Int J Biol Sci. 10:588–595. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li K, Zhan W, Chen Y, Jha RK and Chen X:
Docetaxel and doxorubicin codelivery by nanocarriers for
synergistic treatment of prostate cancer. Front Pharmacol.
10:14362019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pulvirenti L, Monforte F, Lo Presti F, Li
Volti G, Carota G, Sinatra F, Bongiorno C, Mannino G, Cambria MT
and Condorelli GG: Synthesis of MIL-modified
Fe3O4 magnetic nanoparticles for enhancing
uptake and efficiency of temozolomide in glioblastoma treatment.
Int J Mol Sci. 23:28742022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fernández-Acosta R, Iriarte-Mesa C,
Alvarez-Alminaque D, Hassannia B, Wiernicki B, Díaz-García AM,
Vandenabeele P, Vanden Berghe T and Pardo Andreu GL: Novel iron
oxide nanoparticles induce ferroptosis in a panel of cancer cell
lines. Molecules. 27:39702022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu Y, Wu H, Huang J, Qian W, Martinson DE,
Ji B, Li Y, Wang YA, Yang L and Mao H: Probing and enhancing
ligand-mediated active targeting of tumors using sub-5 nm ultrafine
iron oxide nanoparticles. Theranostics. 10:2479–2494. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen H and Wen J: Iron oxide nanoparticles
loaded with paclitaxel inhibits glioblastoma by enhancing
autophagy-dependent ferroptosis pathway. Eur J Pharmacol.
921:1748602022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rakesh R, PriyaDharshini LC, Sakthivel KM
and Rasmi RR: Role and regulation of autophagy in cancer. Biochim
Biophys Acta Mol Basis Dis. 1868:1664002022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ma Q, Long S, Gan Z, Tettamanti G, Li K
and Tian L: Transcriptional and post-transcriptional regulation of
autophagy. Cells. 11:4412022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li X, Yang KB, Chen W, Mai J, Wu XQ, Sun
T, Wu RY, Jiao L, Li DD, Ji J, et al: CUL3 (cullin 3)-mediated
ubiquitination and degradation of BECN1 (beclin 1) inhibit
autophagy and promote tumor progression. Autophagy. 17:4323–4340.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ciołczyk-Wierzbicka D, Krawczyk A,
Zarzycka M, Zemanek G and Wierzbicki K: Three generations of mTOR
kinase inhibitors in the activation of the apoptosis process in
melanoma cells. J Cell Commun Signal. 17:975–989. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zou Z, Tao T, Li H and Zhu X: mTOR
signaling pathway and mTOR inhibitors in cancer: Progress and
challenges. Cell Biosci. 10:312020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang L, Fang Y, Cheng X, Lian Y and Xu H:
Interaction between TRPML1 and p62 in regulating
autophagosome-lysosome fusion and impeding neuroaxonal dystrophy in
Alzheimer's disease. Oxid Med Cell Longev.
2022:80960092022.PubMed/NCBI
|
|
39
|
Hai R, Yang D, Zheng F, Wang W, Han X,
Bode AM and Luo X: The emerging roles of HDACs and their
therapeutic implications in cancer. Eur J Pharmacol.
931:1752162022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tang Q, Li X and Wang J: Tubulin
deacetylase NDST3 modulates lysosomal acidification: Implications
in neurological diseases. Bioessays. 44:e22001102022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li J, Yu M, Fu S, Liu D and Tan Y: Role of
selective histone deacetylase 6 inhibitor ACY-1215 in cancer and
other human diseases. Front Pharmacol. 13:9079812022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Johansen T and Lamark T: Selective
autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J
Mol Biol. 432:80–103. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhou B, Liu J, Kang R, Klionsky DJ,
Kroemer G and Tang D: Ferroptosis is a type of autophagy-dependent
cell death. Semin Cancer Biol. 66:89–100. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang X, Ge H, Ma Y, Song L, Ma Y, Tian G,
Wang L, Meng Q and Sun X: Engineered anti-cancer nanomedicine for
synergistic ferroptosis-immunotherapy. Chem Eng J. 455:1406882023.
View Article : Google Scholar
|
|
45
|
Shen Z, Liu T, Li Y, Lau J, Yang Z, Fan W,
Zhou Z, Shi C, Ke C, Bregadze VI, et al:
Fenton-reaction-acceleratable magnetic nanoparticles for
ferroptosis therapy of orthotopic brain tumors. ACS Nano.
12:11355–11365. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Stockwell BR: Ferroptosis turns 10:
Emerging mechanisms, physiological functions, and therapeutic
applications. Cell. 185:2401–2421. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Song X and Long D: Nrf2 and ferroptosis: A
new research direction for neurodegenerative diseases. Front
Neurosci. 14:2672020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mouri A, Yamaguchi O, Miyauchi S, Shiono
A, Utsugi H, Nishihara F, Murayama Y, Kagamu H and Kobayashi K:
Combination therapy with carboplatin and paclitaxel for small cell
lung cancer. Respir Investig. 57:34–39. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yun T, Kim HT, Han JY, Yoon SJ, Kim HY,
Nam BH and Lee JS: A phase II study of weekly paclitaxel plus
gemcitabine as a second-line therapy in patients with metastatic or
recurrent small cell lung cancer. Cancer Res Treat. 48:465–472.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Oi H, Matsuda T, Kimura T, Morise M,
Yamano Y, Yokoyama T, Kataoka K and Kondoh Y: Weekly nanoparticle
albumin-bound paclitaxel and paclitaxel for relapsed small cell
lung cancer: A retrospective observational study. Medicine
(Baltimore). 101:e288632022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mittal S, Ali J and Baboota S: Overcoming
the challenges in the treatment of glioblastoma via
nanocarrier-based drug delivery approach. Curr Pharm Des.
27:4539–4556. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang L, Wang X, Shen L, Alrobaian M, Panda
SK, Almasmoum HA, Ghaith MM, Almaimani RA, Ibrahim IAA, Singh T, et
al: Paclitaxel and naringenin-loaded solid lipid nanoparticles
surface modified with cyclic peptides with improved tumor targeting
ability in glioblastoma multiforme. Biomed Pharmacother.
138:1114612021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhang DY, Dmello C, Chen L, Arrieta VA,
Gonzalez-Buendia E, Kane JR, Magnusson LP, Baran A, James CD,
Horbinski C, et al: Ultrasound-mediated delivery of paclitaxel for
glioma: A comparative study of distribution, toxicity, and efficacy
of albumin-bound versus cremophor formulations. Clin Cancer Res.
26:477–486. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Liu W, Lin Q, Fu Y, Huang S, Guo C, Li L,
Wang L, Zhang Z and Zhang L: Target delivering paclitaxel by
ferritin heavy chain nanocages for glioma treatment. J Control
Release. 323:191–202. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen T and Gong T, Zhao T, Liu X, Fu Y,
Zhang Z and Gong T: Paclitaxel loaded phospholipid-based gel as a
drug delivery system for local treatment of glioma. Int J Pharm.
528:127–132. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang X, Ye L, He W, Teng C, Sun S, Lu H,
Li S, Lv L, Cao X, Yin H, et al: In situ targeting
nanoparticles-hydrogel hybrid system for combined
chemo-immunotherapy of glioma. J Control Release. 345:786–797.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Que W, Li S and Chen J: NS-398 enhances
the efficacy of bortezomib against RPMI8226 human multiple myeloma
cells. Mol Med Rep. 7:1641–1645. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wen J, Chen H, Ren Z, Zhang P, Chen J and
Jiang S: Ultrasmall iron oxide nanoparticles induced ferroptosis
via Beclin1/ATG5-dependent autophagy pathway. Nano Converg.
8:102021. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sugiyama A, Ohta T, Obata M, Takahashi K,
Seino M and Nagase S: xCT inhibitor sulfasalazine depletes
paclitaxel-resistant tumor cells through ferroptosis in uterine
serous carcinoma. Oncol Lett. 20:2689–2700. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Rudin CM, Brambilla E, Faivre-Finn C and
Sage J: Small-cell lung cancer. Nat Rev Dis Primers. 7:32021.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao
N, Sun B and Wang G: Ferroptosis: Past, present and future. Cell
Death Dis. 11:882020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chang S, Tang M, Zhang B, Xiang D and Li
F: Ferroptosis in inflammatory arthritis: A promising future. Front
Immunol. 13:9550692022. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
He H, Du L, Guo H, An Y, Lu L, Chen Y,
Wang Y, Zhong H, Shen J, Wu J and Shuai X: Redox responsive metal
organic framework nanoparticles induces ferroptosis for cancer
therapy. Small. 16:20012512020. View Article : Google Scholar
|
|
64
|
Xue CC, Li MH, Zhao Y, Zhou J, Hu Y, Cai
KY, Zhao Y, Yu SH and Luo Z: Tumor microenvironment-activatable
Fe-doxorubicin preloaded amorphous CaCO3 nanoformulation
triggers ferroptosis in target tumor cells. Sci Adv.
6:eaax13462020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chen G, Yang Y, Xu Q, Ling M, Lin H, Ma W,
Sun R, Xu Y, Liu X, Li N, et al: Self-amplification of tumor
oxidative stress with degradable metallic complexes for synergistic
cascade tumor therapy. Nano Lett. 20:8141–8150. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Luo S, Ma D, Wei R, Yao W, Pang X, Wang Y,
Xu X, Wei X, Guo Y, Jiang X, et al: A tumor microenvironment
responsive nanoplatform with oxidative stress amplification for
effective MRI-based visual tumor ferroptosis. Acta Biomater.
138:518–527. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Friedmann Angeli JP, Krysko DV and Conrad
M: Ferroptosis at the crossroads of cancer-acquired drug resistance
and immune evasion. Nat Rev Cancer. 19:405–414. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Assi M and Kimmelman AC: Impact of
context-dependent autophagy states on tumor progression. Nat
Cancer. 4:596–607. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhang X, Sui S, Wang L, Li H, Zhang L, Xu
S and Zheng X: Inhibition of tumor propellant glutathione
peroxidase 4 induces ferroptosis in cancer cells and enhances
anticancer effect of cisplatin. J Cell Physiol. 235:3425–3437.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Xia H, Green DR and Zou W: Autophagy in
tumour immunity and therapy. Nat Rev Cancer. 21:281–297. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Yang F, Du L, Song G, Zong X, Jin X, Yang
X and Qi Z: Rapamycin and 3-methyladenine influence the apoptosis,
senescence, and adipogenesis of human adipose-derived stem cells by
promoting and inhibiting autophagy: An in vitro and in vivo study.
Aesthetic Plast Surg. 45:1294–1309. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ding R, Liu Z, Tan J and Sun B: Advanced
oxidation protein products mediate human keratinocytes apoptosis by
inducing cell autophagy through the mTOR-Beclin-1 pathway. Cell
Biochem Funct. 40:880–887. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Rhaman A, Mahmoud E, Abu Alfadl EM,
Mohammed DH and Sheneef A: Expression of autophagy related genes
mTOR, ATG10 and P62 in the peripheral blood mononuclear cells of
systemic lupus erythematosus Egyptian patients. Egypt J Med
Microbiol. 32:133–140. 2023. View Article : Google Scholar
|
|
74
|
Zhang J, Han L, Ma Q, Wang X, Yu J, Xu Y,
Zhang X, Wu X and Deng G: RIP3 impedes Mycobacterium tuberculosis
survival and promotes p62-mediated autophagy. Int Immunopharmacol.
115:1096962023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Schaaf MBE, Keulers TG, Vooijs MA and
Rouschop KMA: LC3/GABARAP family proteins: Autophagy-(un) related
functions. FASEB J. 30:3961–3978. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Gordon MS, Shapiro GI, Sarantopoulos J,
Juric D, Lu B, Zarotiadou A, Connarn JN, Le Bruchec Y, Dumitru CD
and Harvey RD: Phase Ib study of the histone deacetylase 6
inhibitor citarinostat in combination with paclitaxel in patients
with advanced solid tumors. Front Oncol. 11:7861202022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhang N, Sun P, Jin H, Yang Y, Zhao Q,
Zhou L, Guo L, Yang X and Lu L: Chidamide combined with paclitaxel
effectively reverses the expression of histone deacetylase in lung
cancer. Anticancer Drugs. 31:702–708. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Yoo J, Jeon YH, Lee DH, Kim GW, Lee SW,
Kim SY, Park J and Kwon SH: HDAC6-selective inhibitors enhance
anticancer effects of paclitaxel in ovarian cancer cells. Oncol
Lett. 21:2012021. View Article : Google Scholar : PubMed/NCBI
|